Insights into the Pathophysiology of Infertility in Females with Classical Galactosaemia


1
National Centre for Inherited Metabolic Disorders, Adult Services, Mater Misericordiae University Hospital, Dublin, Ireland
2
Department of Paediatrics, Trinity College Dublin, Dublin, Ireland
3
School of Medicine and Medical Sciences, University College Dublin, Dublin, Ireland
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(20), 5236; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205236
Submission received: 27 September 2019
/
Revised: 14 October 2019
/
Accepted: 16 October 2019
/
Published: 22 October 2019
(This article belongs to the Special Issue Molecular Basis of Fertility Preservation and Restoration)
{kind=link}
Abstract
:Classical galactosaemia (CG) (OMIM 230400) is a rare inborn error of galactose metabolism caused by the deficiency of the enzyme galactose-1-phosphate uridylyltransferase (GALT, EC 2.7.7.12). Primary ovarian insufficiency (POI) is the most common long-term complication experienced by females with CG, presenting with hypergonadotrophic hypoestrogenic infertility affecting at least 80% of females despite new-born screening and lifelong galactose dietary restriction. In this review, we describe the hypothesized pathophysiology of POI from CG, implications of timing of the ovarian dysfunction, and the new horizons and future prospects for treatments and fertility preservation.
1. Introduction
Classical galactosaemia (CG) (OMIM 230400) is a rare inborn error of galactose metabolism caused by deficiency of the enzyme galactose-1-phosphate uridylyltransferase (GALT, EC 2.7.7.12), the second enzyme in the Leloir pathway, the pathway for galactose metabolism [1]. Classical galactosaemia is also known as Type 1. Type 2 galactosaemia is caused by mutation of the GALK1 gene, characterized by deficiency of galactose kinase 1 enzyme. Type 3 is caused by mutations involving the GALE gene, characterized by deficiency of the enzyme UDP-galactose-4-epimerase. Recently, Wada et al. [2] described novel finding pathogenic variants of the GALM gene which encodes galactose mutarotase, the enzyme which catalyzes the epimerization between β and α-D-galactose in the first step of the Leloir pathway in patients with ‘unexplained congenital galactosaemia. This has suggested a new Type IV classification for galactosaemia [2].
The prevalence of classical galactosaemia (CG), Type 1, ranges from 1:16,000 to 1:60,000 in Europe and USA [3,4]. The incidence in the Irish population is 1:16,476 and is 1:430 in the Irish Traveller community [4]. Although classical galactosaemia has been described since 1908 and the gene identified in 1992, it is still considered a partially treated rare disease. Restriction of galactose is life-saving in the neonate and improves the neonatal intoxication manifestations of feeding difficulties, failure to thrive, sepsis, hepatocellular damage, renal tubulopathy, and cataracts. Affected individuals develop long term complications affecting the central nervous system, bone density/metabolism, and primary ovarian insufficiency and subfertility in females despite dietary galactose restriction [4,5,6,7,8,9]. Primary ovarian insufficiency in CG, first described in 1978 [10] with ovarian follicular depletion is reported in at least 80% of females [9]. This is a particularly devastating complication for affected females. Moreover, the timing of the ovarian insult and its pathophysiology are poorly understood, which limits interventions. This article aims to clarify recent insights into the pathophysiology of this presentation, and the consideration of new therapeutic interventions.
2. Methods
Literature on the effects of CG on the female reproductive system was reviewed by an extensive Pubmed search (publications from January 1975 to September 2019) using the keywords: galactosaemia; ovarian function/dysfunction; primary ovarian insufficiency/failure; follicle stimulating hormone (FSH); oxidative stress; fertility preservation. In addition, articles cited in the search articles and literature known to the authors were also included in this review. A review of the Cochrane database was also performed with the condition ‘Galactosaemia.’ This did not indicate any review relating to infertility in females with CG.
3. Background
3.1. Galactosaemia and Galactose Metabolism
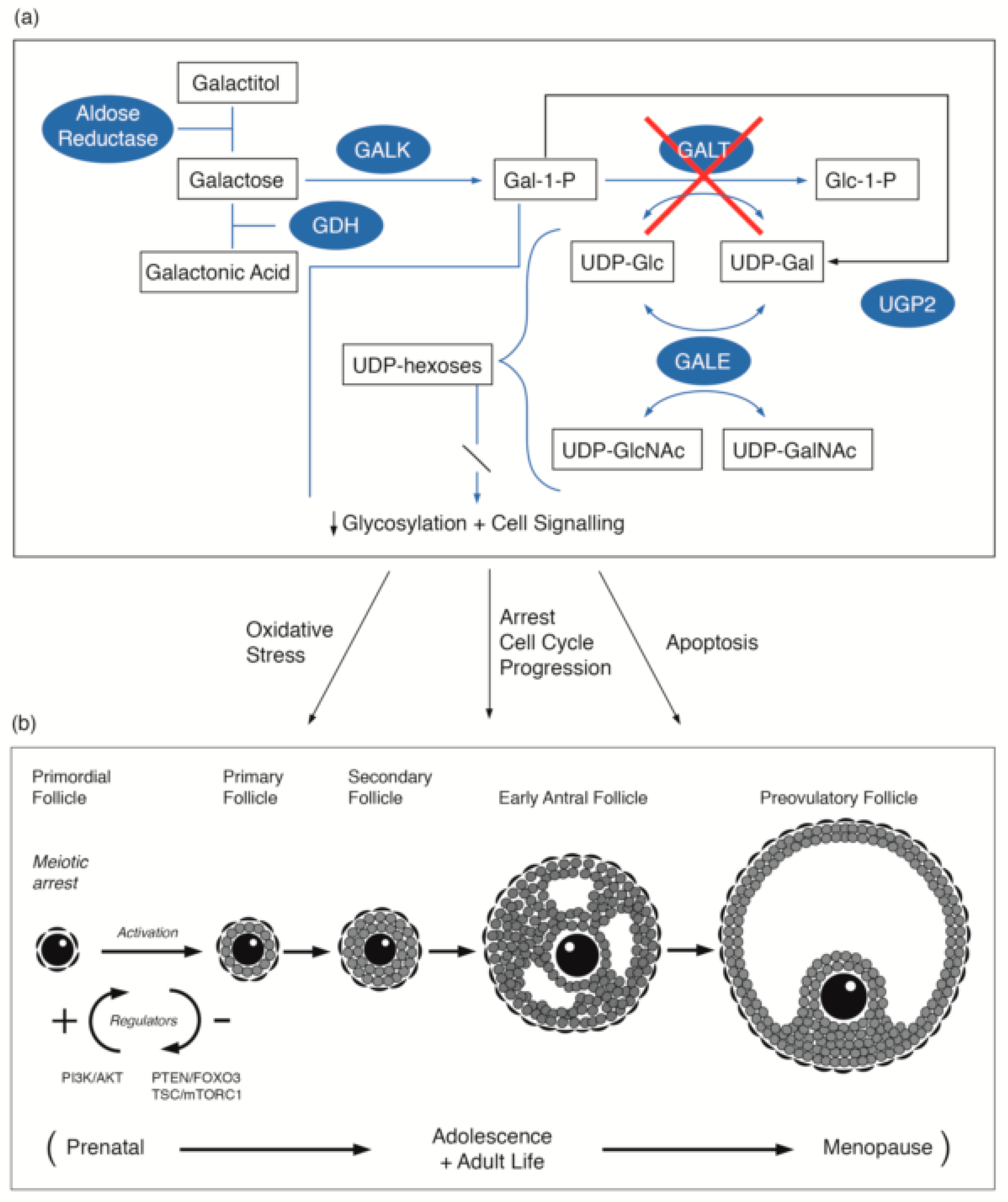
