Ultrafast Processes Occurring in Radiolysis of Highly Concentrated Solutions of Nucleosides/Tides



Abstract
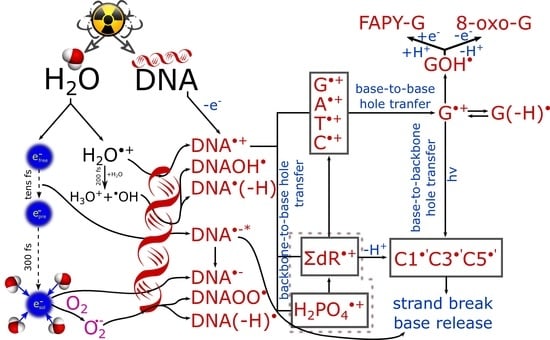
:
1. Introduction
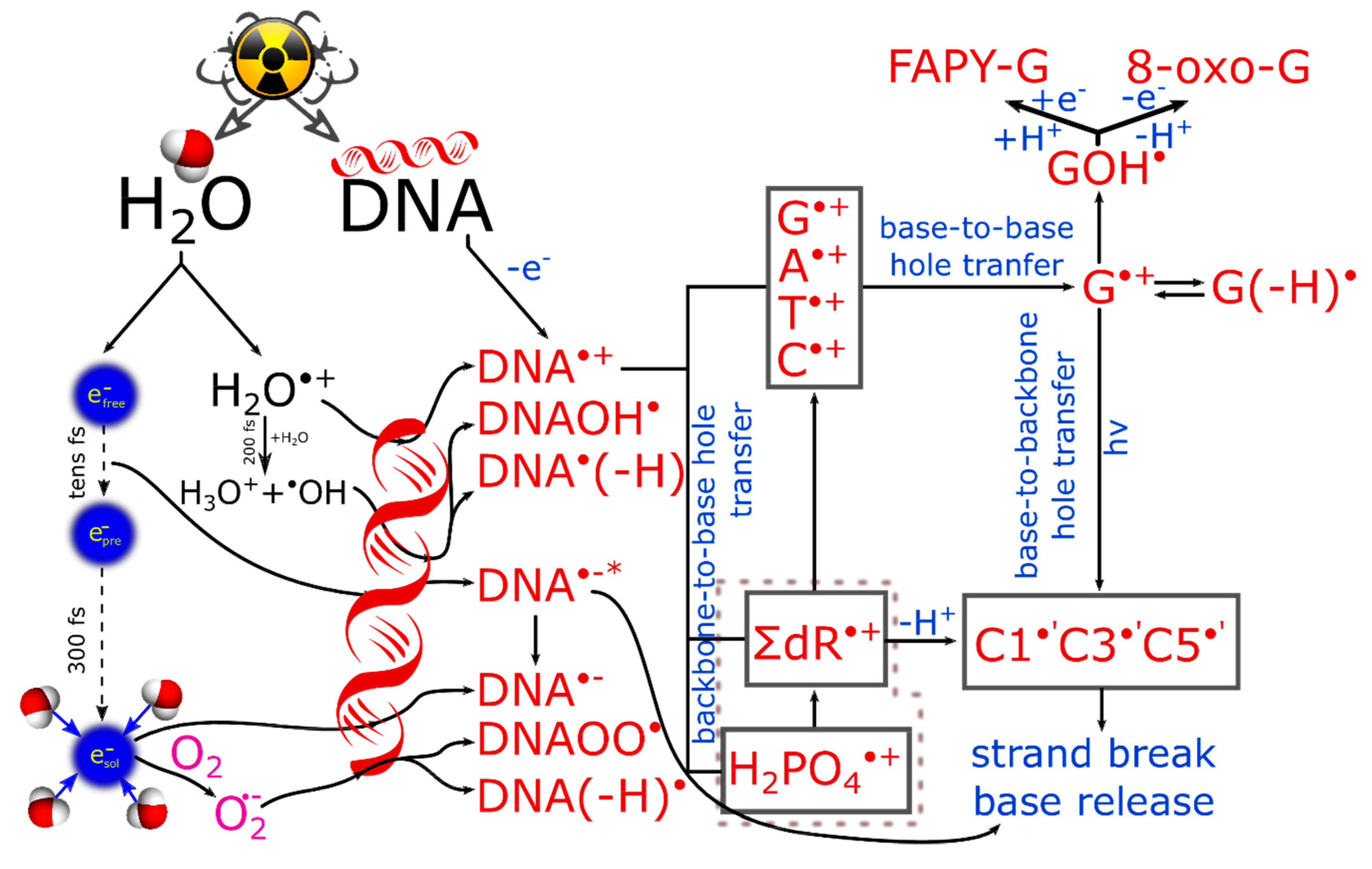
1.1. Ultrafast Processes in Water Radiolysis
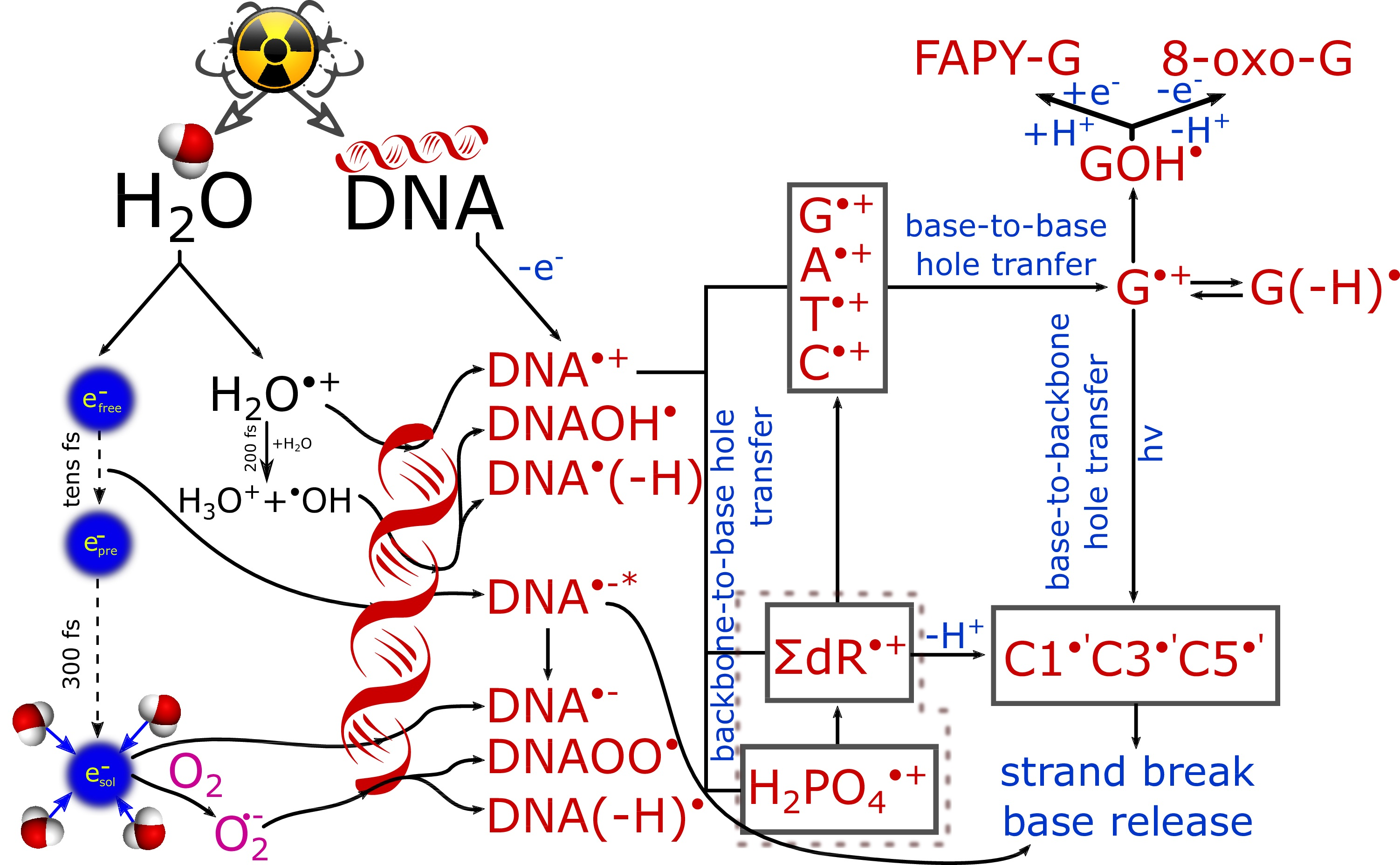
1.2. Effect of Radiation in Concentrated Solutions
1.3. Studies of Biomolecule Model Systems
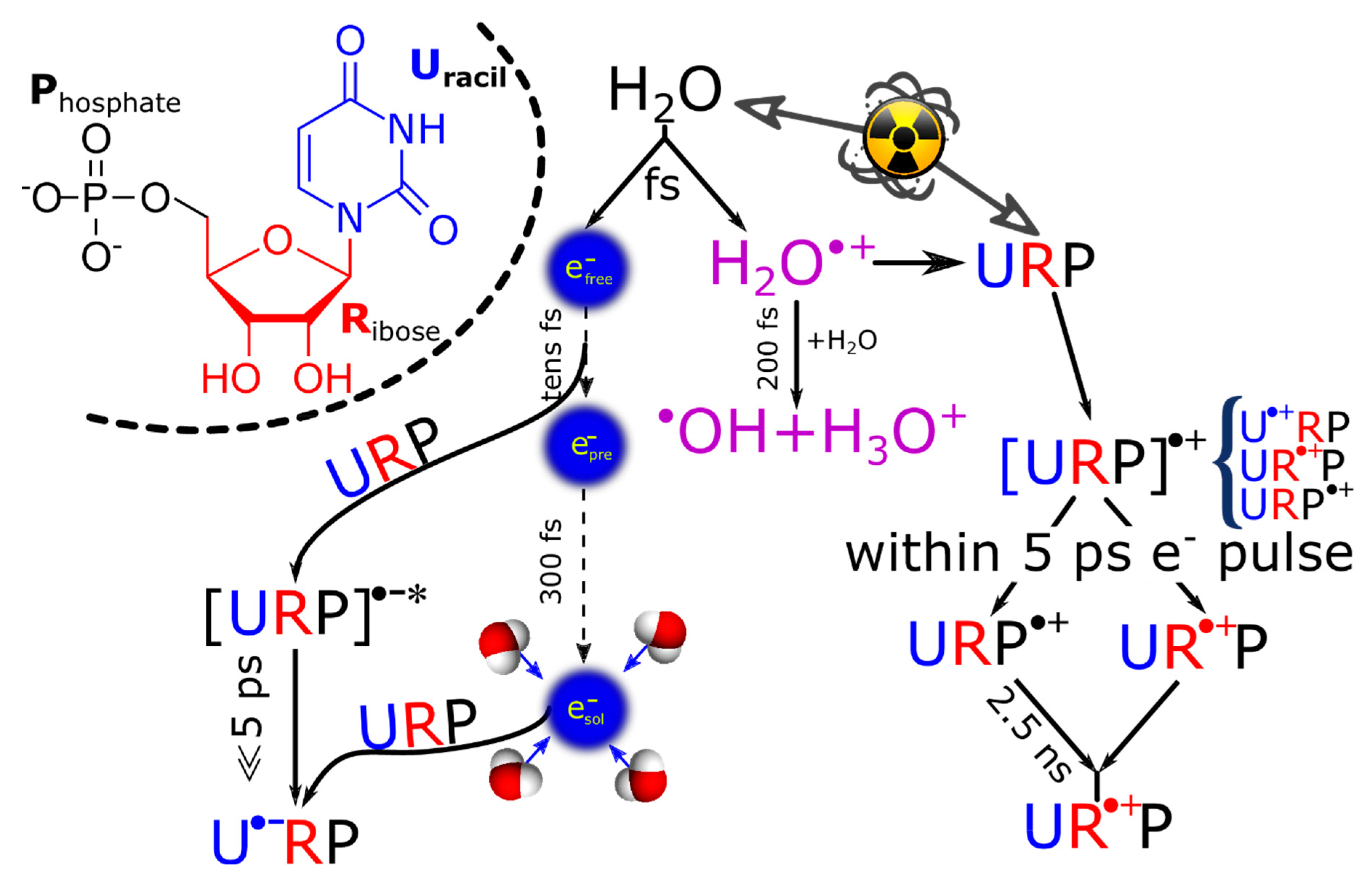
2. Ultrafast Hole and Electron Transfer under Irradiation
2.1. Elucidation of Pathways of Hole Transfer Processes Employing Concentrated Nucleotide Solutions
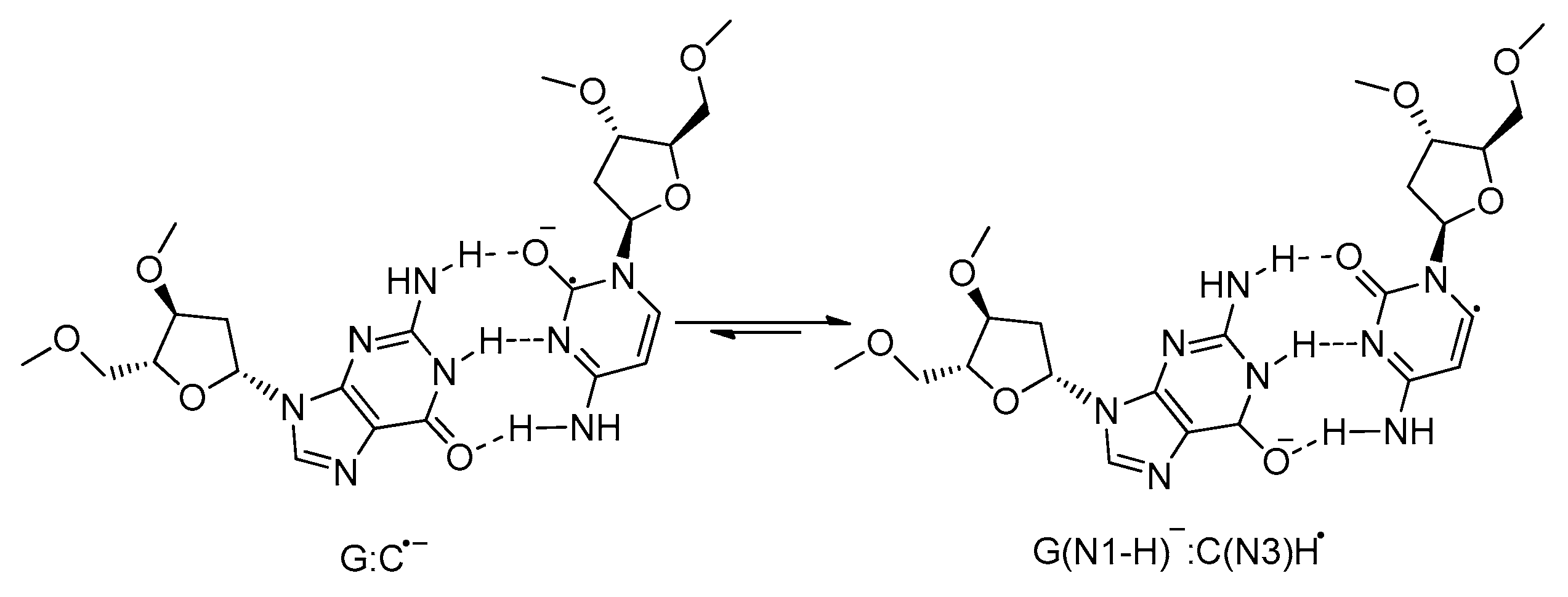
2.1.1. Backbone-to-Base Hole Transfer
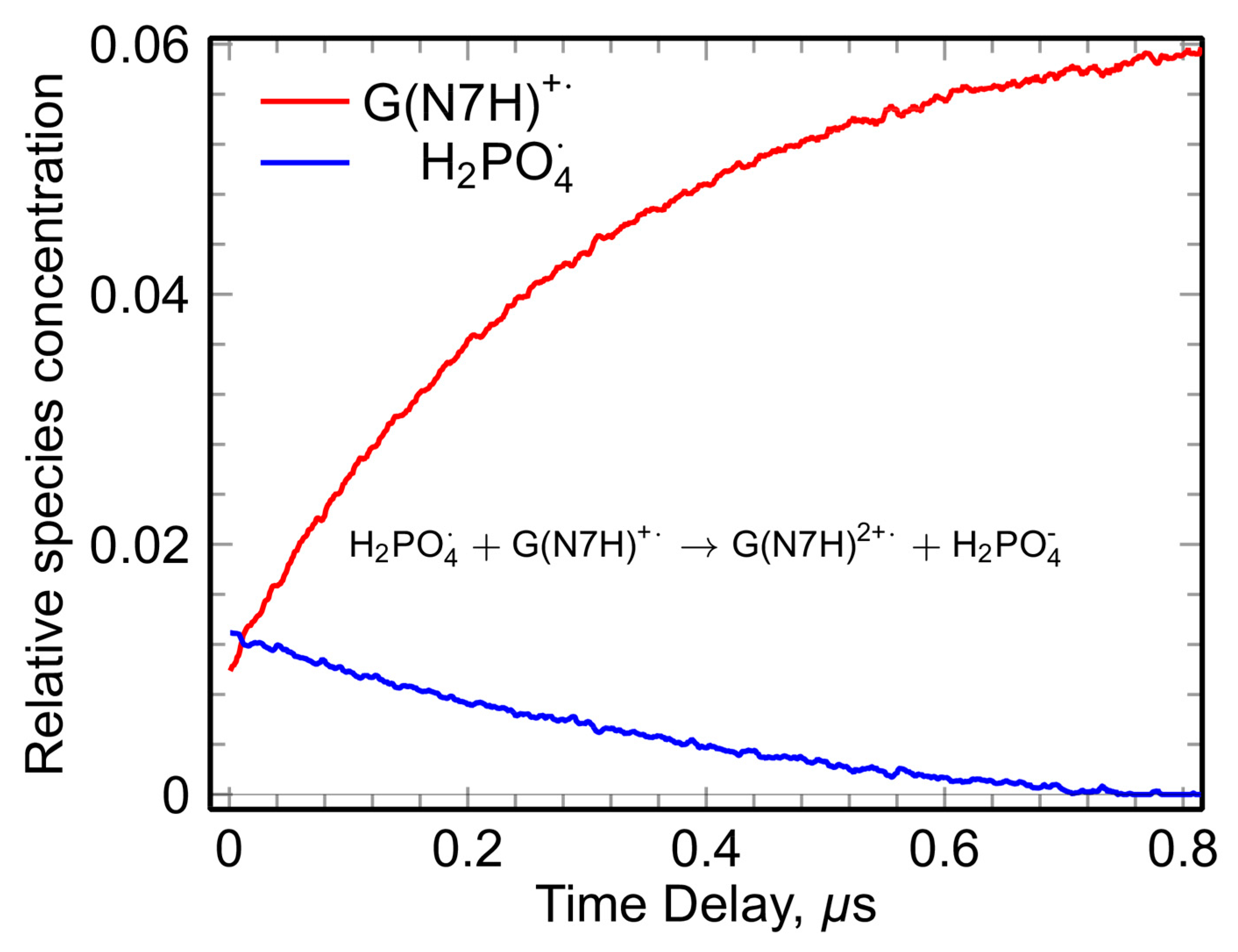
2.1.2. Base-to-Backbone and Phosphate-to-Sugar Hole Transfer Process
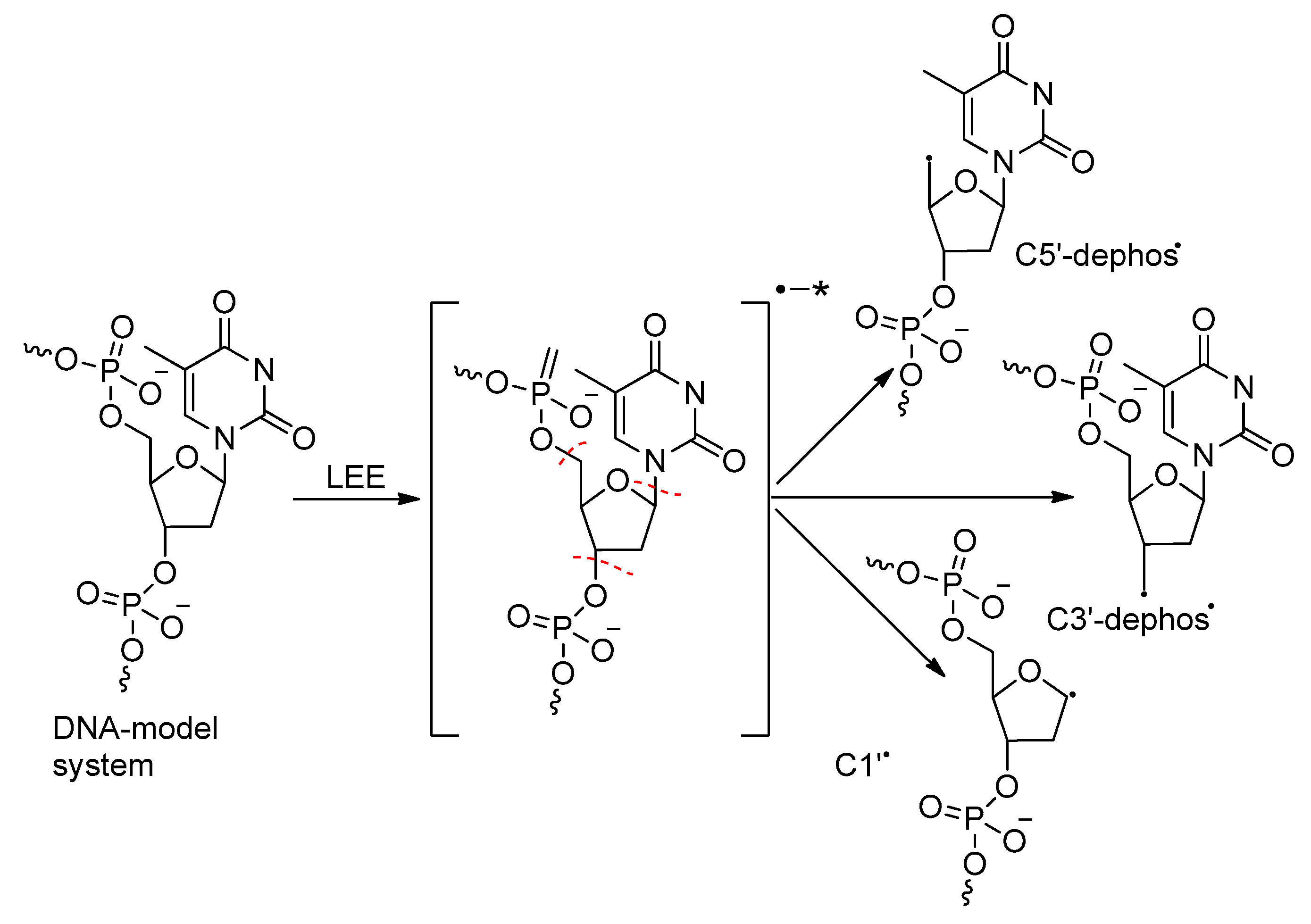
2.2. Excess Electron-Mediated Bond Dissociation in Bulk Solutions
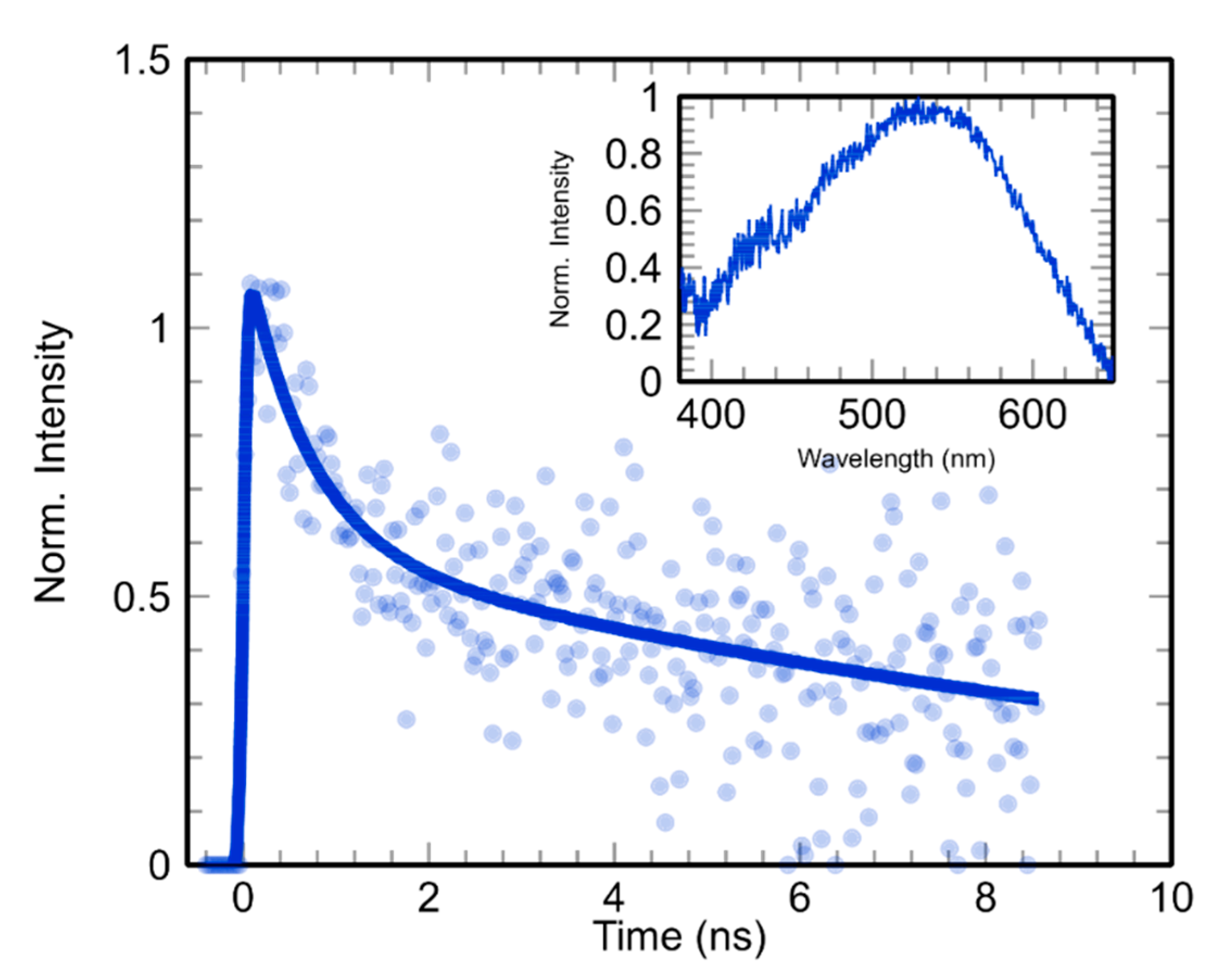
2.2.1. Can Prehydrated Electrons (epre−) Cause Bond Breakage Via DEA?
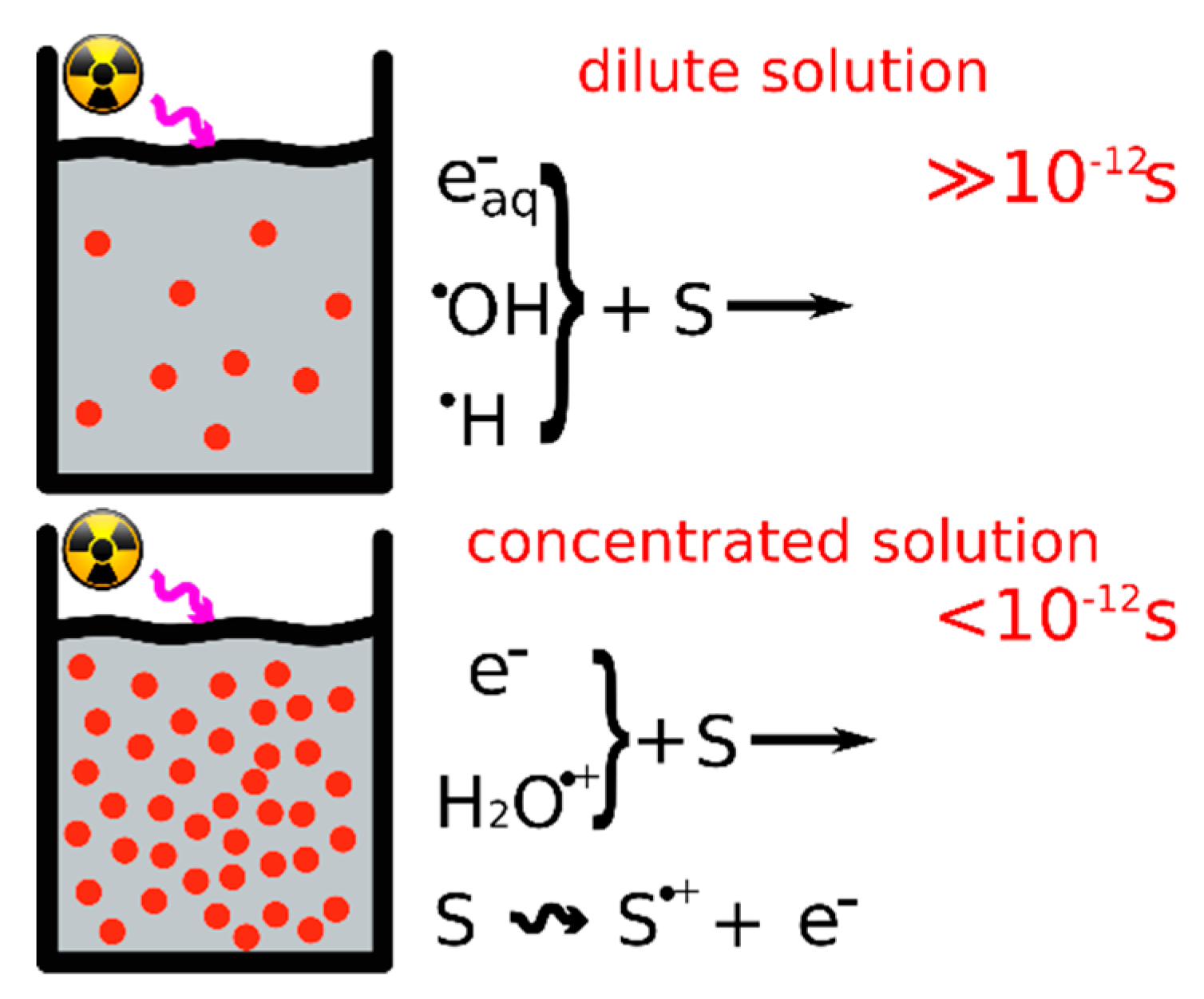
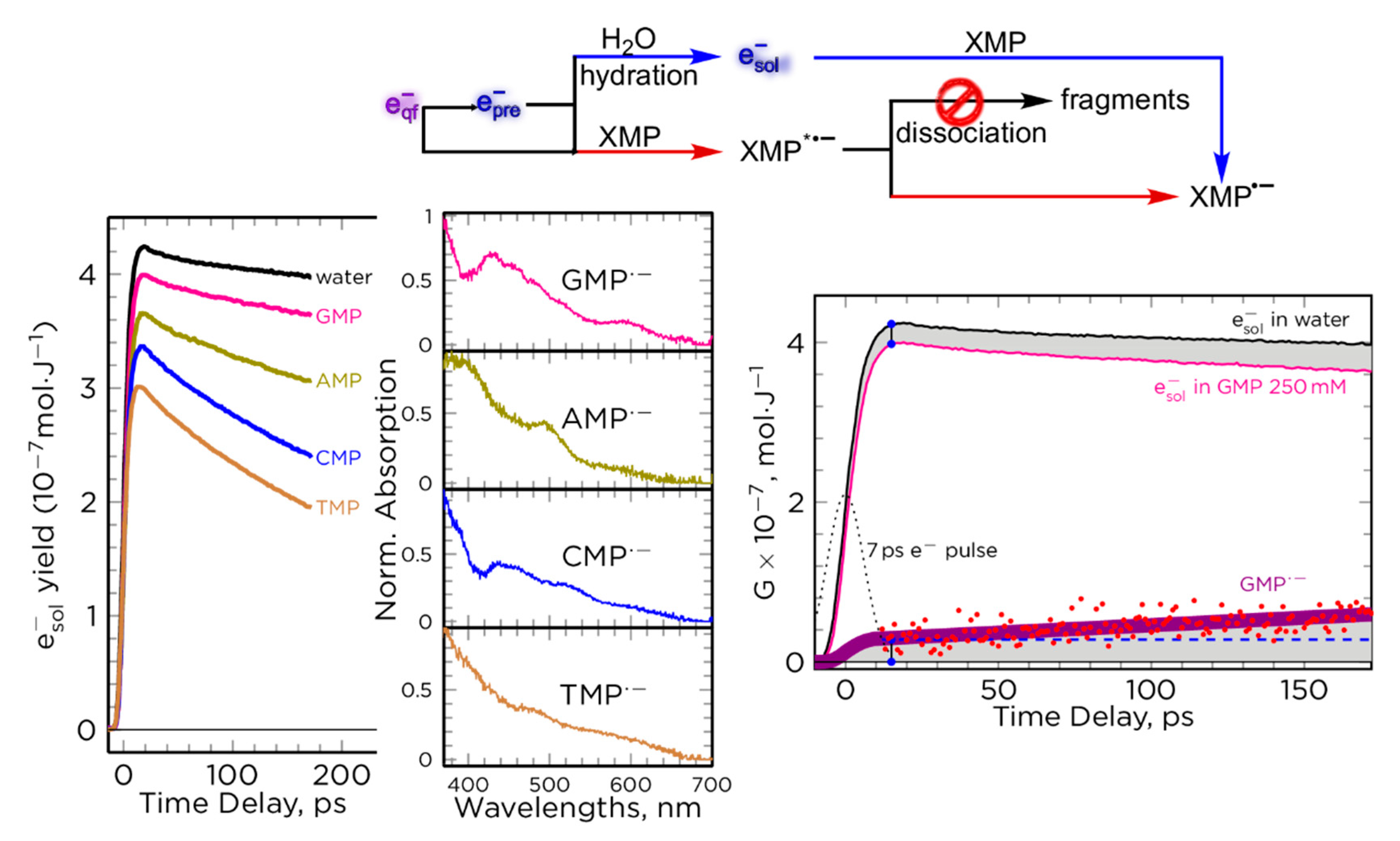
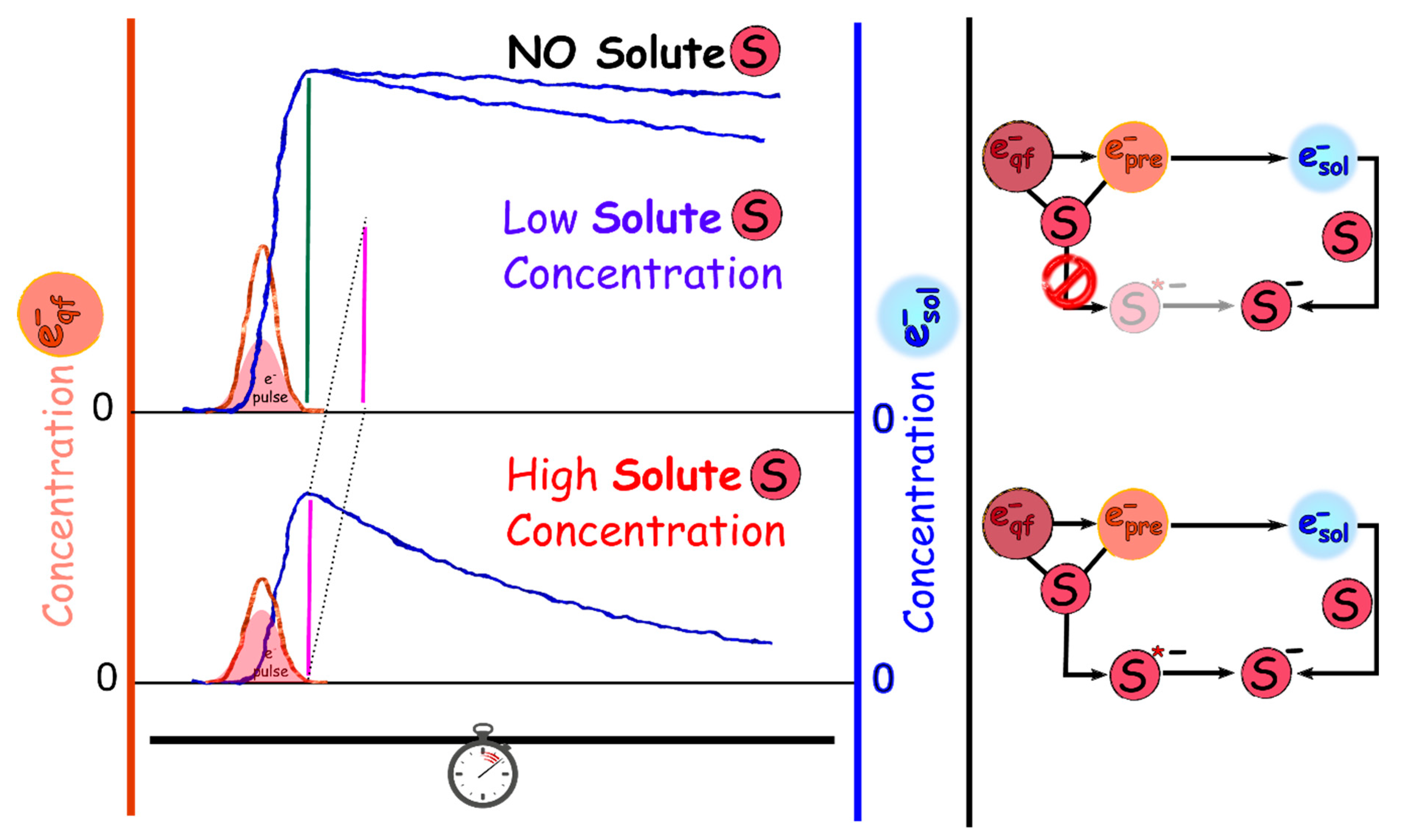
2.2.2. Picosecond Pulse Radiolysis Measurement of the Initial Yield of Formation of esol‒ in a Solution Leads to Study the Reaction of epre‒ with Solute
2.2.3. Reactivity of epre‒ with Nucleobases (X), Nucleosides, and 5′-Nucleotides (XMP) in Water
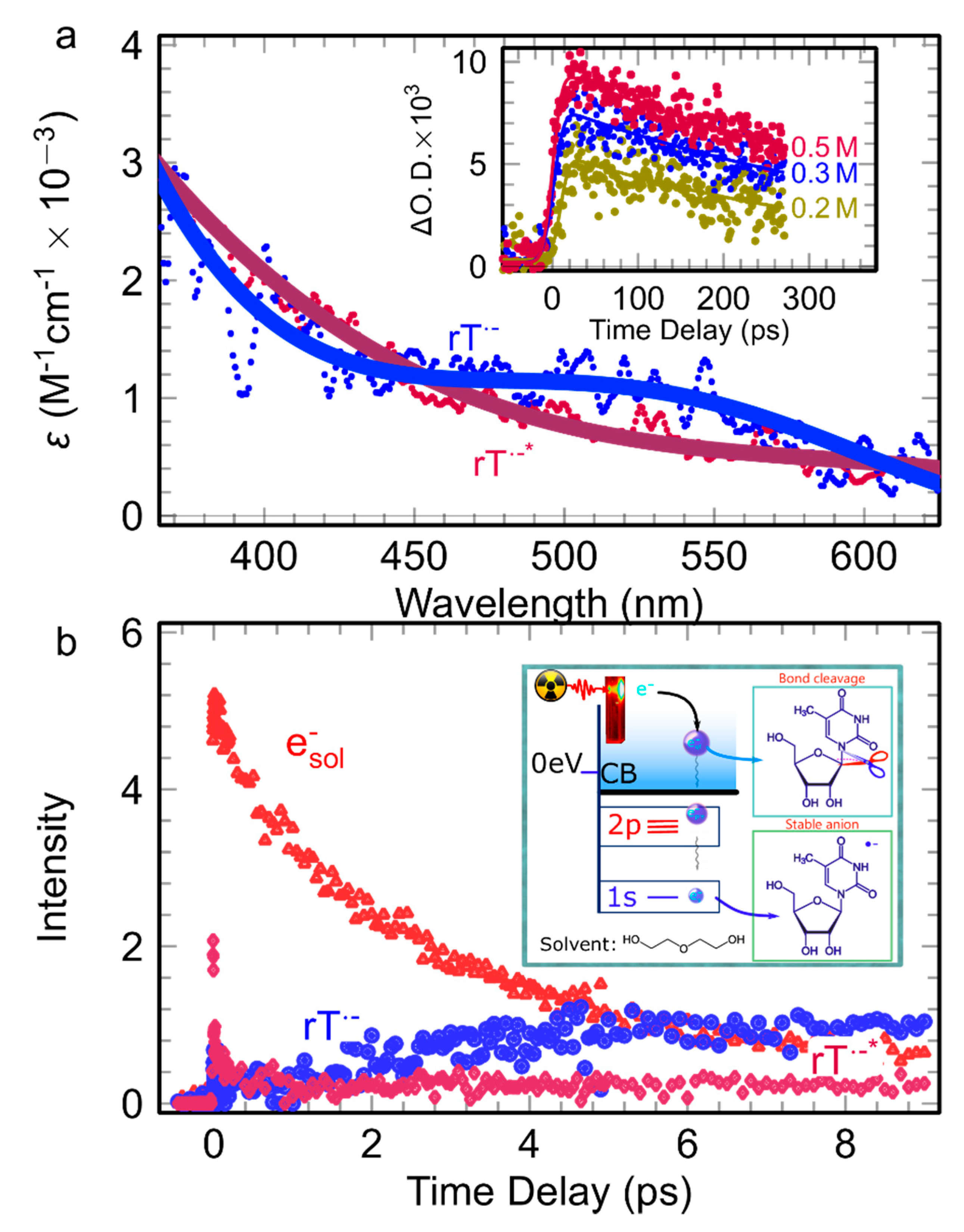
2.2.4. The Reactivity of Quasi-Free Electrons Towards Nucleoside in DEG
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ma, J.; LaVerne, J.A.; Mostafavi, M. Scavenging the Water Cation in Concentrated Acidic Solutions. J. Phys. Chem. A 2015, 119, 10629–10636. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Schmidhammer, U.; Pernot, P.; Mostafavi, M. Reactivity of the strongest oxidizing species in aqueous solutions: The short-lived radical cation H2O+. J. Phys. Chem. Lett. 2014, 5, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Wang, F.; Mostafavi, M. Ultrafast chemistry of water radical cation, H2O•+, in aqueous solutions. Molecules 2018, 23, 244. [Google Scholar]
- Alizadeh, E.; Sanche, L. Precursors of solvated electrons in radiobiological physics and chemistry. Chem. Rev. 2012, 112, 5578–5602. [Google Scholar] [CrossRef] [PubMed]
- Cobut, V.; Frongillo, Y.; Patau, J.P.; Goulet, T.; Fraser, M.J.; Jay-Gerin, J.P. Monte carlo simulation of fast electron and proton tracks in liquid water—i. physical and physicochemical aspects. Radiat. Phys. Chem. 1998, 51, 229–243. [Google Scholar]
- Pimblott, S.M.; Laverne, J.A. Production of low-energy electrons by ionizing radiation. Radiat. Phys. Chem. 2007, 76, 1244–1247. [Google Scholar] [CrossRef]
- Shkrob, I.A. The Structure and Dynamics of Solvated Electrons. In Recent Trends in Radiation Chemistry; Wishart, J.F., Rao, B.S.M., Eds.; World Scientific Publishing Company: Singapore, 2010; pp. 21–58. [Google Scholar]
- Kumar, A.; Sevilla, M.D. Low-Energy Electron (LEE)-Induced DNA Damage: Theoretical Approaches to Modeling Experiment. In Handbook of Computational Chemistry; Leszczynski, J., Ed.; Springer: Dordrecht, The Netherlands, 2015; pp. 1–63. [Google Scholar]
- Migus, A.; Gauduel, Y.; Martin, J.L.; Antonetti, A. Excess Electrons in Liquid Water: First Evidence of a Prehydrated State with Femtosecond Lifetime. Phys. Rev. Lett. 1987, 58, 1559–1562. [Google Scholar] [CrossRef] [PubMed]
- Turi, L.; Holpár, P.; Keszei, E. Alternative mechanisms for solvation dynamics of laser-induced electrons in methanol. J. Phys. Chem. A 1997, 101, 5469–5476. [Google Scholar] [CrossRef]
- Soroushian, B.; Lampre, I.; Bonin, J.; Pernot, P.; Pommeret, S.; Mostafavi, M. Solvation Dynamics of the Electron Produced by Two-Photon Ionization of Liquid Polyols. 1. Ethylene Glycol. J. Phys. Chem. A 2006, 110, 1705–1717. [Google Scholar] [CrossRef] [PubMed]
- Bonin, J.; Lampre, I.; Pernot, P.; Mostafavi, M. Solvation Dynamics of Electron Produced by Two-Photon Ionization of Liquid Polyols. II. Propanediols. J. Phys. Chem. A 2007, 111, 4902–4913. [Google Scholar] [CrossRef]
