Hypoxia Induced Heparan Sulfate Primes the Extracellular Matrix for Endothelial Cell Recruitment by Facilitating VEGF-Fibronectin Interactions


Abstract
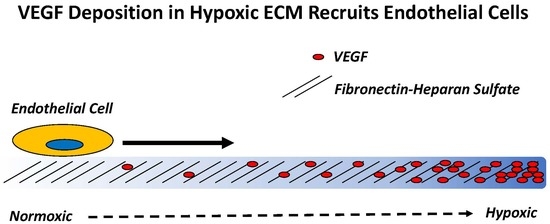
:
1. Introduction
2. Results
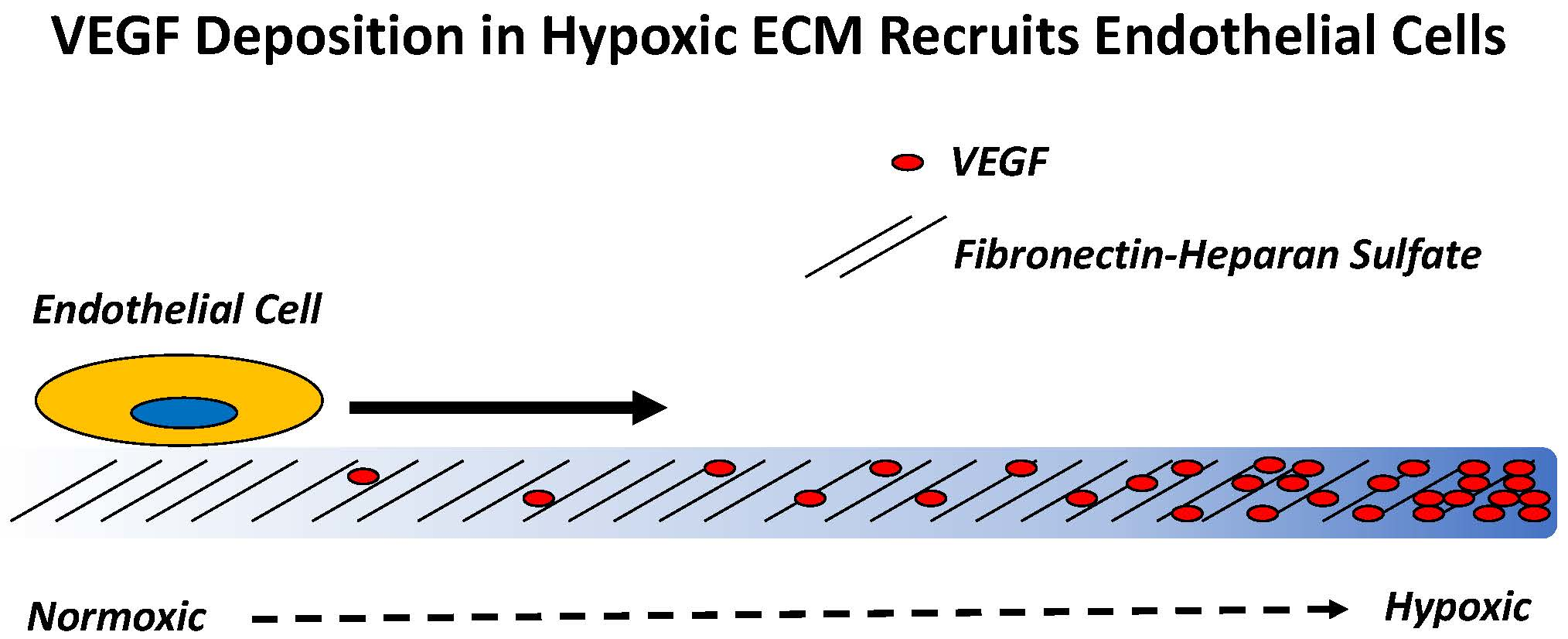
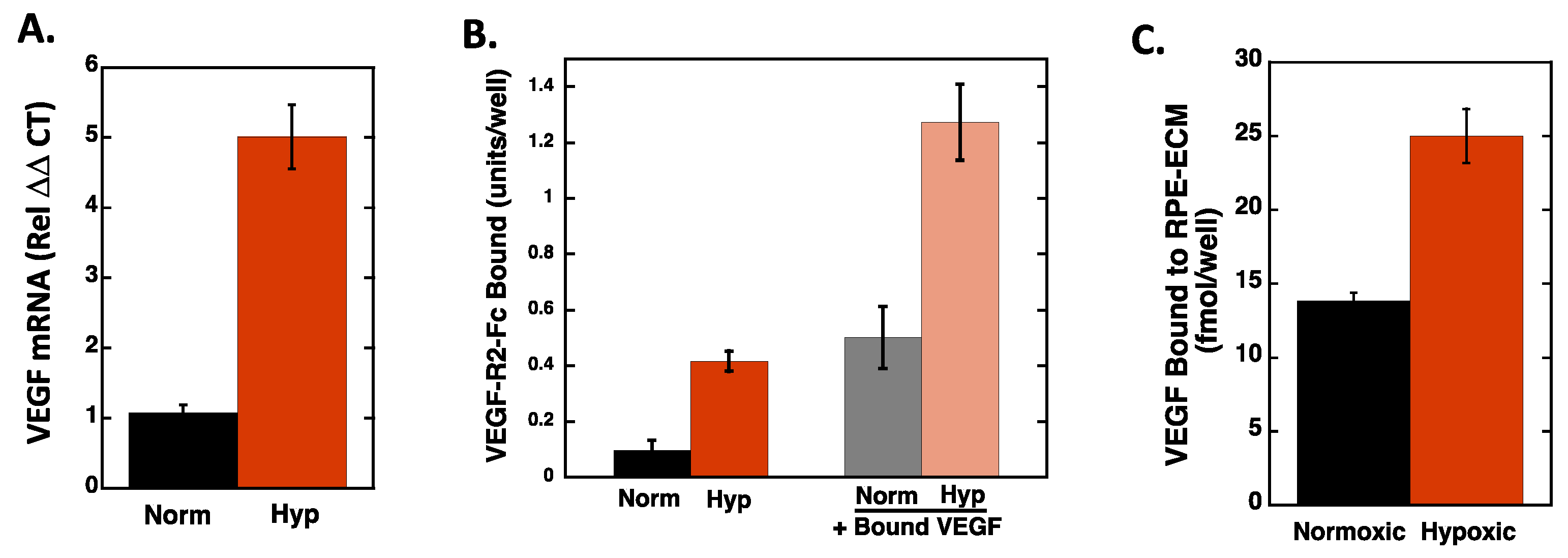
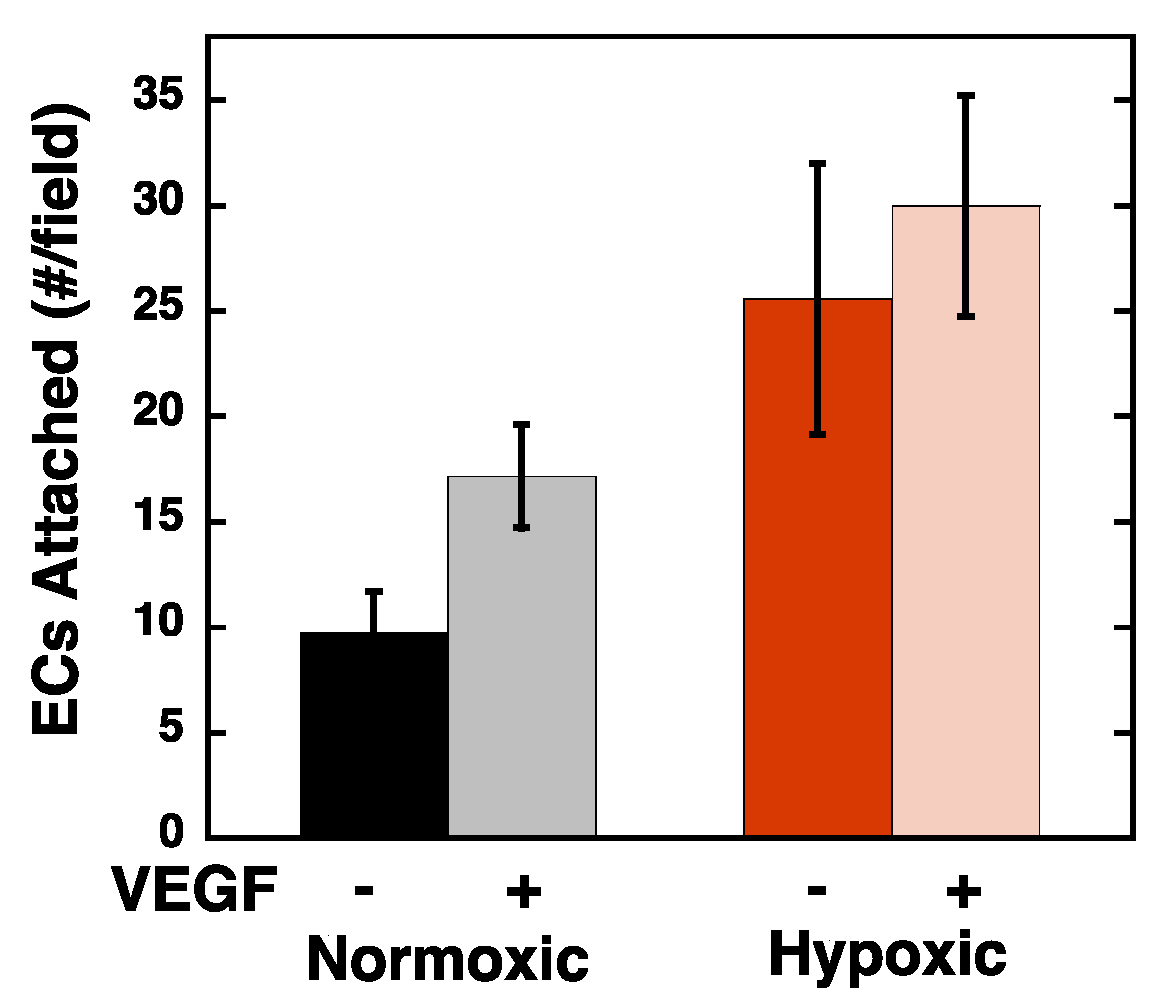
2.1. Endothelial Cell Attachment to Retinal Pigmented Epithelial Cells is Enhanced Under Hypoxic Conditions
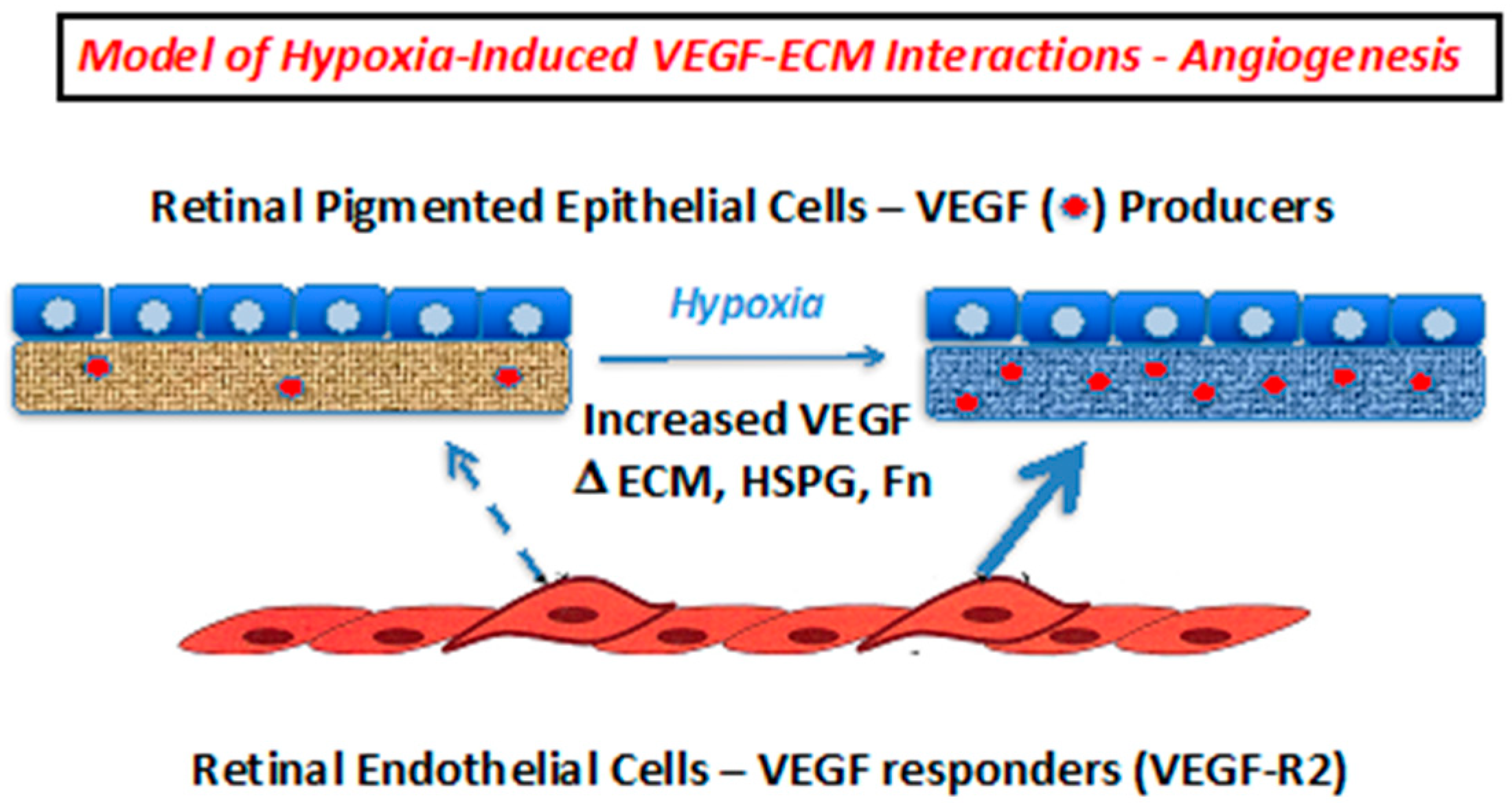
2.2. Hypoxia Enhances Endothelial Cell Attachment by Increasing ECM-Bound VEGF
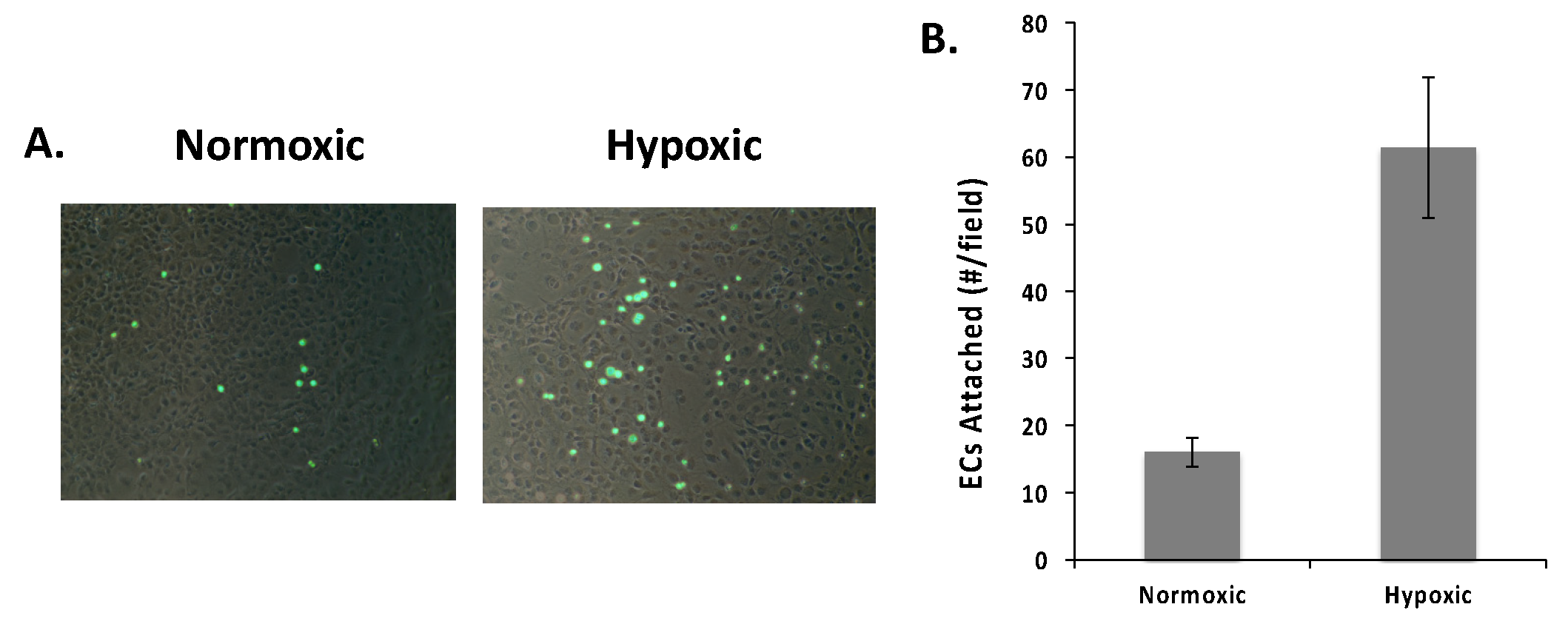
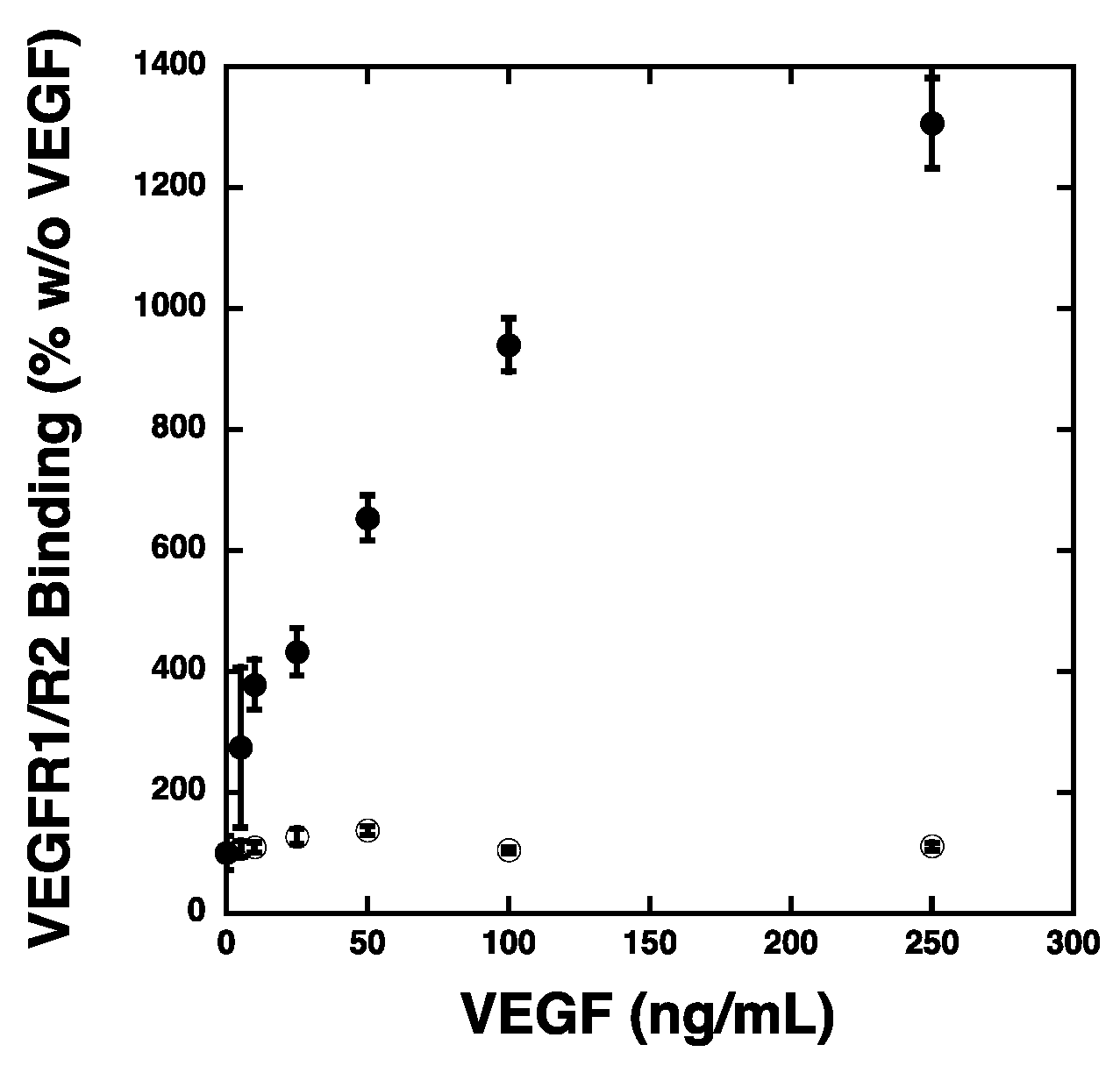
2.3. VEGF Receptor 2 Binds to VEGF-Fibronectin
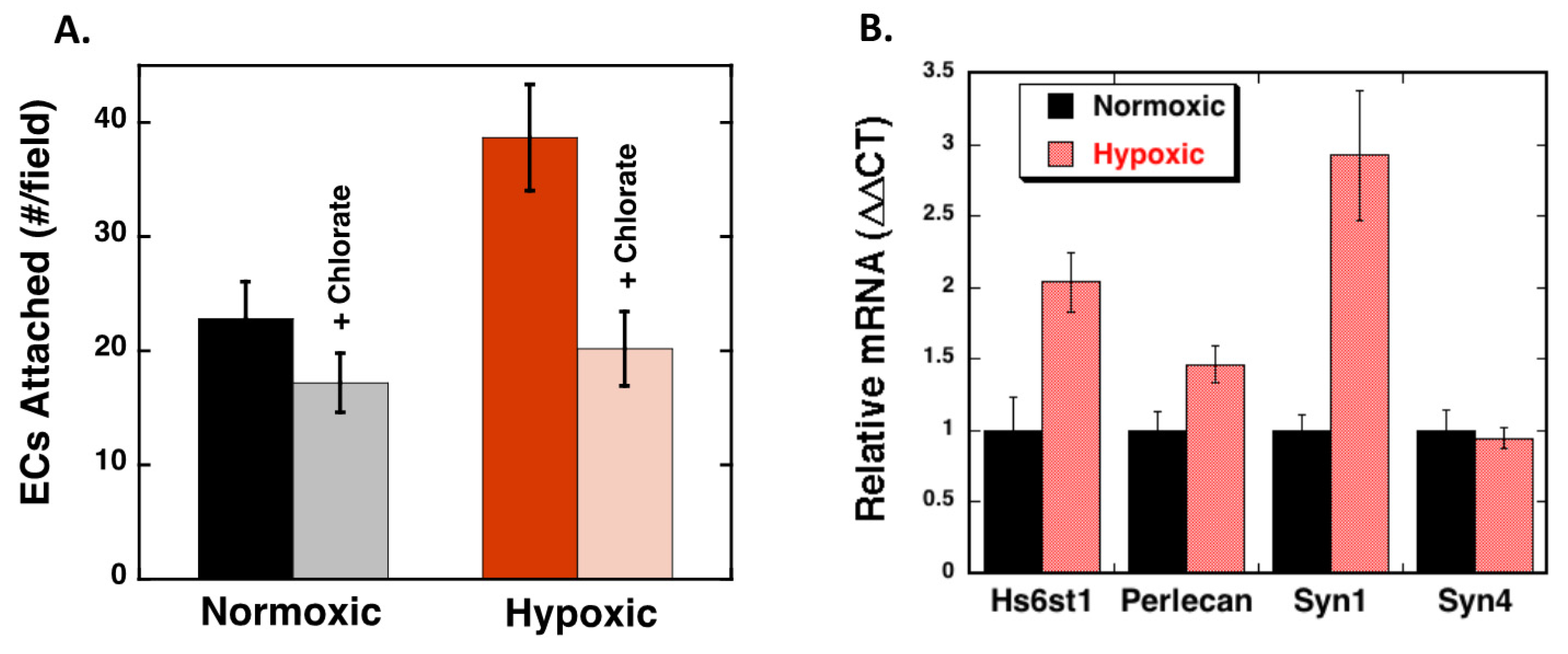
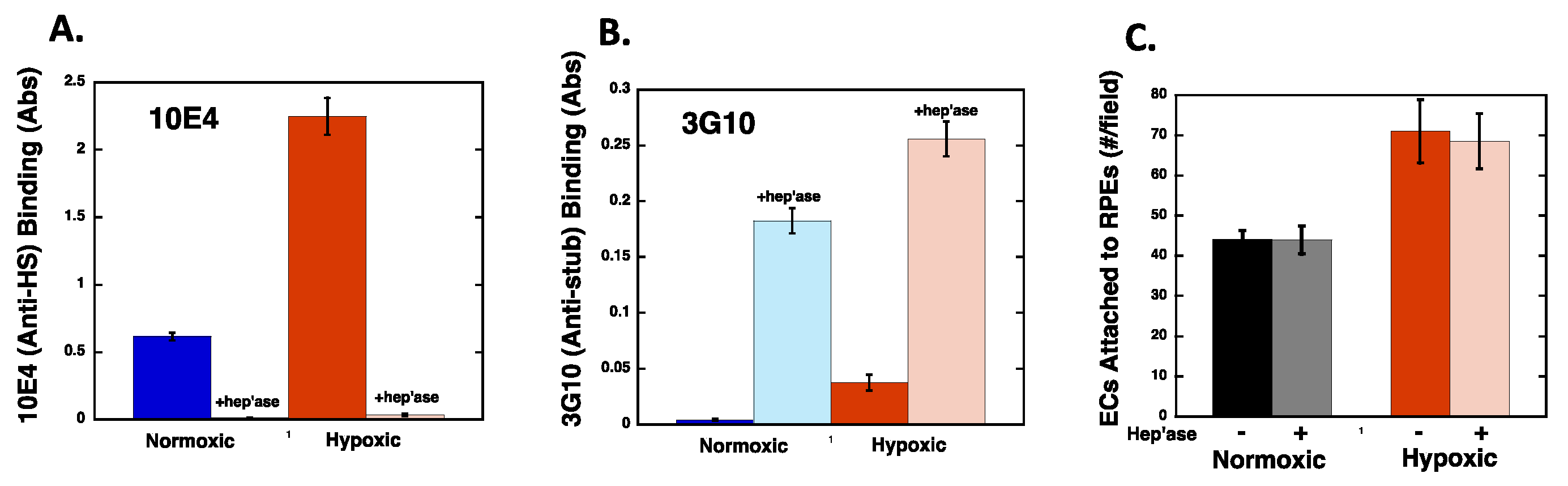
2.4. Hypoxia Alters Heparan Sulfate Proteoglycans Expression
2.5. Heparan Sulfate Chains are Modified by Hypoxia
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
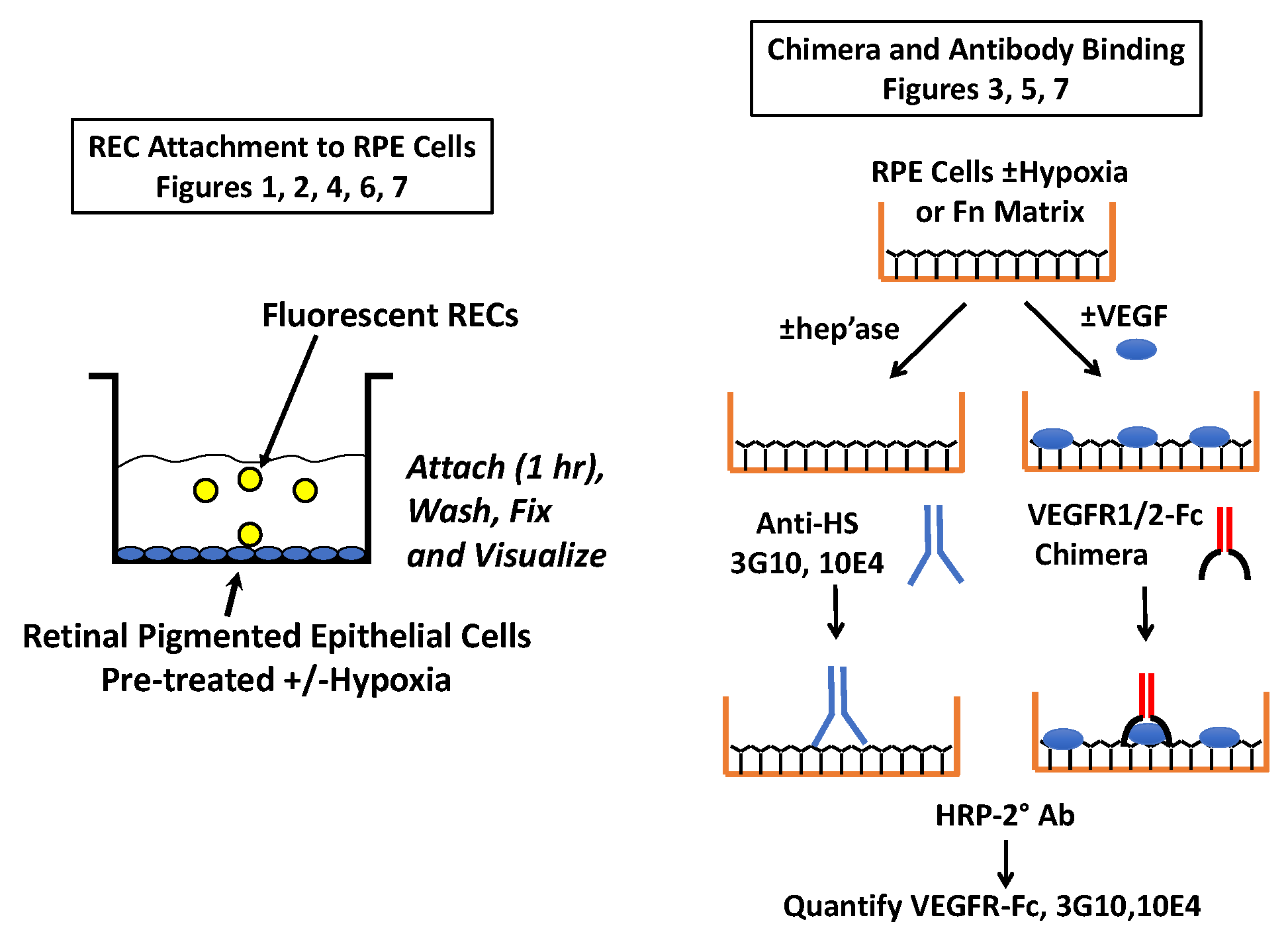
4.3. Cell Attachment Assays
4.4. Real Time Polymerase Chain Reaction (RT-PCR) Analyses
4.5. Proteoglycan ELISAs
4.6. VEGF Receptor Chimera Assays
4.7. VEGF Binding Assays
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| RPE | Retinal pigmented epithelial cells |
| REC | Rat retinal endothelial cells |
| VEGF | Vascular endothelial growth factor-A165 |
| Fc-VEGFR1 | The binding domain of VEGF receptor 1 linked to the fragment crystallizable tail region of human antibody. |
| Fc-VEGFR2 | The binding domain of VEGF receptor 2 linked to the fragment crystallizable tail region of human antibody. |
| Hs6st1 | Heparan sulfate specific 6-O sulfoltransferase-1 |
| Syn1 | Syndecan 1 |
| Perl | Perlecan |
| Syn4 | Syndecan 4 |
| PBS | Phosphate buffered saline |
| BSA | Bovine serum albumin |
| AMD | Age-related macular degeneration |
| PDR | Proliferative diabetic retinopathy |
| HRP | Horse radish peroxidase |
| EC | Endothelial cells |
| HS | Heparan sulfate |
| HSPG | Heparan sulfate proteoglycans |
| N | Biological replicates |
| n | Technical replicates |
