Fetuin-A Inhibits Placental Cell Growth and Ciliogenesis in Gestational Diabetes Mellitus


Abstract
:1. Introduction
2. Results
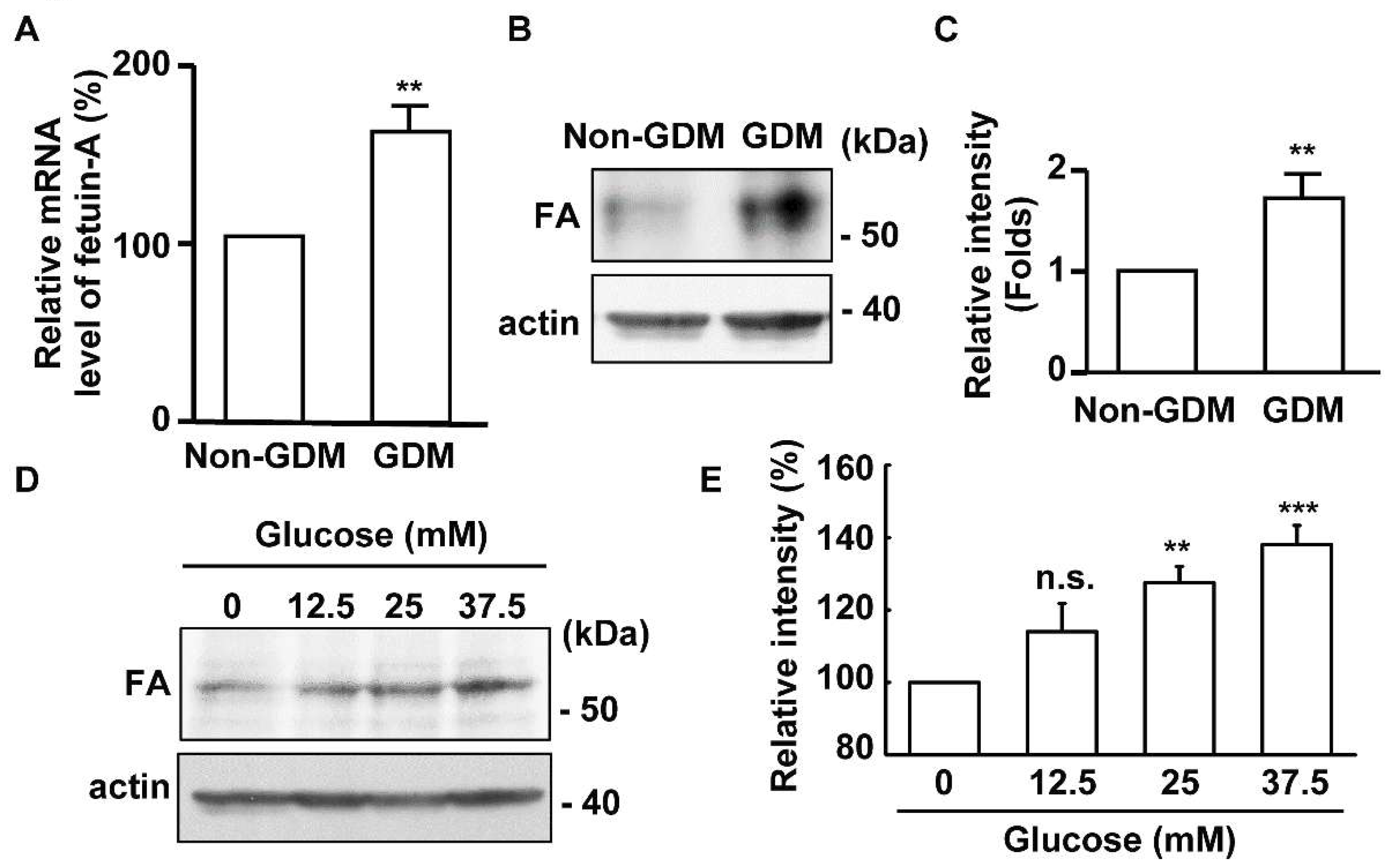
2.1. The Expression of Fetuin-A Is Upregulated in the Placentas of Gestational Diabetes Mellitus (GDM) Patients
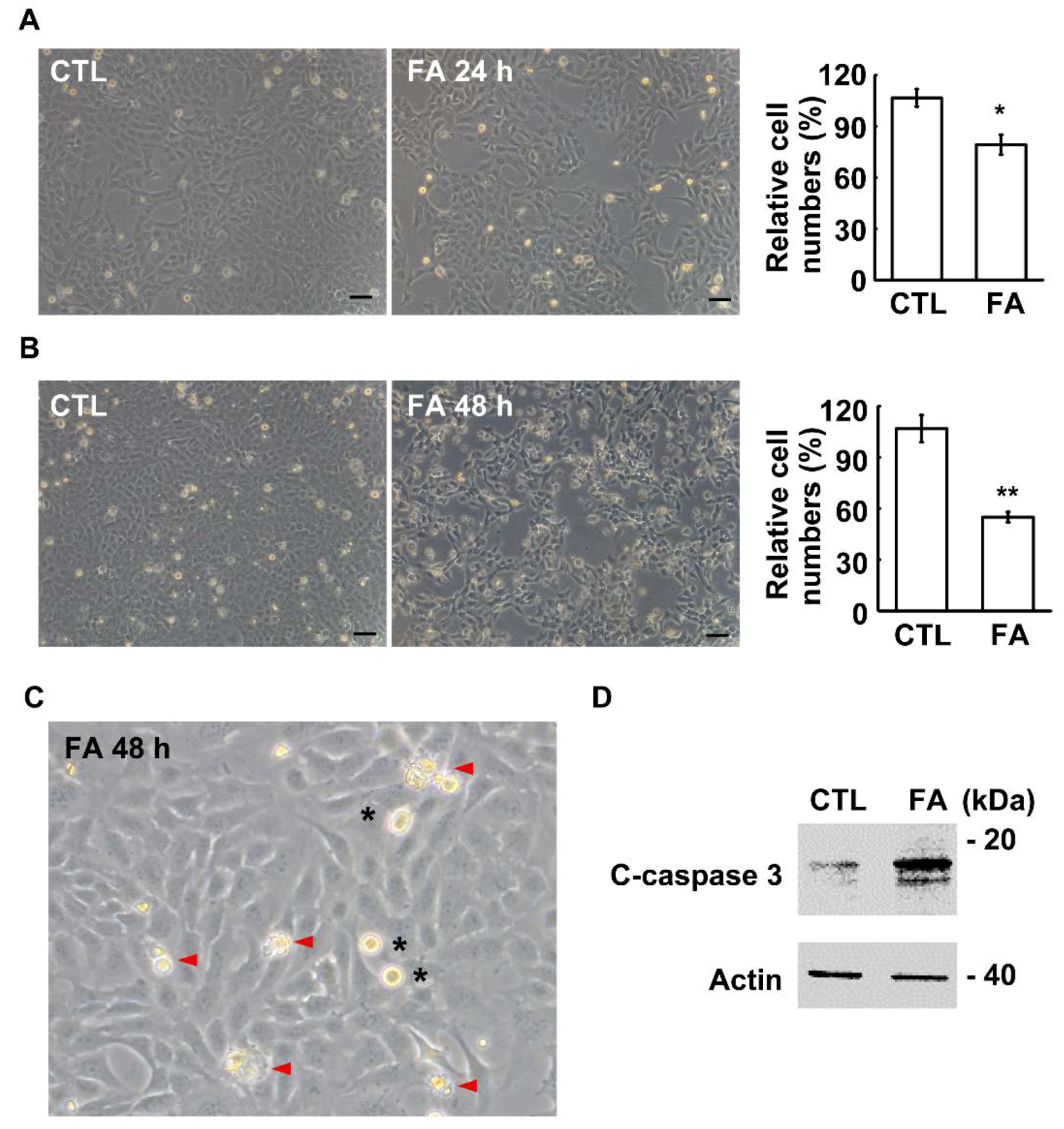
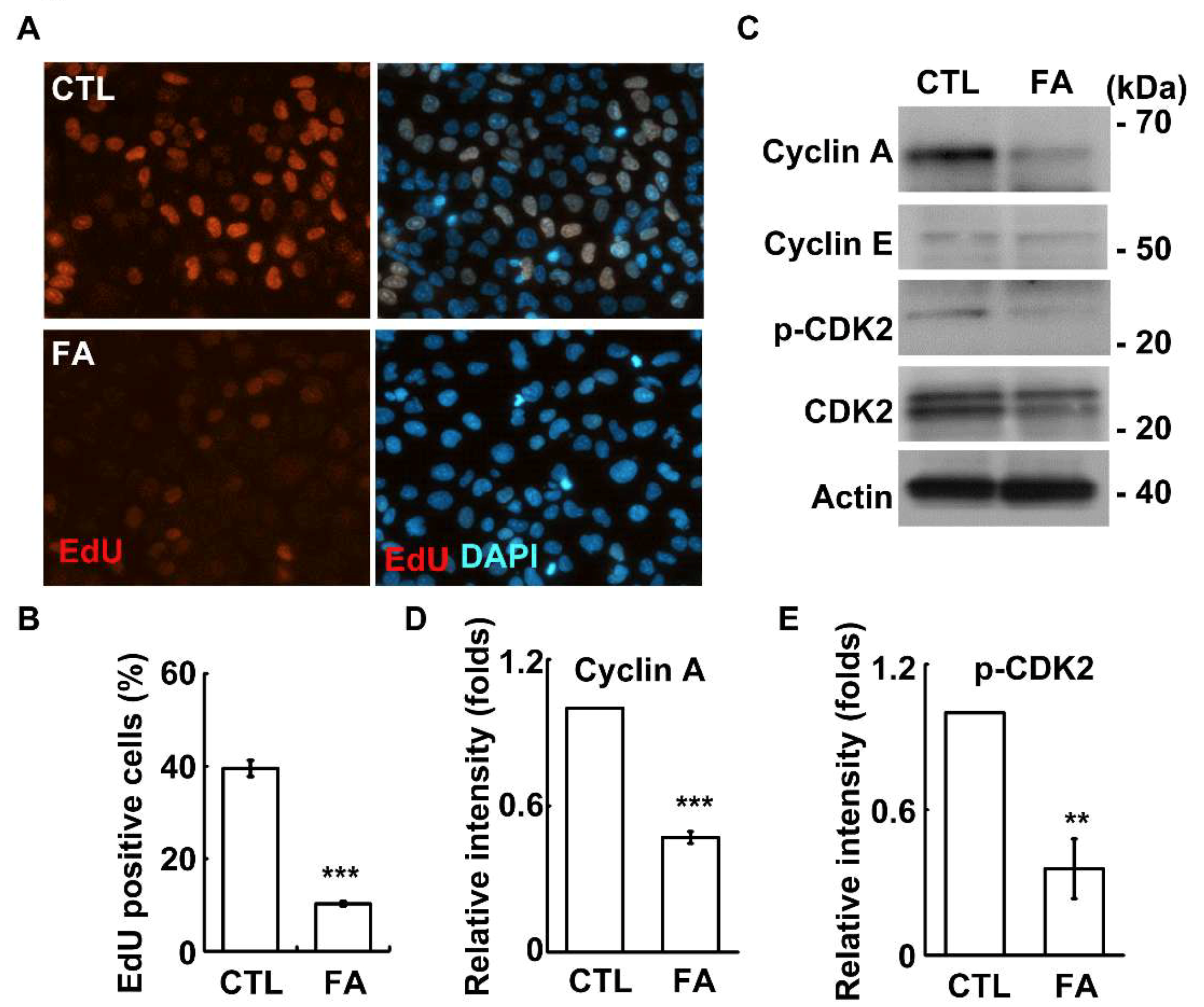
2.2. Fetuin-A Inhibits Placental Cell Growth
2.3. Fetuin-A Induces Centrosome Amplification
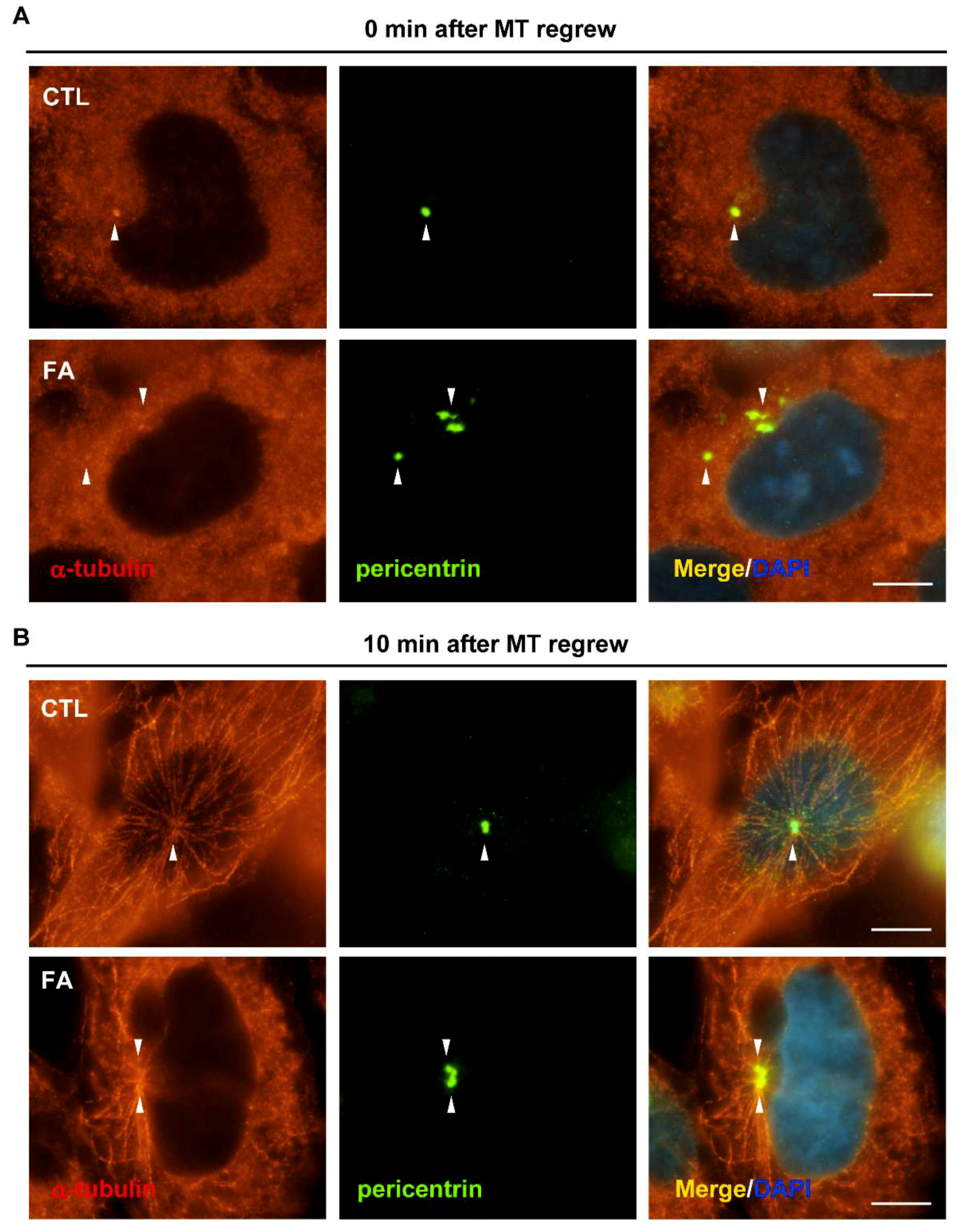
2.4. Fetuin-A Leads to Disorganized Microtubule Nucleation
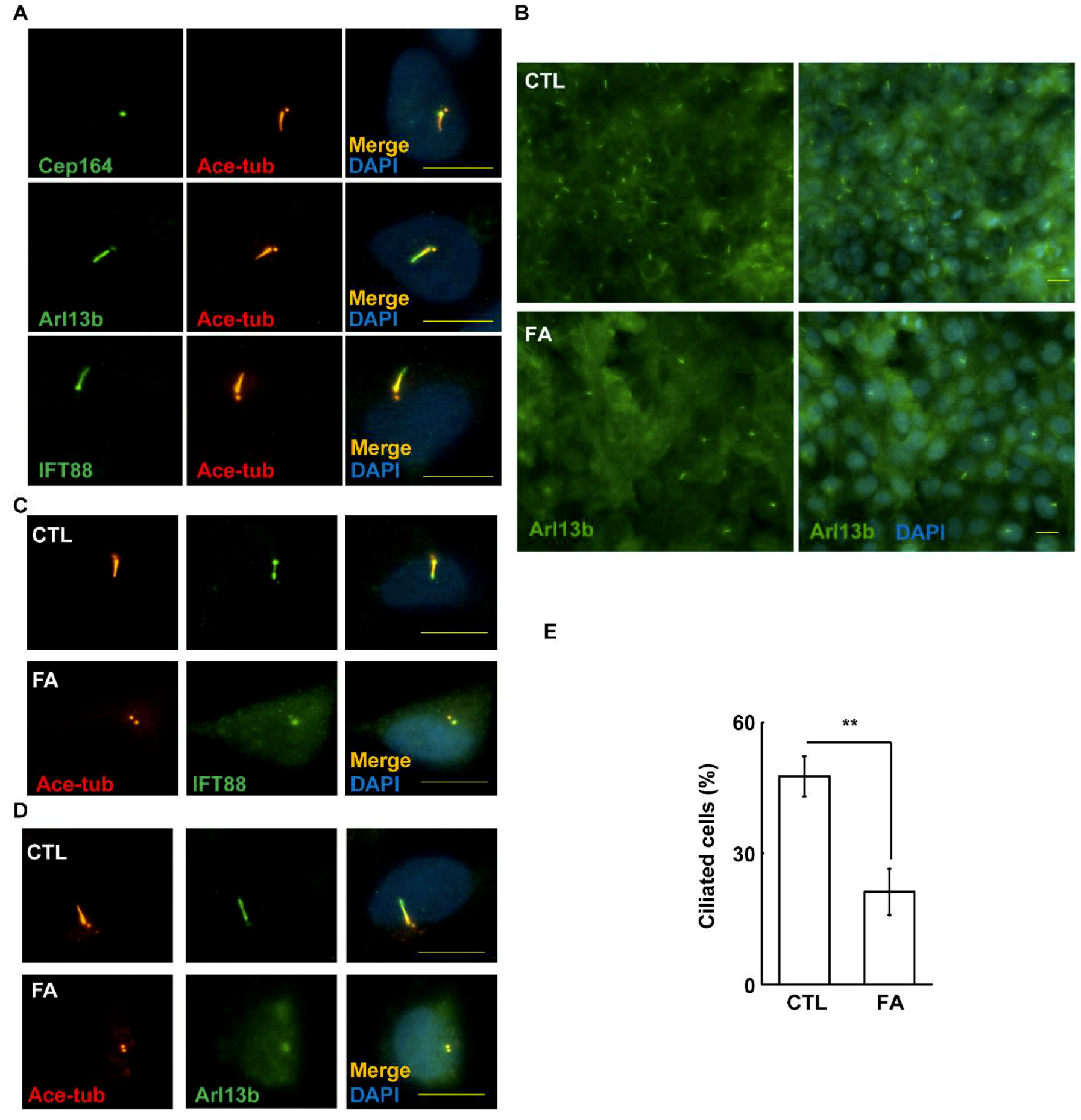
2.5. Fetuin-A Inhibits Primary Cilium Formation in Placental Cells
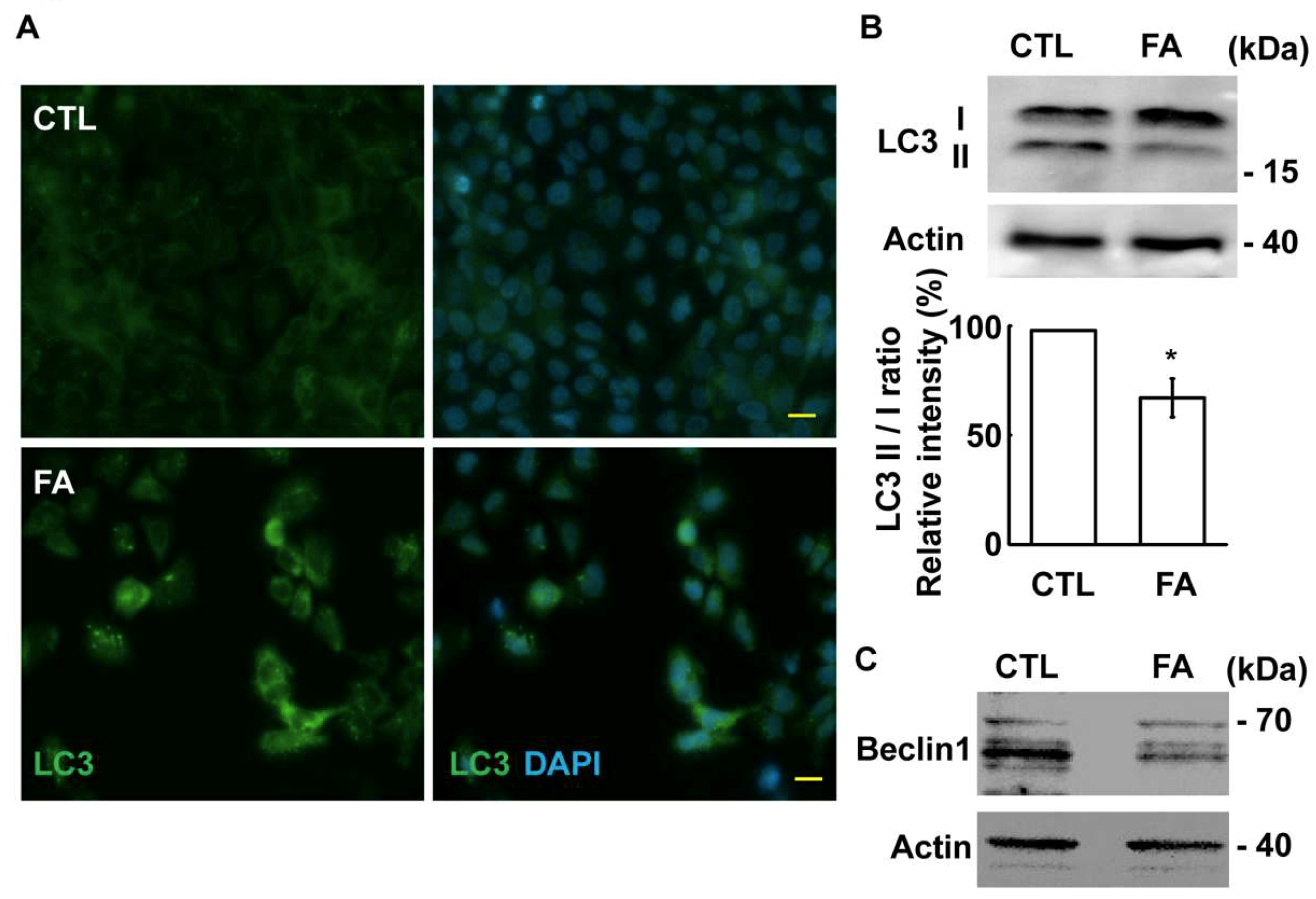
2.6. Fetuin-A Inhibits Autophagic Flux
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Study Population and Sample Collection
4.3. Immunofluorescence Microscopy
4.4. Microtubule Regrowth Assay
4.5. EdU Incorporation Assay
4.6. Cell Growth Assay
4.7. Antibodies
4.8. Statistical Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Melchior, H.; Kurch-Bek, D.; Mund, M. The Prevalence of Gestational Diabetes. Dtsch. Arztebl. Int. 2017, 114, 412–418. [Google Scholar] [PubMed] [Green Version]
