Facets of Theiler’s Murine Encephalomyelitis Virus-Induced Diseases: An Update

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. General Aspects of Theiler’s Murine Encephalomyelitis Virus
2. Virus Tropism and Spread
3. Theiler’s Murine Encephalomyelitis Virus Infection as Model for Multiple Sclerosis
4. Theiler’s Murine Encephalomyelitis Virus Infection as Model for Seizures and Epilepsy
5. Theiler’s Murine Encephalomyelitis Virus Infection as Model for Myocarditis
6. Innate Immunity in Theiler’s Murine Encephalomyelitis Virus Infection
6.1. Mechanisms of the Innate Immune Response
6.2. Expression of Pro-Inflammatory Mediators in Microglia, Macrophages, and Astrocytes
6.3. Innate Immunity Accounts for Seizure Development during Theiler’s Murine Encephalomyelitis Virus Infection of C57BL/6 Mice
6.4. Innate Immunity Participates in Demyelination Processes and Cardiac Damage
6.5. Modulation of the Cannabinoid System
7. Adaptive Immunity in Theiler’s Murine Encephalomyelitis Virus Infection
7.1. CD4+ T Cells
7.1.1. Regulatory T Cells
7.1.2. Th17 Cells
7.2. CD8+ T Cells
7.3. B Cells and Humoral Immunity
7.4. Lymphocyte Apoptosis of Immune Cells
8. Conclusion and Outlook
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AICD | Activation-induced cell death |
| AP-1 | Activator protein 1 |
| β2M | β2-Microglobulin |
| CARD | Caspase activation and recruitment domain |
| CARDIF | CARD adaptor inducing IFNβ |
| CB | Cannabinoid |
| CBD | Cannabidiol |
| CLR | C-type lectin receptor |
| CNS | Central nervous system |
| DAMP | Damage-associated molecular pattern |
| DC | Dendritic cell |
| Δ9-THC | Δ9-Tetrahydrocannabinol |
| DEREG | Depletion of regulatory T cells |
| dsRNA | Double-stranded RNA |
| EEG | Electroencephalogram |
| eIF2α | Eukaryotic translation initiation factor 2α |
| FADD | Fas-associated protein with death domain |
| FasL | Fas ligand |
| GABA | Gamma-aminobutyric acid |
| HIF | Hypoxia-inducible factor |
| ICE | IL1β converting enzyme |
| IFN-I | Type I interferon |
| IL | Interleukin |
| IPS-1 | IFNβ promoter stimulator |
| IL | Interleukin |
| IRES | Internal ribosomal entry site |
| IRF | Interferon regulatory factor |
| ISG | Interferon stimulated gene |
| iTreg | Induced Treg |
| lncRNA | Long noncoding RNA |
| LPS | Lipopolysaccharides |
| MAPK | Mitogen-activated protein kinase |
| MAVS | Mitochondrial antiviral-signaling protein |
| MBP | Myelin basic protein |
| MDA5 | Melanoma differentiation-associated gene 5 |
| MDSC | Myeloid-derived suppressor cell |
| mGluR5 | Metabotropic glutamate receptor 5 |
| MHC | Major histocompatibility complex |
| MOG | Myelin oligodendrocyte glycoprotein |
| MS | Multiple sclerosis |
| MyD88 | Myeloid differentiation primary response gene 88 |
| NF-κB | Nuclear factor κ-light-chain-enhancer of activated B cells |
| NLR | NOD-like receptor |
| Nup | Nuclear pore protein |
| OAS | Oligoadenylate synthetase |
| ORF | Open reading frame |
| PAMP | Pathogen-associated molecular pattern |
| Pik3cg | Phosphoinositide-3-kinase catalytic subunit gamma |
| PKR | dsRNA-Activated protein kinase |
| PLP | Proteolipid protein |
| PPAR | Peroxisome proliferator-activated receptor |
| PRR | Pattern recognition receptor |
| RIG | Retinoic acid-inducible gene |
| RLR | RIG-I-like receptors |
| ROR | Retinoic acid related orphan receptor |
| Th | T helper |
| TIR | Toll/IL1 receptor homology |
| TLR | Toll-like receptor |
| TME | Theiler’s murine encephalomyelitis |
| TMEV | TME virus |
| TMEV-IDD | TMEV-induced demyelinating disease |
| TNF | Tumor necrosis factor |
| TNFR | TNF receptor |
| TRADD | TNFR1-associated death domain protein |
| TRAF | TNFR-associated factor |
| Treg | Regulatory T cells |
| TRIF | TIR domain-containing adaptor inducing IFNβ |
| VCAM | Vascular cell adhesion molecule |
| VEGF | Vascular endothelial growth factor |
| VHEV | Vilyuisk human encephalomyelitis virus |
| VISA | Virus-induced signaling adapter |
References
- Daniels, J.B.; Pappenheimer, A.M.; Richardson, S. Observations on encephalomyelitis of mice (DA strain). J. Exp. Med. 1952, 96, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Libbey, J.E.; Kirkman, N.J.; Smith, M.C.; Tanaka, T.; Wilcox, K.S.; White, H.S.; Fujinami, R.S. Seizures following picornavirus infection. Epilepsia 2008, 49, 1066–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipton, H.L. Theiler’s virus infection in mice: An unusual biphasic disease process leading to demyelination. Infect. Immun. 1975, 11, 1147–1155. [Google Scholar]
- Stewart, K.A.; Wilcox, K.S.; Fujinami, R.S.; White, H.S. Development of postinfection epilepsy after Theiler’s virus infection of C57BL/6 mice. J. Neuropathol. Exp. Neurol. 2010, 69, 1210–1219. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Sato, F.; Omura, S.; Fujita, M.; Sakiyama, N.; Park, A.M. Three immune-mediated disease models induced by Theiler’s virus: Multiple sclerosis, seizures and myocarditis. Clin. Exp. Neuroimmunol. 2016, 7, 330–345. [Google Scholar] [CrossRef]
- Kawai, E.; Sato, F.; Omura, S.; Martinez, N.E.; Reddy, P.C.; Taniguchi, M.; Tsunoda, I. Organ-specific protective role of NKT cells in virus-induced inflammatory demyelination and myocarditis depends on mouse strain. J. Neuroimmunol. 2015, 278, 174–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theiler, M. Spontaneous Encephalomyelitis of Mice, a New Virus Disease. J. Exp. Med. 1937, 65, 705–719. [Google Scholar] [CrossRef]
- Gomez, R.M.; Rinehart, J.E.; Wollmann, R.; Roos, R.P. Theiler’s murine encephalomyelitis virus-induced cardiac and skeletal muscle disease. J. Virol. 1996, 70, 8926–8933. [Google Scholar]
- Omura, S.; Sato, F.; Martinez, N.E.; Range, T.; Ekshyyan, L.; Minagar, A.; Alexander, J.S.; Tsunoda, I. Immunoregulation of Theiler’s virus-induced demyelinating disease by glatiramer acetate without suppression of antiviral immune responses. Arch. Virol. 2018, 163, 1279–1284. [Google Scholar] [CrossRef]
- Liang, Z.; Kumar, A.S.; Jones, M.S.; Knowles, N.J.; Lipton, H.L. Phylogenetic analysis of the species Theilovirus: Emerging murine and human pathogens. J. Virol. 2008, 82, 11545–11554. [Google Scholar] [CrossRef]
- Theiler, M.; Gard, S. Encephalomyelitis of Mice: I. Characteristics and Pathogenesis of the Virus. J. Exp. Med. 1940, 72, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Welsh, C.J.; Tonks, P.; Borrow, P.; Nash, A.A. Theiler’s virus: An experimental model of virus-induced demyelination. Autoimmunity 1990, 6, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.Z.; Tan, M.Z.; Prabakaran, M. Saffold virus, an emerging human cardiovirus. Rev. Med. Virol. 2017, 27. [Google Scholar] [CrossRef] [PubMed]
- Brinkmeyer-Langford, C.L.; Rech, R.; Amstalden, K.; Kochan, K.J.; Hillhouse, A.E.; Young, C.; Welsh, C.J.; Threadgill, D.W. Host genetic background influences diverse neurological responses to viral infection in mice. Sci. Rep. 2017, 7, 12194. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Salinas, F.J.; Mestre, L.; Mecha, M.; Feliu, A.; Del Campo, R.; Villarrubia, N.; Espejo, C.; Montalban, X.; Alvarez-Cermeno, J.C.; Villar, L.M.; et al. Gut dysbiosis and neuroimmune responses to brain infection with Theiler’s murine encephalomyelitis virus. Sci. Rep. 2017, 7, 44377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linsenbardt, H.R.; Cook, J.L.; Young, E.E.; Vichaya, E.G.; Young, C.R.; Reusser, N.M.; Storts, R.; Welsh, C.J.; Meagher, M.W. Social disruption alters pain and cognition in an animal model of multiple sclerosis. J. Neuroimmunol. 2015, 288, 56–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsunoda, I. Lymphatic system and gut microbiota affect immunopathology of neuroinflammatory diseases, including multiple sclerosis, neuromyelitis optica and Alzheimer’s disease. Clin. Exp. Neuroimmunol. 2017, 8, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Dal Canto, M.C.; Kim, B.S.; Miller, S.D.; Melvold, R.W. Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelination: A model for human multiple sclerosis. Methods 1996, 10, 453–461. [Google Scholar] [CrossRef]
- Gerhauser, I.; Alldinger, S.; Baumgärtner, W. Ets-1 represents a pivotal transcription factor for viral clearance, inflammation, and demyelination in a mouse model of multiple sclerosis. J. Neuroimmunol. 2007, 188, 86–94. [Google Scholar] [CrossRef]
- Oleszak, E.L.; Chang, J.R.; Friedman, H.; Katsetos, C.D.; Platsoucas, C.D. Theiler’s virus infection: A model for multiple sclerosis. Clin. Microbiol. Rev. 2004, 17, 174–207. [Google Scholar] [CrossRef]
