BAP1 Status Determines the Sensitivity of Malignant Mesothelioma Cells to Gemcitabine Treatment

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
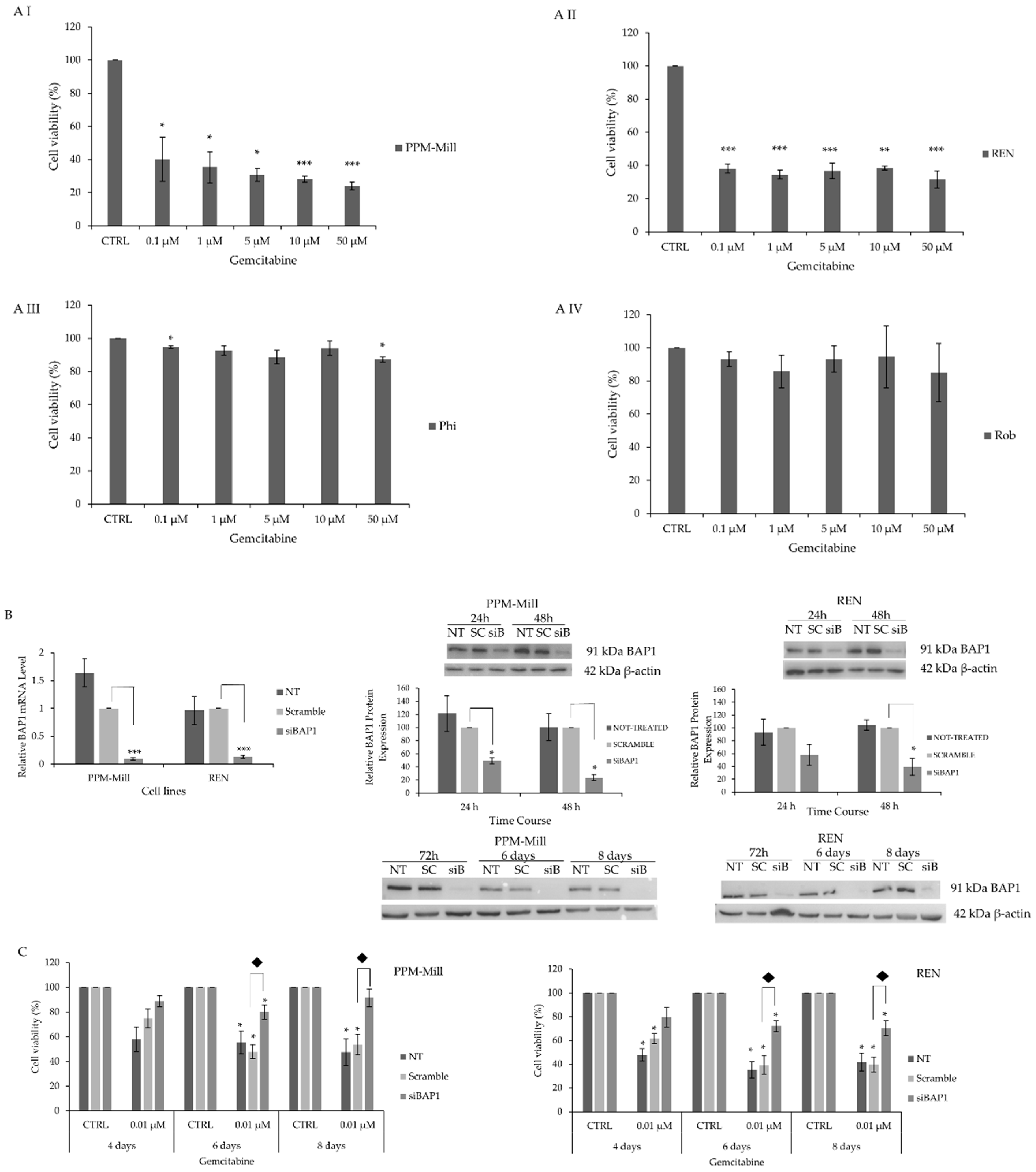
2.1. BAP1 WT MMe Cells Exhibit Higher Sensitivity to Gemcitabine Treatment Comprared to Mutated BAP1 MMe Cells
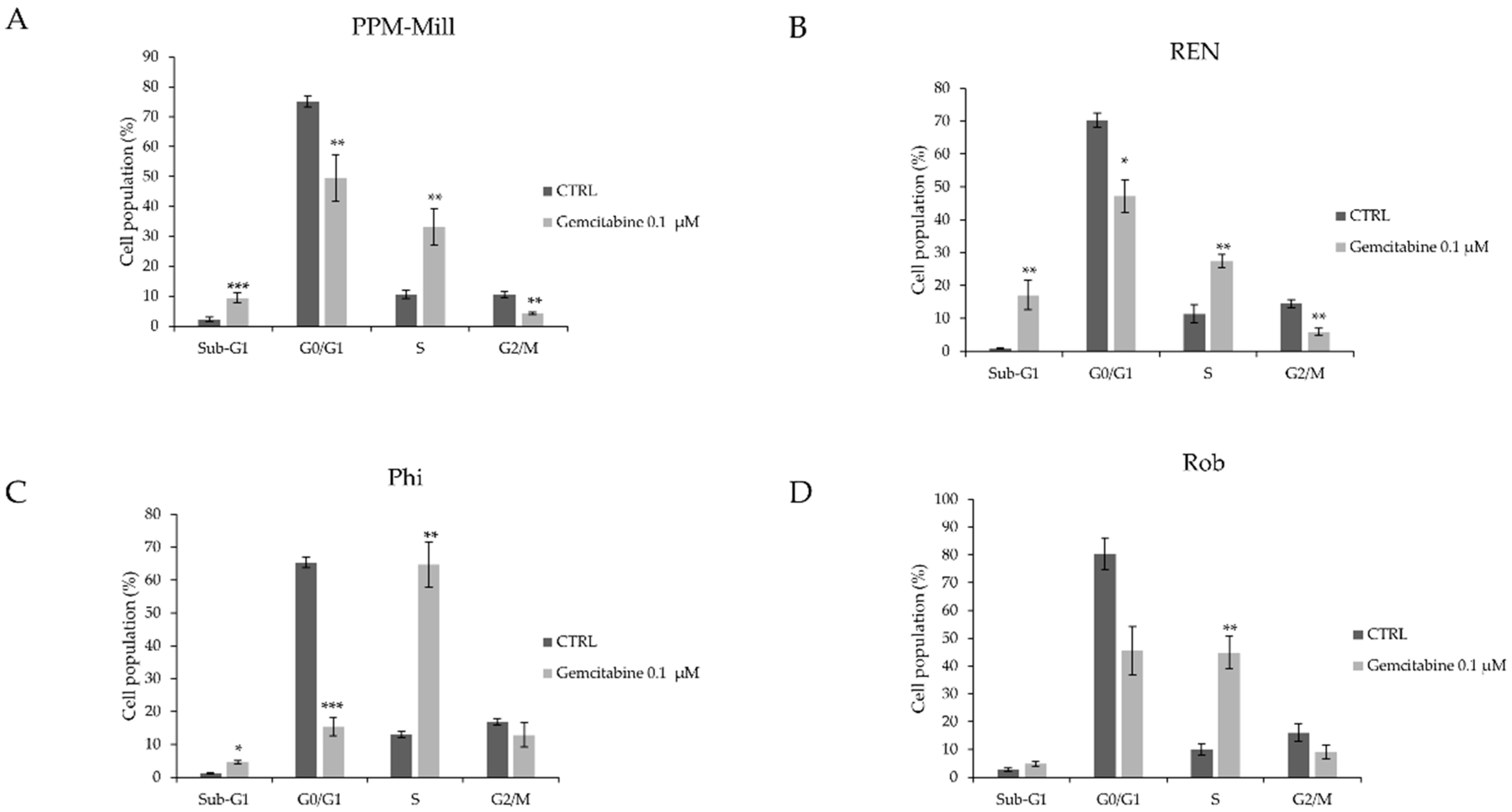
2.2. BAP1 Affects Cell Cycle Progression in MMe Cells Following Gemcitabine Treatment
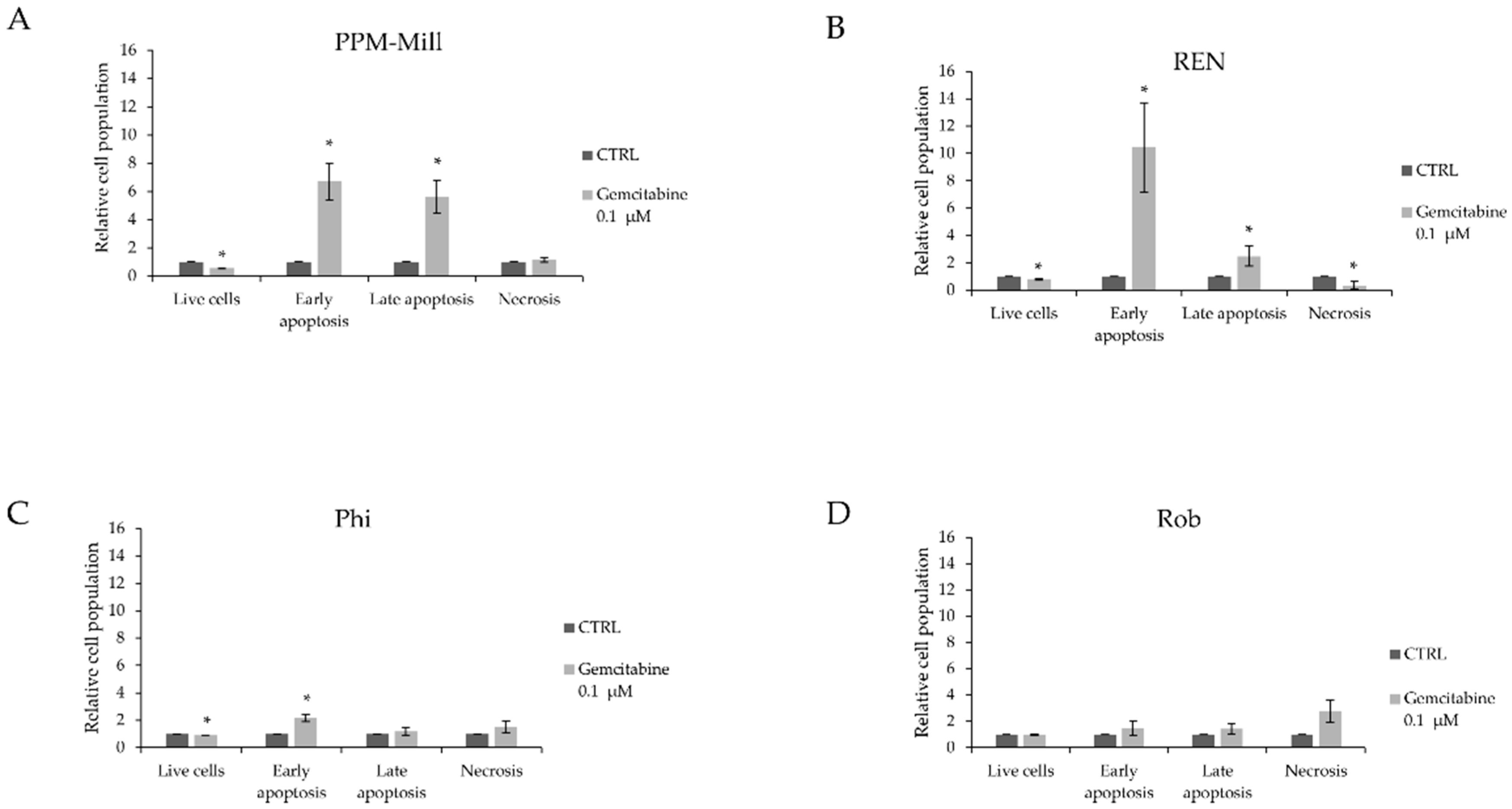
2.3. BAP1 Status Affects Apoptotic Response to Gemcitabine Treatment
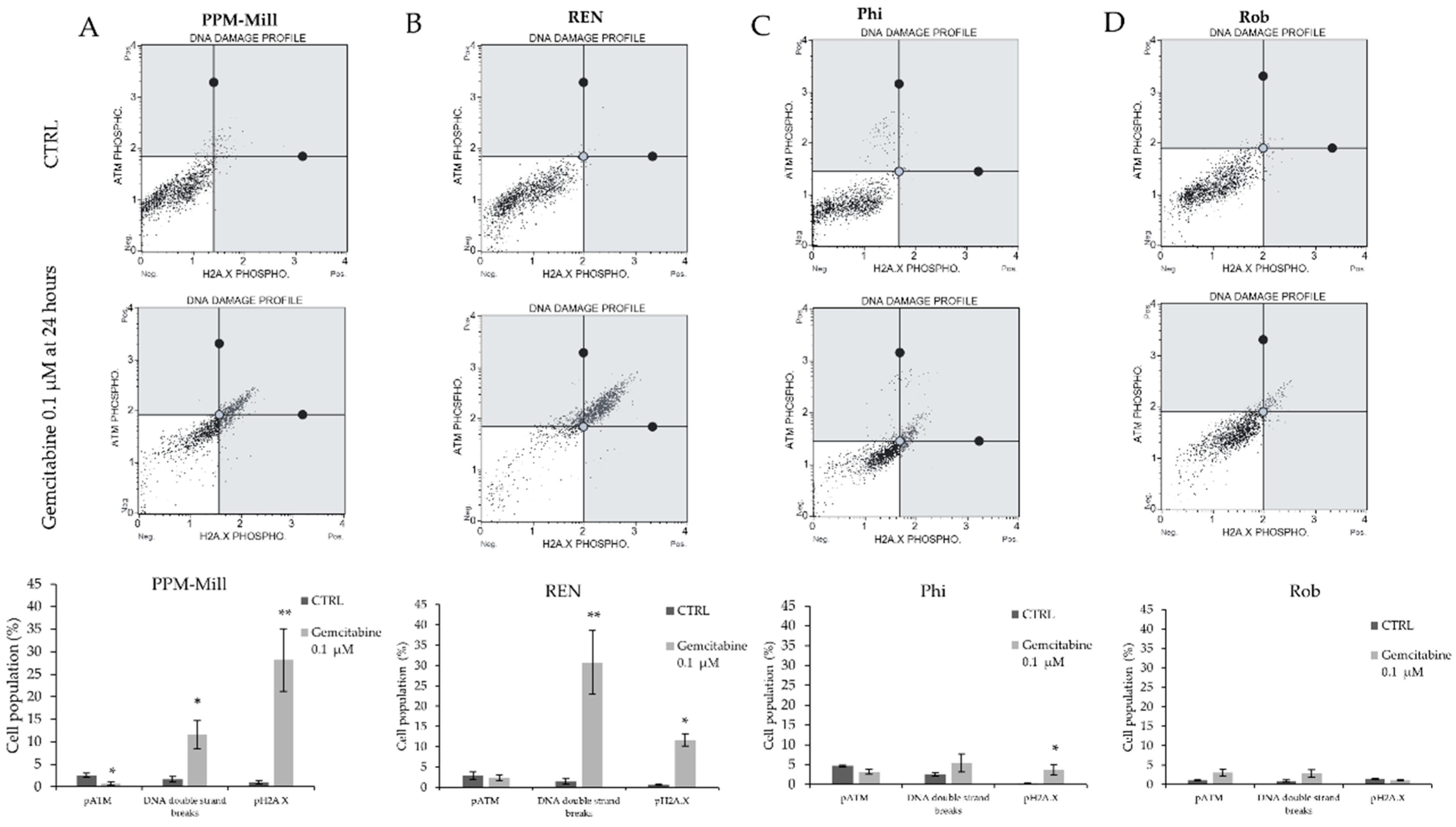
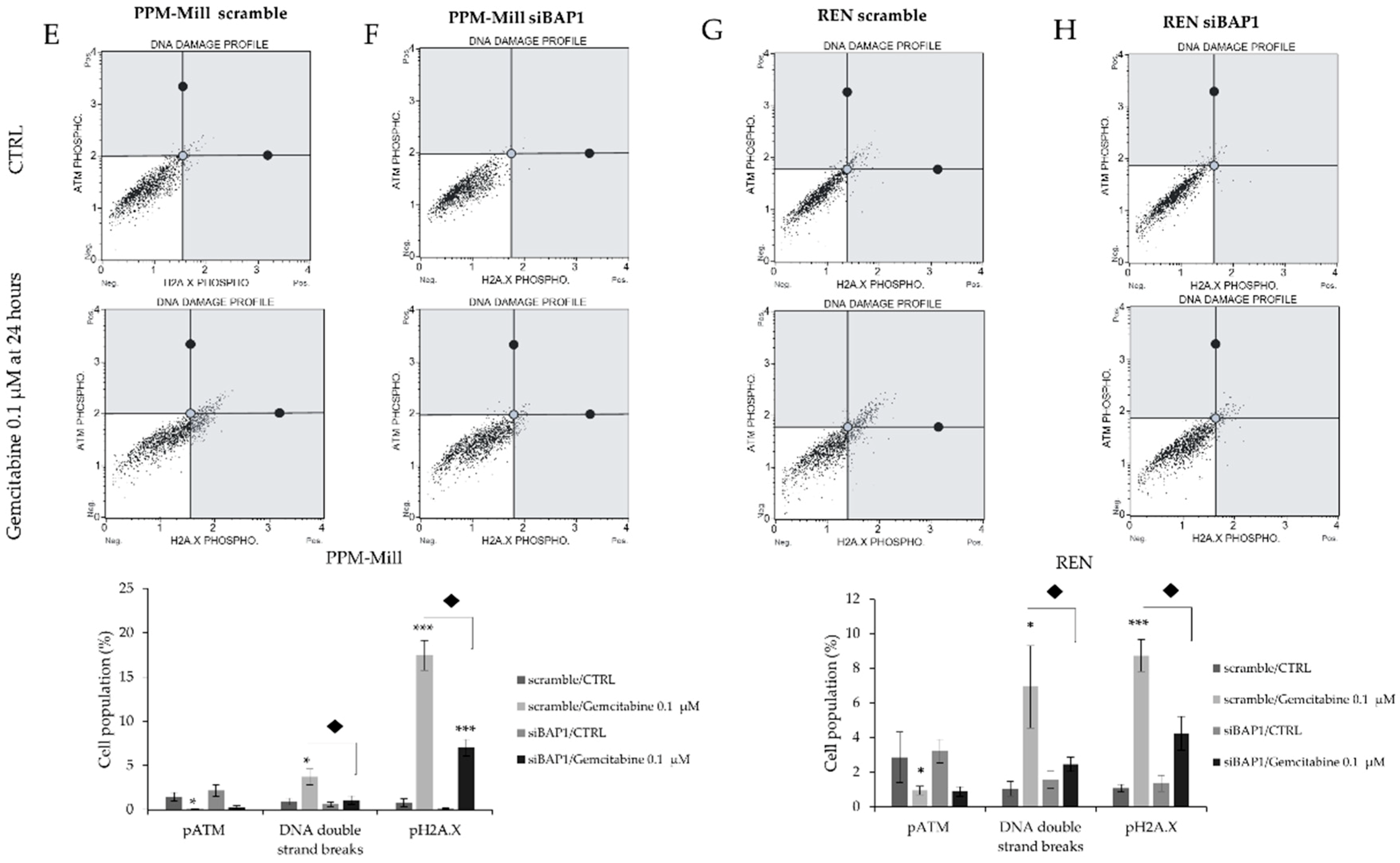
2.4. Gemcitabine Induces More DNA Damage in WT BAP1 than in BAP1-Mutated or -Silenced Cell Lines
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Treatment
4.3. Sulphorhodamine B (SRB) Assay
4.4. BAP1 Silencing
4.5. Quantitative Real Time-Polymerase Chain Reaction (qRT-PCR)
4.6. Western Blot
4.7. Cell Cycle Analysis
4.8. Annexin V Staining
4.9. Multi-Color DNA Damage Assay
4.10. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATM | Ataxia-Telangiectasia-Mutated |
| ATR | Ataxia Telangiectasia and Rad3-related |
| BAP1 | BRCA associated protein 1 |
| DNA-PK | DNA-dependent Protein Kinase |
| E2F1 | E2F transcription factor 1 |
| GEM | Gemcitabine |
| HCF-1 | Host Cell Factor-1 |
| hCNT1 | human Concentrative Nucleoside Transporter |
| KLF5 | Krueppel-Like Factor 5 |
| MMe | Malignant Mesothelioma |
| NF-κB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| OGT | O-linked N-acetylglucosamine transferase |
| PRC1 | Polycomb repressive complex 1 |
| PR-DUB | Polycomb repressive deubiquitinase |
| TCEAL7 | Transcription Elongation factor A-like 7 |
| UCH | Ubiquitin carboxy (C)-terminal Hydrolase |
| WT | Wild Type |
| YY1 | Yin Yang 1 |
References