- Chiefari, E.; Arcidiacono, B.; Foti, D.; Brunetti, A. Gestational diabetes mellitus: An updated overview. J. Endocrinol. Investig. 2017, 40, 899–909. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. 14. Management of Diabetes in Pregnancy: Standards of Medical Care in Diabetes-2019. Diabetes Care 2019, 42, S165–S172. [Google Scholar] [CrossRef] [PubMed]
- Bryson, C.L.; Ioannou, G.N.; Rulyak, S.J.; Critchlow, C. Association between gestational diabetes and pregnancy-induced hypertension. Am. J. Epidemiol. 2003, 158, 1148–1153. [Google Scholar] [CrossRef]
- Zhang, C.; Rawal, S.; Chong, Y.S. Risk factors for gestational diabetes: Is prevention possible? Diabetologia 2016, 59, 1385–1390. [Google Scholar] [CrossRef]
- Holmes, V.A.; Young, I.S.; Patterson, C.C.; Pearson, D.W.; Walker, J.D.; Maresh, M.J.; McCance, D.R. Diabetes and Pre-eclampsia Intervention Trial Study Group. Optimal glycemic control, pre-eclampsia, and gestational hypertension in women with type 1 diabetes in the diabetes and pre-eclampsia intervention trial. Diabetes Care 2011, 34, 1683–1688. [Google Scholar] [CrossRef]
- Dabelea, D.; Hanson, R.L.; Lindsay, R.S.; Pettitt, D.J.; Imperatore, G.; Gabir, M.M.; Roumain, J.; Bennett, P.H.; Knowler, W.C. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: A study of discordant sibships. Diabetes 2000, 49, 2208–2211. [Google Scholar] [CrossRef]