- Ulrich, R.; Baumgärtner, W.; Gerhauser, I.; Seeliger, F.; Haist, V.; Deschl, U.; Alldinger, S. MMP-12, MMP-3, and TIMP-1 are markedly upregulated in chronic demyelinating theiler murine encephalomyelitis. J. Neuropathol. Exp. Neurol. 2006, 65, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Melvold, R.W.; Jokinen, D.M.; Knobler, R.L.; Lipton, H.L. Variations in genetic control of susceptibility to Theiler’s murine encephalomyelitis virus (TMEV)-induced demyelinating disease. I. Differences between susceptible SJL/J and resistant BALB/c strains map near the T cell beta-chain constant gene on chromosome 6. J. Immunol. 1987, 138, 1429–1433. [Google Scholar] [PubMed]
- Bröer, S.; Käufer, C.; Haist, V.; Li, L.; Gerhauser, I.; Anjum, M.; Bankstahl, M.; Baumgärtner, W.; Löscher, W. Brain inflammation, neurodegeneration and seizure development following picornavirus infection markedly differ among virus and mouse strains and substrains. Exp. Neurol. 2016, 279, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Wroblewska, Z.; Gilden, D.H.; Wellish, M.; Rorke, L.B.; Warren, K.G.; Wolinsky, J.S. Virus-specific intracytoplasmic inclusions in mouse brain produced by a newly isolated strain of Theiler virus. I. Virologic and morphologic studies. Lab. Investig. 1977, 37, 595–602. [Google Scholar] [PubMed]
- Lipton, H.L.; Melvold, R. Genetic analysis of susceptibility to Theiler’s virus-induced demyelinating disease in mice. J. Immunol. 1984, 132, 1821–1825. [Google Scholar] [PubMed]
- Rozhon, E.J.; Kratochvil, J.D.; Lipton, H.L. Analysis of genetic variation in Theiler’s virus during persistent infection in the mouse central nervous system. Virology 1983, 128, 16–32. [Google Scholar] [CrossRef]
- Michiels, T.; Jarousse, N.; Brahic, M. Analysis of the leader and capsid coding regions of persistent and neurovirulent strains of Theiler’s virus. Virology 1995, 214, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Monteyne, P.; Bureau, J.F.; Brahic, M. The infection of mouse by Theiler’s virus: From genetics to immunology. Immunol. Rev. 1997, 159, 163–176. [Google Scholar] [CrossRef]
- Ohara, Y.; Stein, S.; Fu, J.; Stillman, L.; Klaman, L.; Roos, R.P. Molecular-Cloning and Sequence Determination of Da Strain of Theiler Murine Encephalomyelitis Viruses. Virology 1988, 164, 245–255. [Google Scholar] [CrossRef]
- Pevear, D.C.; Calenoff, M.; Rozhon, E.; Lipton, H.L. Analysis of the complete nucleotide sequence of the picornavirus Theiler’s murine encephalomyelitis virus indicates that it is closely related to cardioviruses. J. Virol. 1987, 61, 1507–1516. [Google Scholar]
- Brahic, M.; Bureau, J.F.; Michiels, T. The genetics of the persistent infection and demyelinating disease caused by Theiler’s virus. Annu. Rev. Microbiol. 2005, 59, 279–298. [Google Scholar] [CrossRef] [PubMed]
- Dodd, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Poliovirus 3A protein limits interleukin-6 (IL-6), IL-8, and beta interferon secretion during viral infection. J. Virol. 2001, 75, 8158–8165. [Google Scholar] [CrossRef] [PubMed]
- Doedens, J.R.; Kirkegaard, K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 1995, 14, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Neznanov, N.; Kondratova, A.; Chumakov, K.M.; Angres, B.; Zhumabayeva, B.; Agol, V.I.; Gudkov, A.V. Poliovirus protein 3A inhibits tumor necrosis factor (TNF)-induced apoptosis by eliminating the TNF receptor from the cell surface. J. Virol. 2001, 75, 10409–10420. [Google Scholar] [CrossRef] [PubMed]
- De Cock, A.; Michiels, T. Cellular microRNAs Repress Vesicular Stomatitis Virus but Not Theiler’s Virus Replication. Viruses 2016, 8, 75. [Google Scholar] [CrossRef] [PubMed]
- Gomez, J.A.; Wapinski, O.L.; Yang, Y.W.; Bureau, J.F.; Gopinath, S.; Monack, D.M.; Chang, H.Y.; Brahic, M.; Kirkegaard, K. The NeST long ncRNA controls microbial susceptibility and epigenetic activation of the interferon-gamma locus. Cell 2013, 152, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.L.; Rodriguez, M.; Roos, R.P. Strains from both Theilers virus subgroups encode a determinant for demyelination. J. Virol. 1990, 64, 6345–6348. [Google Scholar]
- Jarousse, N.; Martinat, C.; Syan, S.; Brahic, M.; McAllister, A. Role of VP2 amino acid 141 in tropism of Theiler’s virus within the central nervous system. J. Virol. 1996, 70, 8213–8217. [Google Scholar]
- Jarousse, N.; Viktorova, E.G.; Pilipenko, E.V.; Agol, V.I.; Brahic, M. An attenuated variant of the GDVII strain of Theiler’s virus does not persist and does not infect the white matter of the central nervous system. J. Virol. 1999, 73, 801–804. [Google Scholar]
- McCright, I.J.; Tsunoda, I.; Whitby, F.G.; Fujinami, R.S. Theiler’s viruses with mutations in loop I of VP1 lead to altered tropism and pathogenesis. J. Virol. 1999, 73, 2814–2824. [Google Scholar]
- O’Shea, H.; Crang, J.; Tonks, P.; Nash, A.A.; Fazakerley, J.K. The PI capsid region of Theiler’s virus controls replication in mouse glial cell cultures. Arch. Virol. 1997, 142, 1521–1535. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Zhang, L.; Kim, J.; Jakob, J.; Grant, R.A.; Wollmann, R.; Roos, R.P. A neutralization site of DA strain of Theiler’s murine encephalomyelitis virus important for disease phenotype. Virology 1996, 226, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Upfold, N.; Ross, C.; Bishop, O.T.; Luke, G.A.; Knox, C. The generation and characterisation of neutralising antibodies against the Theiler’s murine encephalomyelitis virus (TMEV) GDVII capsid reveals the potential binding site of the host cell co-receptor, heparan sulfate. Virus Res. 2018, 244, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Lipton, H.L.; Kumar, A.S.; Hertzler, S.; Reddi, H.V. Differential usage of carbohydrate co-receptors influences cellular tropism of Theiler’s murine encephalomyelitis virus infection of the central nervous system. Glycoconj. J. 2006, 23, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Aubert, C.; Brahic, M. Early infection of the central nervous system by the GDVII and DA strains of Theiler’s virus. J. Virol. 1995, 69, 3197–3200. [Google Scholar] [PubMed]
- Dal Canto, M.C.; Lipton, H.L. Ultrastructural immunohistochemical localization of virus in acute and chronic demyelinating Theiler’s virus infection. Am. J. Pathol. 1982, 106, 20–29. [Google Scholar] [PubMed]
- Zheng, L.; Calenoff, M.A.; Dal Canto, M.C. Astrocytes, not microglia, are the main cells responsible for viral persistence in Theiler’s murine encephalomyelitis virus infection leading to demyelination. J. Neuroimmunol. 2001, 118, 256–267. [Google Scholar] [CrossRef]
- Njenga, M.K.; Asakura, K.; Hunter, S.F.; Wettstein, P.; Pease, L.R.; Rodriguez, M. The immune system preferentially clears Theiler’s virus from the gray matter of the central nervous system. J. Virol. 1997, 71, 8592–8601. [Google Scholar]
- Buckwalter, M.R.; Nga, P.T.; Gouilh, M.A.; Fiette, L.; Bureau, J.F.; Laird, M.E.; Buchrieser, J.; Ozden, S.; Cheval, J.; Eloit, M.; et al. Identification of a novel neuropathogenic Theiler’s murine encephalomyelitis virus. J. Virol. 2011, 85, 6893–6905. [Google Scholar] [CrossRef]
- Stewart, K.A.; Wilcox, K.S.; Fujinami, R.S.; White, H.S. Theiler’s virus infection chronically alters seizure susceptibility. Epilepsia 2010, 51, 1418–1428. [Google Scholar] [CrossRef]
- Bröer, S.; Hage, E.; Käufer, C.; Gerhauser, I.; Anjum, M.; Li, L.; Baumgärtner, W.; Schulz, T.F.; Löscher, W. Viral mouse models of multiple sclerosis and epilepsy: Marked differences in neuropathogenesis following infection with two naturally occurring variants of Theiler’s virus BeAn strain. Neurobiol. Dis. 2017, 99, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Lipton, H.L.; Twaddle, G.; Jelachich, M.L. The predominant virus antigen burden is present in macrophages in Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J. Virol. 1995, 69, 2525–2533. [Google Scholar] [PubMed]
- Clatch, R.J.; Miller, S.D.; Metzner, R.; Dal Canto, M.C.; Lipton, H.L. Monocytes/macrophages isolated from the mouse central nervous system contain infectious Theiler’s murine encephalomyelitis virus (TMEV). Virology 1990, 176, 244–254. [Google Scholar] [CrossRef]