- Blyth, K.G.; Murphy, D.J. Progress and challenges in Mesothelioma: From bench to bedside. Respir. Med. 2018, 134, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Guazzelli, A.; Bakker, E.; Tian, K.; Demonacos, C.; Krstic-Demonacos, M.; Mutti, L. Promising investigational drug candidates in phase I and phase II clinical trials for mesothelioma. Expert Opin. Investig. Drugs 2017, 26, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Scherpereel, A.; Astoul, P.; Baas, P.; Berghmans, T.; Clayson, H.; de Vuyst, P.; Dienemann, H.; Galateau-Salle, F.; Hennequin, C.; Hillerdal, G.; et al. Guidelines of the European Respiratory Society and the European Society of Thoracic Surgeons for the management of malignant pleural mesothelioma. Eur. Respir. J. 2010, 35, 479–495. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.D.M.; Bedrossian, C. The pathogenesis of mesothelioma. Semin. Diagn. Pathol. 2006, 23, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Amelio, I.; Affar, E.B.; Brugarolas, J.; Cannon-Albright, L.A.; Cantley, L.C.; Cavenee, W.K.; Chen, Z.; Croce, C.M.; Andrea, A. Consensus report of the 8 and 9th Weinman Symposia on Gene x Environment Interaction in carcinogenesis: Novel opportunities for precision medicine. Cell Death Differ 2018, 25, 1885–1904. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Rizzo, P.; Pass, H. Simian virus 40: The link with human malignant mesothelioma is well established. Anticancer Res. 2000, 20, 875–877. [Google Scholar] [PubMed]
- Van Meerbeeck, J.P.; Baas, P.; Debruyne, C.; Groen, H.J.; Manegold, C.; Ardizzoni, A.; Gridelli, C.; van Marck, E.A.; Lentz, M.; Giaccone, G. A Phase II study of gemcitabine in patients with malignant pleural mesothelioma. European Organization for Research and Treatment of Cancer Lung Cancer Cooperative Group. Cancer 1999, 85, 2577–2582. [Google Scholar] [CrossRef]
- Kindler, H.L.; Millard, F.; Herndon, J.E., 2nd; Vogelzang, N.J.; Suzuki, Y.; Green, M.R. Gemcitabine for malignant mesothelioma: A phase II trial by the Cancer and Leukemia Group B. Lung Cancer 2001, 31, 311–317. [Google Scholar] [CrossRef]
