MicroRNA-29a Counteracts Glucocorticoid Induction of Bone Loss through Repressing TNFSF13b Modulation of Osteoclastogenesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
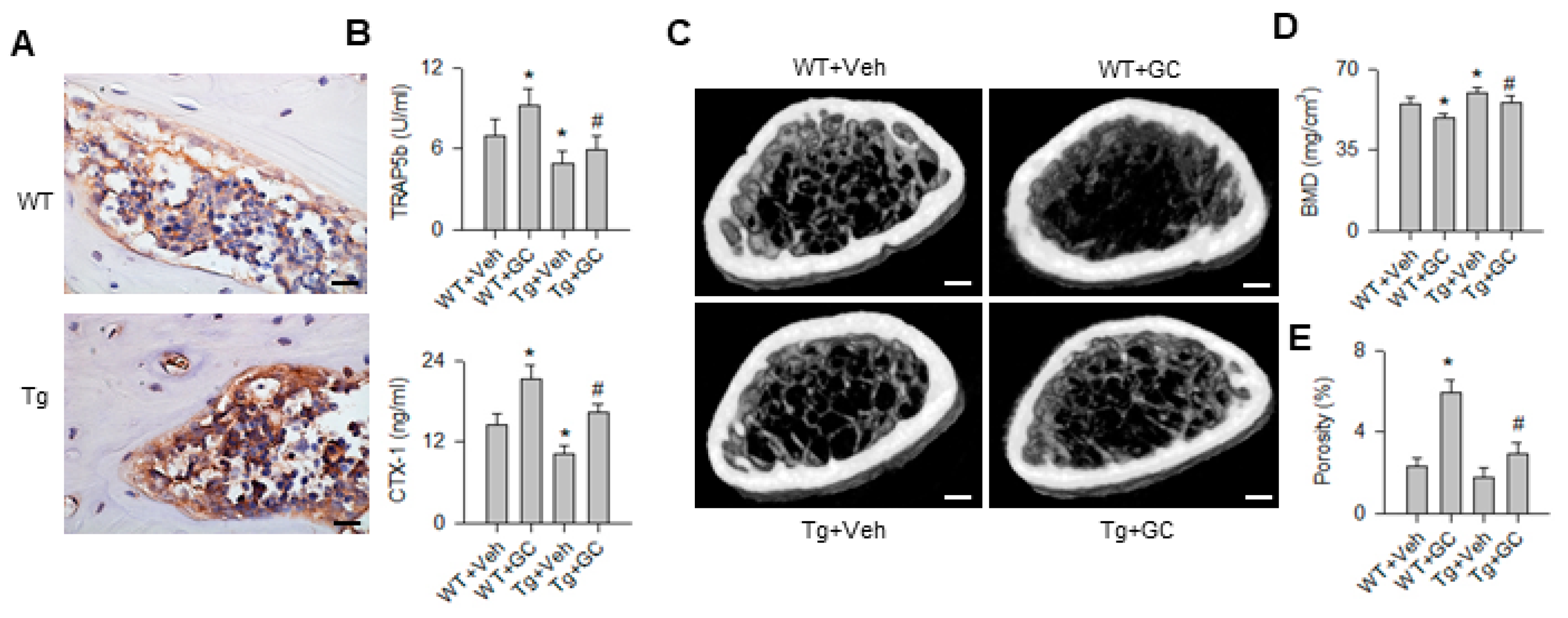
2.1. miR-29 Overexpression Compromised Glucocorticoid-Induced Bone Loss
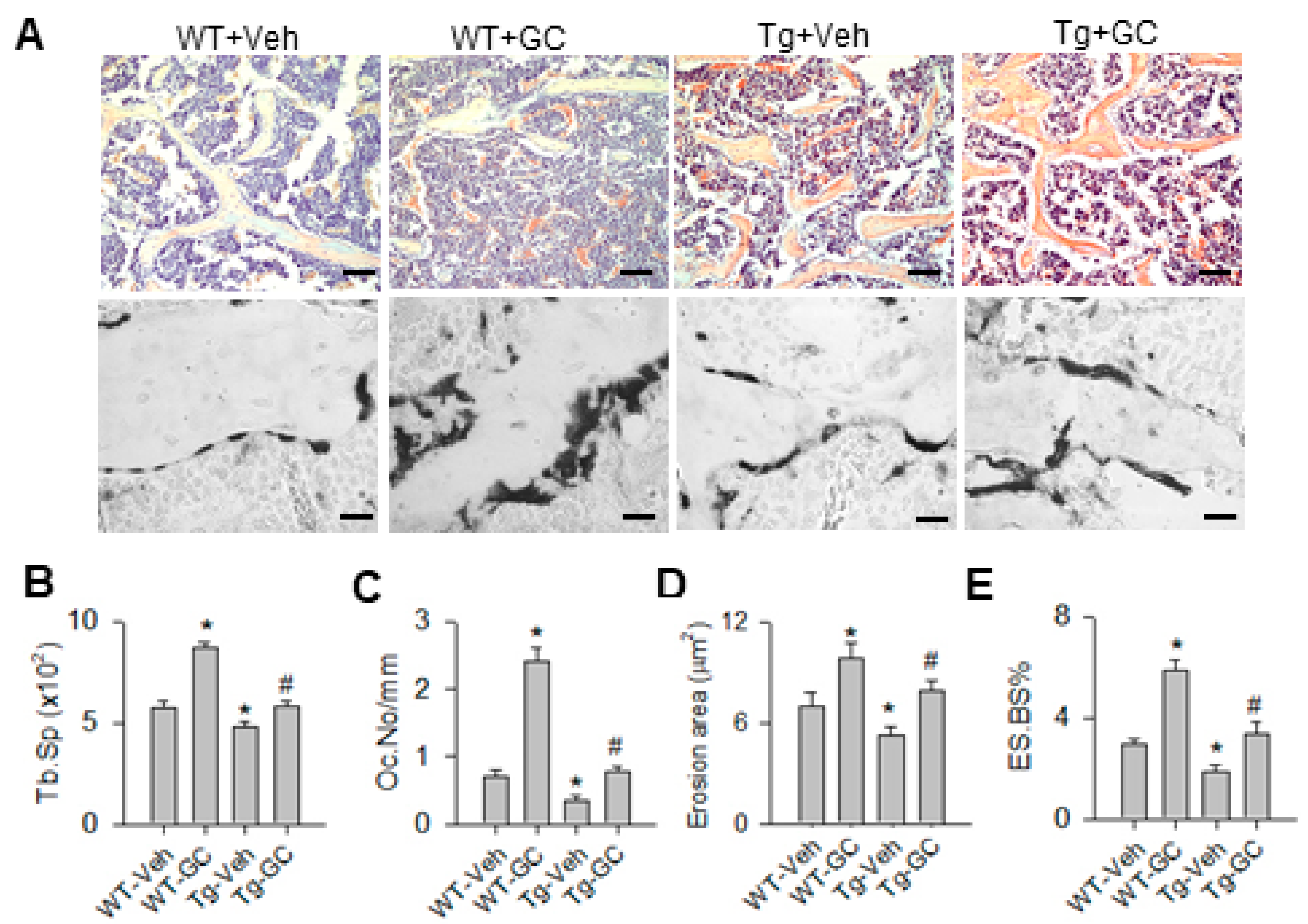
2.2. miR-29 Repressed the Glucocorticoid-Induced Osteoclastic Erosion Histopathology
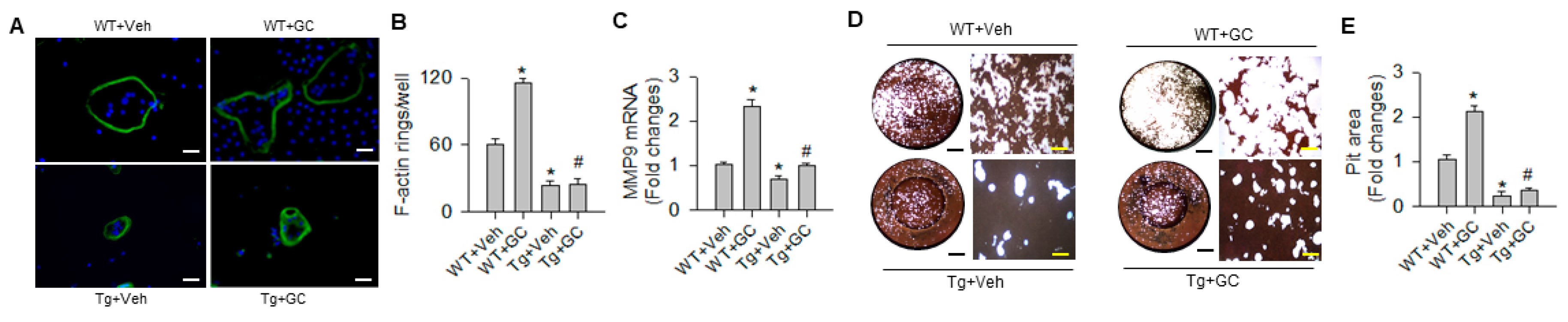
2.3. miR-29a Inhibited Osteoclast Differentiation and Resorption Capacity
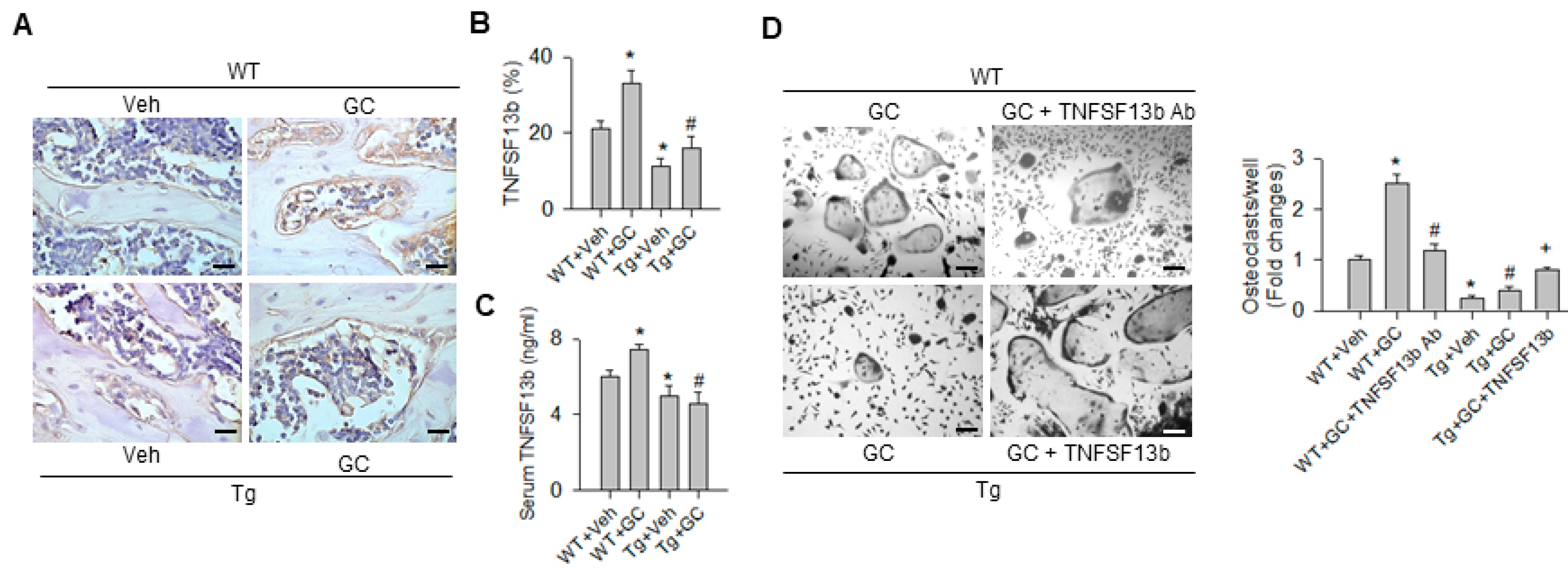
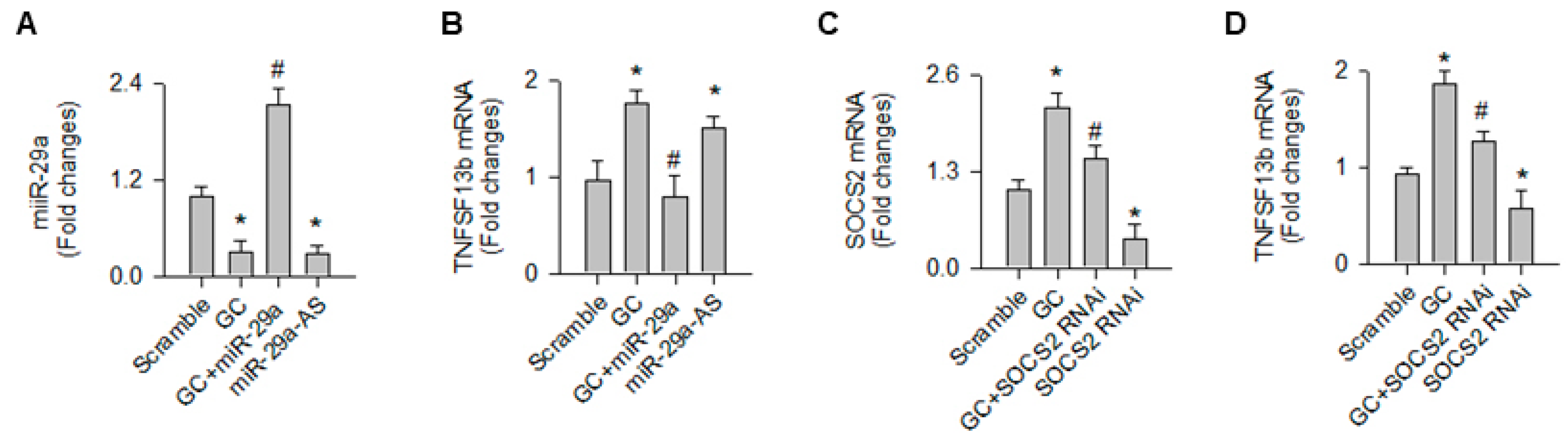
2.4. TNFSF13b-Mediated miR-29a Regulation of Osoteoclast Formation
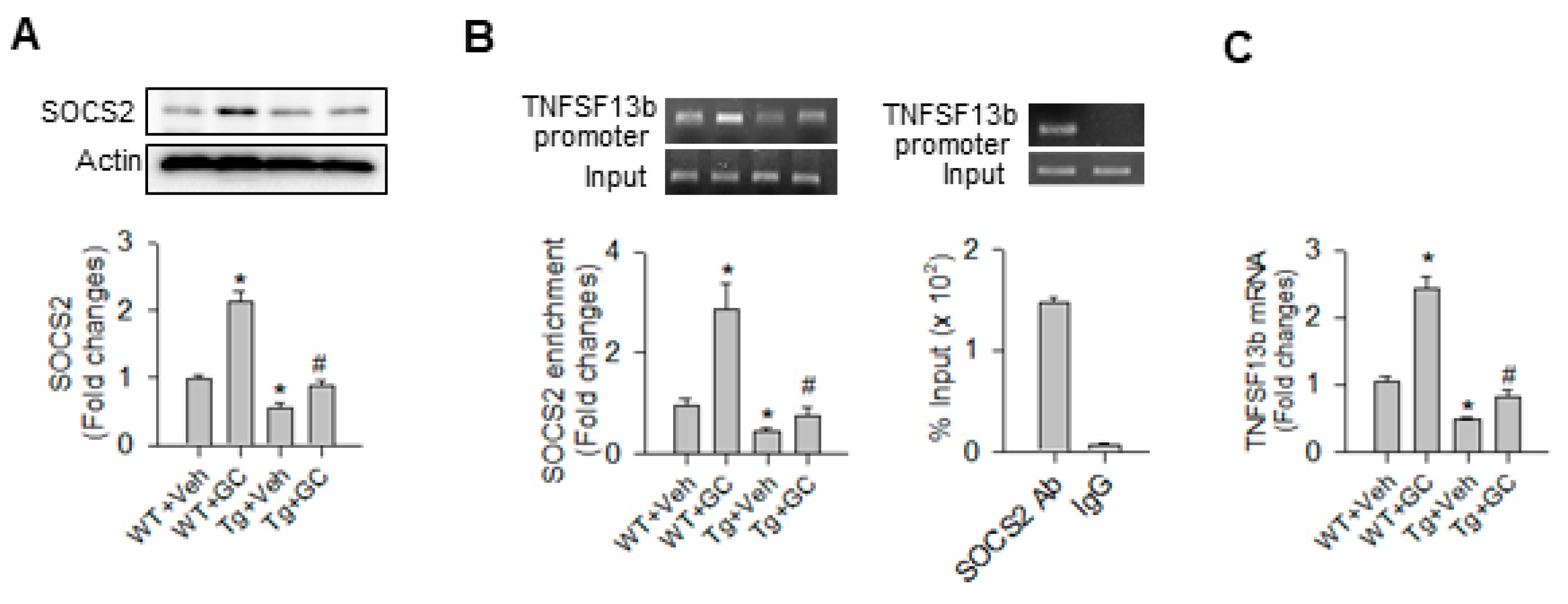
2.5. SOCS2 Controlled the miR-29a Inhibition of TNFSF13b Signaling
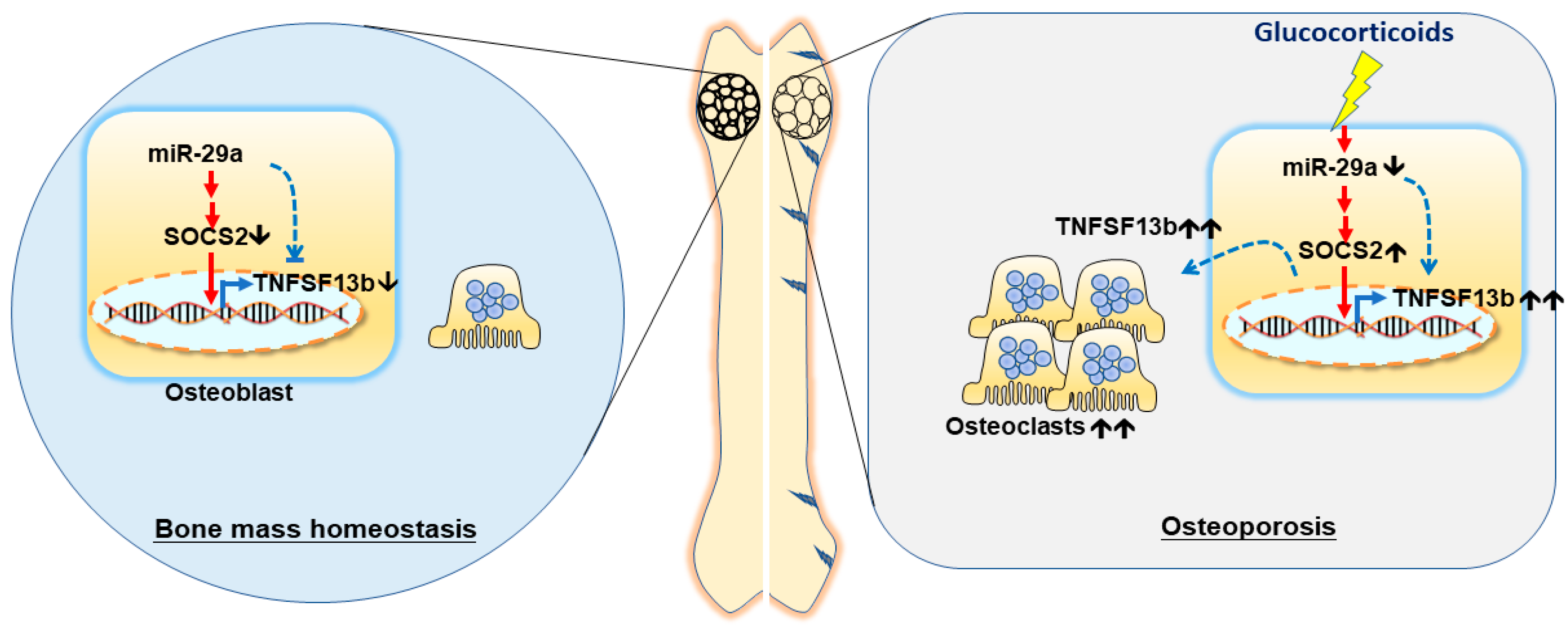
3. Discussion
4. Materials and Methods
4.1. miR-29a Transgenic Mice
4.2. Glucocorticoid-Induced Osteoporosis
4.3. Quantification of Serum Bone Resorption Markers
4.4. Assay of Bone Mass and Microstructure
4.5. In Situ Hybridization and Histomorphometry
4.6. Ex Vivo Osteoclast Differentiation and F-Actin Ring Immunofluorescence Labeling
4.7. Pit Formation
4.8. Quantitative RT-PCR
4.9. Immunoblotting
4.10. Chromatin Immuneprecipitation (ChIP)-PCR
4.11. Transfection
4.12. Statistical Analysis
Author Contributions
Finding
Acknowledgments
Conflicts of Interest
References
- Buckley, L.; Humphrey, M.B. Glucocorticoid-induced osteoporosis. N. Engl. J. Med. 2018, 379, 2547–2556. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.S.; Zhou, H.; Seibel, M.J.; Cooper, M.S. Glucocorticoid and bone: Consequences of endogenous and exogenous excess and replacement therapy. Endocr. Rev. 2018, 39, 519–548. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, R.; Biver, R. Glucocorticoid-induced osteoporosis: Who to treat with what agent. Nat. Rev. Rheumatol. 