Genome-Wide Adductomics Analysis Reveals Heterogeneity in the Induction and Loss of Cyclobutane Thymine Dimers across Both the Nuclear and Mitochondrial Genomes

 , ,
, ,
Abstract
:1. Introduction
2. Results
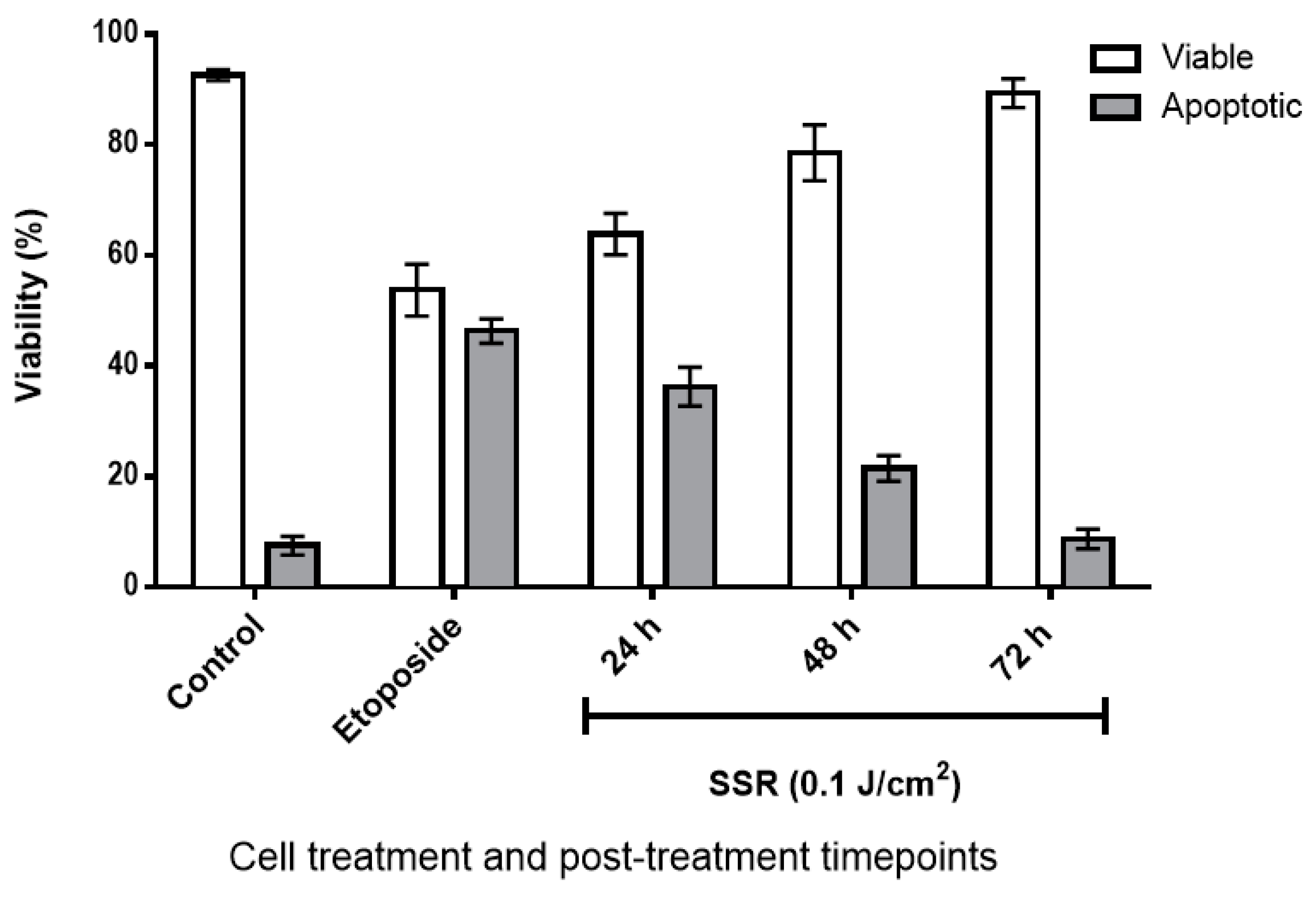
2.1. The Effect of SSR Irradiation on HaCaT Cell Viability
2.2. Optimisation of DNA:Anti-T<>T MAb Ratio
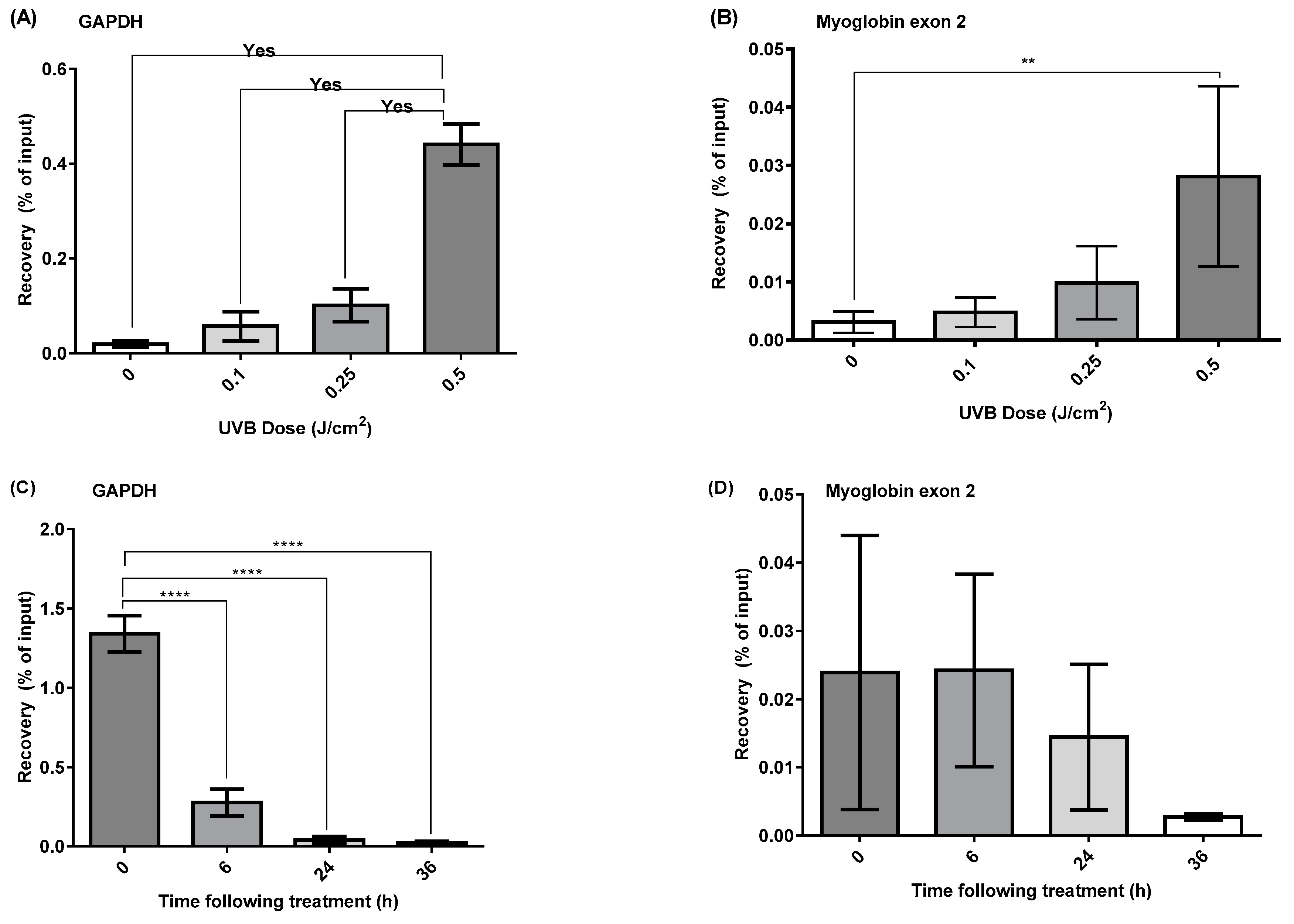
2.3. DDIP-qPCR Quantification of the Induction and Repair of CPDs Induced by UVR
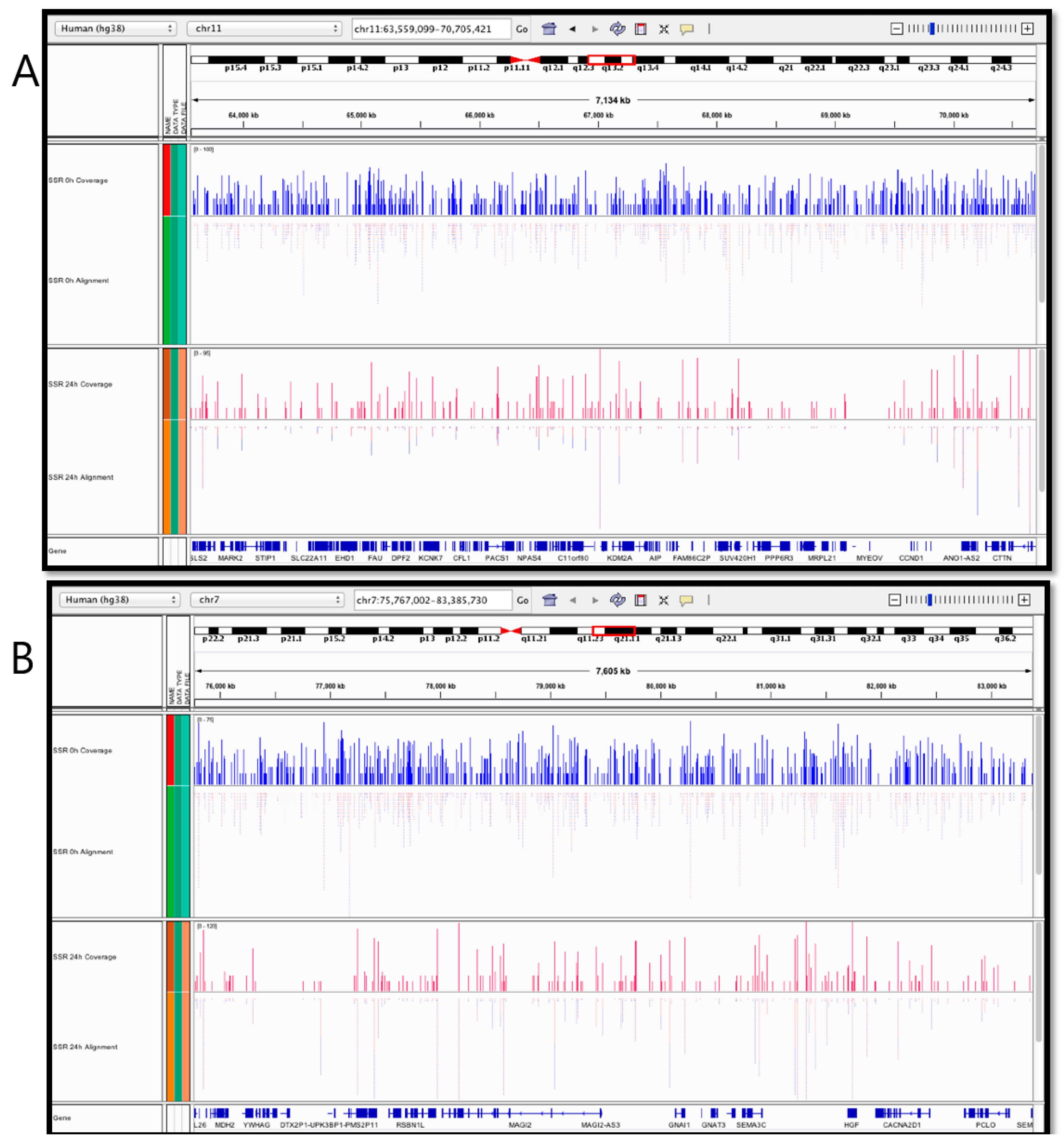
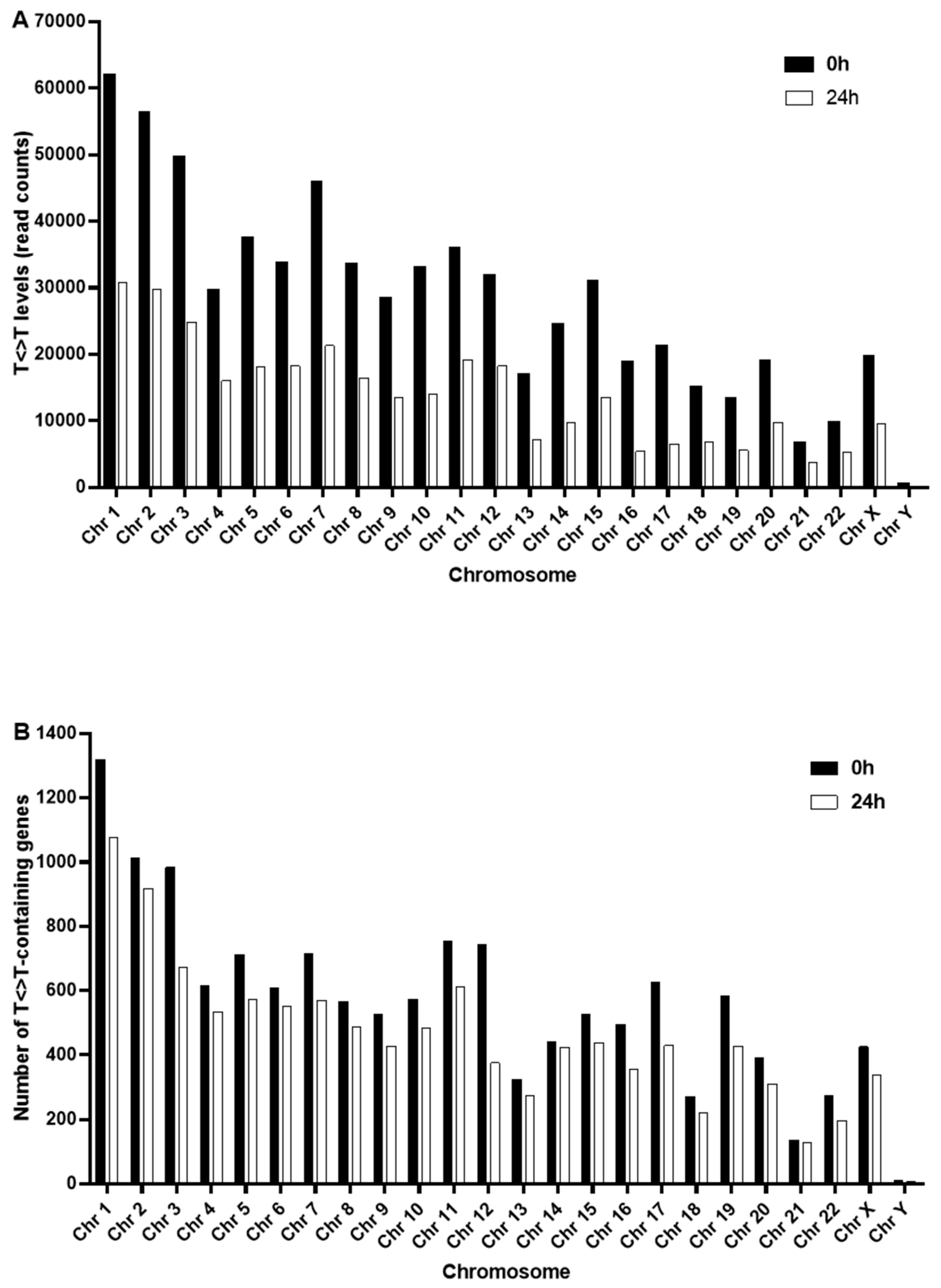
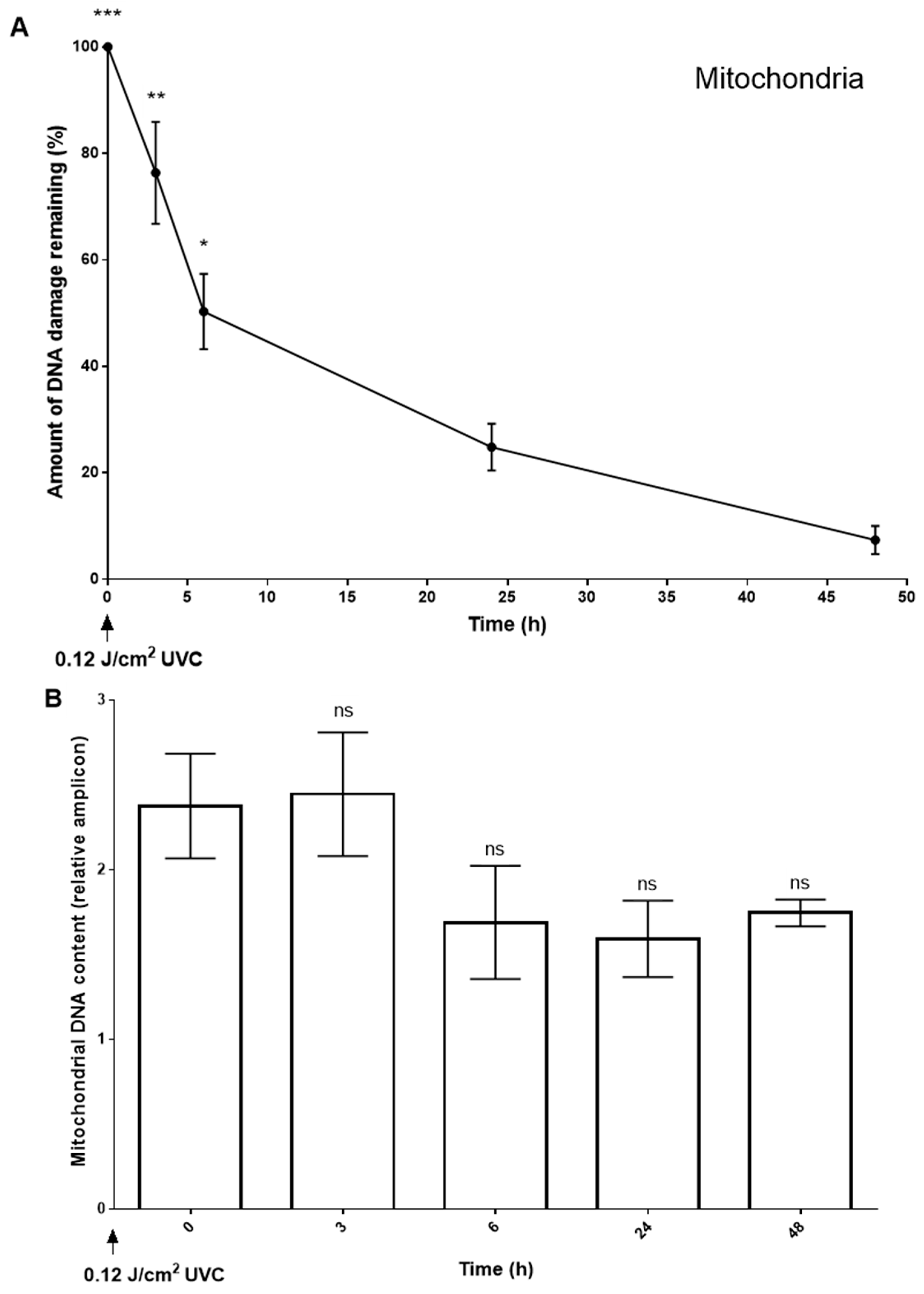
2.4. Nuclear and Mitochondrial Genome-Wide Mapping of T<>T Induction and Repair
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Culture
5.2. Cell Preparation and Treatment
5.3. Cell Viability
5.4. DNA Extraction and Preparation
5.5. DNA Shearing
5.6. DNA Immunoprecipitation (DDIP) Assay
5.7. Removal of CPD Adducts Prior to PCR and Next-Generation Sequencing
5.8. Quantitative PCR Analysis
5.9. MicroPlex Library Preparation™ Kit for Next-Generation Sequencing
5.10. Bioinformatic Analysis of NGS Data
5.11. Quantification of mtDNA Damage
5.12. Quantification of mtDNA Content
5.13. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Availability of Materials and Data
References
- Pfeifer, G.P.; Besaratinia, A. Mutational spectra of human cancer. Hum. Genet. 2009, 125, 493–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvam, E.; Tyrrell, R.M. Induction of oxidative DNA base damage in human skin cells by UV and near visible radiation. Carcinogenesis 1997, 18, 2379–2384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Premi, S.; Wallisch, S.; Mano, C.M.; Weiner, A.B.; Bacchiocchi, A.; Wakamatsu, K.; Bechara, E.J.; Halaban, R.; Douki, T.; Brash, D.E. Chemiexcitation of melanin derivatives induces DNA photoproducts long after UV exposure. Science 2015, 347, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Sancar, A. DNA excision repair. Annu. Rev. Biochem. 1996, 65, 43–81. [Google Scholar] [CrossRef]
- Wood, R.D. Nucleotide excision repair in mammalian cells. J. Biol. Chem. 1997, 272, 23465–23468. [Google Scholar] [CrossRef]