- Gerhauser, I.; Hansmann, F.; Puff, C.; Kumnok, J.; Schaudien, D.; Wewetzer, K.; Baumgärtner, W. Theiler’s murine encephalomyelitis virus induced phenotype switch of microglia in vitro. J. Neuroimmunol. 2012, 252, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Arslan, S.Y.; Son, K.N.; Lipton, H.L. During Infection, Theiler’s Virions Are Cleaved by Caspases and Disassembled into Pentamers. J. Virol. 2016, 90, 3573–3583. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Kurtz, C.I.; Fujinami, R.S. Apoptosis in acute and chronic central nervous system disease induced by Theiler’s murine encephalomyelitis virus. Virology 1997, 228, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Gerhauser, I.; Li, L.; Li, D.; Klein, S.; Elmarabet, S.A.; Deschl, U.; Kalkuhl, A.; Baumgärtner, W.; Ulrich, R.; Beineke, A. Dynamic changes and molecular analysis of cell death in the spinal cord of SJL mice infected with the BeAn strain of Theiler’s murine encephalomyelitis virus. Apoptosis 2018, 23, 170–186. [Google Scholar] [CrossRef]
- Rubio, N.; Sanz-Rodriguez, F. Overexpression of caspase 1 in apoptosis-resistant astrocytes infected with the BeAn Theiler’s virus. J. Neurovirol. 2016, 22, 316–326. [Google Scholar] [CrossRef]
- Rubio, N.; Garcia-Segura, L.M.; Arevalo, M.A. Survivin prevents apoptosis by binding to caspase-3 in astrocytes infected with the BeAn strain of Theiler’s murine encephalomyelitis virus. J. Neurovirol. 2012, 18, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Kummerfeld, M.; Seehusen, F.; Klein, S.; Ulrich, R.; Kreutzer, R.; Gerhauser, I.; Herder, V.; Baumgärtner, W.; Beineke, A. Periventricular demyelination and axonal pathology is associated with subependymal virus spread in a murine model for multiple sclerosis. Intervirology 2012, 55, 401–416. [Google Scholar] [CrossRef]
- Kreutzer, M.; Seehusen, F.; Kreutzer, R.; Pringproa, K.; Kummerfeld, M.; Claus, P.; Deschl, U.; Kalkul, A.; Beineke, A.; Baumgärtner, W.; et al. Axonopathy is associated with complex axonal transport defects in a model of multiple sclerosis. Brain Pathol. 2012, 22, 454–471. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Fujinami, R.S. Inside-Out versus Outside-In models for virus induced demyelination: Axonal damage triggering demyelination. Springer Semin. Immunopathol. 2002, 24, 105–125. [Google Scholar] [CrossRef] [PubMed]
- Martinat, C.; Jarousse, N.; Prevost, M.C.; Brahic, M. The GDVII strain of Theiler’s virus spreads via axonal transport. J. Virol. 1999, 73, 6093–6098. [Google Scholar] [PubMed]
- Tsunoda, I. Axonal degeneration as a self-destructive defense mechanism against neurotropic virus infection. Future Virol. 2008, 3, 579–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palumbo, S.; Pellegrini, S. Experimental In Vivo Models of Multiple Sclerosis: State of the Art. In Multiple Sclerosis: Perspectives in Treatment and Pathogenesis; Zagon, I.S., McLaughlin, P.J., Eds.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar]
- Gilli, F.; Royce, D.B.; Pachner, A.R. Measuring Progressive Neurological Disability in a Mouse Model of Multiple Sclerosis. J. Vis. Exp. 2016, 117. [Google Scholar] [CrossRef] [PubMed]
- Procaccini, C.; De Rosa, V.; Pucino, V.; Formisano, L.; Matarese, G. Animal models of Multiple Sclerosis. Eur. J. Pharmacol. 2015, 759, 182–191. [Google Scholar] [CrossRef]
- Didonna, A. Preclinical Models of Multiple Sclerosis: Advantages and Limitations Towards Better Therapies. Curr. Med. Chem. 2016, 23, 1442–1459. [Google Scholar] [CrossRef]
- Steinman, L.; Zamvil, S.S. How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann. Neurol. 2006, 60, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Pachner, A.R.; Li, L.; Gilli, F. Chemokine biomarkers in central nervous system tissue and cerebrospinal fluid in the Theiler’s virus model mirror those in multiple sclerosis. Cytokine 2015, 76, 577–580. [Google Scholar] [CrossRef]
- Casals, J. Immunological Characterization of Vilyuisk Human Encephalomyelitis Virus. Nature 1963, 200, 339–341. [Google Scholar] [CrossRef]
- Drappier, M.; Opperdoes, F.R.; Michiels, T. Nonstructural Protein L* Species Specificity Supports a Mouse Origin for Vilyuisk Human Encephalitis Virus. J. Virol. 2017, 91, e00573-17. [Google Scholar] [CrossRef] [PubMed]
- Lipton, H.L.; Friedmann, A.; Sethi, P.; Crowther, J.R. Characterization of Vilyuisk virus as a picornavirus. J. Med. Virol. 1983, 12, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, A.E.; Strom, T.; Lipton, H.L. Nucleotide sequence identifies Vilyuisk virus as a divergent Theiler’s virus. Virology 1992, 191, 469–472. [Google Scholar] [CrossRef]
- Gudi, V.; Gai, L.; Herder, V.; Tejedor, L.S.; Kipp, M.; Amor, S.; Suhs, K.W.; Hansmann, F.; Beineke, A.; Baumgärtner, W.; et al. Synaptophysin Is a Reliable Marker for Axonal Damage. J. Neuropathol. Exp. Neurol. 2017, 76, 109–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haist, V.; Ulrich, R.; Kalkuhl, A.; Deschl, U.; Baumgärtner, W. Distinct spatio-temporal extracellular matrix accumulation within demyelinated spinal cord lesions in Theiler’s murine encephalomyelitis. Brain Pathol. 2012, 22, 188–204. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, F.; Herder, V.; Kalkuhl, A.; Haist, V.; Zhang, N.; Schaudien, D.; Deschl, U.; Baumgärtner, W.; Ulrich, R. Matrix metalloproteinase-12 deficiency ameliorates the clinical course and demyelination in Theiler’s murine encephalomyelitis. Acta Neuropathol. 2012, 124, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, R.; Seeliger, F.; Kreutzer, M.; Germann, P.G.; Baumgärtner, W. Limited remyelination in Theiler’s murine encephalomyelitis due to insufficient oligodendroglial differentiation of nerve/glial antigen 2 (NG2)-positive putative oligodendroglial progenitor cells. Neuropathol. Appl. Neurobiol. 2008, 34, 603–620. [Google Scholar] [CrossRef]
- Mack, C.L.; Vanderlugt-Castaneda, C.L.; Neville, K.L.; Miller, S.D. Microglia are activated to become competent antigen presenting and effector cells in the inflammatory environment of the Theiler’s virus model of multiple sclerosis. J. Neuroimmunol. 2003, 144, 68–79. [Google Scholar] [CrossRef]
- Tsunoda, I.; Kuang, L.Q.; Libbey, J.E.; Fujinami, R.S. Axonal injury heralds virus-induced demyelination. Am. J. Pathol. 2003, 162, 1259–1269. [Google Scholar] [CrossRef]
- Dal Canto, M.C.; Calenoff, M.A.; Miller, S.D.; Vanderlugt, C.L. Lymphocytes from mice chronically infected with Theiler’s murine encephalomyelitis virus produce demyelination of organotypic cultures after stimulation with the major encephalitogenic epitope of myelin proteolipid protein. Epitope spreading in TMEV infection has functional activity. J. Neuroimmunol. 2000, 104, 79–84. [Google Scholar]
- Katz-Levy, Y.; Neville, K.L.; Padilla, J.; Rahbe, S.; Begolka, W.S.; Girvin, A.M.; Olson, J.K.; Vanderlugt, C.L.; Miller, S.D. Temporal development of autoreactive Th1 responses and endogenous presentation of self myelin epitopes by central nervous system-resident APCs in Theiler’s virus-infected mice. J. Immunol. 2000, 165, 5304–5314. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.D.; Vanderlugt, C.L.; Begolka, W.S.; Pao, W.; Yauch, R.L.; Neville, K.L.; Katz-Levy, Y.; Carrizosa, A.; Kim, B.S. Persistent infection with Theiler’s virus leads to CNS autoimmunity via epitope spreading. Nat. Med. 1997, 3, 1133–1136. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.K.; Miller, S.D. The Role of T Cells and the Innate Immune System in the Pathogenesis of Theiler’s Virus Demyeliating Disease. In Experimental Models of Multiple Sclerosis; Lavi, E., Constantinescu, C.S., Eds.; Springer US: New York, NY, USA, 2005; pp. 645–657. [Google Scholar]
- Croxford, J.L.; Olson, J.K.; Anger, H.A.; Miller, S.D. Initiation and exacerbation of autoimmune demyelination of the central nervous system via virus-induced molecular mimicry: Implications for the pathogenesis of multiple sclerosis. J. Virol. 2005, 79, 8581–8590. [Google Scholar] [CrossRef] [PubMed]
- Raddatz, B.B.; Sun, W.; Brogden, G.; Sun, Y.; Kammeyer, P.; Kalkuhl, A.; Colbatzky, F.; Deschl, U.; Naim, H.Y.; Baumgärtner, W.; et al. Central Nervous System Demyelination and Remyelination is Independent from Systemic Cholesterol Level in Theiler’s Murine Encephalomyelitis. Brain Pathol. 2016, 26, 102–119. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, R.; Kalkuhl, A.; Deschl, U.; Baumgärtner, W. Machine learning approach identifies new pathways associated with demyelination in a viral model of multiple sclerosis. J. Cell. Mol. Med. 2010, 14, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Trevick, S. The Epidemiology of Global Epilepsy. Neurol. Clin. 2016, 34, 837–847. [Google Scholar] [CrossRef] [PubMed]
- Laxer, K.D.; Trinka, E.; Hirsch, L.J.; Cendes, F.; Langfitt, J.; Delanty, N.; Resnick, T.; Benbadis, S.R. The consequences of refractory epilepsy and its treatment. Epilepsy Behav. 2014, 37, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Vezzani, A.; Fujinami, R.S.; White, H.S.; Preux, P.M.; Blumcke, I.; Sander, J.W.; Loscher, W. Infections, inflammation and epilepsy. Acta Neuropathol. 2016, 131, 211–234. [Google Scholar] [CrossRef]