- Mori, K.; Emoto, M.; Inaba, M. Fetuin-A: A multifunctional protein. Recent Pat. Endocr. Metab. Immune Drug Discov. 2011, 5, 124–146. [Google Scholar] [CrossRef]
- Stefan, N.; Hennige, A.M.; Staiger, H.; Machann, J.; Schick, F.; Krober, S.M.; Machicao, F.; Fritsche, A.; Haring, H.U. Alpha2-Heremans-Schmid glycoprotein/fetuin-A is associated with insulin resistance and fat accumulation in the liver in humans. Diabetes Care 2006, 29, 853–857. [Google Scholar] [CrossRef]
- Ou, H.Y.; Yang, Y.C.; Wu, H.T.; Wu, J.S.; Lu, F.H.; Chang, C.J. Increased fetuin-A concentrations in impaired glucose tolerance with or without nonalcoholic fatty liver disease, but not impaired fasting glucose. J. Clin. Endocrinol. Metab. 2012, 97, 4717–4723. [Google Scholar] [CrossRef]
- Jahnen-Dechent, W.; Heiss, A.; Schafer, C.; Ketteler, M. Fetuin-A regulation of calcified matrix metabolism. Circ. Res. 2011, 108, 1494–1509. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, P.R.; Wagner, A.S.; Reddy, L.V.; Deutsch, D.D.; Leon, M.A.; Goustin, A.S.; Grunberger, G. Serum alpha 2-HS-glycoprotein is an inhibitor of the human insulin receptor at the tyrosine kinase level. Mol. Endocrinol. 1993, 7, 1445–1455. [Google Scholar] [PubMed]