- Gale, J.M.; Nissen, K.A.; Smerdon, M.J. UV-induced formation of pyrimidine dimers in nucleosome core DNA is strongly modulated with a period of 10.3 bases. Proc. Natl. Acad. Sci. USA 1987, 84, 6644–6648. [Google Scholar] [CrossRef]
- Zavala, A.G.; Morris, R.T.; Wyrick, J.J.; Smerdon, M.J. High-resolution characterization of CPD hotspot formation in human fibroblasts. Nucleic Acids Res. 2014, 42, 893–905. [Google Scholar] [CrossRef]
- Waters, R.; van Eijk, P.; Reed, S. Histone modification and chromatin remodeling during NER. DNA Repair 2015, 36, 105–113. [Google Scholar] [CrossRef]
- Schick, S.; Fournier, D.; Thakurela, S.; Sahu, S.K.; Garding, A.; Tiwari, V.K. Dynamics of chromatin accessibility and epigenetic state in response to UV damage. J. Cell Sci. 2015, 128, 4380–4394. [Google Scholar] [CrossRef] [Green Version]
- Polak, P.; Lawrence, M.S.; Haugen, E.; Stoletzki, N.; Stojanov, P.; Thurman, R.E.; Garraway, L.A.; Mirkin, S.; Getz, G.; Stamatoyannopoulos, J.A.; et al. Reduced local mutation density in regulatory DNA of cancer genomes is linked to DNA repair. Nat. Biotechnol. 2014, 32, 71–75. [Google Scholar] [CrossRef]
- Tu, Y.; Tornaletti, S.; Pfeifer, G.P. DNA repair domains within a human gene: Selective repair of sequences near the transcription initiation site. EMBO J. 1996, 15, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Ruven, H.J.; Seelen, C.M.; Lohman, P.H.; van Kranen, H.; van Zeeland, A.A.; Mullenders, L.H. Strand-specific removal of cyclobutane pyrimidine dimers from the p53 gene in the epidermis of UVB-irradiated hairless mice. Oncogene 1994, 9, 3427–3432. [Google Scholar] [PubMed]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Bohr, V.A.; Okumoto, D.S.; Ho, L.; Hanawalt, P.C. Characterization of a DNA repair domain containing the dihydrofolate reductase gene in Chinese hamster ovary cells. J. Biol. Chem. 1986, 261, 16666–16672. [Google Scholar]
- You, Y.H.; Lee, D.H.; Yoon, J.H.; Nakajima, S.; Yasui, A.; Pfeifer, G.P. Cyclobutane pyrimidine dimers are responsible for the vast majority of mutations induced by UVB irradiation in mammalian cells. J. Biol. Chem. 2001, 276, 44688–44694. [Google Scholar] [CrossRef]