- Bonin, J.; Lampre, I.; Pernot, P.; Mostafavi, M. Solvation Dynamics of Electron Produced by Two-Photon Ionization of Liquid Polyols. III. Glycerol. J. Phys. Chem. A 2008, 112, 1880–1886. [Google Scholar] [CrossRef] [PubMed]
- Abramczyk, H.; Kroh, J. Spectroscopic properties of the solvated electron in water, alcohols, amines, ethers and alkanes. Radiat. Phys. Chem. 1994, 43, 291–297. [Google Scholar] [CrossRef]
- Marsalek, O.; Elles, C.G.; Pieniazek, P.A.; Pluhařová, E.; VandeVondele, J.; Bradforth, S.E.; Jungwirth, P. Chasing charge localization and chemical reactivity following photoionization in liquid water. J. Chem. Phys. 2011, 135, 224510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kambhampati, P.; Son, D.H.; Kee, T.W.; Barbara, P.F. Solvation dynamics of the hydrated electron depends on its initial degree of electron delocalization. J. Phys. Chem. A 2002, 106, 2374–2378. [Google Scholar] [CrossRef]
- Thaller, A.; Laenen, R.; Laubereau, A.; Thaller, A.; Laenen, R.; Laubereau, A. The precursors of the solvated electron in methanol studied by femtosecond pump- repump-probe spectroscopy. J. Chem. Phys. 2014, 124, 24515–24524. [Google Scholar] [CrossRef] [PubMed]
- Assel, M.; Laenen, R.; Laubereau, A. Dynamics of Excited Solvated Electrons in Aqueous Solution Monitored with Femtosecond-Time and Polarization Resolution. J. Phys. Chem. A 1998, 102, 2256–2262. [Google Scholar] [CrossRef]
- Scheidt, T.; Laenen, R. Ionization of methanol: Monitoring the trapping of electrons on the fs time scale. Chem. Phys. Lett. 2003, 371, 445–450. [Google Scholar] [CrossRef]
- Pépin, C.; Goulet, T.; Houde, D.; Jay-Gerin, J.P. Observation of a continuous spectral shift in the solvation kinetics of electrons in neat liquid deuterated water. J. Phys. Chem. A 1997, 101, 4351–4360. [Google Scholar] [CrossRef]
- Goulet, T.; Pépin, C.; Houde, D.; Jay-Gerin, J.-P. On the relaxation kinetics following the absorption of light by solvated electrons in polar liquids: Roles of the continuous spectral shifts and of the stepwise transition. Radiat. Phys. Chem. 1999, 54, 441–448. [Google Scholar] [CrossRef]
- Gervais, B.; Beuve, M.; Olivera, G.H.; Galassi, M.E. Numerical simulation of multiple ionization and high LET effects in liquid water radiolysis. Radiat. Phys. Chem. 2006, 75, 493–513. [Google Scholar] [CrossRef]
- Le Caër, S. Water Radiolysis: Influence of Oxide Surfaces on H2 Production under Ionizing Radiation. Water 2011, 3, 235–253. [Google Scholar] [CrossRef]
- Uehara, S.; Nikjoo, H. Monte Carlo Simulation of Water Radiolysis for Low-energy Charged Particles. J. Radiat. Res. 2006, 47, 69–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, M.A.; Mary, Q.; Road, M.E. Calculation of initial and primary yields in the radiolysis of water. Radiat. Phys. Chem. 1994, 43, 265–280. [Google Scholar] [CrossRef]
- Pimblott, S.M.; Laverne, J.A.; Uni, V.; Dame, N. Stochastic Simulation of the Electron Radiolysis of Water and Aqueous Solutions. J. Phys. Chem. A 1997, 5639, 5828–5838. [Google Scholar] [CrossRef]
- El Omar, A.K.; Schmidhammer, U.; Jeunesse, P.; Larbre, J.P.; Lin, M.; Muroya, Y.; Katsumura, Y.; Pernot, P.; Mostafavi, M. Time-dependent radiolytic yield of OH• radical studied by picosecond pulse radiolysis. J. Phys. Chem. A 2011, 115, 12212–12216. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Schmidhammer, U.; Larbre, J.-P.; Zong, Z.; Marignier, J.-L.; Mostafavi, M. Time-dependent yield of the hydrated electron and the hydroxyl radical in D2O: A picosecond pulse radiolysis study. Phys. Chem. Chem. Phys. 2018, 20, 15671–15679. [Google Scholar] [CrossRef] [PubMed]
- von Sonntag, C. Free-Radical-Induced DNA Damage and Its Repair: A Chemical Perspective; Springer: Berlin/Heidelberg, Germany, 2006; ISBN 9783540305927. [Google Scholar]
- Adhikary, A.; Becker, D.; Sevilla, M.D. Electron spin resonance of radicals in irradiated DNA. In Applications of EPR in Radiation Research; Lund, A., Shiotani, M., Eds.; Springer-Verlag: Berlin/Heidelberg, Germany, 2014; pp. 299–352. [Google Scholar]