References
- Dvorak, H.F. Vascular permeability factor/vascular endothelial growth factor: A critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J. Clin. Oncol. 2002, 20, 4368–4380. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.J.; Stringer, S.E. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J. Cell Sci. 2001, 114, 853–865. [Google Scholar] [PubMed]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Nieves, B.J.; D’Amore, P.A.; Bryan, B.A. The function of vascular endothelial growth factor. Biofactors 2009, 35, 332–337. [Google Scholar] [CrossRef]
- Saint-Geniez, M.; Kurihara, T.; Sekiyama, E.; Maldonado, A.E.; D’Amore, P.A. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc. Natl. Acad. Sci. USA 2009, 106, 18751–18756. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Geisen, P.; Wittchen, E.S.; King, B.; Burridge, K.; D’Amore, P.A.; Hartnett, M.E. The role of RPE cell-associated VEGF189 in choroidal endothelial cell transmigration in neovascular age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2010, 52, 570–578. [Google Scholar] [CrossRef]
- Rahimi, N. VEGFR-1 and VEGFR-2: Two non-identical twins with a unique physiognomy. Front. Biosci. 2006, 11, 818–829. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular Endothelial Growth Factor: Basic Science and Clinical Progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef]
- Fong, G.-H.; Rossant, J.; Gertsenstein, M.; Breitman, M.L. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376, 66–70. [Google Scholar] [CrossRef]
- Fong, G.; Klingensmith, J.; Wood, C.R.; Rossant, J.; Breitman, M.L. Regulation of flt1 expression during mouse embryogenesis suggests a role in the establishment of vascular endothelium. Dev. Dyn. 1996, 207, 1–10. [Google Scholar] [CrossRef]