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 2012, 18, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Goustin, A.S.; Derar, N.; Abou-Samra, A.B. Ahsg-fetuin blocks the metabolic arm of insulin action through its interaction with the 95-kD beta-subunit of the insulin receptor. Cell. Signal. 2013, 25, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Ou, H.Y.; Yang, Y.C.; Wu, H.T.; Wu, J.S.; Lu, F.H.; Chang, C.J. Serum fetuin-A concentrations are elevated in subjects with impaired glucose tolerance and newly diagnosed type 2 diabetes. Clin. Endocrinol. 2011, 75, 450–455. [Google Scholar] [CrossRef]
- Iyidir, O.T.; Degertekin, C.K.; Yilmaz, B.A.; Altinova, A.E.; Toruner, F.B.; Bozkurt, N.; Ayvaz, G.; Akturk, M. Serum levels of fetuin A are increased in women with gestational diabetes mellitus. Arch. Gynecol. Obstet. 2015, 291, 933–937. [Google Scholar] [CrossRef]
- Nigg, E.A.; Holland, A.J. Once and only once: Mechanisms of centriole duplication and their deregulation in disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 297–312. [Google Scholar] [CrossRef]
- Chen, T.Y.; Syu, J.S.; Han, T.Y.; Cheng, H.L.; Lu, F.I.; Wang, C.Y. Cell Cycle-Dependent Localization of Dynactin Subunit p150 glued at Centrosome. J. Cell. Biochem. 2015, 116, 2049–2060. [Google Scholar] [CrossRef]