- Bonello, M.; Michael, B.D.; Solomon, T. Infective Causes of Epilepsy. Semin. Neurol. 2015, 35, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Bale, J.F., Jr. Virus and Immune-Mediated Encephalitides: Epidemiology, Diagnosis, Treatment, and Prevention. Pediatr. Neurol. 2015, 53, 3–12. [Google Scholar] [CrossRef]
- Libbey, J.E.; Fujinami, R.S. Neurotropic viral infections leading to epilepsy: Focus on Theiler’s murine encephalomyelitis virus. Future Virol. 2011, 6, 1339–1350. [Google Scholar] [CrossRef] [PubMed]
- DePaula-Silva, A.B.; Hanak, T.J.; Libbey, J.E.; Fujinami, R.S. Theiler’s murine encephalomyelitis virus infection of SJL/J and C57BL/6J mice: Models for multiple sclerosis and epilepsy. J. Neuroimmunol. 2017, 308, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Smeal, R.M.; Fujinami, R.; White, H.S.; Wilcox, K.S. Decrease in CA3 inhibitory network activity during Theiler’s virus encephalitis. Neurosci. Lett. 2015, 609, 210–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libbey, J.E.; Kennett, N.J.; Wilcox, K.S.; White, H.S.; Fujinami, R.S. Lack of correlation of central nervous system inflammation and neuropathology with the development of seizures following acute virus infection. J. Virol. 2011, 85, 8149–8157. [Google Scholar] [CrossRef] [PubMed]
- Bhuyan, P.; Patel, D.C.; Wilcox, K.S.; Patel, M. Oxidative stress in murine Theiler’s virus-induced temporal lobe epilepsy. Exp. Neurol. 2015, 271, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Kirkman, N.J.; Libbey, J.E.; Wilcox, K.S.; White, H.S.; Fujinami, R.S. Innate but not adaptive immune responses contribute to behavioral seizures following viral infection. Epilepsia 2010, 51, 454–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loewen, J.L.; Barker-Haliski, M.L.; Dahle, E.J.; White, H.S.; Wilcox, K.S. Neuronal Injury, Gliosis, and Glial Proliferation in Two Models of Temporal Lobe Epilepsy. J. Neuropathol. Exp. Neurol. 2016, 75, 366–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Sufiani, F.; Ang, L.C. Neuropathology of temporal lobe epilepsy. Epilepsy Res. Treat. 2012, 2012, 624519. [Google Scholar] [CrossRef]
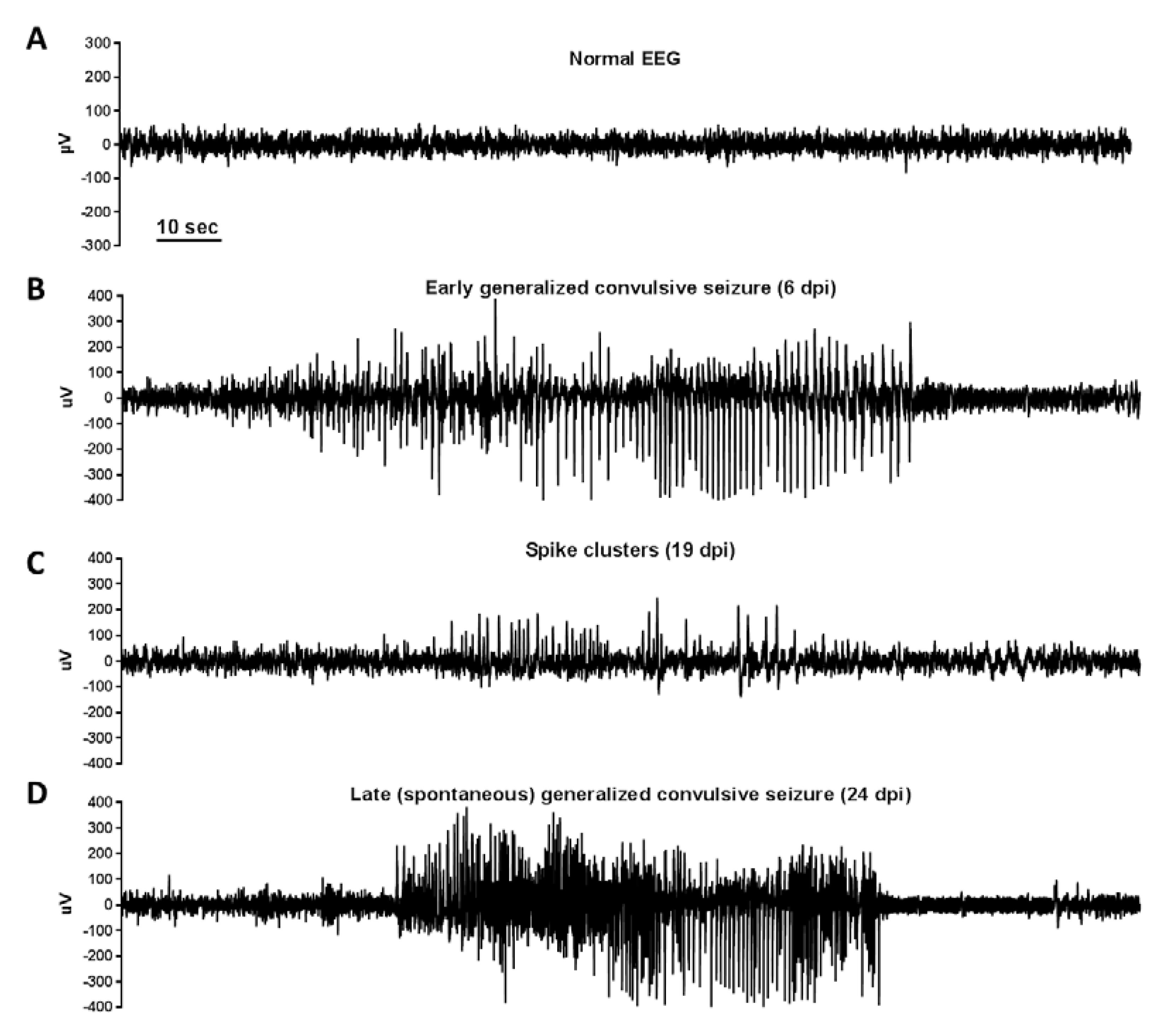
- Anjum, S.M.M.; Käufer, C.; Hopfengärtner, R.; Waltl, I.; Broer, S.; Löscher, W. Automated quantification of EEG spikes and spike clusters as a new read out in Theiler’s virus mouse model of encephalitis-induced epilepsy. Epilepsy Behav. 2018, 88, 189–204. [Google Scholar] [CrossRef]
- Umpierre, A.D.; Remigio, G.J.; Dahle, E.J.; Bradford, K.; Alex, A.B.; Smith, M.D.; West, P.J.; White, H.S.; Wilcox, K.S. Impaired cognitive ability and anxiety-like behavior following acute seizures in the Theiler’s virus model of temporal lobe epilepsy. Neurobiol. Dis. 2014, 64, 98–106. [Google Scholar] [CrossRef]
- Barker-Haliski, M.L.; Heck, T.D.; Dahle, E.J.; Vanegas, F.; Pruess, T.H.; Wilcox, K.S.; White, H.S. Acute treatment with minocycline, but not valproic acid, improves long-term behavioral outcomes in the Theiler’s virus model of temporal lobe epilepsy. Epilepsia 2016, 57, 1958–1967. [Google Scholar] [CrossRef] [PubMed]
- Omura, S.; Kawai, E.; Sato, F.; Martinez, N.E.; Chaitanya, G.V.; Rollyson, P.A.; Cvek, U.; Trutschl, M.; Alexander, J.S.; Tsunoda, I. Bioinformatics multivariate analysis determined a set of phase-specific biomarker candidates in a novel mouse model for viral myocarditis. Circ. Cardiovasc. Genet. 2014, 7, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Omura, S.; Kawai, E.; Sato, F.; Martinez, N.E.; Minagar, A.; Al-Kofahi, M.; Yun, J.W.; Cvek, U.; Trutschl, M.; Alexander, J.S.; et al. Theiler’s Virus-Mediated Immunopathology in the CNS and Heart: Roles of Organ-Specific Cytokine and Lymphatic Responses. Front. Immunol. 2018, 9, 2870. [Google Scholar] [CrossRef] [PubMed]
- Mi, W.; Young, C.R.; Storts, R.W.; Steelman, A.J.; Meagher, M.W.; Welsh, C.J. Restraint stress facilitates systemic dissemination of Theiler’s virus and alters its pathogenecity. Microb. Pathog. 2006, 41, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.R.; Prentice, T.W.; Bridegam, P.; Young, C.R.; Steelman, A.J.; Welsh, T.H.; Welsh, C.J.; Meagher, M.W. Social stress alters the severity and onset of the chronic phase of Theiler’s virus infection. J. Neuroimmunol. 2006, 175, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Takizawa, S.; Kaneyama, T.; Tsugane, S.; Takeichi, N.; Yanagisawa, S.; Ichikawa, M.; Yagita, H.; Kim, B.S.; Koh, C.S. Role of the Programmed Death-1 (PD-1) pathway in regulation of Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J. Neuroimmunol. 2014, 274, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Olson, J.K. Effect of the innate immune response on development of Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J. Neurovirol. 2014, 20, 427–436. [Google Scholar] [CrossRef]
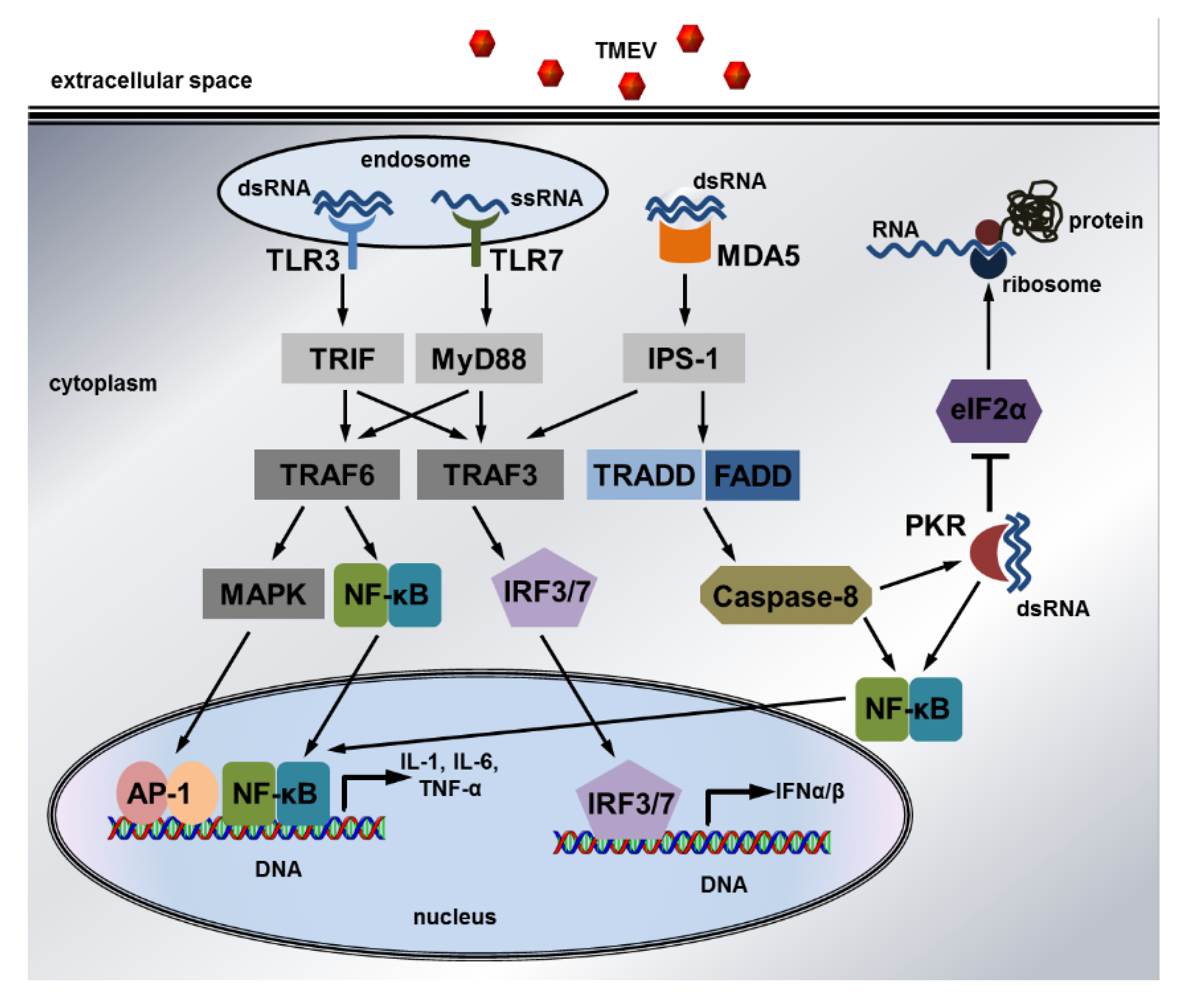
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef]
- Schulz, O.; Pichlmair, A.; Rehwinkel, J.; Rogers, N.C.; Scheuner, D.; Kato, H.; Takeuchi, O.; Akira, S.; Kaufman, R.J.; Reis e Sousa, C. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe 2010, 7, 354–361. [Google Scholar] [CrossRef]
- Satoh, J.; Paty, D.W.; Kim, S.U. Differential effects of beta and gamma interferons on expression of major histocompatibility complex antigens and intercellular adhesion molecule-1 in cultured fetal human astrocytes. Neurology 1995, 45, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.B.; Salazar-Mather, T.P.; Dalod, M.Y.; Van Deusen, J.B.; Wei, X.Q.; Liew, F.Y.; Caligiuri, M.A.; Durbin, J.E.; Biron, C.A. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J. Immunol. 2002, 169, 4279–4287. [Google Scholar] [CrossRef]
- Kolumam, G.A.; Thomas, S.; Thompson, L.J.; Sprent, J.; Murali-Krishna, K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med. 2005, 202, 637–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havenar-Daughton, C.; Kolumam, G.A.; Murali-Krishna, K. Cutting Edge: The direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J. Immunol. 2006, 176, 3315–3319. [Google Scholar] [CrossRef] [PubMed]
- Montoya, M.; Schiavoni, G.; Mattei, F.; Gresser, I.; Belardelli, F.; Borrow, P.; Tough, D.F. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood 2002, 99, 3263–3271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bon, A.; Schiavoni, G.; D’Agostino, G.; Gresser, I.; Belardelli, F.; Tough, D.F. Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 2001, 14, 461–470. [Google Scholar] [CrossRef]
- Stone, L.A.; Frank, J.A.; Albert, P.S.; Bash, C.; Smith, M.E.; Maloni, H.; McFarland, H.F. The effect of interferon-beta on blood-brain barrier disruptions demonstrated by contrast-enhanced magnetic resonance imaging in relapsing-remitting multiple sclerosis. Ann. Neurol. 1995, 37, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Rudick, R.A.; Ransohoff, R.M.; Peppler, R.; VanderBrug Medendorp, S.; Lehmann, P.; Alam, J. Interferon beta induces interleukin-10 expression: Relevance to multiple sclerosis. Ann. Neurol. 1996, 40, 618–627. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.H.; Hou, W.; Kim, S.J.; Fuller, A.C.; Kang, B.; Goings, G.; Miller, S.D.; Kim, B.S. Type I interferon signals control Theiler’s virus infection site, cellular infiltration and T cell stimulation in the CNS. J. Neuroimmunol. 2010, 226, 27–37. [Google Scholar] [CrossRef]
- Kreit, M.; Vertommen, D.; Gillet, L.; Michiels, T. The Interferon-Inducible Mouse Apolipoprotein L9 and Prohibitins Cooperate to Restrict Theiler’s Virus Replication. PLoS ONE 2015, 10, e0133190. [Google Scholar] [CrossRef]
- Delhaye, S.; van Pesch, V.; Michiels, T. The leader protein of Theiler’s virus interferes with nucleocytoplasmic trafficking of cellular proteins. J. Virol. 2004, 78, 4357–4362. [Google Scholar] [CrossRef] [PubMed]
- van Pesch, V.; van Eyll, O.; Michiels, T. The leader protein of Theiler’s virus inhibits immediate-early alpha/beta interferon production. J. Virol. 2001, 75, 7811–7817. [Google Scholar] [CrossRef] [PubMed]
- Ciomperlik, J.J.; Basta, H.A.; Palmenberg, A.C. Three cardiovirus Leader proteins equivalently inhibit four different nucleocytoplasmic trafficking pathways. Virology 2015, 484, 194–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciomperlik, J.J.; Basta, H.A.; Palmenberg, A.C. Cardiovirus Leader proteins bind exportins: Implications for virus replication and nucleocytoplasmic trafficking inhibition. Virology 2016, 487, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drappier, M.; Jha, B.K.; Stone, S.; Elliott, R.; Zhang, R.; Vertommen, D.; Weiss, S.R.; Silverman, R.H.; Michiels, T. A novel mechanism of RNase L inhibition: Theiler’s virus L* protein prevents 2-5A from binding to RNase, L. PLoS Pathog. 2018, 14, e1006989. [Google Scholar] [CrossRef] [PubMed]