- Brash, D.E.; Haseltine, W.A. UV-induced mutation hotspots occur at DNA damage hotspots. Nature 1982, 298, 189–192. [Google Scholar] [CrossRef]
- Doll, R.; Peto, R. The causes of cancer: Quantitative estimates of avoidable risks of cancer in the United States today. J. Natl. Cancer Inst. 1981, 66, 1191–1308. [Google Scholar] [CrossRef]
- Mao, P.; Brown, A.J.; Esaki, S.; Lockwood, S.; Poon, G.M.K.; Smerdon, M.J.; Roberts, S.A.; Wyrick, J.J. ETS transcription factors induce a unique UV damage signature that drives recurrent mutagenesis in melanoma. Nat. Commun. 2018, 9, 2626. [Google Scholar] [CrossRef]
- Pfeifer, G.P.; Denissenko, M.F.; Tang, M.S. PCR-based approaches to adduct analysis. Toxicol. Lett. 1998, 102–103, 447–451. [Google Scholar] [CrossRef]
- Karakoula, A.; Evans, M.D.; Podmore, I.D.; Hutchinson, P.E.; Lunec, J.; Cooke, M.S. Quantification of UVR-induced DNA damage: Global-versus gene-specific levels of thymine dimers. J. Immunol. Methods 2003, 277, 27–37. [Google Scholar] [CrossRef]
- Ohno, M.; Miura, T.; Furuichi, M.; Tominaga, Y.; Tsuchimoto, D.; Sakumi, K.; Nakabeppu, Y. A genome-wide distribution of 8-oxoguanine correlates with the preferred regions for recombination and single nucleotide polymorphism in the human genome. Genome Res. 2006, 16, 567–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akatsuka, S.; Aung, T.T.; Dutta, K.K.; Jiang, L.; Lee, W.H.; Liu, Y.T.; Onuki, J.; Shirase, T.; Yamasaki, K.; Ochi, H.; et al. Contrasting genome-wide distribution of 8-hydroxyguanine and acrolein-modified adenine during oxidative stress-induced renal carcinogenesis. Am. J. Pathol. 2006, 169, 1328–1342. [Google Scholar] [CrossRef] [PubMed]
- Bryan, D.S.; Ransom, M.; Adane, B.; York, K.; Hesselberth, J.R. High resolution mapping of modified DNA nucleobases using excision repair enzymes. Genome Res. 2014, 24, 1534–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, P.; Smerdon, M.J.; Roberts, S.A.; Wyrick, J.J. Chromosomal landscape of UV damage formation and repair at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 2016, 113, 9057–9062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amente, S.; Di