In Silico Insights towards the Identification of NLRP3 Druggable Hot Spots

 , , , , , ,
, , , , , ,
Abstract
:
1. Introduction
2. Results and Discussion
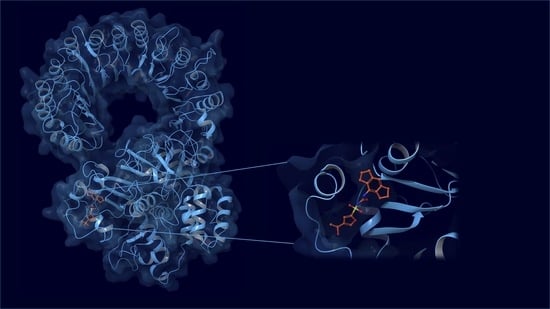
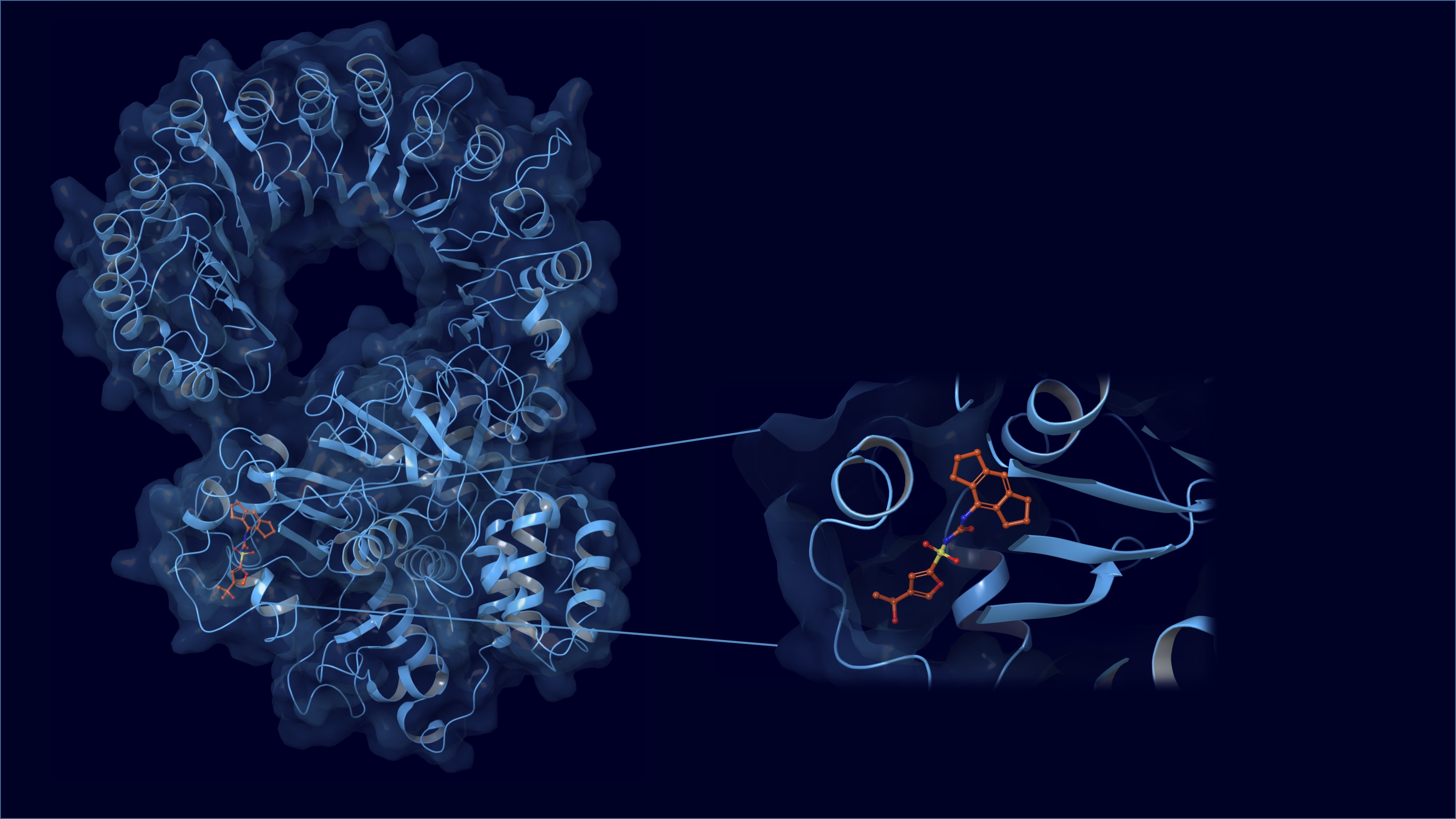
2.1. Homology Modeling
2.2. Binding Site Detection
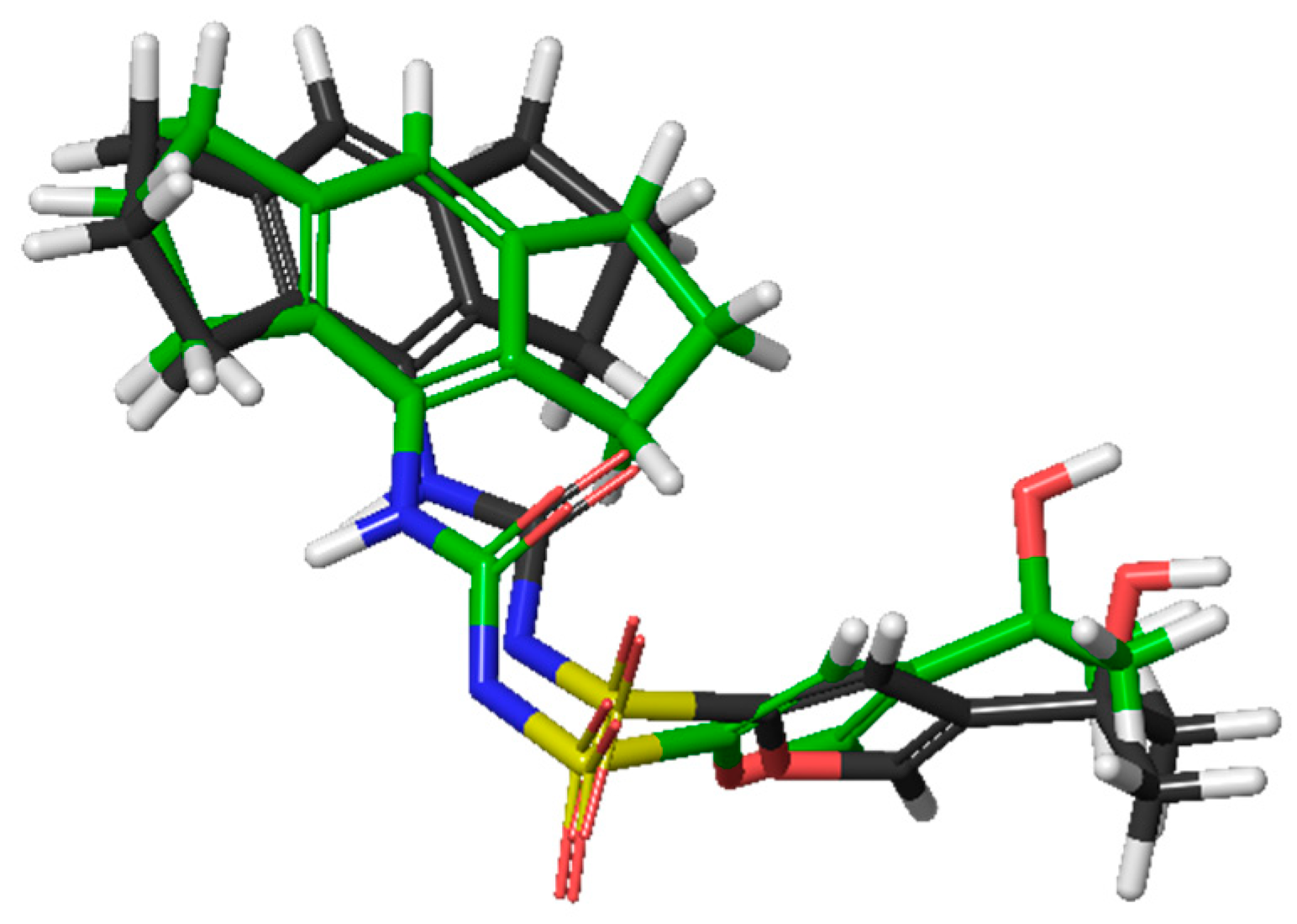
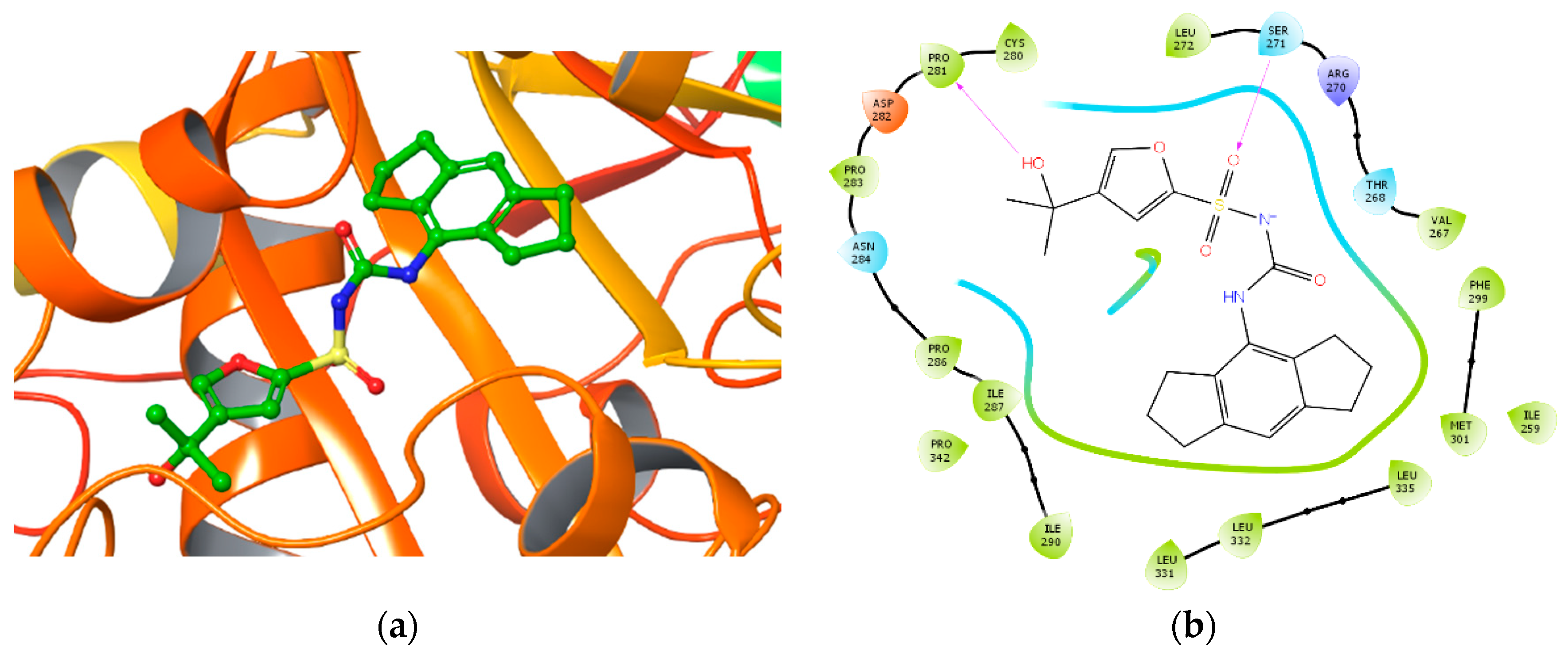
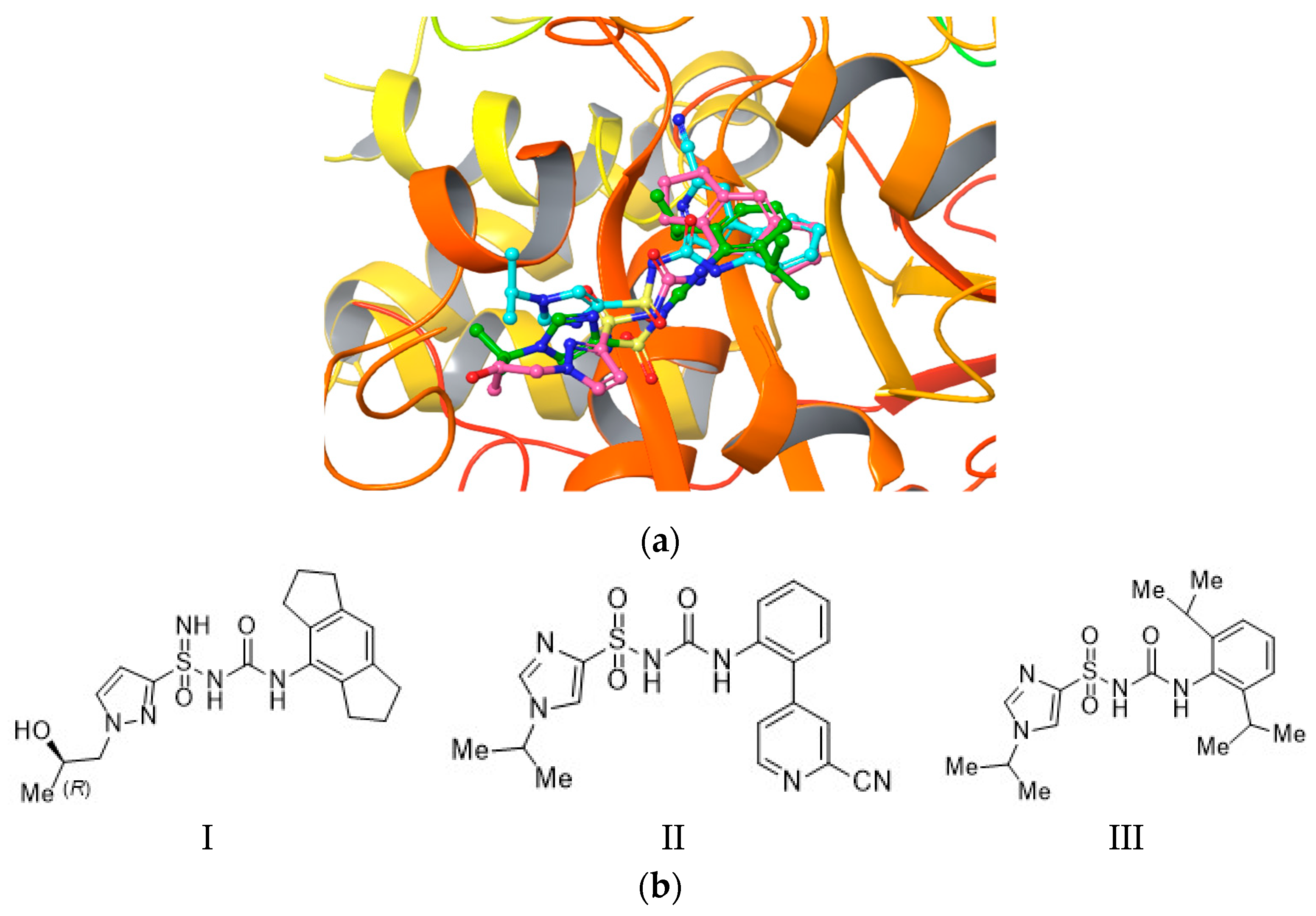
2.3. Molecular Docking and Induced-Fit Docking
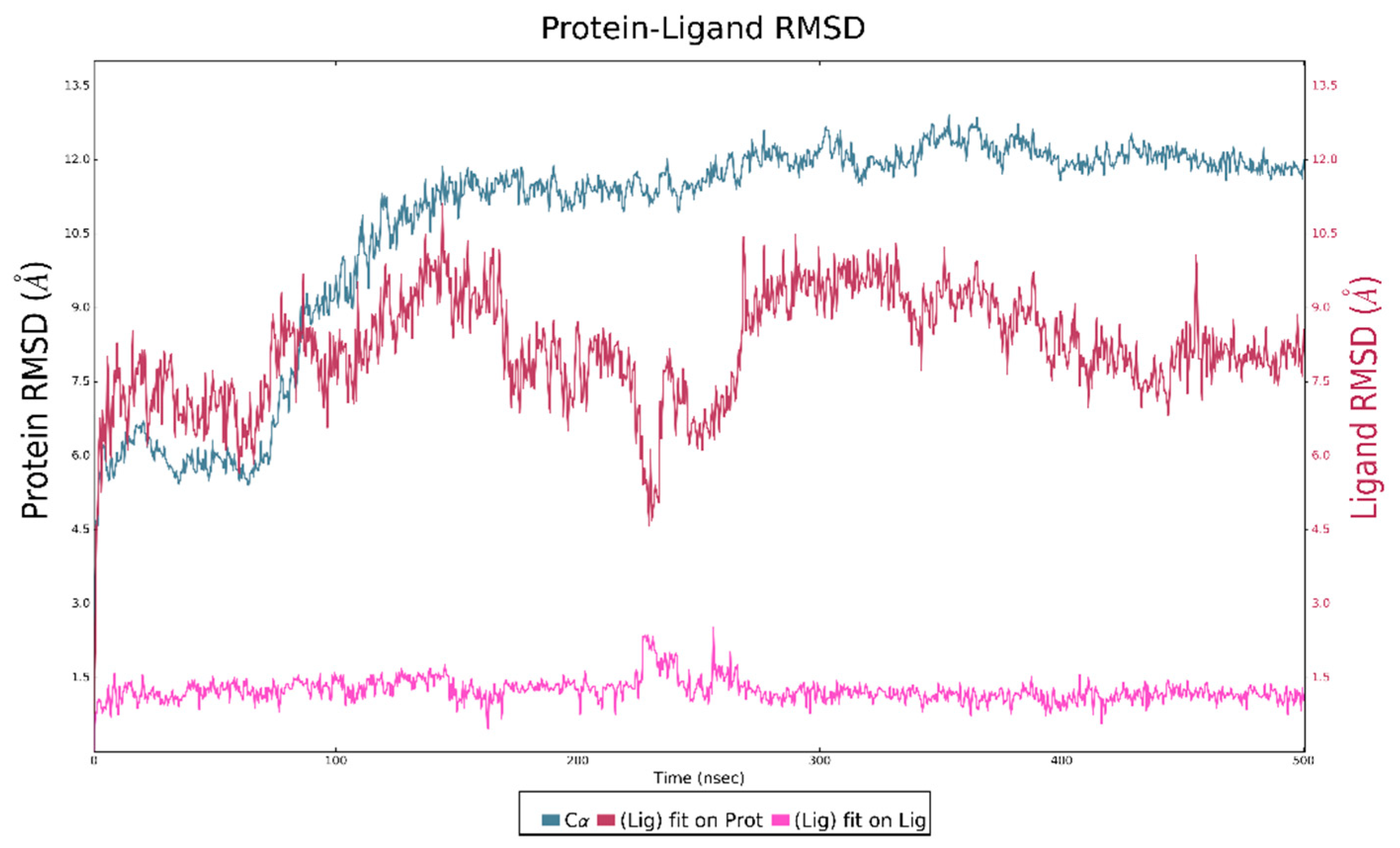
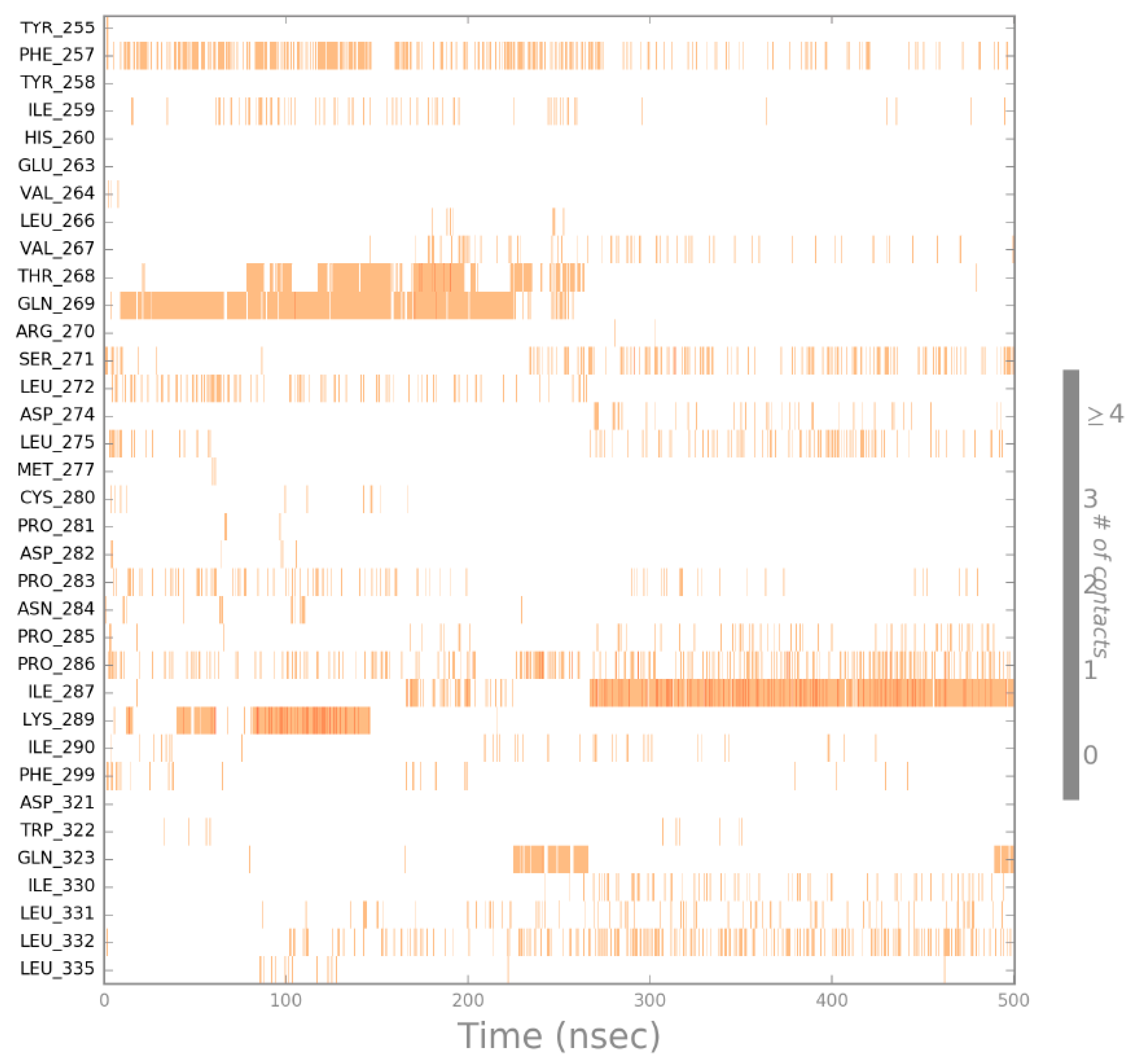
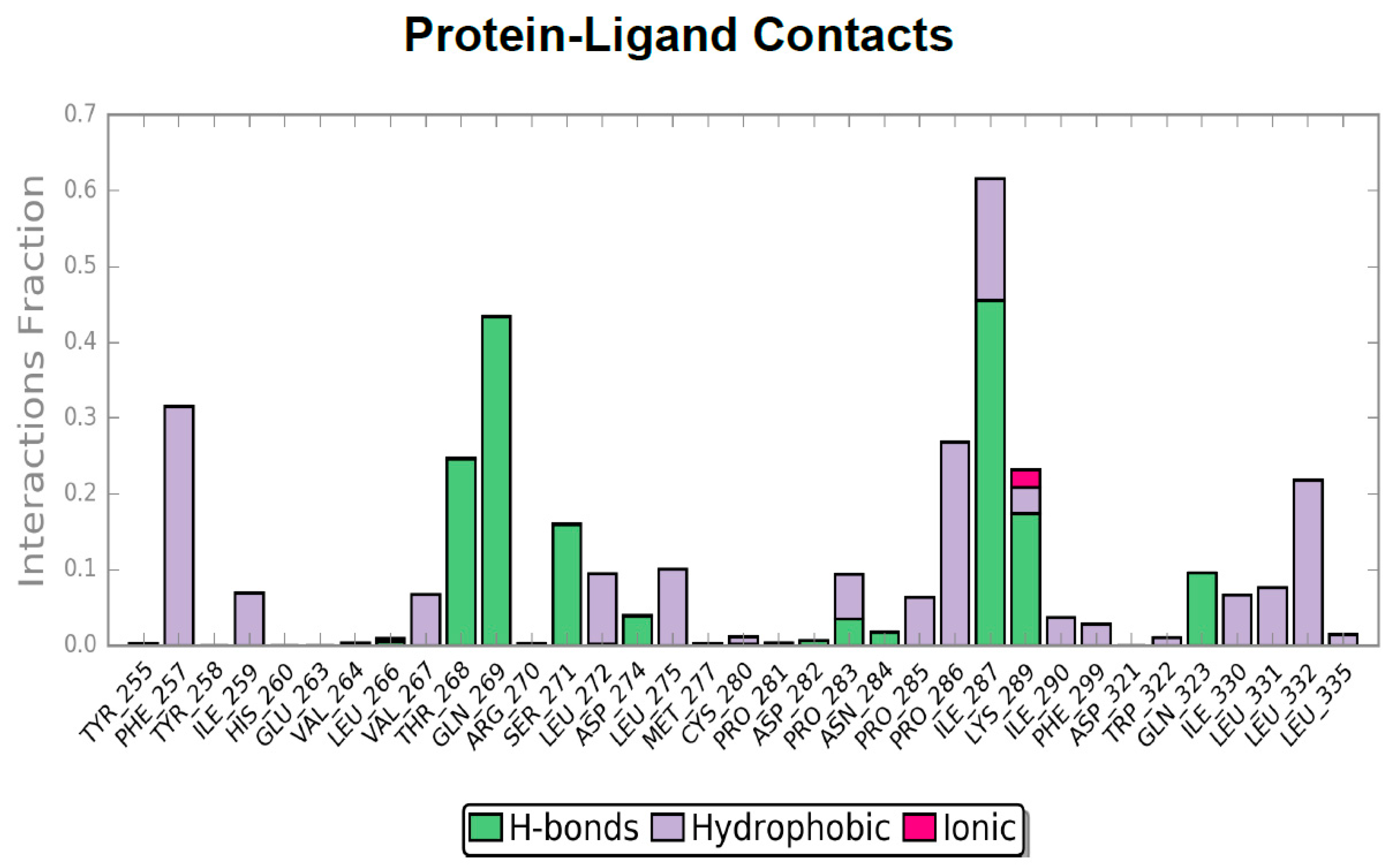
2.4. Molecular Dynamics
3. Materials and Methods
3.1. Homology Modeling
3.2. Binding Site Detection
3.2.1. FTMAP Parameters Description
3.2.2. Sitemap
3.2.3. Fpocket
3.3. Ligand Preparation
3.4. Molecular Docking and Induced-Fit Docking
3.4.1. Molecular Docking
3.4.2. Induced-Fit Docking
3.4.3. MM-GBSA
3.5. Molecular Dynamics
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| IFD | Induced-fit Docking |
| MD | Molecular dynamic simulation |
