Endothelial Dysfunction in Primary Aldosteronism

 and
and
Abstract
:1. Introduction
2. Effect of Aldosterone on Vascular Tone
2.1. Vasomotor Regulation and the eNOS System
2.2. Direct Effect of Aldosterone on eNOS System Regulation and NO Production
2.3. Aldosterone Induces Vasoconstrictors
2.4. Other Pathways Involving Aldosterone-Related Vasomotor Dysregulation
2.4.1. EGFR
2.4.2. GPER
2.4.3. G6PD
2.4.4. RhoA/Rho-Associated Kinase Pathway (ROCK)
2.5. Nongenomic Effects of Aldosterone on Vascular Tones
3. Effect of Aldosterone on Endothelium-Mediated Vascular Inflammation
3.1. ROS Systems in Vascular Inflammation
3.2. MR-Dependent and -Independent Pathways in Inflammation
3.2.1. MR-Dependent Mechanism
3.2.2. MR-Independent Mechanism
3.3. Aldosterone Mediates Interactions between Endothelial and Inflammatory Cells
3.4. Aldosterone Induces Systemic Inflammation Mediated by the Endothelium
4. Effect of Aldosterone on Early Atherosclerosis
5. Effect of Aldosterone on Vascular Remodeling
6. Effect of Aldosterone on Endothelial Progenitor Cells
7. Effect of Aldosterone on Ion Channels in Endothelial Cells
7.1. Epithelial Sodium Channel
7.2. Small Conductance Calcium-Activated Potassium Channels
8. Effect of Aldosterone on Extracellular Vesicles
9. Clinical Data and Treatment among PA and Endothelial Dysfunction
9.1. Circulating Biomarkers Associated with Endothelial Dysfunction
9.2. Flow-Mediated Dilation (FMD)
9.3. Peripheral Arterial Tonometry (PAT) to Evaluate Endothelial Dysfunction
9.4. Pulse Wave Velocity (PWV)
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| PA | Primary aldosteronism |
| EH | Essential hypertension |
| LVH | Left ventricular hypertrophy |
| NO | Nitric oxide |
| BH4 | Tetrahydrobiopterin |
| eNOS | Endothelial NO synthase |
| sGC | Soluble guanylate cyclase |
| cGMP | Cyclic guanosine monophosphate |
| PKG | Protein kinase G |
| VDCC | Voltage-dependent calcium channels |
| IP3 | Inositol 1,4,5-trisphosphate |
| SR | Sarcoplasmic reticulum |
| SERCA | Sarco/endoplasmic reticulum calcium ATPase |
| MLCK | Myosin light chain kinase |
| cIMP | Inosine 3′,5′-cyclic monophosphate |
| ROCK | Rho-associated protein kinase |
| GPCR | G protein-coupled receptors |
| GRK2 | G protein-coupled receptor kinase 2 |
| ROS | Reactive oxygen species |
| MR | Mineralocorticoid receptor |
| COX-2 | Cyclooxygenase-2 |
| EGFR | Epidermal growth factor receptor |
| GPER | G protein-coupled estrogen receptor |
| G6PD | Glucose-6-phosphate dehydrogenase |
| FMD | Flow-mediated vasodilation |
| APA | Aldosterone-producing adenoma |
| IHA | Idiopathic hyperaldosteronism |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| AP | Activator protein |
| NFκB | Nuclear factor kappa B |
| TEMPOL | 4-hydroxy-2,2,6,6-tetramethylpiperidinyl-1-oxyl |
| mitoKATP | Mitochondrial ATP-dependent potassium channels |
| HEK | Human embryonic kidney |
| ERK | Extracellular-signal-regulated kinase |
| JNK | c-Jun N-terminal kinase |
| AT1 | Angiotensin type 1 |
| NOS | NO synthase |
| CCL | Cytokines chemokine ligand |
| IL | Interleukin |
| ICAM | Intercellular adhesion molecule |
| MCP | Monocyte chemoattractant protein |
| VCAM | Vascular cell adhesion molecule |
| VWF | von Willebrand factor |
| MAPK | Mitogen-activated protein kinase |
| DOCA | Deoxycorticosterone acetate |
| ApoE | Apolipoprotein-E |
| ACE | Angiotensin-converting enzyme |
| RAAS | Renin-Angiotensin-Aldosterone system |
| PlGF | Placental growth factor |
| VEGF | Vascular endothelial growth factor |
| ECMROE | Endothelial cell-specific MR overexpression |
| ECMRKO | ECMR knockout |
| EPC | Endothelial progenitor cells |
| ENaC | Epithelial sodium channel |
| SKCa | Small conductance calcium-activated potassium channels |
| EVs | Extracellular vesicles |
| VEGFi | Vascular endothelial growth factor pathway inhibitors |
| ADMA | Asymmetric dimethylarginine |
| ox-LDL | Oxidized low-density lipoprotein |
| FMD | Flow-mediated dilation |
| CFU | Colony forming units |
| ADPKD | Autosomal dominant polycystic kidney disease |
| NMD | Nitrate-mediated dilation |
| IAH | Idiopathic adrenal hyperplasia |
| PAT | Peripheral arterial tonometry |
| RHI | Reactive hyperemic index |
| AI | Augmentation index |
| PWV | Pulse wave velocity |
| IMT | Carotid intima-media thickness |
| KCNJ5 | Potassium voltage-gated channel subfamily J member 5 |
References
- Gyamlani, G.; Headley, C.M.; Naseer, A.; Valaulikar, G.S.; Geraci, S.A. Primary aldosteronism: Diagnosis and management. Am. J. Med Sci. 2016, 352, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.P. Prevalence and diagnosis of primary aldosteronism. Curr. Hypertens. Rep. 2010, 12, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Milliez, P.; Girerd, X.; Plouin, P.F.; Blacher, J.; Safar, M.E.; Mourad, J.J. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J. Am. Coll. Cardiol. 2005, 45, 1243–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catena, C.; Colussi, G.; Nadalini, E.; Chiuch, A.; Baroselli, S.; Lapenna, R.; Sechi, L.A. Cardiovascular outcomes in patients with primary aldosteronism after treatment. Arch. Intern. Med. 2008, 168, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Reincke, M.; Fischer, E.; Gerum, S.; Merkle, K.; Schulz, S.; Pallauf, A.; Quinkler, M.; Hanslik, G.; Lang, K.; Hahner, S.; et al. Observational study mortality in treated primary aldosteronism: The german conn’s registry. Hypertension 2012, 60, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Savard, S.; Amar, L.; Plouin, P.F.; Steichen, O. Cardiovascular complications associated with primary aldosteronism: A controlled cross-sectional study. Hypertension 2013, 62, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Mulatero, P.; Monticone, S.; Bertello, C.; Viola, A.; Tizzani, D.; Iannaccone, A.; Crudo, V.; Burrello, J.; Milan, A.; Rabbia, F.; et al. Long-term cardio- and cerebrovascular events in patients with primary aldosteronism. J. Clin. Endocrinol. Metab. 2013, 98, 4826–4833. [Google Scholar] [CrossRef]
- Monticone, S.; D’Ascenzo, F.; Moretti, C.; Williams, T.A.; Veglio, F.; Gaita, F.; Mulatero, P. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: A systematic review and meta-analysis. Lancet. Diabetes Endocrinol. 2018, 6, 41–50. [Google Scholar] [CrossRef]
- Wu, V.C.; Wang, S.M.; Chang, C.H.; Hu, Y.H.; Lin, L.Y.; Lin, Y.H.; Chueh, S.C.; Chen, L.; Wu, K.D. Long term outcome of aldosteronism after target treatments. Sci. Rep. 2016, 6, 32103. [Google Scholar] [CrossRef]