- Chen, T.Y.; Syu, J.S.; Lin, T.C.; Cheng, H.L.; Lu, F.L.; Wang, C.Y. Chloroquine alleviates etoposide-induced centrosome amplification by inhibiting CDK2 in adrenocortical tumor cells. Oncogenesis 2015, 4, e180. [Google Scholar] [CrossRef]
- Lattao, R.; Kovacs, L.; Glover, D.M. The Centrioles, Centrosomes, Basal Bodies, and Cilia of Drosophila melanogaster. Genetics 2017, 206, 33–53. [Google Scholar] [CrossRef]
- Chen, T.Y.; Lien, W.C.; Cheng, H.L.; Kuan, T.S.; Sheu, S.Y.; Wang, C.Y. Chloroquine inhibits human retina pigmented epithelial cell growth and microtubule nucleation by downregulating p150(glued). J. Cell. Physiol. 2019, 234, 10445–10457. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.A.; Malicki, J.J. The Nuclear Arsenal of Cilia. Dev. Cell 2019, 49, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Tsai, H.L.; Syu, J.S.; Chen, T.Y.; Su, M.T. Primary Cilium-Regulated EG-VEGF Signaling Facilitates Trophoblast Invasion. J. Cell. Physiol. 2017, 232, 1467–1477. [Google Scholar] [CrossRef] [PubMed]