| NLRP3 | NOD-like receptor family, pyrin domain-containing protein 3 |
| vdW DFT | van der Waals Density Functional Theory |
References
- Mangan, M.S.J.; Olhava, E.J.; Roush, W.R.; Seidel, H.M.; Glick, G.D.; Latz, E. Targeting the NLRP3 Inflammasome in Inflammatory Diseases. Nat. Rev. Drug Discov. 2018, 17, 588–606. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A Small-Molecule Inhibitor of the NLRP3 Inflammasome for the Treatment of Inflammatory Diseases. Nat. Med. 2015, 21, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Hill, J.R.; Day, C.J.; Zamoshnikova, A.; Boucher, D.; Massey, N.L.; Chitty, J.L.; Fraser, J.A.; Jennings, M.P.; Robertson, A.A.B.; et al. MCC950 Directly Targets the NLRP3 ATP-Hydrolysis Motif for Inflammasome Inhibition. Nat. Chem. Biol. 2019, 15, 556–559. [Google Scholar] [CrossRef] [PubMed]
- Tapia-Abellán, A.; Angosto-Bazarra, D.; Martínez-Banaclocha, H.; De Torre-Minguela, C.; Cerón-Carrasco, J.P.; Pérez-Sánchez, H.; Arostegui, J.I.; Pelegrin, P. MCC950 Closes the Active Conformation of NLRP3 to an Inactive State. Nat. Chem. Biol. 2019, 15, 560–564. [Google Scholar] [CrossRef] [PubMed]