- Chen, Z.W.; Huang, K.C.; Lee, J.K.; Lin, L.C.; Chen, C.W.; Chang, Y.Y.; Liao, C.W.; Wu, V.C.; Hung, C.S.; Lin, Y.H.; et al. Aldosterone induces left ventricular subclinical systolic dysfunction: A strain imaging study. J. Hypertens. 2018, 36, 353–360. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Lin, L.-Y.; Chen, A.; Wu, X.-M.; Lee, J.-K.; Su, T.-C.; Wu, V.-C.; Chueh, S.-C.; Lin, W.-C.; Lo, M.-T.; et al. Adrenalectomy improves increased carotid intima-media thickness and arterial stiffness in patients with aldosterone producing adenoma. Atherosclerosis 2012, 221, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.W.; Lin, L.Y.; Hung, C.S.; Lin, Y.T.; Chang, Y.Y.; Wang, S.M.; Wu, V.C.; Wu, K.D.; Ho, Y.L.; Satoh, F.; et al. Time course and factors predicting arterial stiffness reversal in patients with aldosterone-producing adenoma after adrenalectomy: Prospective study of 102 patients. Sci. Rep. 2016, 6, 20862. [Google Scholar] [CrossRef] [PubMed]
- Lerman, A.; Zeiher, A.M. Endothelial function: Cardiac events. Circulation 2005, 111, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Hermidorff, M.M.; de Assis, L.V.; Isoldi, M.C. Genomic and rapid effects of aldosterone: What we know and do not know thus far. Heart Fail. Rev. 2017, 22, 65–89. [Google Scholar] [CrossRef] [PubMed]
- Mihailidou, A.S.; Tzakos, A.G.; Ashton, A.W. Non-genomic effects of aldosterone. Vitam. Horm. 2019, 109, 133–149. [Google Scholar] [PubMed]
- Furchgott, R.F. The 1996 albert lasker medical research awards. The discovery of endothelium-derived relaxing factor and its importance in the identification of nitric oxide. JAMA 1996, 276, 1186–1188. [Google Scholar] [CrossRef] [PubMed]
- Murad, F. Nitric oxide and cyclic gmp in cell signaling and drug development. New Engl. J. Med. 2006, 355, 2003–2011. [Google Scholar] [CrossRef]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef]
- Derbyshire, E.R.; Marletta, M.A. Structure and regulation of soluble guanylate cyclase. Annu. Rev. Biochem. 2012, 81, 533–559. [Google Scholar] [CrossRef]
- Carvajal, J.A.; Germain, A.M.; Huidobro-Toro, J.P.; Weiner, C.P. Molecular mechanism of cgmp-mediated smooth muscle relaxation. J. Cell. Physiol. 2000, 184, 409–420. [Google Scholar] [CrossRef]
- Cohen, R.A.; Weisbrod, R.M.; Gericke, M.; Yaghoubi, M.; Bierl, C.; Bolotina, V.M. Mechanism of nitric oxide-induced vasodilatation. Circ. Res. 1999, 84, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.R.; Li, L.; Kitazawa, T. Cyclic gmp causes Ca2+ desensitization in vascular smooth muscle by activating the myosin light chain phosphatase. J. Biol. Chem. 1997, 272, 5063–5068. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Isotani, E.; Huang, J.; Ding, H.; Stull, J.T.; Kamm, K.E. Myosin light chain kinase activation and calcium sensitization in smooth muscle in vivo. Am. J. Physiol. Cell Physiol. 2008, 295, C358–C364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Zhang, X.; Ying, L.; Dou, D.; Li, Y.; Bai, Y.; Liu, J.; Liu, L.; Feng, H.; Yu, X.; et al. Cimp synthesized by sgc as a mediator of hypoxic contraction of coronary arteries. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H328–H336. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, Z.; Leung, S.W.; Vanhoutte, P.M. Hypoxic vasospasm mediated by cimp: When soluble guanylyl cyclase turns bad. J. Cardiovasc. Pharmacol. 2015, 65, 545–548. [Google Scholar] [CrossRef]
- Adachi, T.; Weisbrod, R.M.; Pimentel, D.R.; Ying, J.; Sharov, V.S.; Schöneich, C.; Cohen, R.A. S-glutathiolation by peroxynitrite activates serca during arterial relaxation by nitric oxide. Nat. Med. 2004, 10, 1200–1207. [Google Scholar] [CrossRef]
- Daaka, Y. S-nitrosylation-regulated gpcr signaling. Biochim. Et Biophys. Acta (Bba) Gen. Subj. 2012, 1820, 743–751. [Google Scholar] [CrossRef]
- Ozawa, K.; Whalen, E.J.; Nelson, C.D.; Mu, Y.; Hess, D.T.; Lefkowitz, R.J.; Stamler, J.S. S-nitrosylation of β-arrestin regulates β-adrenergic receptor trafficking. Mol. Cell 2008, 31, 395–405. [Google Scholar] [CrossRef]
- Whalen, E.J.; Foster, M.W.; Matsumoto, A.; Ozawa, K.; Violin, J.D.; Que, L.G.; Nelson, C.D.; Benhar, M.; Keys, J.R.; Rockman, H.A.; et al. Regulation of β-adrenergic receptor signaling by s-nitrosylation of g-protein-coupled receptor kinase 2. Cell 2007, 129, 511–522. [Google Scholar] [CrossRef]
- Kellogg, D.L., Jr.; Zhao, J.L.; Coey, U.; Green, J.V. Acetylcholine-induced vasodilation is mediated by nitric oxide and prostaglandins in human skin. J. Appl. Physiol. 2005, 98, 629–632. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.W.; Kim, H.S.; Cha, Y.N.; Park, Y.S.; Jo, S.A.; Jo, I. Rapid increase in endothelial nitric oxide production by bradykinin is mediated by protein kinase a signaling pathway. Biochem. Biophys. Res. Commun. 2003, 306, 981–987. [Google Scholar] [CrossRef]
- Li, H.; Burkhardt, C.; Heinrich, U.-R.; Brausch, I.; Xia, N.; Förstermann, U. Histamine upregulates gene expression of endothelial nitric oxide synthase in human vascular endothelial cells. Circulation 2003, 107, 2348–2354. [Google Scholar] [CrossRef] [PubMed]
- Kuchan, M.J.; Frangos, J.A. Role of calcium and calmodulin in flow-induced nitric oxide production in endothelial cells. Am. J. Physiol. Cell Physiol. 1994, 266, C628–C636. [Google Scholar] [CrossRef] [PubMed]
- Andrews, A.M.; Jaron, D.; Buerk, D.G.; Kirby, P.L.; Barbee, K.A. Direct, real-time measurement of shear stress-induced nitric oxide produced from endothelial cells in vitro. Nitric Oxide 2010, 23, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, D.G.; Widder, J.; Grumbach, I.; Chen, W.; Weber, M.; Searles, C. Endothelial mechanotransduction, nitric oxide and vascular inflammation. J. Intern. Med. 2006, 259, 351–363. [Google Scholar] [CrossRef] [PubMed]
- Sessa, W.C. Enos at a glance. J. Cell Sci. 2004, 117, 2427–2429. [Google Scholar] [CrossRef]
- Boo, Y.C.; Hwang, J.; Sykes, M.; Michell, B.J.; Kemp, B.E.; Lum, H.; Jo, H. Shear stress stimulates phosphorylation of enos at ser635 by a protein kinase a-dependent mechanism. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1819–H1828. [Google Scholar] [CrossRef]
- Boo, Y.C.; Sorescu, G.; Boyd, N.; Shiojima, I.; Walsh, K.; Du, J.; Jo, H. Shear stress stimulates phosphorylation of endothelial nitric-oxide synthase at ser1179 by akt-independent mechanisms: Role of protein kinase a. J. Biol. Chem. 2002, 277, 3388–3396. [Google Scholar] [CrossRef]
- Fleming, I.; Fisslthaler, B.; Dimmeler, S.; Kemp, B.E.; Busse, R. Phosphorylation of thr(495) regulates ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 2001, 88, E68–E75. [Google Scholar] [CrossRef]