- Zauderer, M.G.; Kass, S.L.; Woo, K.; Sima, C.S.; Ginsberg, M.S.; Krug, L.M. Vinorelbine and gemcitabine as second- or third-line therapy for malignant pleural mesothelioma. Lung Cancer 2014, 84, 271–274. [Google Scholar] [CrossRef] [Green Version]
- Manegold, C.; Symanowski, J.; Gatzemeier, U.; Reck, M.; von Pawel, J.; Kortsik, C.; Nackaerts, K.; Lianes, P.; Vogelzang, N.J. Second-line (post-study) chemotherapy received by patients treated in the phase III trial of pemetrexed plus cisplatin versus cisplatin alone in malignant pleural mesothelioma. Ann. Oncol. 2005, 16, 923–927. [Google Scholar] [CrossRef] [Green Version]
- Zucali, P.A.; Ceresoli, G.L.; Garassino, I.; De Vincenzo, F.; Cavina, R.; Campagnoli, E.; Cappuzzo, F.; Salamina, S.; Soto Parra, H.J.; Santoro, A. Gemcitabine and vinorelbine in pemetrexed-pretreated patients with malignant pleural mesothelioma. Cancer 2008, 112, 1555–1561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carbone, M.; Kanodia, S.; Chao, A.; Miller, A.; Wali, A.; Weissman, D.; Adjei, A.; Baumann, F.; Boffetta, P.; Buck, B.; et al. Consensus Report of the 2015 Weinman International Conference on Mesothelioma. J. Thoracic Oncol. 2016, 11, 1246–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Pak, H.; Hammond-Martel, I.; Ghram, M.; Rodrigue, A.; Daou, S.; Barbour, H.; Corbeil, L.; Hebert, J.; Drobetsky, E.; et al. Tumor suppressor and deubiquitinase BAP1 promotes DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Kee, Y.; Huang, T.T. Role of Deubiquitinating Enzymes in DNA Repair. Mol. Cell. Biol. 2016, 36, 524–544. [Google Scholar] [CrossRef]