- Chen, T.T.; Luque, A.; Lee, S.; Anderson, S.M.; Segura, T.; Iruela-Arispe, M.L. Anchorage of VEGF to the extracellular matrix conveys differential signaling responses to endothelial cells. J. Cell Boil. 2010, 188, 595–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijelath, E.S.; Murray, J.; Rahman, S.; Patel, Y.; Ishida, A.; Strand, K.; Aziz, S.; Cardona, C.; Hammond, W.P.; Savidge, G.F.; et al. Novel vascular endothelial growth factor binding domains of fibronectin enhance vascular endothelial growth factor biological activity. Circ. Res. 2002, 91, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Wijelath, E.S.; Rahman, S.; Murray, J.; Patel, Y.; Savidge, G.; Sobel, M. Fibronectin promotes VEGF-induced CD34+ cell differentiation into endothelial cells. J. Vasc. Surg. 2004, 39, 655–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijelath, E.S.; Rahman, S.; Namekata, M.; Murray, J.; Nishimura, T.; Mostafavi-Pour, Z.; Patel, Y.; Suda, Y.; Humphries, M.J.; Sobel, M. Heparin-II domain of fibronectin is a vascular endothelial growth factor-binding domain: Enhancement of VEGF biological activity by a singular growth factor/matrix protein synergism. Circ. Res. 2006, 99, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Goerges, A.L.; Nugent, M.A. pH Regulates Vascular Endothelial Growth Factor Binding to Fibronectin: A Mechanism for Control of Extracellular Matrix Storage and Release. J. Biol. Chem. 2004, 279, 2307–2315. [Google Scholar] [CrossRef]
- Mitsi, M.; Hong, Z.; Costello, C.E.; Nugent, M.A. Heparin-Mediated Conformational Changes in Fibronectin Expose Vascular Endothelial Growth Factor Binding Sites. Biochemistry 2006, 45, 10319–10328. [Google Scholar] [CrossRef]
- Roy, S.; Sala, R.; Cagliero, E.; Lorenzi, M. Overexpression of fibronectin induced by diabetes or high glucose: Phenomenon with a memory. Proc. Natl. Acad. Sci. USA 1990, 87, 404–408. [Google Scholar] [CrossRef]
- Roy, S.; Cagliero, E.; Lorenzi, M. Fibronectin overexpression in retinal microvessels of patients with diabetes. Investig. Ophthalmol. Vis. Sci. 1996, 37, 258–266. [Google Scholar]
- Polewski, P.; Chadda, M.; Li, A.-F.; Roy, S.; Oshitari, T.; Sato, T. Effect of Combined Antisense Oligonucleotides Against High-Glucose–and Diabetes-Induced Overexpression of Extracellular Matrix Components and Increased Vascular Permeability. Diabetes 2006, 55, 86–92. [Google Scholar]