- Nagata, D.; Takahashi, M.; Sawai, K.; Tagami, T.; Usui, T.; Shimatsu, A.; Hirata, Y.; Naruse, M. Molecular mechanism of the inhibitory effect of aldosterone on endothelial no synthase activity. Hypertension 2006, 48, 165–171. [Google Scholar] [CrossRef]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Martasek, P.; Hogg, N.; Masters, B.S.; Karoui, H.; Tordo, P.; Pritchard, K.A., Jr. Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc. Natl. Acad. Sci. USA 1998, 95, 9220–9225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cachofeiro, V.; Miana, M.; de las Heras, N.; Martín-Fernández, B.; Ballesteros, S.; Fernández-Tresguerres, J.; Lahera, V. Aldosterone and the vascular system. J. Steroid Biochem. Mol. Biol. 2008, 109, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Rivero, J.; Cachofeiro, V.; Lahera, V.; Aras-Lopez, R.; Marquez-Rodas, I.; Salaices, M.; Xavier, F.E.; Ferrer, M.; Balfagon, G. Participation of prostacyclin in endothelial dysfunction induced by aldosterone in normotensive and hypertensive rats. Hypertension 2005, 46, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Xavier, F.E.; Aras-Lopez, R.; Arroyo-Villa, I.; del Campo, L.; Salaices, M.; Rossoni, L.V.; Ferrer, M.; Balfagon, G. Aldosterone induces endothelial dysfunction in resistance arteries from normotensive and hypertensive rats by increasing thromboxane a2 and prostacyclin. Br. J. Pharmacol. 2008, 154, 1225–1235. [Google Scholar] [CrossRef]
- Park, J.B.; Schiffrin, E.L. Et(a) receptor antagonist prevents blood pressure elevation and vascular remodeling in aldosterone-infused rats. Hypertension 2001, 37, 1444–1449. [Google Scholar] [CrossRef]
- Bourque, S.L.; Davidge, S.T.; Adams, M.A. The interaction between endothelin-1 and nitric oxide in the vasculature: New perspectives. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1288–R1295. [Google Scholar] [CrossRef]
- Briet, M.; Schiffrin, E.L. Vascular actions of aldosterone. J. Vasc. Res. 2013, 50, 89–99. [Google Scholar] [CrossRef]
- Griol-Charhbili, V.; Fassot, C.; Messaoudi, S.; Perret, C.; Agrapart, V.; Jaisser, F. Epidermal growth factor receptor mediates the vascular dysfunction but not the remodeling induced by aldosterone/salt. Hypertension 2011, 57, 238–244. [Google Scholar] [CrossRef]
- Gros, R.; Ding, Q.; Sklar, L.A.; Prossnitz, E.E.; Arterburn, J.B.; Chorazyczewski, J.; Feldman, R.D. Gpr30 expression is required for the mineralocorticoid receptor-independent rapid vascular effects of aldosterone. Hypertension 2011, 57, 442–451. [Google Scholar] [CrossRef]
- Gros, R.; Ding, Q.; Liu, B.; Chorazyczewski, J.; Feldman, R.D. Aldosterone mediates its rapid effects in vascular endothelial cells through gper activation. Am. J. Physiol. Cell Physiol. 2013, 304, C532–C540. [Google Scholar] [CrossRef]
- Leopold, J.A.; Dam, A.; Maron, B.A.; Scribner, A.W.; Liao, R.; Handy, D.E.; Stanton, R.C.; Pitt, B.; Loscalzo, J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat. Med. 2007, 13, 189–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Hassona, M.D.; Abouelnaga, Z.A.; Elnakish, M.T.; Awad, M.M.; Alhaj, M.; Goldschmidt-Clermont, P.J.; Hassanain, H. Vascular hypertrophy-associated hypertension of profilin1 transgenic mouse model leads to functional remodeling of peripheral arteries. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H2112–H2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Romero, M.J.; Toque, H.A.; Yang, G.; Caldwell, R.B.; Caldwell, R.W. The role of rhoa/rho kinase pathway in endothelial dysfunction. J. Cardiovasc. Dis. Res. 2010, 1, 165–170. [Google Scholar] [PubMed]
- Matsumoto, T.; Oki, K.; Kajikawa, M.; Nakashima, A.; Maruhashi, T.; Iwamoto, Y.; Iwamoto, A.; Oda, N.; Hidaka, T.; Kihara, Y.; et al. Effect of aldosterone-producing adenoma on endothelial function and rho-associated kinase activity in patients with primary aldosteronism. Hypertension 2015, 65, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, S.; Oki, K.; Maruhashi, T.; Kajikawa, M.; Matsui, S.; Hashimoto, H.; Takaeko, Y.; Kihara, Y.; Chayama, K.; Goto, C.; et al. Eplerenone improves endothelial function and arterial stiffness and inhibits rho-associated kinase activity in patients with idiopathic hyperaldosteronism: A pilot study. J. Hypertens. 2019, 37, 1083–1095. [Google Scholar] [CrossRef]
- Romagni, P.; Rossi, F.; Guerrini, L.; Quirini, C.; Santiemma, V. Aldosterone induces contraction of the resistance arteries in man. Atherosclerosis 2003, 166, 345–349. [Google Scholar] [CrossRef]
- Gunaruwan, P.; Schmitt, M.; Taylor, J.; Lee, L.; Struthers, A.; Frenneaux, M. Lack of rapid aldosterone effects on forearm resistance vasculature in health. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2002, 3, 123–125. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, B.M.W.; Oehmer, S.; Delles, C.; Bratke, R.; Schneider, M.P.; Klingbeil, A.; Fleischmann, E.H.; Schmieder, R.E. Rapid nongenomic effects of aldosterone on human forearm vasculature. Hypertension 2003, 42, 156–160. [Google Scholar] [CrossRef]
- Schmidt, B.M.; Georgens, A.C.; Martin, N.; Tillmann, H.C.; Feuring, M.; Christ, M.; Wehling, M. Interaction of rapid nongenomic cardiovascular aldosterone effects with the adrenergic system. J. Clin. Endocrinol. Metab. 2001, 86, 761–767. [Google Scholar] [CrossRef]
- Schmitt, M.; Gunaruwan, P.; Frenneaux, M.P. Rapid nongenomic aldosterone effects in the human forearm? Hypertension 2004, 43, e1, author reply e2. [Google Scholar] [CrossRef] [PubMed]
- Keane, M.P.; Strieter, R.M. Chemokine signaling in inflammation. Crit. Care Med. 2000, 28, N13–N26. [Google Scholar] [CrossRef] [PubMed]
- Chrissobolis, S.; Drummond, G.R.; Faraci, F.M.; Sobey, C.G. Chronic aldosterone administration causes nox2-mediated increases in reactive oxygen species production and endothelial dysfunction in the cerebral circulation. J. Hypertens. 2014, 32, 1815–1821. [Google Scholar] [CrossRef] [PubMed]
- Iwashima, F.; Yoshimoto, T.; Minami, I.; Sakurada, M.; Hirono, Y.; Hirata, Y. Aldosterone induces superoxide generation via rac1 activation in endothelial cells. Endocrinology 2008, 149, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Taniyama, Y.; Griendling, K.K. Reactive oxygen species in the vasculature: Molecular and cellular mechanisms. Hypertension 2003, 42, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Fiebeler, A.; Schmidt, F.; Muller, D.N.; Park, J.K.; Dechend, R.; Bieringer, M.; Shagdarsuren, E.; Breu, V.; Haller, H.; Luft, F.C. Mineralocorticoid receptor affects ap-1 and nuclear factor-kappab activation in angiotensin ii-induced cardiac injury. Hypertension 2001, 37, 787–793. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, J.; Lu, L.; Chen, S.S.; Quinn, M.T.; Weber, K.T. Aldosterone-induced inflammation in the rat heart: Role of oxidative stress. Am. J. Pathol. 2002, 161, 1773–1781. [Google Scholar] [CrossRef]