2015, 11, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Bellini, G.; Torella, M.; Manzo, I.; Tortora, C.; Luongo, L.; Punzo, F.; Cloacurci, N.; Nobili, B.; Maione, S.; Rossi, F. PKCβII-mediated cross-talk of TRPV1/CB2 modulates the glucocorticoid-induced osteoclast overactivity. Pharmacol. Res. 2017, 115, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, Y.; Kondo, H.; Noguchi, T.; Togari, A. Glucocorticoids mediate circadian timing in peripheral osteoclasts resulting in the circadian expression rhythm of osteoclast-related genes. Bone 2014, 61, 1–9. [Google Scholar] [CrossRef]
- Sabokbar, A.; Mahoney, D.J.; Hemingway, F.; Athanasou, N.A. Non-canonical (RANKL-independent) pathways of osteoclast differentiation and their role in musculoskeletal diseases. Clin. Rev. Allergy Immunol. 2016, 51, 16–26. [Google Scholar] [CrossRef]
- Jiang, P.; Gao, W.; Ma, T.; Wang, R.; Piao, Y.; Dong, X.; Wang, P.; Zhang, X.; Liu, Y.; Su, W.; et al. CD137 promotes bone metastasis of breast cancer by enhancing the migration and osteoclast differentiation of monocytes/macrophages. Theranostics 2019, 9, 2950–2966. [Google Scholar] [CrossRef]
- Brunetti, G.; Faienza, M.F.; Colaianni, G.; Gigante, I.; Oranger, A.; Pignataro, P.; Ingravallo, G.; Di Benedetto, A.; Bortolotti, S.; Di Comite, M.; et al. Impairment of bone remodeling in LIGHT/TNFSF14-deficient mice. J. Bone Miner. Res. 2018, 33, 704–719. [Google Scholar] [CrossRef]
- Hemingway, F.; Taylor, R.; Knowles, H.J.; Athanasou, N.A. RANKL-independent human osteoclast formation with APRIL, BAFF, NGF, IGF-I and IGF-II. Bone 2011, 48, 938–944. [Google Scholar] [CrossRef]