- Bononi, A.; Yang, H.; Giorgi, C.; Patergnani, S.; Pellegrini, L.; Su, M.; Xie, G.; Signorato, V.; Pastorino, S.; Morris, P.; et al. Germline BAP1 mutations induce a Warburg effect. Cell Death Differ. 2017, 24, 1694–1704. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Mashtalir, N.; Daou, S.; Hammond-Martel, I.; Ross, J.; Sui, G.; Hart, G.W.; Rauscher, F.J., 3rd; Drobetsky, E.; Milot, E.; et al. The ubiquitin carboxyl hydrolase BAP1 forms a ternary complex with YY1 and HCF-1 and is a critical regulator of gene expression. Mol. Cell. Biol. 2010, 30, 5071–5085. [Google Scholar] [CrossRef]
- Machida, Y.J.; Machida, Y.; Vashisht, A.A.; Wohlschlegel, J.A.; Dutta, A. The Deubiquitinating Enzyme BAP1 Regulates Cell Growth via Interaction with HCF-1. J. Biol. Chem. 2009, 284, 34179–34188. [Google Scholar] [CrossRef] [Green Version]
- Pena-Llopis, S.; Vega-Rubin-de-Celis, S.; Liao, A.; Leng, N.; Pavia-Jimenez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Shi, Y.; Mulligan, P.; Gay, F.; Landry, J.; Liu, H.; Lu, J.; Qi, H.H.; Wang, W.; Nickoloff, J.A.; et al. A YY1-INO80 complex regulates genomic stability through homologous recombination-based repair. Nat. Struct. Mol. Biol. 2007, 14, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Jackson, S.P. Ubiquitylation, neddylation and the DNA damage response. Open Biol. 2015, 5, 150018. [Google Scholar] [CrossRef]
- Citterio, E. Fine-tuning the ubiquitin code at DNA double-strand breaks: Deubiquitinating enzymes at work. Front. Genet. 2015, 6, 282. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.; Kumar, S.; Marlowe, T.; Chaudhary, A.K.; Kumar, R.; Wang, J.; O’Malley, J.; Boland, P.M.; Jayanthi, S.; Kumar, T.K.; et al. Oxidative phosphorylation-dependent regulation of cancer cell apoptosis in response to anticancer agents. Cell Death Dis. 2015, 6, e1969. [Google Scholar] [CrossRef] [PubMed]