Galactose is converted to glucose-1-phosphate and metabolized to release energy, or alternatively, galactose may be metabolized to UDP-galactose (UDP-GAL) and its derivatives. UDP-GAL is an essential cofactor for the galactose transferases that are involved in the incorporation of galactose into glycoproteins and glycolipids [11]. Galactosaemia is an inborn error of metabolism resulting in impaired activity of one of the four enzymes involved in the main galactose metabolism pathway (known as the Leloir pathway): galactokinase (GALK), galactose-1-phosphate uridylyltransferase (GALT), UDP galactose 4-epimerase (GALE) and the recently described galactose mutarotase (GALM) deficiency, (Figure 1a). Deficiency in GALT causes classical galactosaemia Type 1 (CG). Although the liver is the major organ for galactose metabolism, the enzymes of the Leloir pathway have been found in many cell types and tissues, including the gonads, intestinal mucosa, kidneys, skeletal muscles, fibroblasts, leukocytes, and red cells. Galactose-1-phosphate uridylyltransferase and galactokinase are present in fetal red cells, the liver, the lung, the spleen, and cardiac muscle from at least 10 weeks’ gestation, and their activities are higher in the second and third trimesters than at any time postnatally [12]. This suggests the possibility that damage may occur in utero in GALT deficiency. Studies in rat tissue showed the liver to have the highest GALT mRNA and GALT activity. Kidneys, ovaries, and the heart have similar but lower mRNA and GALT activities, and skeletal muscle and testes have the least [13]. The long-term complications of CG are known to be present in organs with high physiological GALT-expression. Endogenous galactose production occurs in utero and throughout life. It is age-related, with higher levels in children than in adults. The endogenous production of circulating free galactose in adults ranges from 0.53 to 1.05 mg/kg/h [14,15].
Classical galactosaemia represents the most severe form of this disorder as a result of profound impairment in GALT resulting in accumulation of galactose, galactose-1-phosphate, galactitol, and galactonate in body tissues and fluid. Over 300 different mutations are described in the GALT gene [21]. The commonest genotype in European populations is the homozygous GALT Q188R genotype which involves the enzyme’s active catalytic site and is associated with a severe phenotype [22]. The S135L variant, predominantly found in the African and African-American populations, results in significant GALT residual activity and a generally milder phenotype [23].
3.2. Primary Ovarian Insufficiency in Female Galactosaemia Patients
Primary ovarian insufficiency is considered to be present when a woman who is less than 40 years old has had amenorrhea for 4 months or more, with two serum FSH levels (obtained at least 1 month apart) in the menopausal range [24]. This may present as delayed puberty, oligomenorrhoea/amenorrhoea or irregular menstrual cycles, progressing to infertility and complications of hypoestrogenism.
The association between galactosaemia and primary ovarian insufficiency (POI) was first reported in 1979 [25].
The Galactosaemia Consortium (GalNet) Registry outcome study [9] has recently demonstrated that 80% of females (of n = 164) have primary ovarian insufficiency which is consistent with other studies [4,5,6,7,8,26,27].
The onset and timing of damage causing primary ovarian insufficiency (POI) in CG patients remain unknown. It is unclear at which stage of development this occurs, and whether this is ongoing due to galactose exposure or a single event leading to irreversible life-long damage. In depth understanding of the pathophysiology and the mechanism of this process and the implications will lead to the possibility of reversing or even stopping this process. Risk factors for developing ovarian insufficiency in CG include homozygosity for the GALT Q188R mutation, severe whole-body galactose oxidation impairment, and mean Gal-1-P levels in erythrocytes [28]. There were no correlations found between the age of initiation of dietary galactose restriction, compliance, or high erythrocyte Gal-1-P levels with POI from CG.
Females with CG generally have higher concentrations of FSH and reduced concentrations of anti-mullerian hormone (AMH) [29,30]. In total, 90% of patients showed signs of hypergonadotropic hypogonadism over the 4-year study period in the study of Kaufman et al. 1986 [31].