- Neri, P.; Kumar, S.; Fulciniti, M.T.; Vallet, S.; Chhetri, S.; Mukherjee, S.; Tai, Y.; Chauhan, D.; Tassone, P.; Venuta, S.; et al. Neutralizing B-cell factor antibody improves survival and inhibits osteoclastogenesis in a severe combined immunodeficient human multiple myeloma model. Clin. Cancer Res. 2007, 13, 5903–5909. [Google Scholar] [CrossRef]
- Van Meurs, J.B.; Boer, C.G.; Lopez-Delgado, L.; Riancho, J.A. Role of epigenomics in bone and cartilage disease. J. Bone Miner. Res. 2019, 34, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Chen, K.; Yuan, J.; Huang, P.; Xu, X.; Li, C.; Qian, N.; Qi, J.; Shao, Z.; Deng, L.; et al. Estrogen inhibits osteoclasts formation and bone resorption via microRNA-27a targeting PPARγ and APC. J. Cell Physiol 2018, 234, 581–594. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Shen, X.; Si, Y.; Fu, Y.; Zhu, W.; Xiao, T.; Fu, Z.; Zhang, P.; Cheng, J.; Jiang, H. MicroRNA-31a-5p from aging BMSCs links bone formation and resorption in the aged bone marrow microenvironment. Aging Cell 2018, 17, e12794. [Google Scholar] [CrossRef] [PubMed]
- Nakamachi, Y.; Ohnuma, K.; Uto, K.; Noguchi, Y.; Saegusa, J.; Kawano, S. MicroRNA-124 inhibits the progression of adjuvant-induced arthritis in rats. Ann. Rheum. Dis. 2016, 75, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Krzeszinski, J.Y.; Wei, W.; Huynh, H.; Jin, Z.; Wang, X.; Chang, T.C.; Xie, X.J.; He, L.; Mangala, L.S.; Lopez-Berestein, G.; et al. miR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature 2014, 512, 431–435. [Google Scholar] [CrossRef]
- Wei, W.; He, H.B.; Zhang, W.Y.; Zhang, H.X.; Bai, J.B.; Liu, H.Z.; Cao, J.H.; Chang, K.C.; Li, X.Y.; Zhao, S.H. miR-29 targets Akts to reduce proliferation and facilitate differentiation of myoblasts in skeletal muscle development. Cell Death Dis. 2013, 4, e668. [Google Scholar] [CrossRef]