- Drappier, M.; Michiels, T. Inhibition of the OAS/RNase L pathway by viruses. Curr. Opin. Virol. 2015, 15, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Bowen, J.L.; Olson, J.K. IFNγ influences type I interferon response and susceptibility to Theiler’s virus-induced demyelinating disease. Viral Immunol. 2013, 26, 223–238. [Google Scholar] [CrossRef]
- Taki, S.; Sato, T.; Ogasawara, K.; Fukuda, T.; Sato, M.; Hida, S.; Suzuki, G.; Mitsuyama, M.; Shin, E.H.; Kojima, S.; et al. Multistage regulation of Th1-type immune responses by the transcription factor IRF-1. Immunity 1997, 6, 673–679. [Google Scholar] [CrossRef]
- Kimura, T.; Nakayama, K.; Penninger, J.; Kitagawa, M.; Harada, H.; Matsuyama, T.; Tanaka, N.; Kamijo, R.; Vilcek, J.; Mak, T.W.; et al. Involvement of the IRF-1 transcription factor in antiviral responses to interferons. Science 1994, 264, 1921–1924. [Google Scholar] [CrossRef]
- Fiette, L.; Aubert, C.; Müller, U.; Huang, S.; Aguet, M.; Brahic, M.; Bureau, J.F. Theiler’s virus infection of 129Sv mice that lack the interferon alpha/beta or interferon gamma receptors. J. Exp. Med. 1995, 181, 2069–2076. [Google Scholar] [CrossRef]
- Gerhauser, I.; Ulrich, R.; Alldinger, S.; Baumgärtner, W. Induction of activator protein-1 and nuclear factor-kappaB as a prerequisite for disease development in susceptible SJL/J mice after theiler murine encephalomyelitis. J. Neuropathol. Exp. Neurol. 2007, 66, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Jin, Y.H.; Kim, B.S. Prostaglandin E2 produced following infection with Theiler’s virus promotes the pathogenesis of demyelinating disease. PLoS ONE 2017, 12, e0176406. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ulrich, R.; Baumgärtner, W.; Gerhauser, I. Interferon-stimulated genes-essential antiviral effectors implicated in resistance to Theiler’s virus-induced demyelinating disease. J. Neuroinflamm. 2015, 12, 242. [Google Scholar] [CrossRef] [PubMed]
- Turrin, N.P. Central nervous system Toll-like receptor expression in response to Theiler’s murine encephalomyelitis virus-induced demyelination disease in resistant and susceptible mouse strains. Virol. J. 2008, 5, 154. [Google Scholar] [CrossRef]
- Chastain, E.M.; Getts, D.R.; Miller, S.D. Deficient Natural Killer Dendritic Cell Responses Underlay the Induction of Theiler’s Virus-Induced Autoimmunity. mBio 2015, 6, e01175. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; So, E.Y.; Kim, B.S. Role of dendritic cells in differential susceptibility to viral demyelinating disease. PLoS Pathog. 2007, 3, e124. [Google Scholar] [CrossRef] [PubMed]
- Bowen, J.L.; Olson, J.K. Innate immune CD11b + Gr-1+ cells, suppressor cells, affect the immune response during Theiler’s virus-induced demyelinating disease. J. Immunol. 2009, 183, 6971–6980. [Google Scholar] [CrossRef]
- Zhu, B.; Bando, Y.; Xiao, S.; Yang, K.; Anderson, A.C.; Kuchroo, V.K.; Khoury, S.J. CD11b + Ly-6C(hi) suppressive monocytes in experimental autoimmune encephalomyelitis. J. Immunol. 2007, 179, 5228–5237. [Google Scholar] [CrossRef]
- Olson, J.K.; Miller, S.D. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J. Immunol. 2004, 173, 3916–3924. [Google Scholar] [CrossRef]
- Thompson, A.J.; Locarnini, S.A. Toll-like receptors, RIG-I-like RNA helicases and the antiviral innate immune response. Immunol. Cell Biol. 2007, 85, 435–445. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.H.; Kim, S.J.; So, E.Y.; Meng, L.; Colonna, M.; Kim, B.S. Melanoma differentiation-associated gene 5 is critical for protection against Theiler’s virus-induced demyelinating disease. J. Virol. 2012, 86, 1531–1543. [Google Scholar] [CrossRef] [PubMed]
- Son, K.N.; Liang, Z.; Lipton, H.L. SJL bone marrow-derived macrophages do not have IRF3 mutations and are highly susceptible to Theiler’s virus infection. Virology 2017, 512, 21–24. [Google Scholar] [CrossRef]
- Moore, T.C.; Cody, L.; Kumm, P.M.; Brown, D.M.; Petro, T.M. IRF3 helps control acute TMEV infection through IL-6 expression but contributes to acute hippocampus damage following TMEV infection. Virus Res. 2013, 178, 226–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, T.C.; Vogel, A.J.; Petro, T.M.; Brown, D.M. IRF3 deficiency impacts granzyme B expression and maintenance of memory T cell function in response to viral infection. Microbes Infect. 2015, 17, 426–439. [Google Scholar] [CrossRef] [Green Version]
- Moore, T.C.; Al-Salleeh, F.M.; Brown, D.M.; Petro, T.M. IRF3 polymorphisms induce different innate anti-Theiler’s virus immune responses in RAW264.7 macrophages. Virology 2011, 418, 40–48. [Google Scholar] [CrossRef]
- Dahlberg, A.; Auble, M.R.; Petro, T.M. Reduced expression of IL-12 p35 by SJL/J macrophages responding to Theiler’s virus infection is associated with constitutive activation of IRF-3. Virology 2006, 353, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Al-Salleeh, F.; Petro, T.M. Promoter analysis reveals critical roles for SMAD-3 and ATF-2 in expression of IL-23 p19 in macrophages. J. Immunol. 2008, 181, 4523–4533. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Jin, Y.H.; Kang, H.S.; Kim, B.S. Interleukin-6 (IL-6) and IL-17 synergistically promote viral persistence by inhibiting cellular apoptosis and cytotoxic T cell function. J. Virol. 2014, 88, 8479–8489. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Kang, H.S.; Kim, B.S. Th17 cells enhance viral persistence and inhibit T cell cytotoxicity in a model of chronic virus infection. J. Exp. Med. 2009, 206, 313–328. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.H.; Kang, H.S.; Hou, W.; Meng, L.; Kim, B.S. The level of viral infection of antigen-presenting cells correlates with the level of development of Theiler’s murine encephalomyelitis virus-induced demyelinating disease. J. Virol. 2015, 89, 1867–1878. [Google Scholar] [CrossRef]
- Carpentier, P.A.; Begolka, W.S.; Olson, J.K.; Elhofy, A.; Karpus, W.J.; Miller, S.D. Differential activation of astrocytes by innate and adaptive immune stimuli. Glia 2005, 49, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Pfefferkorn, C.; Kallfass, C.; Lienenklaus, S.; Spanier, J.; Kalinke, U.; Rieder, M.; Conzelmann, K.K.; Michiels, T.; Staeheli, P. Abortively Infected Astrocytes Appear to Represent the Main Source of Interferon Beta in the Virus-Infected Brain. J. Virol. 2016, 90, 2031–2038. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, P.A.; Getts, M.T.; Miller, S.D. Pro-inflammatory functions of astrocytes correlate with viral clearance and strain-dependent protection from TMEV-induced demyelinating disease. Virology 2008, 375, 24–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubio, N.; Sanz-Rodriguez, F.; Lipton, H.L. Theiler’s virus induces the MIP-2 chemokine (CXCL2) in astrocytes from genetically susceptible but not from resistant mouse strains. Cell. Immunol. 2006, 239, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Rubio, N.; Sanz-Rodriguez, F. Induction of the CXCL1 (KC) chemokine in mouse astrocytes by infection with the murine encephalomyelitis virus of Theiler. Virology 2007, 358, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, M.H.; Jin, Y.H.; Kim, B.S. Effects of Keratinocyte-Derived Cytokine (CXCL-1) on the Development of Theiler’s Virus-Induced Demyelinating Disease. Front. Cell. Infect. Microbiol. 2018, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- So, E.Y.; Kang, M.H.; Kim, B.S. Induction of chemokine and cytokine genes in astrocytes following infection with Theiler’s murine encephalomyelitis virus is mediated by the Toll-like receptor 3. Glia 2006, 53, 858–867. [Google Scholar] [CrossRef]
- So, E.Y.; Kim, B.S. Theiler’s virus infection induces TLR3-dependent upregulation of TLR2 critical for proinflammatory cytokine production. Glia 2009, 57, 1216–1226. [Google Scholar] [CrossRef]
- Carpentier, P.A.; Williams, B.R.; Miller, S.D. Distinct roles of protein kinase R and toll-like receptor 3 in the activation of astrocytes by viral stimuli. Glia 2007, 55, 239–252. [Google Scholar] [CrossRef]
- Uhde, A.K.; Ciurkiewicz, M.; Herder, V.; Khan, M.A.; Hensel, N.; Claus, P.; Beckstette, M.; Teich, R.; Floess, S.; Baumgärtner, W.; et al. Intact interleukin-10 receptor signaling protects from hippocampal damage elicited by experimental neurotropic virus infection of SJL mice. Sci. Rep. 2018, 8, 6106. [Google Scholar] [CrossRef]