Palo, G.; Scala, G.; Castrignano, T.; Gorini, F.; Cocozza, S.; Moresano, A.; Pucci, P.; Ma, B.; Stepanov, I.; et al. Genome-wide mapping of 8-oxo-7,8-dihydro-2′-deoxyguanosine reveals accumulation of oxidatively-generated damage at DNA replication origins within transcribed long genes of mammalian cells. Nucleic Acids Res. 2019, 47, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.; Bennett, M.; Evans, K.E.; Zhuang-Jackson, H.; Higgs, A.; Reed, S.H.; Waters, R. A novel method for the genome-wide high resolution analysis of DNA damage. Nucleic Acids Res. 2011, 39, e10. [Google Scholar] [CrossRef]
- Hu, J.; Adar, S.; Selby, C.P.; Lieb, J.D.; Sancar, A. Genome-wide analysis of human global and transcription-coupled excision repair of UV damage at single-nucleotide resolution. Genes Dev. 2015, 29, 948–960. [Google Scholar] [CrossRef] [Green Version]
- Powell, J.R.; Bennett, M.R.; Evans, K.E.; Yu, S.; Webster, R.M.; Waters, R.; Skinner, N.; Reed, S.H. 3D-DIP-Chip: A microarray-based method to measure genomic DNA damage. Sci. Rep. 2015, 5, 7975. [Google Scholar] [CrossRef]
- Crosetto, N.; Mitra, A.; Silva, M.J.; Bienko, M.; Dojer, N.; Wang, Q.; Karaca, E.; Chiarle, R.; Skrzypczak, M.; Ginalski, K.; et al. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat. Methods 2013, 10, 361–365. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, M.; Jiang, L.; Akatsuka, S.; Suyama, M.; Toyokuni, S. Genome-wide profiling of 8-oxoguanine reveals its association with spatial positioning in nucleus. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2014, 21, 603–612. [Google Scholar] [CrossRef]
- Powell, J.R.; Bennett, M.; Waters, R.; Skinner, N.; Reed, S.H. Functional genome-wide analysis: A technical review, its developments and its relevance to cancer research. Recent Pat. DNA Gene Seq. 2013, 7, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdottir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimaldi, K.A.; McGurk, C.J.; McHugh, P.J.; Hartley, J.A. PCR-based methods for detecting DNA damage and its repair at the sub-gene and single nucleotide levels in cells. Mol. Biotechnol. 2002, 20, 181–196. [Google Scholar] [CrossRef]