- Becker, D.; Sevilla, M.D. Radiation Damage to DNA and Related Biomolecules. In Specialist Periodical Reports: Electron Paramagnetic Resonance; Gilbert, B.C., Davies, M.J., Murphy, D.M., Eds.; Royal Society of Chemistry: Cambridge, UK, 1998; pp. 79–115. [Google Scholar]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical Review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O−) in Aqueous Solution. J. Phys. Chem. Ref. Data 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Katsumura, Y. Radiation chemistry of concentrated inorganic aqueous solutions. In Radiation Chemistry; Jonah, C.D., Rao, B.S.M., Eds.; Studies in Physical and Theoretical Chemistry; Elsevier: Amsterdam, The Netherlands, 2001; Volume 87, pp. 163–174. [Google Scholar]
- Garaix, G.; Horne, G.P.; Venault, L.; Moisy, P.; Pimblott, S.M.; Marignier, J.L.; Mostafavi, M. Decay Mechanism of NO3• Radical in Highly Concentrated Nitrate and Nitric Acidic Solutions in the Absence and Presence of Hydrazine. J. Phys. Chem. B 2016, 120, 5008–5014. [Google Scholar] [CrossRef] [PubMed]
- Musat, R.; Denisov, S.A.; Marignier, J.L.; Mostafavi, M. Decoding the Three-Pronged Mechanism of NO3• Radical Formation in HNO3 Solutions at 22 and 80 °C Using Picosecond Pulse Radiolysis. J. Phys. Chem. B 2018, 122, 2121–2129. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R. The Nucleus: Volume 1: Nuclei and Subnuclear Components; Methods in Molecular Biology; Springer: Berlin, Germany, 2008; ISBN 9781588299772. [Google Scholar]
- Heslop-Harrison, J.S.; Schwarzacher, T. Nucleosomes and centromeric DNA packaging. Proc. Natl. Acad. Sci. USA 2013, 110, 19974–19975. [Google Scholar] [CrossRef] [Green Version]
- Becker, D.; Adhikary, A.; Sevilla, M.D. Mechanism of Radiation Induced DNA Damage: Direct Effects. In Recent Trends in Radiation Chemistry; Rao, B., Wishart, J.F., Eds.; World Scientific Publishing Company: Singapore, 2010; pp. 509–542. [Google Scholar]
- Becker, D.; Adhikary, A.; Sevilla, M.D. Physicochemical mechanisms of radiation-induced DNA damage. In Charge Particle and Photon Interactions with Matter; Hatano, Y., Katsumura, Y., Mozumder, A., Eds.; CRC Press: Boca Raton, FL, USA, 2010; pp. 503–545. [Google Scholar]
- Kim, K.-J.; Hamill, W.H. Direct and indirect effects in pulse irradiated concentrated aqueous solutions of chloride and sulfate ions. J. Phys. Chem. 1976, 80, 2320–2325. [Google Scholar] [CrossRef]
- Ma, J.; Schmidhammer, U.; Mostafavi, M. Picosecond Pulse Radiolysis of Highly Concentrated Phosphoric Acid Solutions: Mechanism of Phosphate Radical Formation. J. Phys. Chem. B 2015, 119, 7180–7185. [Google Scholar] [CrossRef] [PubMed]
- Belloni, J.; Monard, H.; Gobert, F.; Larbre, J.P.; Demarque, a.; De Waele, V.; Lampre, I.; Marignier, J.L.; Mostafavi, M.; Bourdon, J.C.; et al. ELYSE-A picosecond electron accelerator for pulse radiolysis research. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 2005, 539, 527–539. [Google Scholar] [CrossRef]
- Ma, J.; Denisov, S.A.; Marignier, J.-L.; Pernot, P.; Adhikary, A.; Seki, S.; Mostafavi, M. Ultrafast Electron Attachment and Hole Transfer Following Ionizing Radiation of Aqueous Uridine Monophosphate. J. Phys. Chem. Lett. 2018, 9, 5105–5109. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Marignier, J.-L.; Pernot, P.; Houee-Levin, C.; Kumar, A.; Sevilla, M.D.; Adhikary, A.; Mostafavi, M. Direct observation of the oxidation of DNA bases by phosphate radicals formed under radiation: A model of the backbone-to-base hole transfer. Phys. Chem. Chem. Phys. 2018, 20, 14927–14937. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, A.; Becker, D.; Palmer, B.J.; Heizer, A.N.; Sevilla, M.D. Direct Formation of the C5′-Radical in the Sugar–phosphate Backbone of DNA by High Energy Radiation. J. Phys. Chem. B 2015, 116, 5900–5906. [Google Scholar] [CrossRef]
- Adhikary, A.; Kumar, A.; Palmer, B.J.; Todd, A.D.; Sevilla, M.D. Formation of S-Cl phosphorothioate adduct radicals in dsDNA S-oligomers: Hole transfer to guanine vs disulfide anion radical formation. J. Am. Chem. Soc. 2013, 135, 12827–12838. [Google Scholar] [CrossRef] [PubMed]