It is proposed that the ovarian damage occurs prenatally, suggested by reduced oocyte numbers in rats subjected to prenatal exposure to high levels of galactose [32]. Streak ovaries have been reported on laparoscopy in young females [25,33,34,35,36]. Postnatal damage is supported by the report of normal ovarian tissue in a girl who died from Escherichia coli sepsis at 5 days of age [37], and by the finding that in some patients, gonadal function may initially be normal but can become abnormal with time [31]. Ovarian histology in galactosaemic women with ovarian insufficiency has been variable. The numbers of oocytes have reportedly been absent or low, with fibrous and streaky stroma. In a study of 53 girls with galactosaemia, 77% experienced POI at a mean age of 13 years [28]. A case report of a 17-year-old girl with CG indicated that at the age of seven, her ovaries were normal upon abdominal exploration, but at age 17 the ovaries were described as streaked in appearance [25]. Several other case reports described small, atrophic, streak-like, and sometimes undetectable ovaries in patients between ages of 13 and 30 years [38]. Mamsen et al. studied the pathology of six CG prepubertal girls below age 12 (mean age 3.8 ± 1.7 years; range 0.4–11.7 years)). All five girls less than age five had follicle counts within the normal control limits. In contrast, there were no follicles detected in the sample taken from the 11.7-year-old girl [39].
Although the prevalence of spontaneous conception in POI was previously considered to be low [1], Van Erven et al., 2017 reported that nine out of 21 patients with CG galactosaemia achieved pregnancy (42.9%) [40]. This pregnancy rate was higher than anticipated based on reported cases of POI from any cause (5–10%).
Hypergonadotropic hypogonadism is usually diagnosed in the second decade of life in women with CG. The serum FSH levels may be noted to be elevated, however, as early as 4 months and from early childhood to the onset of puberty. LH levels may be variable; however, oestradiol is usually low [1,24].
The exact mechanism underlying POI from CG is still unknown. There have been a number of possible pathogenic mechanisms proposed. One is the direct toxic effects of galactose and its metabolites to the ovary, and ovarian development. Another hypothesis is atypical function of FSH and/or its receptor secondary due to glycosylation abnormalities. Additionally, cell signalling abnormalities, epigenetic mechanisms, and premature apoptosis have been proposed [41,42,43].
The relatively high physiological GALK, GALT, and GALE activities in the ovaries cause the accumulation of galactose and its metabolites. Direct toxic damage to the ovary through either galactose or its metabolites may occur. Galactitol if not metabolized causes accumulation in ovarian cells, leading to swelling and cell dysfunction, with oxidative stress and premature apoptosis [43].
The activity of UDP-glucose (UDP-Glc) pyrophosphorylase highly expressed in ovarian tissue is critical for nucleotide sugar production and maintenance of germ cell function, follicular maturation and steroidogenesis. UDP galactose (UDP-Gal) is required for the synthesis of glycoproteins and galactolipids that serve many functions; for example, in the ovarian membrane, the support of germ cells, follicular maturation, and steroidogenesis. Therefore, UDP-Gal deficiency might be a pathogenic mechanism [1]. This mechanism has not been studied, however, in regard to ovarian function. Recently, Rubio-Gozalbo et al. 2019, identified abnormal uridine sugars in the zebrafish galactosaemia model. The result from their study showed that CG zebrafish have a decreased UDP-Glc:UDP-Gal ratio in the ovary with a lower UDP-GlcNAc:UDP-GalNAc ratio compared to the wild-type zebrafish. CG fish also showed increased CMP-Neu5Ac levels, suggesting impaired sialyation and increased GDP-fucose levels in early developmental stages and in adulthood, particularly in the ovary, pointing to fucosylation impairments [44].
It was also proposed that auto-ovarian antibodies could cause ovarian insufficiency in CG. This mechanism has not been confirmed, however, in CG.
Earlier studies have reported normal FSH bioactivity], but this does not exclude abnormalities of the FSH-FSH receptor interaction secondary to hypoglycosylation [45,46].
Our group has described significant systemic n-glycosylation deficiencies in CG children and adults in circulating serum glycoproteins and IgG. This is best described as a mixed CDG-I and II defect, and n-glycan assembly and processing defect [47,48,49]. Fundamental findings in the measured glycans are increased fucosylation and branching defects which could potentially affect cell adhesion and cell signalling. Patients with primary congenital disorders of glycosylation (CDG), due to mutations in genes encoding for products involved in glycan assembly also present with learning impairment, speech defects, and ovarian failure with hypergonadotrophic hypogonadism, suggesting a similar pathophysiological aetiology. A similar clinical feature is also seen in female patients with an inactivating mutation of the FSH-receptor showing hypergonadotropic hypogonadism with delayed or no spontaneous puberty [50].
In addition to abnormalities of circulating glycoproteins our group Coman et al., 2010, studying T cell gene microarray expression, identified a major systemic signalling pathway dysfunction in galactosaemia patients involving aberrations of the MAPK signalling pathway, the regulation of the actin cytoskeleton, the calcium signalling pathway, the cell adhesion molecules in addition to the haematopoietic cell lineage, and cytokine-cytokine receptor interactions. This was proposed to be as a consequence of systemic abnormal galactosylation of numerous cellular glycoproteins with possible abnormalities of protein glycan site occupancy, folding, and survival [16]. A further follow up study of a larger patient sample size illustrated systemic dysregulation of numerous gene pathways, including glycosylation, inflammatory, and inositol signalling pathways [17].
In a further study from our group, we studied specific glycan synthesis, the leptin system, and inflammatory gene expression in white blood cells as the potential biomarkers of infertility in 54 adults with CG on a galactose-restricted diet in a multi-site Irish and Dutch study. Systemic dysregulation of genes, including LEP, LEPR, ANXA1, and ICAM1 was evident for CG patients, and combined with abnormalities of IgG glycosylation, hormonal and leptin analyses illustrated the presence of systemic glycosylation, inflammatory, and cell signalling abnormalities [19].
A number of galactose toxicity studies in animal models are informative. The investigation of pregnant rats fed a 50% galactose diet showed a striking reduction in oocyte number in the offspring [32]. Additionally, adult female mice fed a 50% galactose diet demonstrated a decrease in ovulatory response [51].