- Martino, M.M.; Hubbell, J.A. The 12th–14th type III repeats of fibronectin function as a highly promiscuous growth factor-binding domain. FASEB J. 2010, 24, 4711–4721. [Google Scholar] [CrossRef]
- Mitsi, M.; Forsten-Williams, K.; Gopalakrishnan, M.; Nugent, M.A. A catalytic role of heparin within the extracellular matrix. J. Biol. Chem. 2008, 283, 34796–34807. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.; Mitsi, M.; Nugent, M.A.; Symes, K. PDGF-A interactions with fibronectin reveal a critical role for heparan sulfate in directed cell migration during Xenopus gastrulation. Proc. Natl. Acad. Sci. USA 2009, 106, 21683–21688. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Fuster, M.M.; Lawrence, R.; Esko, J.D. Heparan Sulfate Regulates VEGF165- and VEGF121-mediated Vascular Hyperpermeability. J. Biol. Chem. 2010, 286, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Selleck, S.B. Signaling from across the Way: Transactivation of VEGF Receptors by HSPGs. Mol. Cell 2006, 22, 431–432. [Google Scholar] [CrossRef]
- Jakobsson, L.; Kreuger, J.; Holmborn, K.; Lundin, L.; Eriksson, I.; Kjellén, L.; Claesson-Welsh, L. Heparan Sulfate in trans Potentiates VEGFR-Mediated Angiogenesis. Dev. Cell 2006, 10, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Eswarakumar, V.; Lax, I.; Schlessinger, J.; Eswarakumar, J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev. 2005, 16, 139–149. [Google Scholar] [CrossRef]
- Nugent, M.A.; Iozzo, R.V. Fibroblast growth factor-2. Int. J. Biochem. Cell Biol. 2000, 32, 115–120. [Google Scholar] [CrossRef]
- Hirota, K.; Semenza, G.L. Regulation of angiogenesis by hypoxia-inducible factor 1. Crit. Rev. Oncol. 2006, 59, 15–26. [Google Scholar] [CrossRef]
- Moeller, B.J.; Cao, Y.; Vujaskovic, Z.; Li, C.Y.; Haroon, Z.A.; Dewhirst, M.W. The relationship between hypoxia and angiogenesis. Semin. Radiat. Oncol. 2004, 14, 215–221. [Google Scholar] [CrossRef]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef]