- Cadet, J.; Wagner, J.R.; Angelov, D. Biphotonic Ionization of DNA: From Model Studies to Cell. Photochem. Photobiol. 2019, 95, 59–72. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R. Oxidatively generated base damage to cellular DNA by hydroxyl radical and one-electron oxidants: Similarities and differences. Arch. Biochem. Biophys. 2014, 557, 47–54. [Google Scholar] [CrossRef]
- Cadet, J.; Wagner, J.R.; Shafirovich, V.; Geacintov, N.E. One-electron oxidation reactions of purine and pyrimidine bases in cellular DNA. Int. J. Radiat. Biol. 2014, 90, 423–432. [Google Scholar] [CrossRef]
- Sun, H.; Zheng, L.; Greenberg, M.M. Independent Generation of Reactive Intermediates Leads to an Alternative Mechanism for Strand Damage Induced by Hole Transfer in Poly(dA–T) Sequences. J. Am. Chem. Soc. 2018, 140, 11308–11316. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.M. Chapter Three - Reactivity of Nucleic Acid Radicals. Adv. Phys. Org. Chem. 2016, 50, 119–202. [Google Scholar] [PubMed]
- Wu, J.; Sturla, S.J.; Burrows, C.J.; Fleming, A.M. Impact of DNA Oxidation on Toxicology: From Quantification to Genomics. Chem. Res. Toxicol. 2019, 32, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Poonia, V.S.; Fontanesi, C.; Naaman, R.; Fleming, A.M.; Burrows, C.J. Effect of Oxidative Damage on Charge and Spin Transport in DNA. J. Am. Chem. Soc. 2019, 141, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Burrows, C.J.; Muller, J.G. Oxidative Nucleobase Modifications Leading to Strand Scission. Chem. Rev. 1998, 98, 1109–1152. [Google Scholar] [CrossRef] [PubMed]
- Saito, I.; Nakamura, T.; Nakatani, K.; Yoshioka, Y.; Yamaguchi, K.; Sugiyama, H. Mapping of the Hot Spots for DNA Damage by One-Electron Oxidation: Efficacy of GG Doublets and GGG Triplets as a Trap in Long-Range Hole Migration. J. Am. Chem. Soc. 1998, 120, 12686–12687. [Google Scholar] [CrossRef]
- Bernhard, W.A. Radical Reaction Pathways Initiated by Direct Energy Deposition in DNA by Ionizing Radiation. In Radical and Radical Ion Reactivity in Nucleic Acid Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2009; pp. 41–68. [Google Scholar]
- Wagenknecht, H.-A. Reductive Electron Transfer and Transport of Excess Electrons in DNA. Angew. Chem. Int. Ed. 2003, 42, 2454–2460. [Google Scholar] [CrossRef] [PubMed]
- Sagstuen, E.; Hole, E.O. Radiation Produced Radicals. In Electron Paramagnetic Resonance; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2008; pp. 325–338. [Google Scholar]
- Close, D.M. From the Primary Radiation Induced Radicals in DNA Constituents to Strand Breaks: Low Temperature EPR/ENDOR Studies. In Radiation Induced Molecular Phenomena in Nucleic Acids: A Comprehensive Theoretical and Experimental Analysis; Shukla, M.K., Leszczynski, J., Eds.; Springer: Dordrecht, The Netherlands, 2008; pp. 493–529. [Google Scholar]
- Genereux, J.C.; Barton, J.K. Mechanisms for DNA Charge Transport. Chem. Rev. 2010, 110, 1642–1662. [Google Scholar] [CrossRef] [PubMed]
- Giese, B. Long-Distance Electron Transfer Through DNA. Annu. Rev. Biochem. 2002, 71, 51–70. [Google Scholar] [CrossRef]
- Wagenknecht, H.A. Charge Transfer in DNA: From Mechanism to Application; Wiley: Weinheim, Germany, 2005; pp. 1–76. [Google Scholar]
- Schuster, G.B. Long-Range Charge Transfer in DNA I; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2004; pp. 67–137. [Google Scholar]
- Steenken, S. Electron transfer in DNA? Competition by ultra-fast proton transfer? Biol. Chem. 1997, 378, 1293–1297. [Google Scholar]
- Sun, H.; Zheng, L.; Yang, K.; Greenberg, M.M. Positional Dependence of DNA Hole Transfer Efficiency in Nucleosome Core Particles. J. Am. Chem. Soc. 2019, 141, 10154–10158. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.K.K.; Tyagi, R.; Purkayastha, S.; Bernhard, W.A. One-electron oxidation of DNA by ionizing radiation: Competition between base-to-base hole-transfer and hole-trapping. J. Phys. Chem. B 2010, 114, 7672–7680. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Oxidatively induced DNA damage and its repair in cancer. Mutat. Res. Mutat. Res. 2015, 763, 212–245. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M.; Coskun, E.; Jaruga, P. Measurement of oxidatively induced DNA damage and its repair, by mass spectrometric techniques. Free Radic. Res. 2015, 49, 525–548. [Google Scholar] [CrossRef] [PubMed]