Bandyopadhyay et al., 2007 conducted a study of feeding pregnant rats with 35% galactose supplements from day 3 of conception continuing through weaning of the litters. The induced galactose toxicity in the offspring delayed the onset of puberty in the female rat offspring and the offspring rats subsequently developed hypergonadotrophic hypoestrogenism [52].
A GALT knockout zebrafish model has been successfully developed to mimic the human phenotype at both the biochemical and clinical levels of CG [53]. The knockout zebrafish accumulated elevated concentrations of Gal-1-P, which was exacerbated upon exposure to exogenous galactose. Knockout pairs, wildtype females/knockout males, and knockout females/wildtype males exhibited a significantly lower egg quantity per mating as compared to wildtype pairs. The observed gonadal impairments in unexposed GALT knockout zebra fish are in line with the human phenotype of ovarian sequelae. Impaired male infertility in fish is in line with previous reports of possible sub-clinical male infertility [54].
These clinical findings in the zebra fish model are in line with observations in a GALT-deficient mice model, whereas the females showed smaller litter size and longer time to achieve pregnancy [55]. Despite the normal ovaries on autopsy in two neonates [37,56], observations in animal models suggest a prenatal origin of the damage [32,57].
In the GALT deficient mouse model that demonstrated a subfertility phenotype in adult females, Balakrishnan et al. identified that GALT is a positive regulator of the PI3K/Akt signalling pathway [18]. The protein expression of BiP and PTEN (negative regulator) of the PI3K/Akt signalling pathway was higher in GALT deficiency. The up-regulation of PTEN was also demonstrated in the human GC study of Coss et al. (2014), as described earlier [17]. Salubrinal, a chemical compound that alleviates ER stress when administered to mutant mouse fibroblast cell lines normalised the down regulated PI3/Akt signalling pathway and the premature loss of primordial follicles seen in young mutant mice [18].
Interestingly, to date, there is no published data concerning the long-term outcome of children from mothers with classical galactosaemia. There is one case report of a healthy child born to a female CG patient who had documented premature ovarian insufficiency (POI), who conceived following FSH therapy [58].
3.3. Link to Primordial Germ Cell Signalling
Oogenesis, oocyte and follicular growth, and the development and oocyte maturation are complex processes regulated by intra and extra ovarian factors and signalling mechanisms, which, due to the systemic cell signalling abnormalities in galactosaemia, are proposed to be aberrant in galactosaemia. Oocytes originate from primordial germ cells in the extra embryonic mesoderm. Ovarian factors produced by theca/stromal cells, somatic granulosa cells, participate and regulate oocyte and follicle development at each of the developmental stages [20].
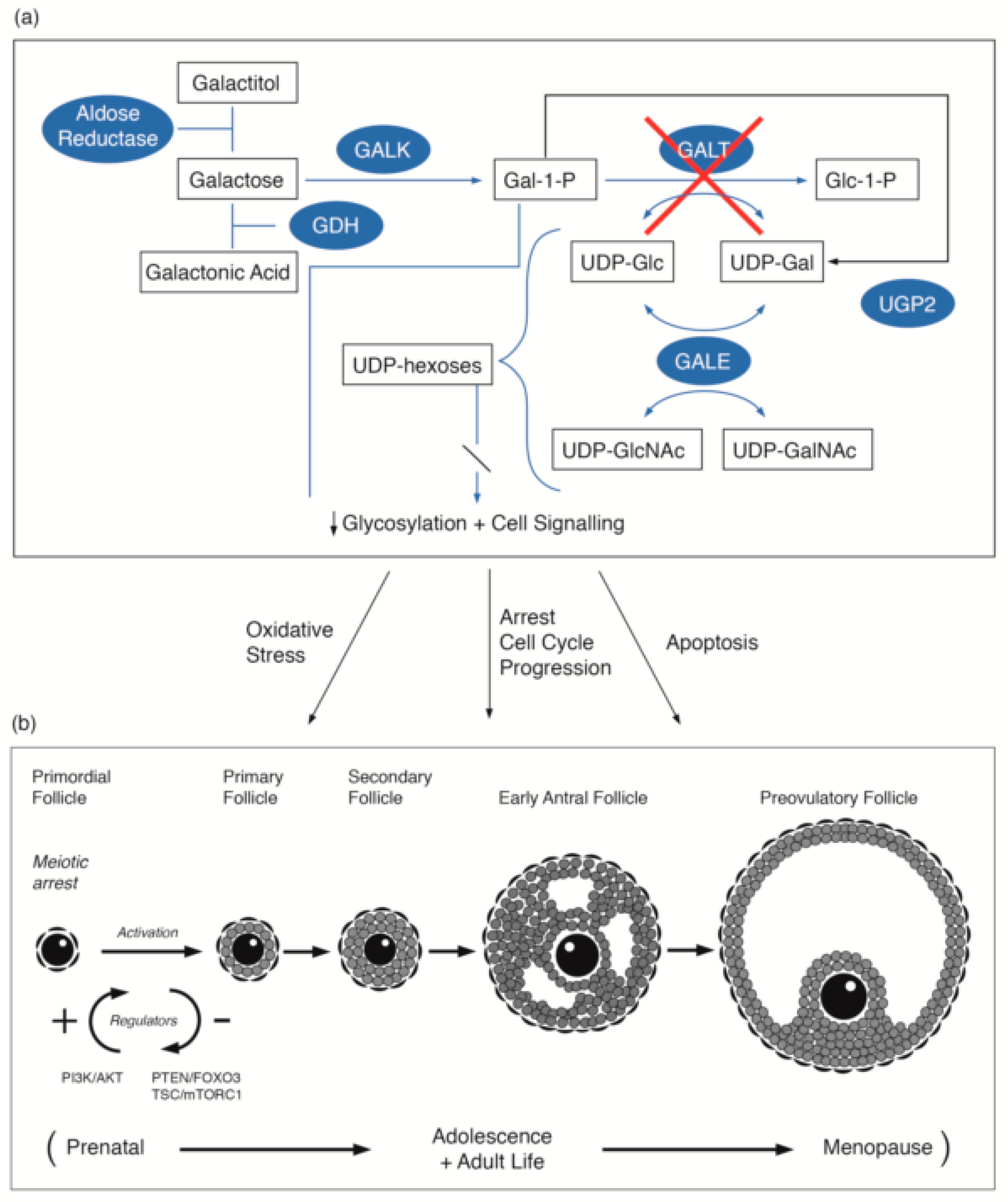
The first production of primordial follicles contributes to the onset of female puberty and fertility. The continuing adult production of primordial follicles provides fertility to usual female menopause. The majority of ovarian primordial follicles are maintained quiescently as a reserve for the reproductive life span. In recent years the molecular and signalling mechanisms that regulate the activation of primordial follicles have been described. Pathways, such as the phosphatidylinositol 3 kinase (PI3K) pathway with its regulator repressor protein PTEN, and the linked MTORC1 pathway and FOXO3 protein, have central roles [59,60]. FOXO3 is considered to have a repressor function. Kim et al., has confirmed the pivotal role of PTEN–PI3K signalling in the activation of primordial follicles [60]. We propose that dysregulation of the PTEN–PI3K signalling pathway in galactosaemia patients was identified in our studies and in the Gal deficient mouse model may be implicated in the primordial follicular dysgenesis observed in galactosaemia (Figure 1b) [17,18].