- Fannon, M.; Forsten-Williams, K.; Zhao, B.; Bach, E.; Parekh, P.P.; Chu, C.L.; Goerges-Wildt, A.L.; Buczek-Thomas, J.A.; Nugent, M.A. Facilitated diffusion of VEGF165 through descemet’s membrane with sucrose octasulfate. J. Cell. Physiol. 2012, 227, 3693–3700. [Google Scholar] [CrossRef] [PubMed]
- Kurtagic, E.; Jedrychowski, M.P.; Nugent, M.A. Neutrophil elastase cleaves VEGF to generate a VEGF fragment with altered activity. Am. J. Physiol. Cell. Mol. Physiol. 2009, 296, L534–L546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtagic, E.; Rich, C.B.; Buczek-Thomas, J.A.; Nugent, M.A. Neutrophil Elastase-Generated Fragment of Vascular Endothelial Growth Factor-A Stimulates Macrophage and Endothelial Progenitor Cell Migration. PLoS ONE 2015, 10, e0145115. [Google Scholar] [CrossRef] [PubMed]
- Teran, M.; Nugent, M.A. Characterization of receptor binding kinetics for vascular endothelial growth factor-A using SPR. Anal. Biochem. 2019, 564, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.M.; Fernig, D.G.; Jesudason, E.C.; Losty, P.D.; van de Westerlo, E.M.; van Kuppevelt, T.H.; Turnbull, J.E. Heparan sulfate phage display antibodies identify distinct epitopes with complex binding characteristics: Insights into protein binding specificities. J. Biol. Chem. 2009, 284, 35621–35631. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar] [CrossRef]
- Crawford, T.N.; Alfaro, D.V., 3rd; Kerrison, J.B.; Jablon, E.P. Diabetic retinopathy and angiogenesis. Curr. Diabetes Rev. 2009, 5, 8–13. [Google Scholar] [CrossRef]
- Folkman, J. Angiogenesis-dependent diseases. Semin. Oncol. 2001, 28, 536–542. [Google Scholar] [CrossRef]
- Qazi, Y.; Maddula, S.; Ambati, B.K. Mediators of ocular angiogenesis. J. Genet. 2009, 88, 495–515. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; McGuire, P.G. Retinal and choroidal angiogenesis: Pathophysiology and strategies for inhibition. Prog. Retin. Eye Res. 2003, 22, 721–748. [Google Scholar] [CrossRef]
- Friedman, D.S.; O’Colmain, B.J.; Munoz, B.; Tomany, S.C.; McCarty, C.; de Jong, P.T.; Nemesure, B.; Mitchell, P.; Kempen, J. Prevalence of age-related macular degeneration in the United States. Arch. Ophthalmol. 2004, 122, 564–572. [Google Scholar] [PubMed]