- Shukla, L.I.; Adhikary, A.; Pazdro, R.; Becker, D.; Sevilla, M.D. Formation of 8-oxo-7,8-dihydroguanine-radicals in γ-irradiated DNA by multiple one-electron oxidations. Nucleic Acids Res. 2004, 32, 6565–6574. [Google Scholar] [CrossRef] [PubMed]
- Fujitsuka, M.; Majima, T. Charge transfer dynamics in DNA revealed by time-resolved spectroscopy. Chem. Sci. 2017, 8, 1752–1762. [Google Scholar] [CrossRef]
- Kawai, K.; Hayashi, M.; Majima, T. HOMO Energy Gap Dependence of Hole-Transfer Kinetics in DNA. J. Am. Chem. Soc. 2012, 134, 4806–4811. [Google Scholar] [CrossRef]
- Fujitsuka, M.; Majima, T. Hole and excess electron transfer dynamics in DNA. Phys. Chem. Chem. Phys. 2012, 14, 11234–11244. [Google Scholar] [CrossRef]
- Kumar, A.; Becker, D.; Adhikary, A.; Sevilla, M.D. Reaction of Electrons with DNA: Radiation Damage to Radiosensitization. Int. J. Mol. Sci. 2019, 20, 3998. [Google Scholar] [CrossRef]
- Ma, J.; Wang, F.; Denisov, S.A.; Adhikary, A.; Mostafavi, M. Reactivity of Prehydrated Electrons towards Nucleobases and Nucleotides in Aqueous Solution. Sci. Adv. 2017, 12, e1701669. [Google Scholar] [CrossRef]
- Ma, J.; Kumar, A.; Muroya, Y.; Yamashita, S.; Sakurai, T.; Denisov, S.A.; Sevilla, M.D.; Adhikary, A.; Seki, S.; Mostafavi, M. Observation of dissociative quasi-free electron attachment to nucleoside via excited anion radical in solution. Nat. Commun. 2019, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Rubio, M.; Roca-Sanjuán, D.; Merchán, M.; Serrano-Andrés, L. Determination of the Lowest-Energy Oxidation Site in Nucleotides: 2‘-Deoxythymidine 5‘-Monophosphate Anion. J. Phys. Chem. B 2006, 110, 10234–10235. [Google Scholar] [CrossRef] [PubMed]
- Rubio, M.; Roca-Sanjuán, D.; Serrano-Andrés, L.; Merchán, M. Determination of the Electron-Detachment Energies of 2′-Deoxyguanosine 5′-Monophosphate Anion: Influence of the Conformation. J. Phys. Chem. B 2009, 113, 2451–2457. [Google Scholar] [CrossRef] [PubMed]
- Swarts, S.G.; Becker, D.; Sevilla, M.; Wheeler, K.T. Radiation-Induced DNA Damage as a Function of Hydration. II. Base Damage from Electron-Loss Centers. Radiat. Res. 1996, 145, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, W.A.; Ezra, F.S. Electron spin resonance of γ-irradiated alkyl phosphates: The C2H5 radical in magnesium diethyl phosphate (MgDEP). J. Chem. Phys. 1974, 60, 1707–1710. [Google Scholar] [CrossRef]
- Bernard, W.A.; Ezra, F.S. Electron spin resonance study of γ-irradiated silver diethyl phosphate. J. Phys. Chem. 1974, 78, 958–961. [Google Scholar] [CrossRef]
- Nelson, D.; Symons, M.C.R. Unstable Intermediates. Part 169. Electron Capture Processes in Organic Phosphates: An Electron Spin Resonance Study. J. Chem. Soc. Perkin II 1977, 286–293. [Google Scholar] [CrossRef]
- Sanderud, A.; Sagstuen, E. EPR study of X-irradiated hydroxyalkyl phosphate esters. Phosphate radical formation in polycrystalline glucose phosphate ribose phosphate and glycerol phosphate salts at 77 and 295 K. J. Chem. Soc. Faraday Trans. 1996, 92, 995–999. [Google Scholar] [CrossRef]
- Celalyan-Berthier, A.; Berclaz, T.; Geoffroy, M. Radiation damage in a phosphated sugar. An electron spin resonance study of phosphorus-centred radicals trapped in an X-irradiated single crystal of a phenoxyphosphoryl xylofuranose derivative. J. Chem. Soc. Faraday Trans. 1 1987, 83, 401–409. [Google Scholar] [CrossRef]
- Armstrong, D.A.; Huie, R.E.; Lymar, S.; Koppenol, W.H.; Merényi, G.; Neta, P.; Stanbury, D.M.; Steenken, S.; Wardman, P. Standard electrode potentials involving radicals in aqueous solution: Inorganic radicals. Bioinorg. React. Mech. 2013, 9, 59–61. [Google Scholar] [CrossRef]
- Steenken, S.; Goldbergerova, L. Photoionization of organic phosphates by 193 nm laser light in aqueous solution: Rapid intramolecular H-transfer to the primarily formed phosphate radical. A model for ionization-induced chain-breakage in DNA? J. Am. Chem. Soc. 1998, 120, 3928–3934. [Google Scholar] [CrossRef]