- Iglarz, M.; Touyz, R.M.; Viel, E.C.; Amiri, F.; Schiffrin, E.L. Involvement of oxidative stress in the profibrotic action of aldosterone. Interaction wtih the renin-angiotension system. Am. J. Hypertens. 2004, 17, 597–603. [Google Scholar] [CrossRef]
- Park, Y.M.; Park, M.Y.; Suh, Y.L.; Park, J.B. Nad(p)h oxidase inhibitor prevents blood pressure elevation and cardiovascular hypertrophy in aldosterone-infused rats. Biochem. Biophys. Res. Commun. 2004, 313, 812–817. [Google Scholar] [CrossRef]
- Datla, S.R.; Griendling, K.K. Reactive oxygen species, nadph oxidases, and hypertension. Hypertension 2010, 56, 325–330. [Google Scholar] [CrossRef]
- Nolly, M.B.; Caldiz, C.I.; Yeves, A.M.; Villa-Abrille, M.C.; Morgan, P.E.; Amado Mondaca, N.; Portiansky, E.L.; Chiappe de Cingolani, G.E.; Cingolani, H.E.; Ennis, I.L. The signaling pathway for aldosterone-induced mitochondrial production of superoxide anion in the myocardium. J. Mol. Cell Cardiol. 2014, 67, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Haseroth, K.; Gerdes, D.; Berger, S.; Feuring, M.; Gunther, A.; Herbst, C.; Christ, M.; Wehling, M. Rapid nongenomic effects of aldosterone in mineralocorticoid-receptor-knockout mice. Biochem. Biophys. Res. Commun. 1999, 266, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, C.; Benesic, A.; Krug, A.W.; Freudinger, R.; Mildenberger, S.; Gassner, B.; Gekle, M. Human mineralocorticoid receptor expression renders cells responsive for nongenotropic aldosterone actions. Mol. Endocrinol. 2005, 19, 1697–1710. [Google Scholar] [CrossRef] [PubMed]
- Hirono, Y.; Yoshimoto, T.; Suzuki, N.; Sugiyama, T.; Sakurada, M.; Takai, S.; Kobayashi, N.; Shichiri, M.; Hirata, Y. Angiotensin ii receptor type 1-mediated vascular oxidative stress and proinflammatory gene expression in aldosterone-induced hypertension: The possible role of local renin-angiotensin system. Endocrinology 2007, 148, 1688–1696. [Google Scholar] [CrossRef]
- Downey, J.M.; Krieg, T.; Cohen, M.V. Mapping preconditioning’s signaling pathways: An engineering approach. Ann. N. Y. Acad. Sci. 2008, 1123, 187–196. [Google Scholar] [CrossRef]
- Brilla, C.G.; Matsubara, L.S.; Weber, K.T. Anti-aldosterone treatment and the prevention of myocardial fibrosis in primary and secondary hyperaldosteronism. J. Mol. Cell Cardiol. 1993, 25, 563–575. [Google Scholar] [CrossRef]
- Lacolley, P.; Labat, C.; Pujol, A.; Delcayre, C.; Benetos, A.; Safar, M. Increased carotid wall elastic modulus and fibronectin in aldosterone-salt-treated rats: Effects of eplerenone. Circulation 2002, 106, 2848–2853. [Google Scholar] [CrossRef]
- Brilla, C.G.; Weber, K.T. Mineralocorticoid excess, dietary sodium, and myocardial fibrosis. J. Lab. Clin. Med. 1992, 120, 893–901. [Google Scholar]
- Young, M.; Fullerton, M.; Dilley, R.; Funder, J. Mineralocorticoids, hypertension, and cardiac fibrosis. J. Clin. Investig. 1994, 93, 2578–2583. [Google Scholar] [CrossRef]
- Benetos, A.; Lacolley, P.; Safar, M.E. Prevention of aortic fibrosis by spironolactone in spontaneously hypertensive rats. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1152–1156. [Google Scholar] [CrossRef]
- Matsui, H.; Ando, K.; Kawarazaki, H.; Nagae, A.; Fujita, M.; Shimosawa, T.; Nagase, M.; Fujita, T. Salt excess causes left ventricular diastolic dysfunction in rats with metabolic disorder. Hypertension 2008, 52, 287–294. [Google Scholar] [CrossRef]
- Yoshida, K.; Kim-Mitsuyama, S.; Wake, R.; Izumiya, Y.; Izumi, Y.; Yukimura, T.; Ueda, M.; Yoshiyama, M.; Iwao, H. Excess aldosterone under normal salt diet induces cardiac hypertrophy and infiltration via oxidative stress. Hypertens. Res. 2005, 28, 447–455. [Google Scholar] [CrossRef] [PubMed]
- Rude, M.K.; Duhaney, T.A.; Kuster, G.M.; Judge, S.; Heo, J.; Colucci, W.S.; Siwik, D.A.; Sam, F. Aldosterone stimulates matrix metalloproteinases and reactive oxygen species in adult rat ventricular cardiomyocytes. Hypertension 2005, 46, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Dinh, Q.N.; Young, M.J.; Evans, M.A.; Drummond, G.R.; Sobey, C.G.; Chrissobolis, S. Aldosterone-induced oxidative stress and inflammation in the brain are mediated by the endothelial cell mineralocorticoid receptor. Brain Res. 2016, 1637, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Rocha, R.; Rudolph, A.E.; Frierdich, G.E.; Nachowiak, D.A.; Kekec, B.K.; Blomme, E.A.; McMahon, E.G.; Delyani, J.A. Aldosterone induces a vascular inflammatory phenotype in the rat heart. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1802–H1810. [Google Scholar] [CrossRef]
- Nishimoto, M.; Fujita, T. Renal mechanisms of salt-sensitive hypertension: Contribution of two steroid receptor-associated pathways. Am. J. Physiol. Ren. Physiol. 2015, 308, F377–F387. [Google Scholar] [CrossRef]
- Martinez, D.V.; Rocha, R.; Matsumura, M.; Oestreicher, E.; Ochoa-Maya, M.; Roubsanthisuk, W.; Williams, G.H.; Adler, G.K. Cardiac damage prevention by eplerenone: Comparison with low sodium diet or potassium loading. Hypertension 2002, 39, 614–618. [Google Scholar] [CrossRef]
- Zhu, C.J.; Wang, Q.Q.; Zhou, J.L.; Liu, H.Z.; Hua, F.; Yang, H.Z.; Hu, Z.W. The mineralocorticoid receptor-p38mapk-nfkappab or erk-sp1 signal pathways mediate aldosterone-stimulated inflammatory and profibrotic responses in rat vascular smooth muscle cells. Acta Pharm. Sin 2012, 33, 873–878. [Google Scholar] [CrossRef]
- Min, L.J.; Mogi, M.; Li, J.M.; Iwanami, J.; Iwai, M.; Horiuchi, M. Aldosterone and angiotensin ii synergistically induce mitogenic response in vascular smooth muscle cells. Circ. Res. 2005, 97, 434–442. [Google Scholar] [CrossRef]
- Mazak, I.; Fiebeler, A.; Muller, D.N.; Park, J.K.; Shagdarsuren, E.; Lindschau, C.; Dechend, R.; Viedt, C.; Pilz, B.; Haller, H.; et al. Aldosterone potentiates angiotensin ii-induced signaling in vascular smooth muscle cells. Circulation 2004, 109, 2792–2800. [Google Scholar] [CrossRef]