- Uhde, A.K.; Herder, V.; Akram Khan, M.; Ciurkiewicz, M.; Schaudien, D.; Teich, R.; Floess, S.; Baumgärtner, W.; Huehn, J.; Beineke, A. Viral Infection of the Central Nervous System Exacerbates Interleukin-10 Receptor Deficiency-Mediated Colitis in SJL Mice. PLoS ONE 2016, 11, e0161883. [Google Scholar] [CrossRef] [PubMed]
- Herder, V.; Gerhauser, I.; Klein, S.K.; Almeida, P.; Kummerfeld, M.; Ulrich, R.; Seehusen, F.; Rohn, K.; Schaudien, D.; Baumgärtner, W.; et al. Interleukin-10 expression during the acute phase is a putative prerequisite for delayed viral elimination in a murine model for multiple sclerosis. J. Neuroimmunol. 2012, 249, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; McCright, I.J.; Kuang, L.Q.; Zurbriggen, A.; Fujinami, R.S. Hydrocephalus in mice infected with a Theiler’s murine encephalomyelitis virus variant. J. Neuropathol. Exp. Neurol. 1997, 56, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Remigio, G.J.; Loewen, J.L.; Heuston, S.; Helgeson, C.; White, H.S.; Wilcox, K.S.; West, P.J. Corneal kindled C57BL/6 mice exhibit saturated dentate gyrus long-term potentiation and associated memory deficits in the absence of overt neuron loss. Neurobiol. Dis. 2017, 105, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochim. Biophys. Acta 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Sheng, W.S.; Ehrlich, L.C.; Peterson, P.K.; Chao, C.C. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation 2000, 7, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Umpierre, A.D.; West, P.J.; White, J.A.; Wilcox, K.S. Conditional Knockout of mGluR5 from Astrocytes during Epilepsy Development Impairs High-Frequency Glutamate Uptake. J. Neurosci. 2018. [Google Scholar] [CrossRef]
- Garcia-Oscos, F.; Salgado, H.; Hall, S.; Thomas, F.; Farmer, G.E.; Bermeo, J.; Galindo, L.C.; Ramirez, R.D.; D’Mello, S.; Rose-John, S.; et al. The stress-induced cytokine interleukin-6 decreases the inhibition/excitation ratio in the rat temporal cortex via trans-signaling. Biol. Psychiatry 2012, 71, 574–582. [Google Scholar] [CrossRef]
- Campbell, I.L.; Abraham, C.R.; Masliah, E.; Kemper, P.; Inglis, J.D.; Oldstone, M.B.; Mucke, L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 10061–10065. [Google Scholar] [CrossRef]
- Heyser, C.J.; Masliah, E.; Samimi, A.; Campbell, I.L.; Gold, L.H. Progressive decline in avoidance learning paralleled by inflammatory neurodegeneration in transgenic mice expressing interleukin 6 in the brain. Proc. Natl. Acad. Sci. USA 1997, 94, 1500–1505. [Google Scholar] [CrossRef] [Green Version]
- Samland, H.; Huitron-Resendiz, S.; Masliah, E.; Criado, J.; Henriksen, S.J.; Campbell, I.L. Profound increase in sensitivity to glutamatergic- but not cholinergic agonist-induced seizures in transgenic mice with astrocyte production of IL-6. J. Neurosci. Res. 2003, 73, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Costello, D.A.; Lynch, M.A. Toll-like receptor 3 activation modulates hippocampal network excitability, via glial production of interferon-beta. Hippocampus 2013, 23, 696–707. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.C.; Wallis, G.; Dahle, E.J.; McElroy, P.B.; Thomson, K.E.; Tesi, R.J.; Szymkowski, D.E.; West, P.J.; Smeal, R.M.; Patel, M.; et al. Hippocampal TNFalpha Signaling Contributes to Seizure Generation in an Infection-Induced Mouse Model of Limbic Epilepsy. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Cusick, M.F.; Libbey, J.E.; Doty, D.J.; DePaula-Silva, A.B.; Fujinami, R.S. The role of peripheral interleukin-6 in the development of acute seizures following virus encephalitis. J. Neurovirol. 2017, 23, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Libbey, J.E.; Kirkman, N.J.; Wilcox, K.S.; White, H.S.; Fujinami, R.S. Role for complement in the development of seizures following acute viral infection. J. Virol. 2010, 84, 6452–6460. [Google Scholar] [CrossRef] [PubMed]
- Libbey, J.E.; Cusick, M.F.; Doty, D.J.; Fujinami, R.S. Complement Components Are Expressed by Infiltrating Macrophages/Activated Microglia Early Following Viral Infection. Viral Immunol. 2017, 30, 304–314. [Google Scholar] [CrossRef]
- DePaula-Silva, A.B.; Sonderegger, F.L.; Libbey, J.E.; Doty, D.J.; Fujinami, R.S. The immune response to picornavirus infection and the effect of immune manipulation on acute seizures. J. Neurovirol. 2018, 24, 464–477. [Google Scholar] [CrossRef]
- Cusick, M.F.; Libbey, J.E.; Patel, D.C.; Doty, D.J.; Fujinami, R.S. Infiltrating macrophages are key to the development of seizures following virus infection. J. Virol. 2013, 87, 1849–1860. [Google Scholar] [CrossRef]
- Waltl, I.; Käufer, C.; Gerhauser, I.; Chhatbar, C.; Ghita, L.; Kalinke, U.; Löscher, W. Microglia have a protective role in viral encephalitis-induced seizure development and hippocampal damage. Brain Behav. Immun. 2018, 74, 186–204. [Google Scholar] [CrossRef]
- Waltl, I.; Käufer, C.; Broer, S.; Chhatbar, C.; Ghita, L.; Gerhauser, I.; Anjum, M.; Kalinke, U.; Löscher, W. Macrophage depletion by liposome-encapsulated clodronate suppresses seizures but not hippocampal damage after acute viral encephalitis. Neurobiol. Dis. 2018, 110, 192–205. [Google Scholar] [CrossRef]
- Howe, C.L.; LaFrance-Corey, R.G.; Goddery, E.N.; Johnson, R.K.; Mirchia, K. Neuronal CCL2 expression drives inflammatory monocyte infiltration into the brain during acute virus infection. J. Neuroinflamm. 2017, 14, 238. [Google Scholar] [CrossRef] [PubMed]
- Käufer, C.; Chhatbar, C.; Bröer, S.; Waltl, I.; Ghita, L.; Gerhauser, I.; Kalinke, U.; Löscher, W. Chemokine receptors CCR2 and CX3CR1 regulate viral encephalitis-induced hippocampal damage but not seizures. Proc. Natl. Acad. Sci. USA 2018, 115, E8929–E8938. [Google Scholar] [CrossRef] [PubMed]
- Herder, V.; Iskandar, C.D.; Kegler, K.; Hansmann, F.; Elmarabet, S.A.; Khan, M.A.; Kalkuhl, A.; Deschl, U.; Baumgärtner, W.; Ulrich, R.; et al. Dynamic Changes of Microglia/Macrophage M1 and M2 Polarization in Theiler’s Murine Encephalomyelitis. Brain Pathol. 2015, 25, 712–723. [Google Scholar] [CrossRef] [PubMed]
- Schneider, K.M.; Watson, N.B.; Minchenberg, S.B.; Massa, P.T. The influence of macrophage growth factors on Theiler’s Murine Encephalomyelitis Virus (TMEV) infection and activation of macrophages. Cytokine 2018, 102, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Gilli, F.; Li, L.; Pachner, A.R. The immune response in the CNS in Theiler’s virus induced demyelinating disease switches from an early adaptive response to a chronic innate-like response. J. Neurovirol. 2016, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Sato, F.; Omura, S.; Kawai, E.; Martinez, N.E.; Acharya, M.M.; Reddy, P.C.; Chaitanya, G.V.; Alexander, J.S.; Tsunoda, I. Distinct kinetics of viral replication, T cell infiltration, and fibrosis in three phases of myocarditis following Theiler’s virus infection. Cell. Immunol. 2014, 292, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feliu, A.; Moreno-Martet, M.; Mecha, M.; Carrillo-Salinas, F.J.; de Lago, E.; Fernandez-Ruiz, J.; Guaza, C. A Sativex((R)) -like combination of phytocannabinoids as a disease-modifying therapy in a viral model of multiple sclerosis. Br. J. Pharmacol. 2015, 172, 3579–3595. [Google Scholar] [CrossRef]
- Navarrete, C.; Carrillo-Salinas, F.; Palomares, B.; Mecha, M.; Jimenez-Jimenez, C.; Mestre, L.; Feliu, A.; Bellido, M.L.; Fiebich, B.L.; Appendino, G.; et al. Hypoxia mimetic activity of VCE-004.8, a cannabidiol quinone derivative: Implications for multiple sclerosis therapy. J. Neuroinflamm. 2018, 15, 64. [Google Scholar] [CrossRef]
- Morales, P.; Gomez-Canas, M.; Navarro, G.; Hurst, D.P.; Carrillo-Salinas, F.J.; Lagartera, L.; Pazos, R.; Goya, P.; Reggio, P.H.; Guaza, C.; et al. Chromenopyrazole, a Versatile Cannabinoid Scaffold with in Vivo Activity in a Model of Multiple Sclerosis. J. Med. Chem. 2016, 59, 6753–6771. [Google Scholar] [CrossRef] [Green Version]
- Feliu, A.; Bonilla Del Rio, I.; Carrillo-Salinas, F.J.; Hernandez-Torres, G.; Mestre, L.; Puente, N.; Ortega-Gutierrez, S.; Lopez-Rodriguez, M.L.; Grandes, P.; Mecha, M.; et al. 2-Arachidonoylglycerol Reduces Proteoglycans and Enhances Remyelination in a Progressive Model of Demyelination. J. Neurosci. 2017, 37, 8385–8398. [Google Scholar] [CrossRef]