- Ding, Y.; Fleming, A.M.; Burrows, C.J. Sequencing the Mouse Genome for the Oxidatively Modified Base 8-Oxo-7,8-dihydroguanine by OG-Seq. J. Am. Chem Soc. 2017, 139, 2569–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Adebali, O.; Adar, S.; Sancar, A. Dynamic maps of UV damage formation and repair for the human genome. Proc. Natl. Acad. Sci. USA 2017, 114, 6758–6763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Lieb, J.D.; Sancar, A.; Adar, S. Cisplatin DNA damage and repair maps of the human genome at single-nucleotide resolution. Proc. Natl. Acad. Sci. USA 2016, 113, 11507–11512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Evans, K.; van Eijk, P.; Bennett, M.; Webster, R.M.; Leadbitter, M.; Teng, Y.; Waters, R.; Jackson, S.P.; Reed, S.H. Global genome nucleotide excision repair is organized into domains that promote efficient DNA repair in chromatin. Genome Res. 2016, 26, 1376–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooke, M.S.; Evans, M.D. Reactive Oxygen Species: From DNA Damage to Disease. Sci. Med. 2010, 12, 98–111. [Google Scholar]
- Poetsch, A.R.; Boulton, S.J.; Luscombe, N.M. Genomic landscape of oxidative DNA damage and repair reveals regioselective protection from mutagenesis. Genome Biol. 2018, 19, 215. [Google Scholar] [CrossRef]
- Mitchell, D.L.; Jen, J.; Cleaver, J.E. Sequence specificity of cyclobutane pyrimidine dimers in DNA treated with solar (ultraviolet B) radiation. Nucleic Acids Res. 1992, 20, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Aruoma, O.I. DNA damage by oxygen-derived species. Its mechanism and measurement in mammalian systems. FEBS Lett. 1991, 281, 9–19. [Google Scholar] [CrossRef]
- Carrell, D.T. The clinical implementation of sperm chromosome aneuploidy testing: Pitfalls and promises. J. Androl. 2008, 29, 124–133. [Google Scholar] [CrossRef] [PubMed]