- Lemarie, C.A.; Simeone, S.M.; Nikonova, A.; Ebrahimian, T.; Deschenes, M.E.; Coffman, T.M.; Paradis, P.; Schiffrin, E.L. Aldosterone-induced activation of signaling pathways requires activity of angiotensin type 1a receptors. Circ. Res. 2009, 105, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J. Aldosterone and vascular inflammation. Hypertension 2008, 51, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Terada, Y.; Ueda, S.; Hamada, K.; Shimamura, Y.; Ogata, K.; Inoue, K.; Taniguchi, Y.; Kagawa, T.; Horino, T.; Takao, T. Aldosterone stimulates nuclear factor-kappa b activity and transcription of intercellular adhesion molecule-1 and connective tissue growth factor in rat mesangial cells via serum- and glucocorticoid-inducible protein kinase-1. Clin. Exp. Nephrol. 2012, 16, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Blasi, E.R.; Rocha, R.; Rudolph, A.E.; Blomme, E.A.; Polly, M.L.; McMahon, E.G. Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int. 2003, 63, 1791–1800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caprio, M.; Newfell, B.G.; la Sala, A.; Baur, W.; Fabbri, A.; Rosano, G.; Mendelsohn, M.E.; Jaffe, I.Z. Functional mineralocorticoid receptors in human vascular endothelial cells regulate intercellular adhesion molecule-1 expression and promote leukocyte adhesion. Circ. Res. 2008, 102, 1359–1367. [Google Scholar] [CrossRef]
- Wong, S.; Brennan, F.E.; Young, M.J.; Fuller, P.J.; Cole, T.J. A direct effect of aldosterone on endothelin-1 gene expression in vivo. Endocrinology 2007, 148, 1511–1517. [Google Scholar] [CrossRef]
- Li, L.; Fink, G.D.; Watts, S.W.; Northcott, C.A.; Galligan, J.J.; Pagano, P.J.; Chen, A.F. Endothelin-1 increases vascular superoxide via endothelin(a)-nadph oxidase pathway in low-renin hypertension. Circulation 2003, 107, 1053–1058. [Google Scholar] [CrossRef]
- Jeong, Y.; Chaupin, D.F.; Matsushita, K.; Yamakuchi, M.; Cameron, S.J.; Morrell, C.N.; Lowenstein, C.J. Aldosterone activates endothelial exocytosis. Proc. Natl. Acad. Sci. USA 2009, 106, 3782–3787. [Google Scholar] [CrossRef] [Green Version]
- Kirsch, T.; Beese, M.; Wyss, K.; Klinge, U.; Haller, H.; Haubitz, M.; Fiebeler, A. Aldosterone modulates endothelial permeability and endothelial nitric oxide synthase activity by rearrangement of the actin cytoskeleton. Hypertension 2013, 61, 501–508. [Google Scholar] [CrossRef]
- Cathcart, M.K. Regulation of superoxide anion production by nadph oxidase in monocytes/macrophages: Contributions to atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 23–28. [Google Scholar] [CrossRef]
- Dragoni, S.; Hudson, N.; Kenny, B.A.; Burgoyne, T.; McKenzie, J.A.; Gill, Y.; Blaber, R.; Futter, C.E.; Adamson, P.; Greenwood, J.; et al. Endothelial mapks direct icam-1 signaling to divergent inflammatory functions. J. Immunol. 2017, 198, 4074–4085. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner-Parzer, S.M.; Waldhausl, W.K. The endothelium as a metabolic and endocrine organ: Its relation with insulin resistance. Exp. Clin. Endocrinol. Diabetes 2001, 109 (Suppl. 2), S166–S179. [Google Scholar] [CrossRef] [PubMed]
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, C.H.; Hung, C.S.; Liao, C.W.; Wei, L.H.; Chen, C.W.; Shun, C.T.; Wen, W.F.; Wan, C.H.; Wu, X.M.; Chang, Y.Y.; et al. Il-6 trans-signalling contributes to aldosterone-induced cardiac fibrosis. Cardiovasc. Res. 2018, 114, 690–702. [Google Scholar] [CrossRef] [PubMed]
- Sturgis, L.C.; Cannon, J.G.; Schreihofer, D.A.; Brands, M.W. The role of aldosterone in mediating the dependence of angiotensin hypertension on il-6. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2009, 297, R1742–R1748. [Google Scholar] [CrossRef] [PubMed]
- Luther, J.M.; Gainer, J.V.; Murphey, L.J.; Yu, C.; Vaughan, D.E.; Morrow, J.D.; Brown, N.J. Angiotensin ii induces interleukin-6 in humans through a mineralocorticoid receptor-dependent mechanism. Hypertension 2006, 48, 1050–1057. [Google Scholar] [CrossRef]
- Lim, J.S.; Park, S.; Park, S.I.; Oh, Y.T.; Choi, E.; Kim, J.Y.; Rhee, Y. Cardiac dysfunction in association with increased inflammatory markers in primary aldosteronism. Endocrinol. Metab. 2016, 31, 567–576. [Google Scholar] [CrossRef]
- Sanz-Rosa, D.; Cediel, E.; de las Heras, N.; Miana, M.; Balfagon, G.; Lahera, V.; Cachofeiro, V. Participation of aldosterone in the vascular inflammatory response of spontaneously hypertensive rats: Role of the nfkappab/ikappab system. J. Hypertens. 2005, 23, 1167–1172. [Google Scholar] [CrossRef]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III–27–III–32. [Google Scholar] [CrossRef]
- Chhabra, R.K. Endothelial Dysfunction—A Predictor of Atherosclerosis. Internet J. Med. Update 2009, 4. [Google Scholar] [CrossRef]
- Hillaert, M.A.; Lentjes, E.G.; Beygui, F.; Kemperman, H.; Asselbergs, F.W.; Nathoe, H.M.; Agostoni, P.; Voskuil, M.; Ivanes, F.; Jude, B.; et al. Measuring and targeting aldosterone and renin in atherosclerosis-a review of clinical data. Am. Heart J. 2011, 162, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, K.C.; Brown, N.J. Aldosterone and inflammation. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudau, M.; Genis, A.; Lochner, A.; Strijdom, H. Endothelial dysfunction: The early predictor of atherosclerosis. Cardiovasc. J. Afr. 2012, 23, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Van Belle, E.; Bauters, C.; Wernert, N.; Hamon, M.; McFadden, E.P.; Racadot, A.; Dupuis, B.; Lablanche, J.M.; Bertrand, M.E. Neointimal thickening after balloon denudation is enhanced by aldosterone and inhibited by spironolactone, and aldosterone antagonist. Cardiovasc. Res. 1995, 29, 27–32. [Google Scholar] [CrossRef]
- Keidar, S.; Hayek, T.; Kaplan, M.; Pavlotzky, E.; Hamoud, S.; Coleman, R.; Aviram, M. Effect of eplerenone, a selective aldosterone blocker, on blood pressure, serum and macrophage oxidative stress, and atherosclerosis in apolipoprotein e-deficient mice. J. Cardiovasc. Pharmacol. 2003, 41, 955–963. [Google Scholar] [CrossRef]
- Keidar, S.; Kaplan, M.; Pavlotzky, E.; Coleman, R.; Hayek, T.; Hamoud, S.; Aviram, M. Aldosterone administration to mice stimulates macrophage nadph oxidase and increases atherosclerosis development: A possible role for angiotensin-converting enzyme and the receptors for angiotensin ii and aldosterone. Circulation 2004, 109, 2213–2220. [Google Scholar] [CrossRef]
- Suzuki, J.; Iwai, M.; Mogi, M.; Oshita, A.; Yoshii, T.; Higaki, J.; Horiuchi, M. Eplerenone with valsartan effectively reduces atherosclerotic lesion by attenuation of oxidative stress and inflammation. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 917–921. [Google Scholar] [CrossRef]
- Imanishi, T.; Ikejima, H.; Tsujioka, H.; Kuroi, A.; Kobayashi, K.; Muragaki, Y.; Mochizuki, S.; Goto, M.; Yoshida, K.; Akasaka, T. Addition of eplerenone to an angiotensin-converting enzyme inhibitor effectively improves nitric oxide bioavailability. Hypertension 2008, 51, 734–741. [Google Scholar] [CrossRef]