- Cai, Z.; Sevilla, M.D. Studies of Excess Electron and Hole Transfer in DNA at Low Temperatures. In Long-Range Charge Transfer in DNA II; Schuster, G.B., Ed.; Springer: Berlin/Heidelberg, Germany, 2004; pp. 103–128. [Google Scholar]
- Adhikary, A.; Kumar, A.; Rayala, R.; Hindi, R.M.; Adhikary, A.; Wnuk, S.F.; Sevilla, M.D. One-electron oxidation of gemcitabine and analogs: Mechanism of formation of C3 and C2 sugar radicals. J. Am. Chem. Soc. 2014, 136, 15646–15653. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, M.D.; Becker, D.; Kumar, A.; Adhikary, A. Gamma and ion-beam irradiation of DNA: Free radical mechanisms, electron effects, and radiation chemical track structure. Radiat. Phys. Chem. 2016, 128, 60–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildenbrand, K. The SO4- induced Oxidation of 2′-Deoxyuridine-5′-phosphate, Uridine-5′-phosphate and Thymidine-5′-phosphate. An ESR Study in Aqueous Solution. Zeitschrift für Naturforsch. C 1990, 45, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, A.; Malkhasian, A.Y.S.; Collins, S.; Koppen, J.; Becker, D.; Sevilla, M.D. UVA-visible photo-excitation of guanine radical cations produces sugar radicals in DNA and model structures. Nucleic Acids Res. 2005, 33, 5553–5564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenberg, M.M. Elucidating DNA damage and repair processes by independently generating reactive and metastable intermediates. Org. Biomol. Chem. 2007, 5, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.F. Complexity of damage produced by ionizing radiation. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, A.; Kumar, A.; Palmer, B.J.; Todd, A.D.; Heizer, A.N.; Sevilla, M.D. Reactions of 5-methylcytosine cation radicals in DNA and model systems: Thermal deprotonation from the 5-methyl group vs. excited state deprotonation from sugar. Int. J. Radiat. Biol. 2014, 90, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Khanduri, D.; Adhikary, A.; Sevilla, M.D. Highly Oxidizing Excited States of One-Electron-Oxidized Guanine in DNA: Wavelength and pH Dependence. J. Am. Chem. Soc. 2011, 133, 4527–4537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikary, A.; Collins, S.; Khanduri, D.; Sevilla, M.D. Sugar Radicals Formed by Photoexcitation of Guanine Cation Radical in Oligonucleotides. J. Phys. Chem. B 2007, 111, 7415–7421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomun, T.; Hultschig, C.; Illenberger, E. Microarray technology for the study of DNA damage by low-energy electrons. Eur. Phys. J. D-At. Mol. Opt. Plasma Phys. 2005, 35, 437–441. [Google Scholar] [CrossRef]
- Simons, J. How Do Low-Energy (0.1−2 eV) Electrons Cause DNA-Strand Breaks? Acc. Chem. Res. 2006, 39, 772–779. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Leszczynski, J.; Schaefer, H.F. Interactions of electrons with bare and hydrated biomolecules: From nucleic acid bases to DNA segments. Chem. Rev. 2012, 112, 5603–5640. [Google Scholar] [CrossRef] [PubMed]
- Baccarelli, I.; Bald, I.; Gianturco, F.A.; Illenberger, E.; Kopyra, J. Electron-induced damage of DNA and its components: Experiments and theoretical models. Phys. Rep. 2011, 508, 1–44. [Google Scholar] [CrossRef]
- Arumainayagam, C.R.; Lee, H.-L.; Nelson, R.B.; Haines, D.R.; Gunawardane, R.P. Low-energy electron-induced reactions in condensed matter. Surf. Sci. Rep. 2010, 65, 1–44. [Google Scholar] [CrossRef]
- Wang, C.R.; Nguyen, J.; Lu, Q.B. Bond breaks of nucleotides by dissociative electron transfer of nonequilibrium prehydrated electrons: A new molecular mechanism for reductive DNA damage. J. Am. Chem. Soc. 2009, 131, 11320–11322. [Google Scholar] [CrossRef]
- Zheng, Y.; Sanche, L. Clustered DNA Damages induced by 0.5 to 30 eV Electrons. Int. J. Mol. Sci. 2019, 20, 3749. [Google Scholar] [CrossRef]
- Bald, I.; Denifl, S. The Role of Low-Energy Electrons in DNA Radiation Damage. In Low-Energy Electrons: Fundamentals and Applications; Ingolfsson, O., Ed.; Pan Stanford Publishing: New York, NY, USA, 2019; pp. 285–340. [Google Scholar]