- McLachlan, R.I.; O’Bryan, M.K. Clinical Review#: State of the art for genetic testing of infertile men. J. Clin. Endocrinol. Metab. 2010, 95, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Hotaling, J.; Carrell, D.T. Clinical genetic testing for male factor infertility: Current applications and future directions. Andrology 2014, 2, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Stahl, P.J.; Masson, P.; Mielnik, A.; Marean, M.B.; Schlegel, P.N.; Paduch, D.A. A decade of experience emphasizes that testing for Y microdeletions is essential in American men with azoospermia and severe oligozoospermia. Fertil. Steril. 2010, 94, 1753–1756. [Google Scholar] [CrossRef] [PubMed]
- Bohr, V.A.; Smith, C.A.; Okumoto, D.S.; Hanawalt, P.C. DNA repair in an active gene: Removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell 1985, 40, 359–369. [Google Scholar] [CrossRef]
- Denissenko, M.F.; Pao, A.; Tang, M.; Pfeifer, G.P. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science 1996, 274, 430–432. [Google Scholar] [CrossRef]
- Strand, J.M.; Scheffler, K.; Bjoras, M.; Eide, L. The distribution of DNA damage is defined by region-specific susceptibility to DNA damage formation rather than repair differences. DNA Repair 2014, 18, 44–51. [Google Scholar] [CrossRef]
- Wauchope, O.R.; Mitchener, M.M.; Beavers, W.N.; Galligan, J.J.; Camarillo, J.M.; Sanders, W.D.; Kingsley, P.J.; Shim, H.N.; Blackwell, T.; Luong, T.; et al. Oxidative stress increases M1dG, a major peroxidation-derived DNA adduct, in mitochondrial DNA. Nucleic Acids Res. 2018, 46, 3458–3467. [Google Scholar] [CrossRef]
- Rothfuss, O.; Gasser, T.; Patenge, N. Analysis of differential DNA damage in the mitochondrial genome employing a semi-long run real-time PCR approach. Nucleic Acids Res. 2010, 38, e24. [Google Scholar] [CrossRef]
- Chimienti, G.; Picca, A.; Sirago, G.; Fracasso, F.; Calvani, R.; Bernabei, R.; Russo, F.; Carter, C.S.; Leeuwenburgh, C.; Pesce, V.; et al. Increased TFAM binding to mtDNA damage hot spots is associated with mtDNA loss in aged rat heart. Free Radic. Biol. Med. 2018, 124, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Lehle, S.; Hildebrand, D.G.; Merz, B.; Malak, P.N.; Becker, M.S.; Schmezer, P.; Essmann, F.; Schulze-Osthoff, K.; Rothfuss, O. LORD-Q: A long-run real-time PCR-based DNA-damage quantification method for nuclear and mitochondrial genome analysis. Nucleic Acids Res. 2014, 42, e41. [Google Scholar] [CrossRef] [PubMed]