- Kalabay, L.; Cseh, K.; Pajor, A.; Baranyi, E.; Csakany, G.M.; Melczer, Z.; Speer, G.; Kovacs, M.; Siller, G.; Karadi, I.; et al. Correlation of maternal serum fetuin/alpha2-HS-glycoprotein concentration with maternal insulin resistance and anthropometric parameters of neonates in normal pregnancy and gestational diabetes. Eur. J. Endocrinol. 2002, 147, 243–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez, L.M.; Anton, L.; Srinivas, S.K.; Elovitz, M.A.; Parry, S. Effects of increased fetuin-A in human trophoblast cells and associated pregnancy outcomes. Am. J. Obstet. Gynecol. 2012, 207, 484.e1–484.e8. [Google Scholar] [CrossRef] [PubMed]
- Kralisch, S.; Hoffmann, A.; Lossner, U.; Kratzsch, J.; Bluher, M.; Stumvoll, M.; Fasshauer, M.; Ebert, T. Regulation of the novel adipokines/hepatokines fetuin A and fetuin B in gestational diabetes mellitus. Metab. Clin. Exp. 2017, 68, 88–94. [Google Scholar] [CrossRef]
- Wang, C.Y.; Tsai, P.Y.; Chen, T.Y.; Tsai, H.L.; Kuo, P.L.; Su, M.T. Elevated miR-200a and miR-141 inhibit endocrine gland-derived vascular endothelial growth factor expression and ciliogenesis in preeclampsia. J. Physiol. 2019, 597, 3069–3083. [Google Scholar] [CrossRef]
- Goetz, S.C.; Liem, K.F., Jr.; Anderson, K.V. The spinocerebellar ataxia-associated gene Tau tubulin kinase 2 controls the initiation of ciliogenesis. Cell 2012, 151, 847–858. [Google Scholar] [CrossRef]