Recent studies have reported successful in vitro activation of mammalian primordial follicles with synthetic PTEN inhibitors [60,61]. The use of existing primordial follicles as a source for obtaining fertilizable oocytes offers a new, exciting addition for the treatment of females diagnosed with POI. Successful in vitro activation of mouse and human primordial follicles with the synergistic use of an MTORI activator and a PTEN inhibitor has now been reported [62,63].
4. Approaches to Treatment
Fertility Preservation
The Galactosemia Network (GalNet) makes recommendations for girls and women with CG, for annual monitoring for menstrual abnormalities, secondary amenorrhoea, and symptoms of POI, including FSH levels. Women with hypergonadotropic hypogonadism or POI should have counselling and support on their reproductive options and management of their irregular or absent menses. A referral to a reproductive endocrinologist can be offered to women who desire pregnancy but are unable to conceive naturally or who would like to know other available fertility treatment [64].
One of the key challenges to prevention of POI from CG remains the uncertainty of the timing of the irreversible changes, which may be within the first 10 years of life with a likely prenatal effect. The current approach to treatment for galactosaemic women with POI mainly involves hormonal replacement with oestrogen supplementation and added progesterone to reduce the risk of endometrial hyperplasia or cancer.
Despite the high prevalence of POI and infertility in women with galactosaemia, spontaneous unassisted pregnancies are possible, even repeatedly. They have been reported and are not as rare as previously assumed as demonstrated by the recent study referred to earlier [40]. This should be emphasized to patients and they should not be discouraged from trying conceiving naturally. The chances of conceiving are higher in patients who reached puberty spontaneously and in younger patients, as in any woman, fertility declines with age.
A conservative approach to achieve pregnancy in POI is through ovarian stimulation. If this fails or is not possible, the next available treatment option is oocyte donation. Female CG patients have been reported to have successful pregnancy after this procedure [40]. Even though this procedure has been proven to be effective with higher pregnancy rates; the cost, availability of donor eggs, and need for psychological counselling are limiting factors. Embryo freezing after in vitro fertilization (IVF) is another available option with good outcomes. However, a major limiting factor for this procedure in this cohort of patients is that a large number of eggs would need to be retrieved from controlled ovarian hyperstimulation which may not be effective in CG females with diminished ovarian reserve.
Ovarian tissue cryopreservation and transplantation (OTCP-TP) has dramatically evolved over recent years from experimental breakthrough to a well-accepted treatment. Unlike the conservative treatment approach to POI. This procedure does not require ovarian stimulation. The techniques involve ovarian tissue extraction, freezing/thawing, and in vitro maturation or transplantation back into the same patient. This procedure is remarkably successful, with studies reporting return of ovarian function in up to 95% of graft recipients and pregnancy rates of between 30–50%. The most significant limitation of OTCP-TP is the massive loss of follicles that occurs following transplantation, which is primarily attributed to ischaemic damage and follicle activation [65].
Cutting-edge technologies for in vitro follicle growth, and tissue engineering principles to bioengineer artificial ovaries are currently being explored [66,67].
Currently, there are no defined recommendations for fertility preservation for patients with CG [68]. As the onset of ovarian reserve depletion is still unclear, this complicates the timing to offer fertility preservation. Most of the girls who would benefit from fertility preservation are too young to make the decision and to consent. Counselling female patients with CG regarding their fertility is complex with the evidence of spontaneous pregnancies and it is difficult to predict the ideal candidates for fertility preservation. The publication of van Erven et al. 2017 proposed a number of recommendations regarding fertility preservation in CG females [68]. These included encouraging patients to recognise that spontaneous pregnancies can occur in women with CG with POI, recommendations regarding ovarian cryopreservation at an early pre-pubertal age, aspects of fertility preservation, and other options.
5. New Horizons and Future Prospects for Fertility Treatments in Classical Galactosaemia
A major breakthrough in infertility is the activation of dormant ovarian follicles using phosphatidylinositol-3-kinase activators and the suppression of the Hippo signalling pathway—with successful pregnancies reported using this method [69,70]. This may be applicable to galactosaemia given that this pathway is involved. However, an overall approach to timely stabilisation of the function of the mutant GALT enzyme during the critical early childhood period (or indeed prenatal period) may be required to stabilise not only the galactose and its products’ intoxication effects, but also the dysfunctional systemic glycosylation defects, and subsequent systemic signalling abnormalities.
Therapeutic strategies for intervention (new treatments in development for CG), such as chaperones, antioxidants, substrate inhibitors, aldose reductase inhibitors, gene modification strategies, proteostasis regulator, or mRNA approaches will also need to consider the prenatal period, although ethical and practical concerns would accompany such intervention. It will be important also to consider at which stage in the postnatal period that any given form of intervention may have the greatest chance for success [58,71,72].