- Boesch, P.; Weber-Lotfi, F.; Ibrahim, N.; Tarasenko, V.; Cosset, A.; Paulus, F.; Lightowlers, R.N.; Dietrich, A. DNA repair in organelles: Pathways, organization, regulation, relevance in disease and aging. Biochim. Biophys. Acta 2011, 1813, 186–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakouri, N.B.; Hou, Y.; Demarest, T.G.; Christiansen, L.S.; Okur, M.N.; Mohanty, J.G.; Croteau, D.L.; Bohr, V.A. Towards Understanding Genomic Instability, Mitochondrial Dysfunction and Aging. FEBS J. 2018. [Google Scholar] [CrossRef]
- Vasileiou, P.V.S.; Mourouzis, I.; Pantos, C. Principal Aspects Regarding the Maintenance of Mammalian Mitochondrial Genome Integrity. Int. J. Mol. Sci. 2017, 18, 1821. [Google Scholar] [CrossRef]
- Meyer, J.N.; Leuthner, T.C.; Luz, A.L. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 2017, 391, 42–53. [Google Scholar] [CrossRef]
- Karbaschi, M.; Macip, S.; Mistry, V.; Abbas, H.H.; Delinassios, G.; Evans, M.D.; Young, A.R.; Cooke, M.S. Rescue of cells from apoptosis increases repair in UVB exposed cells: Implications for the DNA damage response. Toxicol. Res. 2015, 4, 725–738. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]
- Ji, Y.; Karbaschi, M.; Cooke, M.S. Mycoplasma infection of cultured cells induces oxidative stress and attenuates cellular base excision repair activity. Mutat. Res. 2019, 845, 403054. [Google Scholar] [CrossRef]
- Cooke, M.S.; Podmore, I.D.; Mistry, N.; Evans, M.D.; Herbert, K.E.; Griffiths, H.R.; Lunec, J. Immunochemical detection of UV-induced DNA damage and repair. J. Immunol. Methods 2003, 280, 125–133. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.H.; Meyer, J.N.; Mandavilli, B.S.; Van Houten, B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 2006, 314, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Alrumaihi, F.A. Assessment of UVR-Induced DNA Damage and Repair in Nuclear Genome versus Mitochondrial Genome. Ph.D. Thesis, Department of Cancer Studies & Molecular Medicine, University of Leicester, Leicester, UK, 2016. [Google Scholar]