- Pugacheva, E.N.; Jablonski, S.A.; Hartman, T.R.; Henske, E.P.; Golemis, E.A. HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 2007, 129, 1351–1363. [Google Scholar] [CrossRef]
- Guarino, E.; Delli Poggi, C.; Grieco, G.E.; Cenci, V.; Ceccarelli, E.; Crisci, I.; Sebastiani, G.; Dotta, F. Circulating MicroRNAs as Biomarkers of Gestational Diabetes Mellitus: Updates and Perspectives. Int. J. Endocrinol. 2018, 2018, 6380463. [Google Scholar] [CrossRef]
- Kong, L.; Zhu, J.; Han, W.; Jiang, X.; Xu, M.; Zhao, Y.; Dong, Q.; Pang, Z.; Guan, Q.; Gao, L.; et al. Significance of serum microRNAs in pre-diabetes and newly diagnosed type 2 diabetes: A clinical study. Acta Diabetol. 2011, 48, 61–69. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal Control (n = 20) | GDM (n = 20) | |
|---|---|---|
| Maternal age (years) | 32.78 ± 4.2 | 33.56 ± 4.93 |
| Nulliparity (%) | 41.94 | 48 |
| Gestational age at delivery (weeks) | 38.43 ± 1.14 | 38 ± 1.2 |
| Chinese Han ethnicity (%) | 100 | 100 |
| BMI 1st trimester (Kg/m2) | 23.4 ± 3.48 | 27.06 ± 5.85 ** |
| BMI 3rd trimester (Kg/m2) | 26.8 ± 3.39 | 30.38 ± 6 ** |
| Systolic blood pressure (mmHg) | 123.04 ± 12.55 | 133.43 ± 19.73 * |
| Diastolic blood pressure (mmHg) | 73.83 ± 10.01 | 81.71 ± 11.56 * |
| Glucose-AC (mg/dL) | 78 ± 7.01 | 97.74 ± 28.22 |