- Eletr, Z.M.; Yin, L.; Wilkinson, K.D. BAP1 is phosphorylated at serine 592 in S-phase following DNA damage. FEBS Lett. 2013, 587, 3906–3911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.; Papneja, A.; Hyrcza, M.; Al-Habeeb, A.; Ghazarian, D. Gene of the month: BAP1. J. Clin. Pathol. 2016, 69, 750–753. [Google Scholar] [CrossRef] [PubMed]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesner, T.; Obenauf, A.C.; Murali, R.; Fried, I.; Griewank, K.G.; Ulz, P.; Windpassinger, C.; Wackernagel, W.; Loy, S.; Wolf, I.; et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat. Genet. 2011, 43, 1018–1021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochel, M.C.; Piris, A.; Nose, V.; Hoang, M.P. Loss of BAP1 Expression in Basal Cell Carcinomas in Patients With Germline BAP1 Mutations. Am. J. Clin. Pathol. 2015, 143, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, M.; Kadariya, Y.; Talarchek, J.; Pei, J.; Ohar, J.A.; Kayaleh, O.R.; Testa, J.R. Germline BAP1 mutation in a family with high incidence of multiple primary cancers and a potential gene-environment interaction. Cancer Lett. 2015, 369, 261–265. [Google Scholar] [CrossRef]
- Cheung, M.; Testa, J.R. BAP1, a tumor suppressor gene driving malignant mesothelioma. Transl. Lung Cancer Res. 2017, 6, 270–278. [Google Scholar] [CrossRef] [Green Version]
- Nasu, M.; Emi, M.; Pastorino, S.; Tanji, M.; Powers, A.; Luk, H.; Baumann, F.; Zhang, Y.A.; Gazdar, A.; Kanodia, S.; et al. High Incidence of Somatic BAP1 alterations in sporadic malignant mesothelioma. J. Thorac. Oncol. 2015, 10, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, Y.; Emi, M.; Hashimoto-Tamaoki, T.; Ohmuraya, M.; Sato, A.; Tsujimura, T.; Hasegawa, S.; Nakano, T.; Nasu, M.; Pastorino, S.; et al. High-density array-CGH with targeted NGS unmask multiple noncontiguous minute deletions on chromosome 3p21 in mesothelioma. Proc. Natl. Acad. Sci. USA 2016, 113, 13432–13437. [Google Scholar] [CrossRef] [PubMed]