- Guêrit, D.; Brondello, J.M.; Chuchana, P.; Philipot, D.; Toupet, K.; Bony, C.; Jorgensen, C.; Noël, D. FoxO3a regulation by miRNA-29a controls chondrogenic differentiation of mesenchymal stem cells and cartilage formation. Stem Cells Dev. 2014, 23, 1195–1205. [Google Scholar] [CrossRef]
- Parker, M.W.; Rossi, D.; Peterson, M.; Smith, K.; Sikstrom, K.; White, E.S.; Connett, J.E.; Henke, C.A.; Larsson, O.; Bitterman, P.B. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. J. Clin. Invest. 2014, 124, 1622–1635. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.M.; Guerau-de-Arellano, M.; Costinean, S.; Williams, J.L.; Bottoni, A.; Mayrikis Cox, G.; Satoskar, A.R.; Croce, C.M.; Racke, M.K.; Lovett-Racke, A.E.; et al. miR-29ab1 deficiency identifies a negative feedback loop controlling Th1 bias that is dysregulated in multiple sclerosis. J. Immunol. 2012, 189, 1567–1576. [Google Scholar] [CrossRef]
- Ma, F.; Xu, S.; Liu, X.; Zhang, Q.; Xu, X.; Liu, M.; Hua, M.; Li, N.; Yao, H.; Cao, X. The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-γ. Nat. Immunol. 2011, 12, 861–869. [Google Scholar] [CrossRef]
- Fukata, T.; Mizushima, T.; Nishimura, J.; Okuzaki, D.; Wu, X.; Hirose, H.; Yokoyama, Y.; Kubota, Y.; Nagata, K.; Tsujimura, N.; et al. The supercarbonate apatite-microRNA complex inhibits dextran sodium sulfate-induced colitis. Mol. Ther. Nucleic Acids 2018, 12, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.Y.; Chuang, P.C.; Su, W.H.; Ke, H.C.; Chen, Y.S.; Sun, Y.C.; Wang, F.S. MicroRNA-29a mitigates glucocorticoid induction of bone loss and fatty marrow by rescuing Runx2 acetylation. Bone 2015, 81, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Søe, K.; Dalaissé, J.M. Glucocorticoids maintain human osteoclasts in the active mode of their resorption cycle. J. Bone Mineral. Res. 2010, 25, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.Y.; Cregor, M.; McAndrews, K.; Li, T.; Condon, K.W.; Plotkin, L.; Bellido, T. Glucocorticoid-induced bone fragility is prevented in female mice by blocking Pyk2/Anoikis signaling. Endocrinology 2019, 160, 1659–1673. [Google Scholar] [CrossRef] [PubMed]