- Gorka, O.; Neuwirt, E.; Groß, O. Walking Over The Inflammasome. Nat. Chem. Biol. 2019, 15, 552–553. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Szilagyi, A.; Roy, A.; Zhang, Y. Genome-Wide Protein Structure Prediction. In Multiscale Approaches to Protein Modeling; Kolinski, A., Ed.; Springer: New York, NY, USA, 2011; pp. 255–279. [Google Scholar]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical Assessment of Methods of Protein Structure Prediction (CASP)—Round XII. Proteins Struct. Funct. Bioinform. 2018, 86, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Kozakov, D.; Grove, L.E.; Hall, D.R.; Bohnuud, T.; Mottarella, S.E.; Luo, L.; Xia, B.; Beglov, D.; Vajda, S. The FTMap Family of Web Servers for Determining and Characterizing Ligand-Binding Hot Spots of Proteins. Nat. Protoc. 2015, 10, 733–755. [Google Scholar] [CrossRef]
- Halgren, T.A. Identifying and Characterizing Binding Sites and Assessing Druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef]
- Rooklin, D.; Wang, C.; Katigbak, J.; Arora, P.S.; Zhang, Y. Alphaspace: Fragment-Centric Topographical Mapping to Target Protein-Protein Interaction Interfaces. J. Chem. Inf. Model. 2015, 55, 1585–1599. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An Open Source Platform for Ligand Pocket Detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A High-Performance Quantum Chemistry Software Program with Strengths in Life and Materials Sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating A Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Click, G.; Ghosh, S.; Roush, W.R. Compounds and Compositions for Treating Conditions Associated with NLRP Activity. Patent WO 2017/184624, 26 October 2017. [Google Scholar]
- Click, G.; Roush, W.; Venkatraman, S.; Shen, D.-M.; Ghosh, S.; Katz, J.; Seidel, H.M.; Franchi, L.; Winkler, D.G.; Opipari, J.R.A.W. Compounds and Compositions for Treating Conditions Associated with NLRP Activity. Patent WO 2019/023147, 31 January 2019. [Google Scholar]