- The Eye Diseases Prevalence Research Group. The Prevalence of Diabetic Retinopathy Among Adults in the United States. Arch. Ophthalmol. 2004, 122, 552–563. [Google Scholar]
- Congdon, N.; O’Colmain, B.; Klaver, C.C.; Klein, R.; Munoz, B.; Friedman, D.S.; Kempen, J.; Taylor, H.R.; Mitchell, P. Causes and prevalence of visual impairment among adults in the United States. Arch. Ophthalmol. 2004, 122, 477–485. [Google Scholar] [PubMed]
- Maharaj, A.S.; D’Amore, P.A. Roles for VEGF in the adult. Microvasc. Res. 2007, 74, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Jardeleza, M.S.R.; Miller, J.W. Review of Anti-VEGF Therapy in Proliferative Diabetic Retinopathy. Semin. Ophthalmol. 2009, 24, 87–92. [Google Scholar] [CrossRef]
- Nicholson, B.P.; Schachat, A.P. A review of clinical trials of anti-VEGF agents for diabetic retinopathy. Graefe’s Arch. Clin. Exp. Ophthalmol. 2010, 248, 915–930. [Google Scholar] [CrossRef]
- Ozkiris, A. Anti-VEGF agents for age-related macular degeneration. Expert Opin. Ther. Pat. 2010, 20, 103–118. [Google Scholar] [CrossRef]
- Regatieri, C.; Dreyfuss, J.; Melo, G.; Lavinsky, D.; Hossaka, S.; Rodrigues, E.; Farah, M.; Maia, M.; Nader, H. Quantitative evaluation of experimental choroidal neovascularization by confocal scanning laser ophthalmoscopy: Fluorescein angiogram parallels heparan sulfate proteoglycan expression. Braz. J. Med Biol. Res. 2010, 43, 627–633. [Google Scholar] [CrossRef]
- Dreyfuss, J.L.; Regatieri, C.V.; Lima, M.A.; Brito, A.S.; Chavante, S.F.; Belfort, R., Jr.; Farah, M.E.; Nader, H.B.; Paredes-Gamero, E.J.; Paredes-Gamero, E.J.; et al. A heparin mimetic isolated from a marine shrimp suppresses neovascularization. J. Thromb. Haemost. 2010, 8, 1828–1837. [Google Scholar] [CrossRef]
- Sasisekharan, R.; Moses, M.; Nugent, M.A.; Cooney, C.; Langer, R. Heparinase inhibits neovascularization. Proc. Natl. Acad. Sci. USA 1993, 91, 1524–1528. [Google Scholar] [CrossRef]