- Conway, H.H.; Henning, P.; Lie, A.; Tuckermann, J.; Lemer, U.H. Activation of dimer glucocorticoid receptors in osteoclast progenitors potentiates RANKL induced mature osteoclast bone resorbing activity. Bone 2016, 93, 43–54. [Google Scholar] [CrossRef]
- Lozano, C.; Duroux-Richard, I.; Firat, H.; Schordan, E.; Apparailly, F. MicroRNAs: Key regulators to understand osteoclast differentiation? Front. Immunol. 2019, 10, 375. [Google Scholar] [CrossRef]
- Liu, J.; Li, D.; Dang, L.; Liang, C.; Guo, B.; Lu, C.; He, X.; Cheung, H.Y.; He, B.; Liu, B.; et al. Osteoclastic miR-214 targets TRAF3 to contribute to osteolytic bone metastasis of breast cancer. Sci. Rep. 2017, 17, 40487. [Google Scholar] [CrossRef]
- Alam, I.; Oakes, D.K.; Reilly AMBillingsley, C.; Sbeta, S.; Gerard-O’Riley, R.L.; Acton, D.; Sato, A.; Bellido, T.; Econs, M.J. Overexpression of WNT16 does not prevent cortical bone loss due to glucocorticoid treatment in mice. JBMR Plus 2018, 3, e10084. [Google Scholar] [CrossRef]
- Piemontese, M.; Xiong, J.; Fujiwara, Y.; Thostenson, J.D.; O’Brien, C.A. Cortical bone loss caused by glucocorticoid excess requires RANKL production by osteocytes and is associated with reduced OPG expression in mice. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E587–E593. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Pitari, M.R.; Amodio, N.; Di Martino, M.T.; Conforti, F.; Leone, E.; Botta, C.; Paolino, F.M.; Del Giudice, T.; Iuliano, E.; et al. miR-29b negatively regulates human osteoclastic cell differentiation and function: Implications for the treatment of multiple myeloma-related bone disease. J. Cell Physiol. 2013, 228, 1506–1515. [Google Scholar] [CrossRef]