Other treatment options on the horizon include tissue or organ transplantation; for example, liver and in vivo or ex vivo gene therapy. The prospect of utilizing ovarian stem-cell based therapeutics for ovarian regeneration is also interesting. Such advances in reproductive medicine in the pipeline are bringing new hope to women diagnosed with POI from CG [73,74].
6. Conclusion
Despite several studies, the underlying pathophysiological mechanism and the timing of the ovarian dysfunction of POI from CG remain unclear. Substantial progress accomplished through exponential increase in knowledge in the field of infertility with better understanding of key signalling pathways and biomarkers offer exciting new discoveries and methods to preserve fertility in CG patients.
Author Contributions
The authors Z.A. and E.P.T. were involved in the conceptualisation of the review. Z.A. performed the literature review and first draft of the article. Both Z.A. and E.P.T. were involved in writing and reviewing the manuscript.
Funding
The authors acknowledge an Irish HRB funding award (HRA-POR-2014-623) to EPT which supported these studies.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| Akt | Protein kinase B |
| AMH | Anti-Mullerian hormone |
| CG | Classical Galactosaemia |
| FOX03 | Forkhead box 03 gene |
| FSH | Follicle stimulating hormone |
| LH | Luteinizing hormone |
| Gal | Galactosaemia |
| Gal-1-P | Galactose-1-phosphate |
| GALT | Galactose-1-phosphate uridylytransferase |
| GALE | Galactose epimerase |
| GALK | Galactokinase |
| GALM | Galactose mutarotase |
| MTORC1 | Mammalian target of rapamycin complex 1 |
| P13K | Phosphatidylinositol 3-kinase |
| POI | Primary Ovarian Insufficiency |
| PTEN | Phosphatase and tensin homolog |
| UDP-Gal | Uridine diphosphate galactose |
| UGP | UDP-glucose pyrophosphorylase |
References
- Valle, D.; Antonarakis, S.; Ballabio, A.; Beaudet, A.; Mitchell, G.A. The Online Metabolic and Molecular Bases of Inherited Disease; The McGraw-Hill Companies: New York, NY, USA, 2008. [Google Scholar]
- Wada, Y.; Kikuchi, A.; Arai-Ichinoi, N.; Sakamoto, O.; Takezawa, Y.; Iwasawa, S.; Niihori, T.; Nyuzuki, H.; Nakajima, Y.; Ogawa, E.; et al. Biallelic GALM pathogenic variants cause a novel type of galactosemia. Genet. Med. 2019, 21, 1286–1294. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.T. Classic Galactosemia and Clinical Variant Galactosemia. Gene Rev. 2017. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK1518/ (accessed on 10 September 2019).
- Coss, K.P.; Doran, P.P.; Owoeye, C.; Codd, M.B.; Hamid, N.; Mayne, P.D.; Crushell, E.; Knerr, I.; Monavari, A.A.; Treacy, E.P. Classical galactosaemia in Ireland: Incidence, complications and outcomes of treatment. J. Inherit. Metab. Dis. 2013, 36, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Waggoner, D.D.; Buist, N.R.; Donnell, G.N. Long-term prognosis in galactosaemia: Results of a survey of 350 cases. J. Inherit. Metab. Dis. 1990, 13, 802–818. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, S.; Shin, Y.; Jakobs, C.; Brodehl, J. Long-term outcome in 134 patients with galactosaemia. Eur. J. Pediatr. 1993, 152, 36–43. [Google Scholar] [CrossRef]
- Waisbren, S.E.; Potter, N.L.; Gordon, C.M.; Green, R.C.; Greenstein, P.; Gubbels, C.S.; Rubio-Gozalbo, E.; Schomer, D.; Welt, C.; Anastasoaie, V.; et al. The adult galactosemic phenotype. J. Inherit. Metab. Dis. 2012, 35, 279–286. [Google Scholar] [CrossRef]
- Jumbo-Lucioni, P.P.; Garber, K.; Kiel, J.; Baric, I.; Berry, G.T.; Bosch, A.; Burlina, A.; Chiesa, A.; Pico, M.L.; Estrada, S.C.; et al. Diversity of approaches to classic galactosemia around the world: A comparison of diagnosis, intervention, and outcomes. J. Inherit. Metab. Dis. 2012, 35, 1037–1049. [Google Scholar] [CrossRef]
- Rubio-Gozalbo, M.E.; Haskovic, M.; Bosch, A.M.; Burnyte, B.; Coelho, A.I.; Cassiman, D.; Couce, M.L.; Dawson, C.; Demirbas, D.; Derks, T.; et al. The natural history of classic galactosaemia: Lessons from the GalNet registry. Orphanet J. Rare Dis. 2019, 14, 86. [Google Scholar] [CrossRef]
- Kaufman, F.; Kogut, M.D.; Donnell, G.N.; Koch, H.; Goebelsmann, U. Ovarian failure in galactosaemia. Lancet 1979, 314, 737–738. [Google Scholar] [CrossRef]
- Roth, S.; McGuire, E.J.; Roseman, S. Evidence for cell-surface glycosyltransferases: Their potential role in cellular recognition. J. Cell Biol. 1971, 51, 536. [Google Scholar] [CrossRef]
- Shin-Buehring, Y.S.; Beier, T.; Tan, A.; Osang, M.; Schaub, J. The activity of galactose-1-phosphate uridyltransferase and galactokinase in human fetal organs. Pediatr. Res. 1977, 11, 1045. [Google Scholar] [CrossRef]