- Ahluwalia, A.; Tarnawski, A.S. Critical role of hypoxia sensor-HIF-1alpha in VEGF gene activation. Implications for angiogenesis and tissue injury healing. Curr. Med. Chem. 2012, 19, 90–97. [Google Scholar] [CrossRef]
- Chen, L.; Endler, A.; Shibasaki, F. Hypoxia and angiogenesis: Regulation of hypoxia-inducible factors via novel binding factors. Exp. Mol. Med. 2009, 41, 849–857. [Google Scholar] [CrossRef] [PubMed]
- Germain, S.; Monnot, C.; Muller, L.; Eichmann, A. Hypoxia-driven angiogenesis: Role of tip cells and extracellular matrix scaffolding. Curr. Opin. Hematol. 2010, 17, 1. [Google Scholar] [CrossRef] [PubMed]
- Myllyharju, J.; Schipani, E. Extracellular matrix genes as hypoxia-inducible targets. Cell Tissue Res. 2010, 339, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Onochie, O.E.; Onyejose, A.J.; Rich, C.B.; Trinkaus-Randall, V. The Role of Hypoxia in Corneal Extracellular Matrix Deposition and Cell Motility. Anat. Rec. 2019. [Google Scholar] [CrossRef]
- Wiafe, B.; Adesida, A.; Churchill, T.; Adewuyi, E.E.; Li, Z.; Metcalfe, P. Hypoxia-increased expression of genes involved in inflammation, dedifferentiation, pro-fibrosis, and extracellular matrix remodeling of human bladder smooth muscle cells. In Vitro Cell Dev. Biol. Anim. 2017, 53, 58–66. [Google Scholar] [CrossRef]
- Sack, K.D.; Teran, M.; Nugent, M.A. Extracellular Matrix Stiffness Controls VEGF Signaling and Processing in Endothelial Cells. J. Cell. Physiol. 2016, 231, 2026–2039. [Google Scholar] [CrossRef]
- Derricks, K.E.; Trinkaus-Randall, V.; Nugent, M.A. Extracellular matrix stiffness modulates VEGF calcium signaling in endothelial cells: Individual cell and population analysis. Integr. Boil. 2015, 7, 1011–1025. [Google Scholar] [CrossRef]
- Hubbard, B.; Buczek-Thomas, J.A.; Nugent, M.A.; Smith, M.L. Heparin-dependent regulation of fibronectin matrix conformation. Matrix Biol. J. Int. Soc. Matrix Biol. 2014, 34, 124–131. [Google Scholar] [CrossRef]
- Sazonova, O.V.; Lee, K.L.; Isenberg, B.C.; Rich, C.B.; Nugent, M.A.; Wong, J.Y. Cell-Cell Interactions Mediate the Response of Vascular Smooth Muscle Cells to Substrate Stiffness. Biophys. J. 2011, 101, 622–630. [Google Scholar] [CrossRef] [Green Version]