- Mecha, M.; Feliu, A.; Machin, I.; Cordero, C.; Carrillo-Salinas, F.; Mestre, L.; Hernandez-Torres, G.; Ortega-Gutierrez, S.; Lopez-Rodriguez, M.L.; de Castro, F.; et al. 2-AG limits Theiler’s virus induced acute neuroinflammation by modulating microglia and promoting MDSCs. Glia 2018, 66, 1447–1463. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, N.; Ueno, H. Regulation of human helper T cell subset differentiation by cytokines. Curr. Opin. Immunol. 2015, 34, 130–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.D.; Pavelko, K.D.; Leibowitz, J.; Lin, X.; Rodriguez, M. CD4(+) and CD8(+) T cells make discrete contributions to demyelination and neurologic disease in a viral model of multiple sclerosis. J. Virol. 1998, 72, 7320–7329. [Google Scholar] [PubMed]
- Jin, Y.H.; Kang, B.; Kim, B.S. Theiler’s virus infection induces a predominant pathogenic CD4+ T cell response to RNA polymerase in susceptible SJL/J mice. J. Virol. 2009, 83, 10981–10992. [Google Scholar] [CrossRef] [PubMed]
- McMahon, E.J.; Bailey, S.L.; Castenada, C.V.; Waldner, H.; Miller, S.D. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat. Med. 2005, 11, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Fujinami, R.S. Neuropathogenesis of Theiler’s murine encephalomyelitis virus infection, an animal model for multiple sclerosis. J. Neuroimmune Pharmacol. 2010, 5, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Monteyne, P.; Bihl, F.; Levillayer, F.; Brahic, M.; Bureau, J.F. The Th1/Th2 balance does not account for the difference of susceptibility of mouse strains to Theiler’s virus persistent infection. J. Immunol. 1999, 162, 7330–7334. [Google Scholar] [PubMed]
- Jin, Y.H.; Kaneyama, T.; Kang, M.H.; Kang, H.S.; Koh, C.S.; Kim, B.S. TLR3 signaling is either protective or pathogenic for the development of Theiler’s virus-induced demyelinating disease depending on the time of viral infection. J. Neuroinflamm. 2011, 8, 178. [Google Scholar] [CrossRef]
- Richards, M.H.; Getts, M.T.; Podojil, J.R.; Jin, Y.H.; Kim, B.S.; Miller, S.D. Virus expanded regulatory T cells control disease severity in the Theiler’s virus mouse model of MS. J. Autoimmun. 2011, 36, 142–154. [Google Scholar] [CrossRef] [Green Version]
- Perlman, S.; Zhao, J. Roles of regulatory T cells and IL-10 in virus-induced demyelination. J. Neuroimmunol. 2017, 308, 6–11. [Google Scholar] [CrossRef]
- Martinez, N.E.; Karlsson, F.; Sato, F.; Kawai, E.; Omura, S.; Minagar, A.; Grisham, M.B.; Tsunoda, I. Protective and detrimental roles for regulatory T cells in a viral model for multiple sclerosis. Brain Pathol. 2014, 24, 436–451. [Google Scholar] [CrossRef] [PubMed]
- Vignali, D. How many mechanisms do regulatory T cells need? Eur. J. Immunol. 2008, 38, 908–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahl, K.; Loddenkemper, C.; Drouin, C.; Freyer, J.; Arnason, J.; Eberl, G.; Hamann, A.; Wagner, H.; Huehn, J.; Sparwasser, T. Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J. Exp. Med. 2007, 204, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Prajeeth, C.K.; Beineke, A.; Iskandar, C.D.; Gudi, V.; Herder, V.; Gerhauser, I.; Haist, V.; Teich, R.; Huehn, J.; Baumgärtner, W.; et al. Limited role of regulatory T cells during acute Theiler virus-induced encephalitis in resistant C57BL/6 mice. J. Neuroinflam. 2014, 11, 180. [Google Scholar] [CrossRef] [PubMed]
- Ciurkiewicz, M.; Herder, V.; Khan, M.A.; Uhde, A.K.; Teich, R.; Floess, S.; Baumgärtner, W.; Huehn, J.; Beineke, A. Cytotoxic CD8(+) T cell ablation enhances the capacity of regulatory T cells to delay viral elimination in Theiler’s murine encephalomyelitis. Brain Pathol. 2018, 28, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Levillayer, F.; Mas, M.; Levi-Acobas, F.; Brahic, M.; Bureau, J.F. Interleukin 22 is a candidate gene for Tmevp3, a locus controlling Theiler’s virus-induced neurological diseases. Genetics 2007, 176, 1835–1844. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.H.; Kang, H.S.; Mohindru, M.; Kim, B.S. Preferential induction of protective T cell responses to Theiler’s virus in resistant (C57BL/6 × SJL)F1 mice. J. Virol. 2011, 85, 3033–3040. [Google Scholar] [CrossRef] [PubMed]
- Martinez, N.E.; Sato, F.; Kawai, E.; Omura, S.; Takahashi, S.; Yoh, K.; Tsunoda, I. Th17-biased RORγt transgenic mice become susceptible to a viral model for multiple sclerosis. Brain Behav. Immun. 2015, 43, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Getts, M.T.; Kim, B.S.; Miller, S.D. Differential outcome of tolerance induction in naive versus activated Theiler’s virus epitope-specific CD8+ cytotoxic T cells. J. Virol. 2007, 81, 6584–6593. [Google Scholar] [CrossRef]
- Getts, M.T.; Richards, M.H.; Miller, S.D. A critical role for virus-specific CD8(+) CTLs in protection from Theiler’s virus-induced demyelination in disease-susceptible SJL mice. Virology 2010, 402, 102–111. [Google Scholar] [CrossRef]
- Miller, D.J.; Rivera-Quinones, C.; Njenga, M.K.; Leibowitz, J.; Rodriguez, M. Spontaneous CNS remyelination in beta 2 microglobulin-deficient mice following virus-induced demyelination. J. Neurosci. 1995, 15, 8345–8352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huseby Kelcher, A.M.; Atanga, P.A.; Gamez, J.D.; Cumba Garcia, L.M.; Teclaw, S.J.; Pavelko, K.D.; Macura, S.I.; Johnson, A.J. Brain atrophy in picornavirus-infected FVB mice is dependent on the H-2D(b) class I molecule. FASEB J. 2017, 31, 2267–2275. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Quinones, C.; McGavern, D.; Schmelzer, J.D.; Hunter, S.F.; Low, P.A.; Rodriguez, M. Absence of neurological deficits following extensive demyelination in a class I-deficient murine model of multiple sclerosis. Nat. Med. 1998, 4, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, M.; Dunkel, A.J.; Thiemann, R.L.; Leibowitz, J.; Zijlstra, M.; Jaenisch, R. Abrogation of resistance to Theiler’s virus-induced demyelination in H-2b mice deficient in beta 2-microglobulin. J. Immunol. 1993, 151, 266–276. [Google Scholar] [PubMed]
- Johnson, A.J.; Njenga, M.K.; Hansen, M.J.; Kuhns, S.T.; Chen, L.; Rodriguez, M.; Pease, L.R. Prevalent class I-restricted T-cell response to the Theiler’s virus epitope Db:VP2121-130 in the absence of endogenous CD4 help, tumor necrosis factor alpha, gamma interferon, perforin, or costimulation through CD28. J. Virol. 1999, 73, 3702–3708. [Google Scholar] [PubMed]
- Borson, N.D.; Paul, C.; Lin, X.; Nevala, W.K.; Strausbauch, M.A.; Rodriguez, M.; Wettstein, P.J. Brain-infiltrating cytolytic T lymphocytes specific for Theiler’s virus recognize H2Db molecules complexed with a viral VP2 peptide lacking a consensus anchor residue. J. Virol. 1997, 71, 5244–5250. [Google Scholar] [PubMed]
- Lyman, M.A.; Lee, H.G.; Kang, B.S.; Kang, H.K.; Kim, B.S. Capsid-specific cytotoxic T lymphocytes recognize three distinct H-2D(b)-restricted regions of the BeAn strain of Theiler’s virus and exhibit different cytokine profiles. J. Virol. 2002, 76, 3125–3134. [Google Scholar] [CrossRef]
- Lyman, M.A.; Myoung, J.; Mohindru, M.; Kim, B.S. Quantitative, not qualitative, differences in CD8(+) T cell responses to Theiler’s murine encephalomyelitis virus between resistant C57BL/6 and susceptible SJL/J mice. Eur. J. Immunol. 2004, 34, 2730–2739. [Google Scholar] [CrossRef]
- Pavelko, K.D.; Bell, M.P.; Harrington, S.M.; Dong, H. B7-H1 Influences the Accumulation of Virus-Specific Tissue Resident Memory T Cells in the Central Nervous System. Front. Immunol. 2017, 8, 1532. [Google Scholar] [CrossRef]
- McDole, J.R.; Danzer, S.C.; Pun, R.Y.; Chen, Y.; Johnson, H.L.; Pirko, I.; Johnson, A.J. Rapid formation of extended processes and engagement of Theiler’s virus-infected neurons by CNS-infiltrating CD8 T cells. Am. J. Pathol. 2010, 177, 1823–1833. [Google Scholar] [CrossRef]
- Pirko, I.; Chen, Y.; Lohrey, A.K.; McDole, J.; Gamez, J.D.; Allen, K.S.; Pavelko, K.D.; Lindquist, D.M.; Dunn, R.S.; Macura, S.I.; et al. Contrasting roles for CD4 vs. CD8 T-cells in a murine model of virally induced “T1 black hole” formation. PLoS ONE 2012, 7, e31459. [Google Scholar] [CrossRef] [PubMed]
- Willenbring, R.C.; Jin, F.; Hinton, D.J.; Hansen, M.; Choi, D.S.; Pavelko, K.D.; Johnson, A.J. Modulatory effects of perforin gene dosage on pathogen-associated blood-brain barrier (BBB) disruption. J. Neuroinflamm. 2016, 13, 222. [Google Scholar] [CrossRef] [PubMed]
- Johnson, H.L.; Chen, Y.; Suidan, G.L.; McDole, J.R.; Lohrey, A.K.; Hanson, L.M.; Jin, F.; Pirko, I.; Johnson, A.J. A hematopoietic contribution to microhemorrhage formation during antiviral CD8 T cell-initiated blood-brain barrier disruption. J. Neuroinflamm. 2012, 9, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, H.L.; Willenbring, R.C.; Jin, F.; Manhart, W.A.; LaFrance, S.J.; Pirko, I.; Johnson, A.J. Perforin competent CD8 T cells are sufficient to cause immune-mediated blood-brain barrier disruption. PLoS ONE 2014, 9, e111401. [Google Scholar] [CrossRef]