- Sul, O.J.; Rajasekaran, M.; Park, H.J.; Suh, J.H.; Choi, H.S. MicroRNA-29b enhances osteoclast survival by targeting BCL-2-modifying factor after lipopolysaccharide stimulation. Oxid. Med. Cell. Longev. 2019, 2019, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Franceschetti, T.; Kessler, C.B.; Lee, S.K.; Delany, A.M. miR-29 promotes murine osteoclastogenesis by regulating osteoclast commitment and migration. J. Biol. Chem. 2013, 288, 33347–33360. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.L.; Williams, J.O.; Bloom, A.C.; Singh, R.K.; Jordan, L.; Stone, M.D.; McCabe, L.R.; Wang, E.C.Y.; Williams, A.S. CCL3 and MMP-9 are induced by TL1A during death receptor 3 (TNFRSF25)-dependent osteoclast function and systemic bone loss. Bone 2017, 97, 94–104. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.T.; Acharya, C.; An, G.; Moschetta, M.; Zhong, M.Y.; Feng, X.; Cea, M.; Cagnetta, A.; Wen, K.; van Eenennaam, H.; et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood 2016, 127, 3225–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spences, S.; Fitzsimons, A.; Boyd, C.R.; Kessler, J.; Fitzgerald, D.; Elliott, J.; Gabhann, J.N.; Smith, S.; Sica, A.; Hams, E.; et al. Suppression of cytokine signaling 2 and 3 diametrically control macrophage polarization. Immunity 2013, 38, 66–78. [Google Scholar] [CrossRef]
- Pass, C.; MacRae, V.E.; Huesa, C.; Faisal Ahmed, S.; Farquharson, C. SOCS2 is the critical regulator GH action in murine growth plate chondrogenesis. J. Bone Miner. Res. 2012, 27, 1055–1066. [Google Scholar] [CrossRef]
- Dobie, R.; Ahmed, S.F.; Staines, K.A.; Pass, C.; Jasim, S.; MacRae, V.E.; Farquharson, C. Increased linear bone growth by GH in the absence of SOCS2 is independent of IGF-1. J. Cell Physiol. 2015, 230, 2796–2806. [Google Scholar] [CrossRef] [Green Version]