- Marzolla, V.; Armani, A.; Mammi, C.; Moss, M.E.; Pagliarini, V.; Pontecorvo, L.; Antelmi, A.; Fabbri, A.; Rosano, G.; Jaffe, I.Z.; et al. Essential role of icam-1 in aldosterone-induced atherosclerosis. Int. J. Cardiol. 2017, 232, 233–242. [Google Scholar] [CrossRef]
- McGraw, A.P.; Bagley, J.; Chen, W.S.; Galayda, C.; Nickerson, H.; Armani, A.; Caprio, M.; Carmeliet, P.; Jaffe, I.Z. Aldosterone increases early atherosclerosis and promotes plaque inflammation through a placental growth factor-dependent mechanism. J. Am. Heart Assoc. 2013, 2, e000018. [Google Scholar] [CrossRef]
- McCurley, A.; Jaffe, I.Z. Mineralocorticoid receptors in vascular function and disease. Mol. Cell Endocrinol. 2012, 350, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakabayashi, K.; Suzuki, H.; Sato, T.; Iso, Y.; Katagiri, T.; Takeyama, Y. Eplerenone suppresses neointimal formation after coronary stent implantation in swine. Int. J. Cardiol. 2006, 107, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Pu, Q.; Neves, M.F.; Virdis, A.; Touyz, R.M.; Schiffrin, E.L. Endothelin antagonism on aldosterone-induced oxidative stress and vascular remodeling. Hypertension 2003, 42, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Dinh Cat, A.; Griol-Charhbili, V.; Loufrani, L.; Labat, C.; Benjamin, L.; Farman, N.; Lacolley, P.; Henrion, D.; Jaisser, F. The endothelial mineralocorticoid receptor regulates vasoconstrictor tone and blood pressure. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 2454–2463. [Google Scholar] [Green Version]
- Schafer, N.; Lohmann, C.; Winnik, S.; van Tits, L.J.; Miranda, M.X.; Vergopoulos, A.; Ruschitzka, F.; Nussberger, J.; Berger, S.; Luscher, T.F.; et al. Endothelial mineralocorticoid receptor activation mediates endothelial dysfunction in diet-induced obesity. Eur. Heart J. 2013, 34, 3515–3524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickard, A.J.; Morgan, J.; Chrissobolis, S.; Miller, A.A.; Sobey, C.G.; Young, M.J. Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity but not blood pressure. Hypertension 2014, 63, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Werner, N.; Kosiol, S.; Schiegl, T.; Ahlers, P.; Walenta, K.; Link, A.; Böhm, M.; Nickenig, G. Circulating endothelial progenitor cells and cardiovascular outcomes. New Engl. J. Med. 2005, 353, 999–1007. [Google Scholar] [CrossRef]
- Schmidt-Lucke, C.; Rossig, L.; Fichtlscherer, S.; Vasa, M.; Britten, M.; Kamper, U.; Dimmeler, S.; Zeiher, A.M. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: Proof of concept for the clinical importance of endogenous vascular repair. Circulation 2005, 111, 2981–2987. [Google Scholar] [CrossRef]
- Verhovez, A.; Zeoli, A.; Williams, T.A.; Morello, F.; Brizzi, M.F.; Veglio, F.; Mulatero, P. Primary aldosteronism (pa) and endothelial progenitor cell (epc) bioavailability. Clin. Endocrinol. 2008, 69, 528–534. [Google Scholar] [CrossRef]
- Thum, T.; Schmitter, K.; Fleissner, F.; Wiebking, V.; Dietrich, B.; Widder, J.D.; Jazbutyte, V.; Hahner, S.; Ertl, G.; Bauersachs, J. Impairment of endothelial progenitor cell function and vascularization capacity by aldosterone in mice and humans. Eur. Heart J. 2011, 32, 1275–1286. [Google Scholar] [CrossRef]
- Wu, V.C.; Lo, S.C.; Chen, Y.L.; Huang, P.H.; Tsai, C.T.; Liang, C.J.; Kuo, C.C.; Kuo, Y.S.; Lee, B.C.; Wu, E.L.; et al. Endothelial progenitor cells in primary aldosteronism: A biomarker of severity for aldosterone vasculopathy and prognosis. J. Clin. Endocrinol. Metab. 2011, 96, 3175–3183. [Google Scholar] [CrossRef] [PubMed]
- DuPont, J.J.; Hill, M.A.; Bender, S.B.; Jaisser, F.; Jaffe, I.Z. Aldosterone and vascular mineralocorticoid receptors: Regulators of ion channels beyond the kidney. Hypertension 2014, 63, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, V.; Dahlmann, A.; Krueger, B.; Bertog, M.; Loffing, J.; Korbmacher, C. Aldosterone-dependent and -independent regulation of the epithelial sodium channel (enac) in mouse distal nephron. Am. J. Physiol. Ren. Physiol. 2012, 303, F1289–F1299. [Google Scholar] [CrossRef] [PubMed]
- Kusche-Vihrog, K.; Sobczak, K.; Bangel, N.; Wilhelmi, M.; Nechyporuk-Zloy, V.; Schwab, A.; Schillers, H.; Oberleithner, H. Aldosterone and amiloride alter enac abundance in vascular endothelium. Pflug. Arch. Eur. J. Physiol. 2008, 455, 849–857. [Google Scholar] [CrossRef]
- Kusche-Vihrog, K.; Jeggle, P.; Oberleithner, H. The role of enac in vascular endothelium. Pflug. Arch. Eur. J. Physiol. 2014, 466, 851–859. [Google Scholar] [CrossRef]
- Druppel, V.; Kusche-Vihrog, K.; Grossmann, C.; Gekle, M.; Kasprzak, B.; Brand, E.; Pavenstadt, H.; Oberleithner, H.; Kliche, K. Long-term application of the aldosterone antagonist spironolactone prevents stiff endothelial cell syndrome. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 3652–3659. [Google Scholar] [CrossRef]
- Jia, G.; Habibi, J.; Aroor, A.R.; Hill, M.A.; Yang, Y.; Whaley-Connell, A.; Jaisser, F.; Sowers, J.R. Epithelial sodium channel in aldosterone-induced endothelium stiffness and aortic dysfunction. Hypertension 2018, 72, 731–738. [Google Scholar] [CrossRef]
- Wulff, H.; Kohler, R. Endothelial small-conductance and intermediate-conductance kca channels: An update on their pharmacology and usefulness as cardiovascular targets. J. Cardiovasc. Pharmacol. 2013, 61, 102–112. [Google Scholar] [CrossRef]
- Dora, K.A.; Gallagher, N.T.; McNeish, A.; Garland, C.J. Modulation of endothelial cell kca3.1 channels during endothelium-derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ. Res. 2008, 102, 1247–1255. [Google Scholar] [CrossRef]
- Ledoux, J.; Taylor, M.S.; Bonev, A.D.; Hannah, R.M.; Solodushko, V.; Shui, B.; Tallini, Y.; Kotlikoff, M.I.; Nelson, M.T. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc. Natl. Acad. Sci. USA 2008, 105, 9627–9632. [Google Scholar] [CrossRef] [Green Version]
- Saliez, J.; Bouzin, C.; Rath, G.; Ghisdal, P.; Desjardins, F.; Rezzani, R.; Rodella, L.F.; Vriens, J.; Nilius, B.; Feron, O.; et al. Role of caveolar compartmentation in endothelium-derived hyperpolarizing factor-mediated relaxation: Ca2+ signals and gap junction function are regulated by caveolin in endothelial cells. Circulation 2008, 117, 1065–1074. [Google Scholar] [CrossRef] [PubMed]