- Toyokawa, G.; Takenoyama, M.; Hirai, F.; Toyozawa, R.; Inamasu, E.; Kojo, M.; Morodomi, Y.; Shiraishi, Y.; Takenaka, T.; Yamaguchi, M.; et al. Gemcitabine and vinorelbine as second-line or beyond treatment in patients with malignant pleural mesothelioma pretreated with platinum plus pemetrexed chemotherapy. Int. J. Clin. Oncol. 2014, 19, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Cappella, P.; Tomasoni, D.; Faretta, M.; Lupi, M.; Montalenti, F.; Viale, F.; Banzato, F.; D’Incalci, M.; Ubezio, P. Cell cycle effects of gemcitabine. Int. J. Cancer 2001, 93, 401–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montano, R.; Khan, N.; Hou, H.; Seigne, J.; Ernstoff, M.S.; Lewis, L.D.; Eastman, A. Cell cycle perturbation induced by gemcitabine in human tumor cells in cell culture, xenografts and bladder cancer patients: Implications for clinical trial designs combining gemcitabine with a Chk1 inhibitor. Oncotarget 2017, 8, 67754–67768. [Google Scholar] [CrossRef]
- Mori, T.; Sumii, M.; Fujishima, F.; Ueno, K.; Emi, M.; Nagasaki, M.; Ishioka, C.; Chiba, N. Somatic alteration and depleted nuclear expression of BAP1 in human esophageal squamous cell carcinoma. Cancer Sci. 2015, 106, 1118–1129. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Azuma, A.; Sampath, D.; Li, Y.X.; Huang, P.; Plunkett, W. S-Phase arrest by nucleoside analogues and abrogation of survival without cell cycle progression by 7-hydroxystaurosporine. Cancer Res. 2001, 61, 1065–1072. [Google Scholar]
- Eletr, Z.M.; Wilkinson, K.D. An emerging model for BAP1′s role in regulating cell cycle progression. Cell Biochem. Biophys. 2011, 60, 3–11. [Google Scholar] [CrossRef]
- Qin, J.; Zhou, Z.; Chen, W.; Wang, C.; Zhang, H.; Ge, G.; Shao, M.; You, D.; Fan, Z.; Xia, H.; et al. BAP1 promotes breast cancer cell proliferation and metastasis by deubiquitinating KLF5. Nat. Commun. 2015, 6, 8471. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Taylor, M.; Miao, B.; Ji, Z.; Njauw, J.C.; Jonsson, G.; Frederick, D.T.; Tsao, H. BAP1 has a survival role in cutaneous melanoma. J. Investig. Dermatol. 