- Rackwitz, J.; Bald, I. Low-Energy Electron-Induced Strand Breaks in Telomere-Derived DNA Sequences—Influence of DNA Sequence and Topology. Chem. Eur. J. 2018, 24, 4680–4688. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.; Ebel, K.; Schürmann, R.M.; Heck, C.; Meiling, T.; Milosavljevic, A.R.; Giuliani, A.; Bald, I. Vacuum-UV and Low-Energy Electron-Induced DNA Strand Breaks–Influence of the DNA Sequence and Substrate. ChemPhysChem 2019, 20, 823–830. [Google Scholar] [CrossRef]
- Zheng, Y.; Cloutier, P.; Hunting, D.J.; Wagner, J.R.; Sanche, L. Glycosidic Bond Cleavage of Thymidine by Low-Energy Electrons. J. Am. Chem. Soc. 2004, 126, 1002–1003. [Google Scholar] [CrossRef]
- Ptasińska, S.; Denifl, S.; Gohlke, S.; Scheier, P.; Illenberger, E.; Märk, T.D. Decomposition of Thymidine by Low-Energy Electrons: Implications for the Molecular Mechanisms of Single-Strand Breaks in DNA. Angew. Chem. Int. Ed. 2006, 45, 1893–1896. [Google Scholar] [CrossRef] [PubMed]
- Panajotovic, R.; Martin, F.; Cloutier, P.; Hunting, D.; Sanche, L. Effective Cross Sections for Production of Single-Strand Breaks in Plasmid DNA by 0.1 to 4.7 eV Electrons. Radiat. Res. 2006, 165, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Boudaı̈ffa, B.; Cloutier, P.; Hunting, D.; Huels, M.A.; Sanche, L. Resonant Formation of DNA Strand Breaks by Low-Energy (3 to 20 eV) Electrons. Science 2000, 287, 1658–1660. [Google Scholar] [PubMed]
- Woldhuis, J.; Verberne, J.B.; Lafleur, M.V.M.; Retel, J.; Blok, J.; Loman, H. γ-rays inactivate phiX174 DNA in frozen anoxic solutions at −200 °C mainly by reactions of dry electrons. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1984, 46, 329–330. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, M.; Sanche, L.; Hunting, D.J. Cisplatin Enhances the Formation of DNA Single- and Double-Strand Breaks by Hydrated Electrons and Hydroxyl Radicals. Radiat. Res. 2013, 179, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, G.K.; Lafleur, M.V.M. Characterization of DNA damage induced by gamma-radiationderived water radicals, using DNA repair enzymes. Int. J. Radiat. Biol. 1998, 74, 511–519. [Google Scholar] [CrossRef]
- Nabben, F.J.; Karman, J.P.; Loman, H. Inactivation of Biologically Active DNA by Hydrated Electrons. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1982, 42, 23–30. [Google Scholar] [CrossRef]
- Schuster, G.B. Long-range charge transfer in DNA: Transient structural distortions control the distance dependence. Acc. Chem. Res. 2000, 33, 253–260. [Google Scholar] [CrossRef]
- Wang, W.; Sevilla, M.D. Protonation of Nucleobase Anions in Gamma-Irradiated DNA and Model Systems. Which DNA Base Is the Ultimate Sink for the Electron? Radiat. Res. 1994, 138, 9–17. [Google Scholar] [CrossRef]
- Turi, L.; Rossky, P.J. Theoretical Studies of Spectroscopy and Dynamics of Hydrated Electrons. Chem. Rev. 2012, 112, 5641–5674. [Google Scholar] [CrossRef] [Green Version]
- Savolainen, J.; Uhlig, F.; Ahmed, S.; Hamm, P.; Jungwirth, P. Direct observation of the collapse of the delocalized excess electron in water. Nat. Chem. 2014, 6, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Silva, C.; Son, D.H.; Walhout, P.K.; Barbara, P.F. Detailed Investigation of the Femtosecond Pump - Probe Spectroscopy of the Hydrated Electron. J. Phys. Chem. A 1998, 5639, 6957–6966. [Google Scholar] [CrossRef]
- Aldrich, J.E.; Lam, K.Y.; Shragge, P.C.; Hunt, J.W. Fast Electron Reactions in Concentrated Solutions of Amino Acids and Nucleotides. Radiat. Res. 1975, 52, 42–52. [Google Scholar] [CrossRef]
- Tisler, J.; Balasubramanian, G.; Naydenov, B.; Kolesov, R.; Grotz, B.; Reuter, R.; Boudou, J.-P.; Curmi, P.A.; Sennour, M.; Thorel, A.; et al. Fluorescence and Spin Properties of Defects in Single Digit Nanodiamonds. ACS Nano 2009, 3, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Adhikary, A.; Shamoun, L.; Sevilla, M.D. Do Solvated Electrons (eaq−) Reduce DNA Bases? A Gaussian 4 and Density Functional Theory- Molecular Dynamics Study. J. Phys. Chem. B 2016, 120, 2115–2123. [Google Scholar] [CrossRef] [PubMed]