- Koch, H.; Wittern, K.P.; Bergemann, J. In human keratinocytes the Common Deletion reflects donor variabilities rather than chronologic aging and can be induced by ultraviolet A irradiation. J. Investig. Dermatol. 2001, 117, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.N.; Shahni, R.; Rodriguez-de-Ledesma, A.; Laftah, A.; Cunningham, P. Mitochondrial DNA as a non-invasive biomarker: Accurate quantification using real time quantitative PCR without co-amplification of pseudogenes and dilution bias. Biochem. Biophys. Res. Commun. 2011, 412, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.; Crowther, J.M.; Bulsara, P.A.; Wang, X.; Moore, D.J.; Birch-Machin, M.A. Optimised detection of mitochondrial DNA strand breaks. Mitochondrion 2018. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GAPDH Primer Sequence | |
| Forward | 5′-GCCCCCGGTTTCTATAAATTG-3′ |
| Reverse | 5′-GTCGAACAGGAGGAGCAGAGA-3′ |
| Myoglobin exon 2 Primer Sequence | |
| Forward | 5′-AAGTTTGACAAGTTCAAGCACCTG-3′ |
| Reverse | 5′-TGGCACCATGCTTCTTTAAGTC-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhegaili, A.S.; Ji, Y.; Sylvius, N.; Blades, M.J.; Karbaschi, M.; Tempest, H.G.; Jones, G.D.D.; Cooke, M.S. Genome-Wide Adductomics Analysis Reveals Heterogeneity in the Induction and Loss of Cyclobutane Thymine Dimers across Both the Nuclear and Mitochondrial Genomes. Int. J. Mol. Sci. 2019, 20, 5112. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205112
Alhegaili AS, Ji Y, Sylvius N, Blades MJ, Karbaschi M, Tempest HG, Jones GDD, Cooke MS. Genome-Wide Adductomics Analysis Reveals Heterogeneity in the Induction and Loss of Cyclobutane Thymine Dimers across Both the Nuclear and Mitochondrial Genomes. International Journal of Molecular Sciences. 2019; 20(20):5112. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205112
Chicago/Turabian StyleAlhegaili, Alaa S., Yunhee Ji, Nicolas Sylvius, Matthew J. Blades, Mahsa Karbaschi, Helen G. Tempest, George D. D. Jones, and Marcus S. Cooke. 2019. "Genome-Wide Adductomics Analysis Reveals Heterogeneity in the Induction and Loss of Cyclobutane Thymine Dimers across Both the Nuclear and Mitochondrial Genomes" International Journal of Molecular Sciences 20, no. 20: 5112. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205112