- Derricks, K.E.; Rich, C.B.; Buczek-Thomas, J.A.; Nugent, M.A. Ascorbate enhances elastin synthesis in 3D tissue-engineered pulmonary fibroblasts constructs. Tissue Cell 2013, 45, 253–260. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.-T.; Sinai, P.; Kain, S.R. An Acid Phosphatase Assay for Quantifying the Growth of Adherent and Nonadherent Cells. Anal. Biochem. 1996, 241, 103–108. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent | Mode of Action | Expected Impact |
|---|---|---|
| Fc-VEGF-R2 | Binds to VEGF in the ECM. | Block VEGF-R2 binding site on VEGF. Specifically prevents VEGF-R2 on RECs from engaging VEGF in the ECM. |
| sFn | Competes for VEGF specifically bound to Fn. | Binds and releases VEGF from Fn and into soluble VEGF-sFn form. Reduces sites for VEGF-R2 on RECs to bind in the ECM. |
| SOS | Binds to VEGF and blocks its binding to Fn. | Reduces VEGF-Fn complexes. Reduces sites for VEGF-R2 on RECs to bind in the ECM. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buczek-Thomas, J.A.; Rich, C.B.; Nugent, M.A. Hypoxia Induced Heparan Sulfate Primes the Extracellular Matrix for Endothelial Cell Recruitment by Facilitating VEGF-Fibronectin Interactions. Int. J. Mol. Sci. 2019, 20, 5065. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205065
Buczek-Thomas JA, Rich CB, Nugent MA. Hypoxia Induced Heparan Sulfate Primes the Extracellular Matrix for Endothelial Cell Recruitment by Facilitating VEGF-Fibronectin Interactions. International Journal of Molecular Sciences. 2019; 20(20):5065. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205065
Chicago/Turabian StyleBuczek-Thomas, Jo Ann, Celeste B. Rich, and Matthew A. Nugent. 2019. "Hypoxia Induced Heparan Sulfate Primes the Extracellular Matrix for Endothelial Cell Recruitment by Facilitating VEGF-Fibronectin Interactions" International Journal of Molecular Sciences 20, no. 20: 5065. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205065