- Moghaddam, A.S.; Afshari, J.T.; Esmaeili, S.A.; Saburi, E.; Joneidi, Z.; Momtazi-Borojeni, A.A. Cardioprotective microRNAs: Lessons from stem cell-derived exosomal microRNAs to treat cardiovascular disease. Atherosclerosis 2019, 285, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Oiao, B.; Gao, N.; Lin, N.; He, W. Oral squamous cell carcinoma-derived exosomes promote M2 subtype macrophage polarization mediated by exosome-enclosed miR-29a-3p. Am. J. Physiol. Cell Physiol. 2019, 316, C731–C740. [Google Scholar] [CrossRef]
- Korabecna, M.; Koutova, L.; Tesarova, P. The potential roles of vesicle-enclosed miRNAs in communication between macrophages and cancer cells in tumor microenvironment. Neoplasma 2017, 64, 406–411. [Google Scholar] [CrossRef]
- Asano, J.; Nakano, A.; Oda, A.; Amou, H.; Hiasa, M.; Takeuchi, K.; Miki, H.; Nakamura, S.; Harada, T.; Fuji, S.; et al. The serine/threonine kinase Pim-2 is a novel anti-apoptotic mediator in myeloma cells. Leukemia 2011, 25, 1182–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Zhao, H.; Kitaura, H.; Bhattacharyya, S.; Brewer, J.A.; Muglia, L.J.; Ross, F.P.; Teitelbaum, S.L. Glucocorticoids suppress bone formation via the osteoclast. J. Clin. Invest. 2006, 116, 2152–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dempster, D.W.; Compston, J.E.; Drezner, M.K.; Glorieux, F.H.; Kanis, J.A.; Malluche, H.; Meunier, P.J.; Ott, S.M.; Recker, R.R.; Parfitt, A.M. Standardized nomenclature, symbols, and units for bone histomorphometry: A 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner Res. 2013, 28, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.M.; Lee, S.Y.; Her, Y.M.; Ryu, J.G.; Kim, E.K.; Son, H.J.; Kwok, S.K.; Ju, J.H.; Yang, C.W.; Park, S.H.; et