- Cooper, M.; Miller, D.; Macleod, A.; Thom, S.; St-Gallay, S.; Shannon, J. Sulfonylureas and Sulfonylthioureas as NLRP3 Inhibitors. Patent WO 2019/034693, 21 February 2019. [Google Scholar]
- Miller, D.; Thom, S.; St-Gallay, S.; Shannon, J.; Leeson, P. Novel Compounds Nouveaux Composés. Patent WO 2019/068772, 11 April 2019. [Google Scholar]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 Model: A Next Generation Energy Model for High Resolution Protein Structure Modeling. Proteins Struct. Funct. Bioinf. 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel Procedure for Modeling Ligand/Receptor Induced Fit Effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE SC|06 Conference (SC’06), Tampa, FL, USA, 11–17 November 2006; D. E. Shaw Research, LLC: New York, NY, USA, 2006. [Google Scholar]
- Perricone, U.; Gulotta, M.R.; Lombino, J.; Parrino, B.; Cascioferro, S.; Diana, P.; Cirrincione, G.; Padova, A. An Overview of Recent Molecular Dynamics Applications as Medicinal Chemistry Tools for the Undruggable Site Challenge. Medchemcomm 2018, 9, 920–936. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A Software Program for Pk(A) Prediction And Protonation State Generation for Drug-Like Molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the Comprehensive, Rapid, and Accurate Prediction of the Favorable Tautomeric States of Drug-like Molecules in Aqueous Solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Izadi, S.; Onufriev, A.V. Accuracy limit of rigid 3-point water models. J. Chem. Phys. 2016, 145, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Merz, K.M. Application of the Nosé-Hoover chain algorithm to the study of protein dynamics. J. Phys. Chem. 1996, 100, 1927–1937. [Google Scholar] [CrossRef]
- Fogolari, F.; Corazza, A.; Toppo, S.; Tosatto, S.C.E.; Viglino, P.; Ursini, F.; Esposito, G. Studying interactions by molecular dynamics simulations at high concentration. J. Biomed. Biotechnol. 2012, 2012, 303190. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Olsson, S.; Wehmeyer, C.; Pérez, A.; Charron, N.E.; De Fabritiis, G.; Noé, F.; Clementi, C. Machine Learning of Coarse-Grained Molecular Dynamics Force Fields. ACS Cent. Sci. 2019, 5, 755–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchko, G.W.; Pulavarti, S.V.S.R.K.; Ovchinnikov, V.; Shaw, E.A.; Rettie, S.A.; Myler, P.J.; Karplus, M.; Szyperski, T.; Baker, D.; Bahl, C.D. Cytosolic expression, solution structures, and molecular dynamics simulation of genetically encodable disulfide-rich de novo designed peptides. Protein Sci. 2018, 27, 1611–1623. [Google Scholar] [CrossRef] [PubMed]
- Dalle Vedove, A.; Falchi, F.; Donini, S.; Dobric, A.; Germain, S.; Di Martino, G.P.; Prosdocimi, T.; Vettraino, C.; Torretta, A.; Cavalli, A.; et al. Structure-Based Virtual Screening Allows the Identification of Efficient Modulators of E-Cadherin-Mediated Cell–Cell Adhesion. Int. J. Mol. Sci. 2019, 20, 3404–3421. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PROTEIN | Docking Score (Kcal/mol) | MM-GBSA (Kcal/mol) |
|---|---|---|
| NLRP3 model | −8.85 | −41.58 |
| NLRC4 (pdbid:4KXF) | −4.01 | −28.62 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mekni, N.; De Rosa, M.; Cipollina, C.; Gulotta, M.R.; De Simone, G.; Lombino, J.; Padova, A.; Perricone, U. In Silico Insights towards the Identification of NLRP3 Druggable Hot Spots. Int. J. Mol. Sci. 2019, 20, 4974. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20204974
Mekni N, De Rosa M, Cipollina C, Gulotta MR, De Simone G, Lombino J, Padova A, Perricone U. In Silico Insights towards the Identification of NLRP3 Druggable Hot Spots. International Journal of Molecular Sciences. 2019; 20(20):4974. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20204974
Chicago/Turabian StyleMekni, Nedra, Maria De Rosa, Chiara Cipollina, Maria Rita Gulotta, Giada De Simone, Jessica Lombino, Alessandro Padova, and Ugo Perricone. 2019. "In Silico Insights towards the Identification of NLRP3 Druggable Hot Spots" International Journal of Molecular Sciences 20, no. 20: 4974. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20204974