| Glucose-PC 1 hour (mg/dL) | 134.83 ± 6.34 | 195.67 ± 55.1 * |
| Glucose-PC 2 hours (mg/dL) | 126.67 ± 26.07 | 193.47 ± 71.5 * |
| Neonatal outcome | ||
| Birth weight (g) | 3033.49 ± 643.81 | 3244.7 ± 562.75 |
| Female sex (%) | 44.12 | 37.04 |
| 1 min Apgar score | 8.7 ± 0.61 | 8.75 ± 0.56 |
| 5 min Apgar score | 9.81 ± 0.4 | 9.79 ± 0.42 |
| Placenta weight | 662 ± 128.69 | 695 ± 167.06 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-Y.; Su, M.-T.; Cheng, H.-l.; Kuo, P.-L.; Tsai, P.-Y. Fetuin-A Inhibits Placental Cell Growth and Ciliogenesis in Gestational Diabetes Mellitus. Int. J. Mol. Sci. 2019, 20, 5207. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205207
Wang C-Y, Su M-T, Cheng H-l, Kuo P-L, Tsai P-Y. Fetuin-A Inhibits Placental Cell Growth and Ciliogenesis in Gestational Diabetes Mellitus. International Journal of Molecular Sciences. 2019; 20(20):5207. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205207
Chicago/Turabian StyleWang, Chia-Yih, Mei-Tsz Su, Hui-ling Cheng, Pao-Lin Kuo, and Pei-Yin Tsai. 2019. "Fetuin-A Inhibits Placental Cell Growth and Ciliogenesis in Gestational Diabetes Mellitus" International Journal of Molecular Sciences 20, no. 20: 5207. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205207