- Heidenreich, R.A.; Mallee, J.; Rogers, S.; Segal, S. Developmental and tissue-specific modulation of rat galactose-1-phosphate uridyltransferase steady state messenger RNA and specific activity levels. Pediatr. Res. 1993, 34, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Berry, G.T.; Moate, P.J.; Reynolds, R.A.; Yager, C.T.; Ning, C.; Boston, R.C.; Segal, S. The rate of de novo galactose synthesis in patients with galactose-1-phosphate uridyltransferase deficiency. Mol. Genet. Metab. 2004, 81, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Schadewaldt, P.; Kamalanathan, L.; Hammen, H.W.; Wendel, U. Age dependence of endogenous galactose formation in Q188R homozygous galactosemic patients. Mol. Genet. Metab. 2004, 81, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Coman, D.J.; Murray, D.W.; Byrne, J.C.; Rudd, P.M.; Bagaglia, P.M.; Doran, P.D.; Treacy, E.P. Galactosemia, a single gene disorder with epigenetic consequences. Pediatr. Res. 2010, 67, 286–292. [Google Scholar] [CrossRef]
- Coss, K.P.; Treacy, E.P.; Cotter, E.J.; Knerr, I.; Murray, D.W.; Shin, Y.S.; Doran, P.P. Systemic gene dysregulation in classical Galactosaemia: Is there a central mechanism? Mol. Genet. Metab. 2014, 113, 177–187. [Google Scholar] [CrossRef]
- Balakrishnan, B.; Nicholas, C.; Siddiqi, A.; Chen, W.; Bales, E.; Feng, M.; Johnson, J.; Lai, K. Reversal of abberant PI3K/Akt signalling by Salubrinal in a GaltT-deficient mouse model. Biochem. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3286–3293. [Google Scholar] [CrossRef]
- Colhoun, H.O.; Rubio-Gozalbo, E.M.; Bosch, A.M.; Knerr, I.; Dawson, C.; Brady, J.; Galligan, M.; Stepien, K.; O’Flaherty, R.; Catherine Moss, C.; et al. Fertility in classical galactosaemia, a study of n-glycan, hormonal and inflammatory gene interactions. Orphanet. J. Rare Dis. 2018, 13, 164. [Google Scholar] [CrossRef]
- Sanchez, F.; Smitz, J. Molecular control of oogenesis. Biochim. Biophys. Acta 2012, 1822, 1896–1912. [Google Scholar] [CrossRef] [Green Version]
- Calderon, F.R.O.; Phansalkar, A.R.; Crockett, D.K.; Miller, M.; Mao, R. Mutation database for the galactose-1-phosphate uridylytransferase (GALT) gene. Hum. Mutat. 2007, 28, 939–943. [Google Scholar] [CrossRef]
- Lai, K.; Willis, A.C.; Elsas, L.J. The biochemical role of glutamine 188 in human galactose-1-phosphate uridyltransferase. J. Biol. Chem. 1999, 274, 6559–6566. [Google Scholar] [CrossRef]
- Lai, K.; Langley, S.; Singh, R.H.; Dembure, P.P.; Hjelm, L.; Elsas, L.J. A prevalent mutation for galactosaemia among black Americans. J. Pediatr. 1996, 128, 89–95. [Google Scholar] [CrossRef]
- Nelson, L.M. Clinical Practice. Primary ovarian insufficiency. N. Engl. J. Med. 2009, 360, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, F.R.; Kogut, M.D.; Donnell, G.N.; Goebelsmann, U.; March, C.; Koch, R. Hypergonadotropic hypogonadism in female patients with galactosemia. N. Engl. J. Med. 1981, 304, 994–998. [Google Scholar] [CrossRef]
- Gubbels, C.S.; Land, J.A.; Rubio-Gozalbo, M.E. Fertility and impact of pregnancies on the mother and child in classic galactosemia. Obstet. Gynecol. Surv. 2008, 63, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Fridovich-Keil, J.L.; Gubbles, C.S.; Spencer, J.B.; Sanders, R.D.; Land, J.A.; Rubio-Gozalbo, E. Ovarian function in girls and women with GALT-deficiency galactosemia. J. Inherit. Metab. Dis. 2011, 34, 357–366. [Google Scholar] [CrossRef]
- Guerrero, N.V.; Singh, R.H.; Manatunga, A.; Berry, G.T.; Steiner, R.D.; Elsas, L.J.I. Risk factors for premature ovarian failure in females with galactosemia. J. Pediatr. 2000, 137, 833–841. [Google Scholar] [CrossRef]
- Sanders, R.D.; Spencer, J.B.; Epstein, M.P.; Pollak, S.V.; Vardhana, P.A.; Lustbader, J.W.; Fridovich-Keil, J.L. Biomarkers of ovarian function in girls and women with classic galactosaemia. Fertil. Steril. 2009, 92, 344–351. [Google Scholar] [CrossRef]
- Spencer, J.B.; Badik, J.R.; Ryan, E.L.; Gleason, T.J.; Broadaway, K.A.; Epstein, M.P.; Fridovich-Keil, J.L. Modifiers of ovarian function in girls and women with classic galactosaemia. J. Clin. Endocrinol. Metab. 2013, 98, E1257–1265. [Google Scholar] [CrossRef]
- Kaufman, F.R.; Donnell, G.N.; Roe, T.F.; Kogut, M.D. Gonadal function in patients with galactosaemia. J. Inherit. Metab. Dis. 1986, 9, 140–146. [Google Scholar] [CrossRef]
- Chen, Y.T.; Mattison, D.R.; Feigenbaum, L.; Fukui, H.; Schulman, J.D. Reduction in oocyte number following prenatal exposure to a diet high in galactose. Science 1981, 214, 1145–1147. [Google Scholar] [CrossRef]
- Hoefnagel, D.; Wurster-Hill, D.; Child, E.L. Ovarian failure in galactosaemia. Lancet 1979, 314, 1197. [Google Scholar] [CrossRef]