al. Gene associated with retinoid-interferon-induced mortality 19 attenuates murine autoimmune arthritis by regulation of th17 and treg cells. Arthritis Rheumatol 2014, 66, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Ferron, M.; Settembre, C.; Shimazu, J.; Lacombe, J.; Kato, S.; Rawlings, D.J.; Ballabio, A.; Karsenty, G. A RANKL-PKCβ-RFEB signaling cascade is necessary for lysosomal biogenesis in osteoclasts. Gene. Dev. 2013, 27, 955–969. [Google Scholar] [CrossRef]
- Lian, W.S.; Ko, J.Y.; Wu, R.W.; Sun, Y.C.; Chen, Y.S.; Wu, S.L.; Weng, L.H.; Jahr, H.; Wang, F.S. MicroRNA-128 repressed chondrocyte autophagy exacerbates knee osteoarthritis by disrupting Atg12. Cell Death Dis. 2018, 9, 919. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, R.-W.; Lian, W.-S.; Chen, Y.-S.; Kuo, C.-W.; Ke, H.-C.; Hsieh, C.-K.; Wang, S.-Y.; Ko, J.-Y.; Wang, F.-S. MicroRNA-29a Counteracts Glucocorticoid Induction of Bone Loss through Repressing TNFSF13b Modulation of Osteoclastogenesis. Int. J. Mol. Sci. 2019, 20, 5141. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205141
Wu R-W, Lian W-S, Chen Y-S, Kuo C-W, Ke H-C, Hsieh C-K, Wang S-Y, Ko J-Y, Wang F-S. MicroRNA-29a Counteracts Glucocorticoid Induction of Bone Loss through Repressing TNFSF13b Modulation of Osteoclastogenesis. International Journal of Molecular Sciences. 2019; 20(20):5141. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205141
Chicago/Turabian StyleWu, Re-Wen, Wei-Shiung Lian, Yu-Shan Chen, Chung-Wen Kuo, Huei-Ching Ke, Chin-Kuei Hsieh, Shao-Yu Wang, Jih-Yang Ko, and Feng-Sheng Wang. 2019. "MicroRNA-29a Counteracts Glucocorticoid Induction of Bone Loss through Repressing TNFSF13b Modulation of Osteoclastogenesis" International Journal of Molecular Sciences 20, no. 20: 5141. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205141