2015, 135, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Matatall, K.A.; Agapova, O.A.; Onken, M.D.; Worley, L.A.; Bowcock, A.M.; Harbour, J.W. BAP1 deficiency causes loss of melanocytic cell identity in uveal melanoma. BMC Cancer 2013, 13, 371. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Wu, W.; Koike, A.; Kojima, R.; Gomi, H.; Fukuda, M.; Ohta, T. BRCA1-associated protein 1 interferes with BRCA1/BARD1 RING heterodimer activity. Cancer Res. 2009, 69, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Zarrizi, R.; Menard, J.A.; Belting, M.; Massoumi, R. Deubiquitination of γ-Tubulin by BAP1 Prevents Chromosome Instability in Breast Cancer Cells. Cancer Res. 2014, 74, 6499–6508. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Ventii, K.H.; Devi, N.S.; Friedrich, K.L.; Chernova, T.A.; Tighiouart, M.; Van Meir, E.G.; Wilkinson, K.D. BRCA1-associated protein-1 is a tumor suppressor that requires deubiquitinating activity and nuclear localization. Cancer Res. 2008, 68, 6953–6962. [Google Scholar] [CrossRef]
- Vandenabeele, P.; Grootjans, S.; Callewaert, N.; Takahashi, N. Necrostatin-1 blocks both RIPK1 and IDO: Consequences for the study of cell death in experimental disease models. Cell Death Differ. 2013, 20, 185–187. [Google Scholar] [CrossRef]
- Dai, F.; Lee, H.; Zhang, Y.; Zhuang, L.; Yao, H.; Xi, Y.; Xiao, Z.D.; You, M.J.; Li, W.; Su, X.; et al. BAP1 inhibits the ER stress gene regulatory network and modulates metabolic stress response. Proc. Natl. Acad. Sci. USA 2017, 114, 3192–3197. [Google Scholar] [CrossRef] [Green Version]
- Affar, E.B.; Carbone, M. BAP1 regulates different mechanisms of cell death. Cell Death Dis. 2018, 9, 1151. [Google Scholar] [CrossRef]
- Sime, W.; Niu, Q.; Abassi, Y.; Masoumi, K.C.; Zarrizi, R.; Køhler, J.B.; Kjellström, S.; Lasorsa, V.A.; Capasso, M.; Fu, H.; et al. BAP1 induces cell death via interaction with 14-3-3 in neuroblastoma. Cell Death Dis. 2018, 9, 458. [Google Scholar] [CrossRef] [PubMed]