- Brahler, S.; Kaistha, A.; Schmidt, V.J.; Wolfle, S.E.; Busch, C.; Kaistha, B.P.; Kacik, M.; Hasenau, A.L.; Grgic, I.; Si, H.; et al. Genetic deficit of sk3 and ik1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation 2009, 119, 2323–2332. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.S.; Bonev, A.D.; Gross, T.P.; Eckman, D.M.; Brayden, J.E.; Bond, C.T.; Adelman, J.P.; Nelson, M.T. Altered expression of small-conductance ca2+-activated k+ (sk3) channels modulates arterial tone and blood pressure. Circ. Res. 2003, 93, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Kohler, R.; Ruth, P. Endothelial dysfunction and blood pressure alterations in k+-channel transgenic mice. Pflug. Arch. Eur. J. Physiol. 2010, 459, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Celerier, I.; Bousquet, E.; Jeanny, J.C.; Jonet, L.; Savoldelli, M.; Offret, O.; Curan, A.; Farman, N.; Jaisser, F.; et al. Mineralocorticoid receptor is involved in rat and human ocular chorioretinopathy. J. Clin. Investig. 2012, 122, 2672–2679. [Google Scholar] [CrossRef]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [Green Version]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef]
- Gould, S.J.; Raposo, G. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef]
- Neves, K.B.; Rios, F.J.; Jones, R.; Evans, T.R.J.; Montezano, A.C.; Touyz, R.M. Microparticles from vascular endothelial growth factor pathway inhibitor-treated cancer patients mediate endothelial cell injury. Cardiovasc. Res. 2019, 115, 978–988. [Google Scholar] [CrossRef] [Green Version]
- Lopez Andres, N.; Tesse, A.; Regnault, V.; Louis, H.; Cattan, V.; Thornton, S.N.; Labat, C.; Kakou, A.; Tual-Chalot, S.; Faure, S.; et al. Increased microparticle production and impaired microvascular endothelial function in aldosterone-salt-treated rats: Protective effects of polyphenols. PLoS ONE 2012, 7, e39235. [Google Scholar] [CrossRef]
- Burger, D.; Kwart, D.G.; Montezano, A.C.; Read, N.C.; Kennedy, C.R.J.; Thompson, C.S.; Touyz, R.M. Microparticles induce cell cycle arrest through redox-sensitive processes in endothelial cells: Implications in vascular senescence. J. Am. Heart Assoc. 2012, 1, e001842. [Google Scholar] [CrossRef] [PubMed]
- Burrello, J.; Gai, C.; Tetti, M.; Lopatina, T.; Deregibus, M.C.; Veglio, F.; Mulatero, P.; Camussi, G.; Monticone, S. Characterization and gene expression analysis of serum-derived extracellular vesicles in primary aldosteronism. Hypertension 2019, 74, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Neves, K.B.; Touyz, R.M. Extracellular vesicles as biomarkers and biovectors in primary aldosteronism. Hypertension 2019, 74, 250–252. [Google Scholar] [CrossRef] [PubMed]
- Taddei, S.; Virdis, A.; Mattei, P.; Salvetti, A. Vasodilation to acetylcholine in primary and secondary forms of human hypertension. Hypertension 1993, 21, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Farquharson, C.A.; Struthers, A.D. Aldosterone induces acute endothelial dysfunction in vivo in humans: Evidence for an aldosterone-induced vasculopathy. Clin. Sci. 2002, 103, 425–431. [Google Scholar] [CrossRef]
- Farquharson, C.A.; Struthers, A.D. Spironolactone increases nitric oxide bioactivity, improves endothelial vasodilator dysfunction, and suppresses vascular angiotensin i/angiotensin ii conversion in patients with chronic heart failure. Circulation 2000, 101, 594–597. [Google Scholar] [CrossRef]
- Macdonald, J.E.; Kennedy, N.; Struthers, A.D. Effects of spironolactone on endothelial function, vascular angiotensin converting enzyme activity, and other prognostic markers in patients with mild heart failure already taking optimal treatment. Heart 2004, 90, 765–770. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Al-Qaisi, M.; Kharbanda, R.K.; Mittal, T.K.; Donald, A.E. Measurement of endothelial function and its clinical utility for cardiovascular risk. Vasc. Health Risk Manag. 2008, 4, 647–652. [Google Scholar] [CrossRef]
- Flammer, A.J.; Anderson, T.; Celermajer, D.S.; Creager, M.A.; Deanfield, J.; Ganz, P.; Hamburg, N.M.; Luscher, T.F.; Shechter, M.; Taddei, S.; et al. The assessment of endothelial function: From research into clinical practice. Circulation 2012, 126, 753–767. [Google Scholar] [CrossRef]
- Matrozova, J.; Vasilev, V.; Vandeva, S.; Elenkova, A.; Kirilov, G.; Zaharieva, S. Asymmetric dimethylarginin (adma) as a marker of endothelial dysfunction in primary aldosteronism. Int. J. Endocrinol. Metab. 2016, 14, e30324. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Yin, G.S.; Tang, J.Y.; Ma, D.J.; Ru, J.; Huang, X.H. Endothelial dysfunction in patients with primary aldosteronism: A biomarker of target organ damage. J. Hum. Hypertens. 2014, 28, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Nishizaka, M.K.; Zaman, M.A.; Green, S.A.; Renfroe, K.Y.; Calhoun, D.A. Impaired endothelium-dependent flow-mediated vasodilation in hypertensive subjects with hyperaldosteronism. Circulation 2004, 109, 2857–2861. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.; Petramala, L.; Mastroluca, D.; Petraglia, E.; Di Gaeta, A.; Indino, E.; Panebianco, V.; Ciccariello, M.; Shahabadi, H.H.; Galani, A.; et al. Hyperaldosteronism and cardiovascular risk in patients with autosomal dominant polycystic kidney disease. Medicine 2016, 95, e4175. [Google Scholar] [CrossRef] [PubMed]
- Chou, C.H.; Chen, Y.H.; Hung, C.S.; Chang, Y.Y.; Tzeng, Y.L.; Wu, X.M.; Wu, V.C.; Tsai, C.T.; Wu, C.K.; Ho, Y.L.; et al. Aldosterone impairs vascular smooth muscle function: From clinical to bench research. J. Clin. Endocrinol. Metab. 2015, 100, 4339–4347. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, S.; Matsumoto, T.; Oki, K.; Maruhashi, T.; Kajikawa, M.; Matsui, S.; Hashimoto, H.; Kihara, Y.; Yusoff, F.M.; Higashi, Y. Microvascular endothelial function is impaired in patients with idiopathic hyperaldosteronism. Hypertens Res 2018, 41, 932–938. [Google Scholar] [CrossRef]
- Chang, Y.Y.; Chen, A.; Chen, Y.H.; Hung, C.S.; Wu, V.C.; Wu, X.M.; Lin, Y.H.; Ho, Y.L.; Wu, K.D.; Group, T.S. Hypokalemia correlated with arterial stiffness but not microvascular endothelial function in patients with primary aldosteronism. J. Renin-Angiotensin-Aldosterone Syst. JRAAS 2015, 16, 353–359. [Google Scholar] [CrossRef]
- Bernini, G.; Galetta, F.; Franzoni, F.; Bardini, M.; Taurino, C.; Bernardini, M.; Ghiadoni, L.; Bernini, M.; Santoro, G.; Salvetti, A. Arterial stiffness, intima-media thickness and carotid artery fibrosis in patients with primary aldosteronism. J Hypertens. 2008, 26, 2399–2405. [Google Scholar] [CrossRef]
- Strauch, B.; Petrak, O.; Wichterle, D.; Zelinka, T.; Holaj, R.; Widimsky, J., Jr. Increased arterial wall stiffness in primary aldosteronism in comparison with essential hypertension. Am. J. Hypertens. 2006, 19, 909–914. [Google Scholar] [CrossRef]