- Borrow, P.; Tonks, P.; Welsh, C.J.; Nash, A.A. The role of CD8+T cells in the acute and chronic phases of Theiler’s murine encephalomyelitis virus-induced disease in mice. J. Gen. Virol. 1992, 73 Pt 7, 1861–1865. [Google Scholar] [CrossRef]
- Rodriguez, M.; Lindsley, M.D.; Pierce, M.L. Role of T cells in resistance to Theiler’s virus infection. Microb. Pathog. 1991, 11, 269–281. [Google Scholar] [CrossRef]
- Mendez-Fernandez, Y.V.; Johnson, A.J.; Rodriguez, M.; Pease, L.R. Clearance of Theiler’s virus infection depends on the ability to generate a CD8+ T cell response against a single immunodominant viral peptide. Eur. J. Immunol. 2003, 33, 2501–2510. [Google Scholar] [CrossRef]
- Begolka, W.S.; Haynes, L.M.; Olson, J.K.; Padilla, J.; Neville, K.L.; Dal Canto, M.; Palma, J.; Kim, B.S.; Miller, S.D. CD8-deficient SJL mice display enhanced susceptibility to Theiler’s virus infection and increased demyelinating pathology. J. Neurovirol. 2001, 7, 409–420. [Google Scholar]
- Johnson, A.J.; Upshaw, J.; Pavelko, K.D.; Rodriguez, M.; Pease, L.R. Preservation of motor function by inhibition of CD8+ virus peptide-specific T cells in Theiler’s virus infection. FASEB J. 2001, 15, 2760–2762. [Google Scholar] [CrossRef]
- Howe, C.L.; Ure, D.; Adelson, J.D.; LaFrance-Corey, R.; Johnson, A.; Rodriguez, M. CD8+ T cells directed against a viral peptide contribute to loss of motor function by disrupting axonal transport in a viral model of fulminant demyelination. J. Neuroimmunol. 2007, 188, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Deb, C.; Lafrance-Corey, R.G.; Schmalstieg, W.F.; Sauer, B.M.; Wang, H.; German, C.L.; Windebank, A.J.; Rodriguez, M.; Howe, C.L. CD8+ T cells cause disability and axon loss in a mouse model of multiple sclerosis. PLoS ONE 2010, 5, e12478. [Google Scholar] [CrossRef] [PubMed]
- Deb, C.; Lafrance-Corey, R.G.; Zoecklein, L.; Papke, L.; Rodriguez, M.; Howe, C.L. Demyelinated axons and motor function are protected by genetic deletion of perforin in a mouse model of multiple sclerosis. J. Neuropathol. Exp. Neurol. 2009, 68, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Howe, C.L.; Adelson, J.D.; Rodriguez, M. Absence of perforin expression confers axonal protection despite demyelination. Neurobiol. Dis. 2007, 25, 354–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denic, A.; Wootla, B.; Zoecklein, L.; Rodriguez, M. Deletion of Virus-specific T-cells Enhances Remyelination in a Model of Multiple Sclerosis. J. Neurol. Transl. Neurosci. 2014, 2, 1032. [Google Scholar]
- Denic, A.; Pirko, I.; Wootla, B.; Bieber, A.; Macura, S.; Rodriguez, M. Deletion of beta-2-microglobulin ameliorates spinal cord lesion load and promotes recovery of brainstem NAA levels in a murine model of multiple sclerosis. Brain Pathol. 2012, 22, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Tsunoda, I.; Kuang, L.Q.; Fujinami, R.S. Induction of autoreactive CD8+ cytotoxic T cells during Theiler’s murine encephalomyelitis virus infection: Implications for autoimmunity. J. Virol. 2002, 76, 12834–12844. [Google Scholar] [CrossRef]
- Tsunoda, I.; Kuang, L.Q.; Kobayashi-Warren, M.; Fujinami, R.S. Central nervous system pathology caused by autoreactive CD8+ T-cell clones following virus infection. J. Virol. 2005, 79, 14640–14646. [Google Scholar] [CrossRef] [PubMed]
- Libbey, J.E.; Cusick, M.F.; Tsunoda, I.; Fujinami, R.S. Antiviral CD8(+) T cells cause an experimental autoimmune encephalomyelitis-like disease in naive mice. J. Neurovirol. 2012, 18, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Myoung, J.; Kang, H.S.; Hou, W.; Meng, L.; Dal Canto, M.C.; Kim, B.S. Epitope-specific CD8+ T cells play a differential pathogenic role in the development of a viral disease model for multiple sclerosis. J. Virol. 2012, 86, 13717–13728. [Google Scholar] [CrossRef] [PubMed]
- Myoung, J.; Bahk, Y.Y.; Kang, H.S.; Dal Canto, M.C.; Kim, B.S. Anticapsid immunity level, not viral persistence level, correlates with the progression of Theiler’s virus-induced demyelinating disease in viral P1-transgenic mice. J. Virol. 2008, 82, 5606–5617. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.S.; Lyman, M.A.; Kim, B.S. Differences in avidity and epitope recognition of CD8(+) T cells infiltrating the central nervous systems of SJL/J mice infected with BeAn and DA strains of Theiler’s murine encephalomyelitis virus. J. Virol. 2002, 76, 11780–11784. [Google Scholar] [CrossRef] [PubMed]
- Gilli, F.; Li, L.; Campbell, S.J.; Anthony, D.C.; Pachner, A.R. The effect of B-cell depletion in the Theiler’s model of multiple sclerosis. J. Neurol. Sci. 2015, 359, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Kang, B.S.; Palma, J.P.; Lyman, M.A.; Dal Canto, M.; Kim, B.S. Antibody response is required for protection from Theiler’s virus-induced encephalitis in C57BL/6 mice in the absence of CD8+ T cells. Virology 2005, 340, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Talloni, M.J.; Kalkuhl, A.; Deschl, U.; Ulrich, R.; Kummerfeld, M.; Rohn, K.; Baumgärtner, W.; Beineke, A. Transient peripheral immune response and central nervous system leaky compartmentalization in a viral model for multiple sclerosis. Brain Pathol. 2010, 20, 890–901. [Google Scholar] [CrossRef] [PubMed]
- Pachner, A.R.; Brady, J.; Narayan, K. Antibody-secreting cells in the central nervous system in an animal model of MS: Phenotype, association with disability, and in vitro production of antibody. J. Neuroimmunol. 2007, 190, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Pachner, A.R.; Li, L.; Lagunoff, D. Plasma cells in the central nervous system in the Theiler’s virus model of multiple sclerosis. J. Neuroimmunol. 2011, 232, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Brück, W.; Lucchinetti, C.F. The immunopathology of multiple sclerosis: An overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef]
- Oleszak, E.L.; Hoffman, B.E.; Chang, J.R.; Zaczynska, E.; Gaughan, J.; Katsetos, C.D.; Platsoucas, C.D.; Harvey, N. Apoptosis of infiltrating T cells in the central nervous system of mice infected with Theiler’s murine encephalomyelitis virus. Virology 2003, 315, 110–123. [Google Scholar] [CrossRef]
- Schlitt, B.P.; Felrice, M.; Jelachich, M.L.; Lipton, H.L. Apoptotic cells, including macrophages, are prominent in Theiler’s virus-induced inflammatory, demyelinating lesions. J. Virol. 2003, 77, 4383–4388. [Google Scholar] [CrossRef]
- Hansmann, F.; Jungwirth, N.; Zhang, N.; Skripuletz, T.; Stein, V.M.; Tipold, A.; Stangel, M.; Baumgärtner, W. Beneficial and detrimental impact of transplanted canine adipose-derived stem cells in a virus-induced demyelinating mouse model. Vet. Immunol. Immunopathol. 2018, 202, 130–140. [Google Scholar] [CrossRef]
- Fernandez, O.; Izquierdo, G.; Fernandez, V.; Leyva, L.; Reyes, V.; Guerrero, M.; Leon, A.; Arnaiz, C.; Navarro, G.; Paramo, M.D.; et al. Adipose-derived mesenchymal stem cells (AdMSC) for the treatment of secondary-progressive multiple sclerosis: A triple blinded, placebo controlled, randomized phase I/II safety and feasibility study. PLoS ONE 2018, 13, e0195891. [Google Scholar] [CrossRef] [PubMed]
- Bateman, M.E.; Strong, A.L.; Gimble, J.M.; Bunnell, B.A. Concise Review: Using Fat to Fight Disease: A Systematic Review of Nonhomologous Adipose-Derived Stromal/Stem Cell Therapies. Stem Cells 2018, 36, 1311–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanak, T.J.; Libbey, J.E.; Doty, D.J.; Sim, J.T.; DePaula-Silva, A.B.; Fujinami, R.S. Positive modulation of mGluR5 attenuates seizures and reduces TNF-alpha(+) macrophages and microglia in the brain in a murine model of virus-induced temporal lobe epilepsy. Exp. Neurol. 2019, 311, 194–204. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gerhauser, I.; Hansmann, F.; Ciurkiewicz, M.; Löscher, W.; Beineke, A. Facets of Theiler’s Murine Encephalomyelitis Virus-Induced Diseases: An Update. Int. J. Mol. Sci. 2019, 20, 448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020448
Gerhauser I, Hansmann F, Ciurkiewicz M, Löscher W, Beineke A. Facets of Theiler’s Murine Encephalomyelitis Virus-Induced Diseases: An Update. International Journal of Molecular Sciences. 2019; 20(2):448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020448
Chicago/Turabian StyleGerhauser, Ingo, Florian Hansmann, Malgorzata Ciurkiewicz, Wolfgang Löscher, and Andreas Beineke. 2019. "Facets of Theiler’s Murine Encephalomyelitis Virus-Induced Diseases: An Update" International Journal of Molecular Sciences 20, no. 2: 448. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020448