- Parasramka, M.; Yan, I.K.; Wang, X.; Nguyen, P.; Matsuda, A.; Maji, S.; Foye, C.; Asmann, Y.; Patel, T. BAP1 dependent expression of long non-coding RNA NEAT-1 contributes to sensitivity to gemcitabine in cholangiocarcinoma. Mol. Cancer 2017, 16, 22. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, S.; Yoshikawa, Y.; Pass, H.I.; Emi, M.; Nasu, M.; Pagano, I.; Takinishi, Y.; Yamamoto, R.; Minaai, M.; Hashimoto-Tamaoki, T.; et al. A Subset of Mesotheliomas With Improved Survival Occurring in Carriers of BAP1 and Other Germline Mutations. J. Clin. Oncol. 2018, 36, 3485–3494. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pass, H.I.; Stevens, E.J.; Oie, H.; Tsokos, M.G.; Abati, A.D.; Fetsch, P.A.; Mew, D.J.; Pogrebniak, H.W.; Matthews, W.J. Characteristics of nine newly derived mesothelioma cell lines. Ann. Thorac. Surg. 1995, 59, 835–844. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guazzelli, A.; Meysami, P.; Bakker, E.; Demonacos, C.; Giordano, A.; Krstic-Demonacos, M.; Mutti, L. BAP1 Status Determines the Sensitivity of Malignant Mesothelioma Cells to Gemcitabine Treatment. Int. J. Mol. Sci. 2019, 20, 429. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020429
Guazzelli A, Meysami P, Bakker E, Demonacos C, Giordano A, Krstic-Demonacos M, Mutti L. BAP1 Status Determines the Sensitivity of Malignant Mesothelioma Cells to Gemcitabine Treatment. International Journal of Molecular Sciences. 2019; 20(2):429. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020429
Chicago/Turabian StyleGuazzelli, Alice, Parisa Meysami, Emyr Bakker, Constantinos Demonacos, Antonio Giordano, Marija Krstic-Demonacos, and Luciano Mutti. 2019. "BAP1 Status Determines the Sensitivity of Malignant Mesothelioma Cells to Gemcitabine Treatment" International Journal of Molecular Sciences 20, no. 2: 429. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020429