- Strauch, B.; Petrak, O.; Zelinka, T.; Wichterle, D.; Holaj, R.; Kasalicky, M.; Safarik, L.; Rosa, J.; Widimsky, J., Jr. Adrenalectomy improves arterial stiffness in primary aldosteronism. Am. J. Hypertens. 2008, 21, 1086–1092. [Google Scholar] [CrossRef]
- Rosa, J.; Somloova, Z.; Petrak, O.; Strauch, B.; Indra, T.; Senitko, M.; Zelinka, T.; Holaj, R.; Widimsky, J., Jr. Peripheral arterial stiffness in primary aldosteronism. Physiol. Res. 2012, 61, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Hu, Y.H.; Tsai, Y.C.; Wu, C.H.; Wang, S.M.; Lin, L.Y.; Lin, Y.H.; Satoh, F.; Wu, K.D.; Wu, V.C. Arterial stiffness and blood pressure improvement in aldosterone-producing adenoma harboring kcnj5 mutations after adrenalectomy. Oncotarget 2017, 8, 29984–29995. [Google Scholar] [CrossRef] [PubMed]
- Moerland, M.; Kales, A.J.; Schrier, L.; van Dongen, M.G.J.; Bradnock, D.; Burggraaf, J. Evaluation of the endopat as a tool to assess endothelial function. Int. J. Vasc. Med. 2012, 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Node, K. Microvascular and macrovascular endothelial function in two different types of primary aldosteronism. Hypertens Res 2019, 42, 739–740. [Google Scholar] [CrossRef]
- Verdecchia, P.; Angeli, F.; Taddei, S. At the beginning of stiffening: Endothelial dysfunction meets “pulsology”. Hypertension 2006, 48, 541–542. [Google Scholar] [CrossRef]
- McEniery, C.M.; Wallace, S.; Mackenzie, I.S.; McDonnell, B.; Yasmin; Newby, D.E.; Cockcroft, J.R.; Wilkinson, I.B. Endothelial function is associated with pulse pressure, pulse wave velocity, and augmentation index in healthy humans. Hypertension 2006, 48, 602–608. [Google Scholar] [CrossRef]
- Naka, K.K.; Tweddel, A.C.; Doshi, S.N.; Goodfellow, J.; Henderson, A.H. Flow-mediated changes in pulse wave velocity: A new clinical measure of endothelial function. Eur. Heart J. 2006, 27, 302–309. [Google Scholar] [CrossRef]
- Williams, T.A.; Monticone, S.; Mulatero, P. Kcnj5 mutations are the most frequent genetic alteration in primary aldosteronism. Hypertension 2015, 65, 507–509. [Google Scholar] [CrossRef]
- Lenzini, L.; Rossitto, G.; Maiolino, G.; Letizia, C.; Funder, J.W.; Rossi, G.P. A meta-analysis of somatic kcnj5 k(+) channel mutations in 1636 patients with an aldosterone-producing adenoma. J. Clin. Endocrinol. Metab. 2015, 100, E1089–E1095. [Google Scholar] [CrossRef]
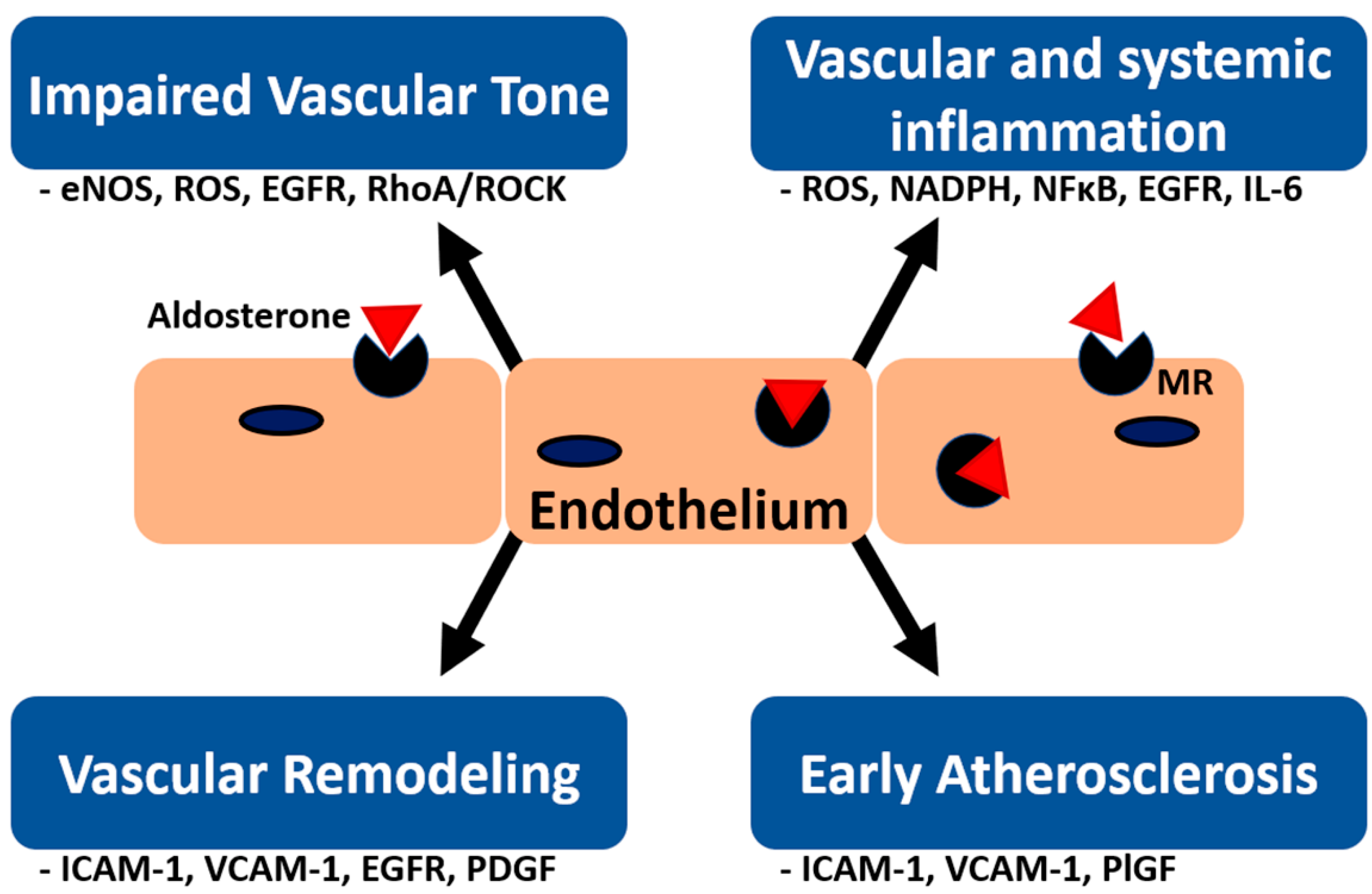

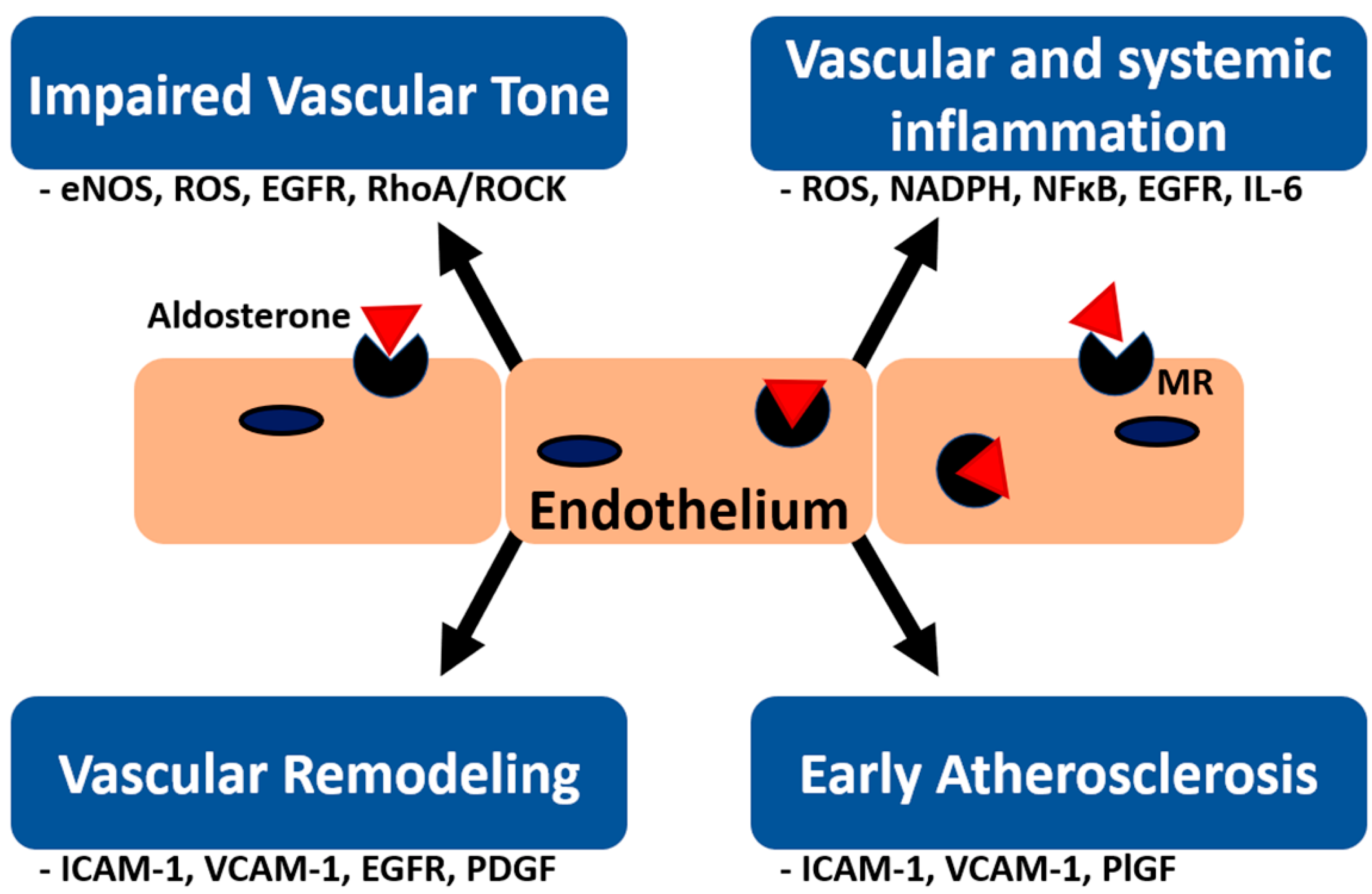{kind=link}
| Author and Year | Assessment Method | Study Group | Control Group | Treatment | Change and Effect on Endothelial Function |
|---|---|---|---|---|---|
| Biomarker | |||||
| Verhovez et al. (2008) [129] | Biomarkers: EPC | PA | Healthy control | - | No difference between high aldosterone treated EPCs with healthy EPCs |
| Thum et al. (2011) [130] | Biomarkers: EPC PAT | PA | Healthy control Mouse EPCs | Spironolactone | EPCs from PA showed reduced migratory potential and reduced RHI. EPCs treated with aldosterone in vitro showed impaired multiple cellular functions through MR-dependent pathway. |
| Wu et al. (2011) [131] | Biomarkers: EPC PWV | PA | EH | Adrenalectomy or spironolactone | Decreased circulating EPCs and endothelial CFUs, improved after treatment |
| Matrozova et al. (2016) [161] | Biomarkers: ADMA | PA | EH and healthy control | - | No difference between PA and EH |
| Liu et al. (2014) [162] | Biomarkers: vWF | PA | EH | - | Increased |
| Biomarkers: ICAM-1 | PA | EH | - | Increased | |
| Biomarkers: ox-LDL | PA | EH | - | Increased | |
| Chou et al. (2018) [104] | Biomarkers: IL-6 | PA | EH | Adrenalectomy | Elevated IL-6 among PA using mineralocorticoid receptor/PI3K/Akt/NF-kB pathway |
| FMD/PAT | |||||
| Nishizaka et al. (2004) [163] | FMD + NMD | Resistant hypertension with hyperaldosteronism | Resistant hypertension without hyperaldosteronism | Spironolactone | Resistant hypertension with hyperaldosteronism showed lower FMD and improved after spironolactone. |
| Lai et al. (2016) [164] | FMD | ADPKD with PA | ADPKD without PA | - | ADPKD with PA shows lower FMD. |
| Chou et al. (2015) [165] | FMD + NMD | PA | EH | - | FMD and NMD are both decreased in PA SERCA2a suppression is observed in vascular smooth muscle. |
| Matsumoto et al. (2015) [55] | FMD + NMD | APA IHA | EH | Adrenalectomy on APA | FMD lower in APA than IHA and EH NMD no significant difference |
| Kishimoto et al. (2018) [166] | FMD + NMD PAT | APA IHA | EH | - | FMD lower in APA NMD lower in APA RHI lower in APA and IHA than EH. |
| Kishimoto et al. (2019) [56] | FMD + NMD PAT PWV | IHA | - | Eplerenone | RHI, NMD improved. PWV, ROCK activity decreased. No change in FMD and IMT. |
| Chang et al. (2015) [167] | PAT | PA | EH | - | PA had significantly higher AI but not RHI than EH |
| PWV | |||||
| Bernini et al. (2008) [168] | PWV | PA | EH and normotensives | - | PA showed more dysfunction and thicker IMT than EH and normotensives. |
| Strauch et al. (2006) [169] | PWV | PA | EH and normotensives | - | PA showed more dysfunction than EH and normotensives. |
| Strauch et al. (2008) [170] | PWV | PA receiving adrenalectomy | PA receiving spironolactone | Adrenalectomy or spironolactone | Endothelial dysfunction improved after adrenalectomy; not seen in spironolactone. |
| Rosa et al. (2012) [171] | PWV | PA | EH | - | PA showed more dysfunction |
| Wu et al. (2011) [131] | PWV | PA | EH | Adrenalectomy or spironolactone | Increased PWV in PA |
| Lin et al. (2012) [11] | PWV | APA | EH | Adrenalectomy | IMT and dysfunction improved after operation |
| Liao et al. (2016) [12] | PWV | APA | - | Adrenalectomy | Dysfunction improved after operation |
| Chang et al. (2017) [172] | PWV | APA with KCNJ5 (+) | APA with KCNJ5 (−) | Adrenalectomy | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.-W.; Tsai, C.-H.; Pan, C.-T.; Chou, C.-H.; Liao, C.-W.; Hung, C.-S.; Wu, V.-C.; Lin, Y.-H.; TAIPAI Study Group. Endothelial Dysfunction in Primary Aldosteronism. Int. J. Mol. Sci. 2019, 20, 5214. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205214
Chen Z-W, Tsai C-H, Pan C-T, Chou C-H, Liao C-W, Hung C-S, Wu V-C, Lin Y-H, TAIPAI Study Group. Endothelial Dysfunction in Primary Aldosteronism. International Journal of Molecular Sciences. 2019; 20(20):5214. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205214
Chicago/Turabian StyleChen, Zheng-Wei, Cheng-Hsuan Tsai, Chien-Ting Pan, Chia-Hung Chou, Che-Wei Liao, Chi-Sheng Hung, Vin-Cent Wu, Yen-Hung Lin, and TAIPAI Study Group. 2019. "Endothelial Dysfunction in Primary Aldosteronism" International Journal of Molecular Sciences 20, no. 20: 5214. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205214