- Francés-Monerris, A.; Segarra-Martí, J.; Merchán, M.; Roca-Sanjuán, D. Complete-active-space second-order perturbation theory (CASPT2//CASSCF) study of the dissociative electron attachment in canonical DNA nucleobases caused by low-energy electrons (0–3 eV). J. Chem. Phys. 2015, 143, 215101. [Google Scholar] [CrossRef]
- Abdoul-Carime, H.; Gohlke, S.; Illenberger, E. Site-Specific Dissociation of DNA Bases by Slow Electrons at Early Stages of Irradiation. Phys. Rev. Lett. 2004, 92, 168103. [Google Scholar] [CrossRef]
- Kheir, J.F.; Chomicz, L.; Rak, J.; Bowen, K.H.; Sevilla, M.D. Radicals Formed in N-Acetylproline by Electron Attachment: Electron Spin Resonance Spectroscopy and Computational Studies. J. Phys. Chem. B 2011, 115, 14846–14851. [Google Scholar] [CrossRef] [Green Version]
- Kheir, J.; Chomicz, L.; Engle, A.; Rak, J.; Sevilla, M.D. Presolvated Low Energy Electron Attachment to Peptide Methyl Esters in Aqueous Solution: C–O Bond Cleavage at 77 K. J. Phys. Chem. B 2013, 117, 2872–2877. [Google Scholar] [CrossRef]
- Sevilla, M.D.; Morehouse, K.M.; Swarts, S. An ESR study of electron reactions with esters and triglycerides. J. Phys. Chem. 1981, 85, 923–927. [Google Scholar] [CrossRef]
- Petrovici, A.; Adhikary, A.; Kumar, A.; Sevilla, M.D. Presolvated Electron Reactions with Methyl Acetoacetate: Electron Localization, Proton-Deuteron Exchange, and H-Atom Abstraction. Molecules 2014, 19, 13486–13497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slinker, J.; Bernards, D.; Houston, P.L.; Abruna, H.D.; Bernhard, S.; Malliaras, G.G. Solid-state electroluminescent devices based on transition metal complexes. Chem. Commun. 2003, 2392–2399. [Google Scholar] [CrossRef] [PubMed]
- Terazono, Y.; Kodis, G.; Liddell, P.A.; Garg, V.; Moore, T.A.; Moore, A.L.; Gust, D. Multiantenna Artificial Photosynthetic Reaction Center Complex. J. Phys. Chem. B 2009, 113, 7147–7155. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cai, Z.; Sevilla, M.D. DFT Calculations of the Electron Affinities of Nucleic Acid Bases: Dealing with Negative Electron Affinities. J. Phys. Chem. A 2002, 106, 1596–1603. [Google Scholar] [CrossRef]
- Roca-Sanjuán, D.; Merchán, M.; Serrano-Andrés, L.; Rubio, M. Ab initio determination of the electron affinities of DNA and RNA nucleobases. J. Chem. Phys. 2008, 129, 95104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chase, W.J.; Hunt, J.W. Solvation Time of the Electron in Polar Liquids. Water and Alcohols. J. Phys. Chem. 1975, 79, 2835–2845. [Google Scholar] [CrossRef]
- Kenney-Wallace, G.A.; Jonah, C.D. Picosecond Spectroscopy and Solvation Clusters. The Dynamics of Localizing Electrons in Polar Fluids. J. Phys. Chem. 1982, 329, 2572–2586. [Google Scholar] [CrossRef]
- Eliasson, R.; Hammarsten, E.; Lindahl, T.; Björk, I.; Laurent, T.C. The stability of deoxyribonucleic acid in glycol solution. Biochim. Biophys. Acta Spec. Sect. Nucleic Acids Relat. Subj. 1963, 68, 234–239. [Google Scholar] [CrossRef]
- Mcallister, M.; Kazemigazestane, N.; Henry, L.T.; Gu, B.; Fabrikant, I.; Tribello, G.A.; Kohano, J. Solvation Effects on Dissociative Electron Attachment to Thymine. J. Phys. Chem. B 2019, 123, 1537–1544. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Bases | k (H2PO4•) L mol−1s−1 | E0/V (dB•+/dB) |
|---|---|---|
| G | 6.9 × 108 | 1.47 |
| A | 2.4 × 108 | 1.94 |
| T | 1.1 × 109 | 2.09 |
| C | <5 × 107 | 2.12 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MA, J.; Denisov, S.A.; Adhikary, A.; Mostafavi, M. Ultrafast Processes Occurring in Radiolysis of Highly Concentrated Solutions of Nucleosides/Tides. Int. J. Mol. Sci. 2019, 20, 4963. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194963
MA J, Denisov SA, Adhikary A, Mostafavi M. Ultrafast Processes Occurring in Radiolysis of Highly Concentrated Solutions of Nucleosides/Tides. International Journal of Molecular Sciences. 2019; 20(19):4963. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194963
Chicago/Turabian StyleMA, Jun, Sergey A. Denisov, Amitava Adhikary, and Mehran Mostafavi. 2019. "Ultrafast Processes Occurring in Radiolysis of Highly Concentrated Solutions of Nucleosides/Tides" International Journal of Molecular Sciences 20, no. 19: 4963. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194963