- Fraser, I.S.; Russell, P.; Greco, S.; Robertson, D.M. Resistant ovary syndrome and premature ovarian failure in young women with galactosaemia. Clin. Reprod. Fertil. 1986, 4, 133–138. [Google Scholar] [PubMed]
- Morrow, R.J.; Atkinson, A.B.; Carson, D.J.; Carson, N.A.; Sloan, J.M.; Traub, A.I. Ovarian failure in a young woman with galactosaemia. Ulster Med. J. 1985, 54, 218. [Google Scholar] [PubMed]
- Robinson, A.C.; Dockeray, C.J.; Cullen, M.J.; Sweeney, E.C. Hypergonadotrophic hypogonadism in classical galactosaemia: Evidence for defective oogenesis. Case report. Br. J. Obstet. Gynaecol. 1984, 91, 199–200. [Google Scholar] [CrossRef]
- Levy, H.L.; Driscoll, S.G.; Porensky, R.S.; Wender, D.F. Ovarian failure in galactosemia (letter). N. Eng. J. Med. 1984, 310, 50. [Google Scholar]
- Rubio-Gozalbo, M.E.; Gubbels, C.S.; Bakker, J.A.; Menheere, P.P.; Wodzig, W.K.; Land, J.A. Gonadal function in male and female patients with classic galactosemia. Hum. Reprod. Update 2010, 16, 177–188. [Google Scholar] [CrossRef]
- Mamsen, L.S.; Kelsey, T.; Ernst, E.; Macklon, K.T.; Lund, A.M.; Andersen, C.Y. Cryopreservation of ovarian tissue may be considered in young girls with galactosaemia. J. Assis. Reprod. Genet. 2018, 2018 35, 1209–1217. [Google Scholar] [CrossRef]
- Van Erven, B.; Berry, G.T.; Cassiman, D.; Connolly, G.; Forga, M.; Gautschi, M.; Gubbels, C.S.; Hollak, C.E.M.; Janssen, M.C.; Knerr, I.; et al. Fertility in adult women with classic galactosaemia and primary ovarian insufficiency. Fertil. Steril. 2017, 108, 168–174. [Google Scholar] [CrossRef]
- Thakur, M.; Feldman, G.; Puscheck, E.E. Primary ovarian insufficiency in classic galactosaemia: Current understanding and future research opportunities. J. Assist. Reprod. Genet. 2018, 35, 3–16. [Google Scholar] [CrossRef]
- Gibson, J.B. Gonadal function in galactosemics and in galactose-intoxicated animals. Eur. J. Pediatr. 1995, 154, S14. [Google Scholar] [CrossRef]
- Meyer, W.R.; Doyle, M.B.; Grifo, J.A.; Lipetz, K.J.; Oates, P.J.; DeCherney, A.H.; Diamond, M.P. Aldose reductase inhibition prevents galactose-induced ovarian dysfunction in the Sprague-Dawley rat. Am. J. Obstet. Gynecol. 1992, 167, 1837–1843. [Google Scholar] [CrossRef]
- Rubio-Gozalbo, M.E.; Coelho, A.I.; Haskovic, M.; Van Scherpenzeel, M.; Lindhout, M.; Zijlstra, F.; Vanoevelen, J.M.; Bierau, J.; lefeber, D.J. Nucleotide sugar profile in the galactosaemia zebrafish model reveals new pathogenic mechanisms and potential readouts. J. Inherit. Metab. Dis. 2019, 42, S1. [Google Scholar]
- Davis, D.; Liu, X.; Segaloff, D.L. Identification of the sites of N-linked glycosylation on the follicle-stimulating hormone (FSH) receptor and assessment of their role in FSH receptor function. Mol. Endocrinol. 1995, 9, 159–170. [Google Scholar] [PubMed]
- Gubbels, C.S.; Land, J.A.; Evers, J.L.; Bierau, J.; Menheere, P.P.; Robben, S.G.; Rubio-Gozalbo, M.E. Primary ovarian insufficiency in classic galactosemia: Role of FSH dysfunction and timing of the lesion. J. Inherit. Metab. Dis. 2013, 36, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Coss, K.P.; Byrne, J.C.; Coman, D.J.; Adamczyk, B.; Abrahams, J.L.; Saldova, R.; Brown, A.Y.; Walsh, O.; Hendroff, U.; CArolan, C.; et al. IgG N-glycans as potential biomarkers for determining galactose tolerance in Classical Galactosaemia. Mol. Genet. Metab. 2012, 105, 212–220. [Google Scholar] [CrossRef]
- Coss, K.P.; Hawkes, C.P.; Crushell, E.; Adamczyk, B.; Saldova, R.; Knerr, I.; Monavari, A.A.; Rudd, P.M.; Treacy, E.P. IgG N-glycan abnormalities in children with Galactosaemia. J. Proteome Res. 2014, 13, 385–394. [Google Scholar] [CrossRef]
- Maratha, A.; Stockmann, H.; Coss, K.P.; Rubio-Gozalbo, E.M.; Knerr, I.; Fitzgibbon, M.; McVeigh, T.P.; Foley, P.; Moss, C.; Colhoun, H.-E.; et al. Classical galactosaemia: Novel insights in IgG n-glycosylation and n-glycan biosynthesis. Eur. J. Hum. Genet. 2016, 24, 976–984. [Google Scholar] [CrossRef]
- Aittomäki, K.; Lucena, J.L.; Pakarinen, P.; Sistonen, P.; Tapanainen, J.; Gromoll, J.; Kaskikari, R.; Sankila, E.M.; Lehväslaiho, H.; Engel, A.R.; et al. Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell 1995, 82, 959–968. [Google Scholar] [CrossRef] [Green Version]
- Swartz, W.J.; Mattison, D.R. Galactose inhibition of ovulation in mice. Fertil. Steril. 1988, 49, 522–526. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Chakrabarti, J.; Banerjee, S.; Pal, A.K.; Goswami, S.K.; Chakravarty, B.N.; Kabir, S.N. Galactose toxicity in the rat as a model for premature ovarian failure: An experimental approach readdressed. Hum. Reprod. 2003, 18, 2031–2038. [Google Scholar] [CrossRef]
- Vanoevelen, J.M.; van Erven, B.; Bierau, J.; Huang, X.; Berry, G.T.; Vos, R.; Coelo, A.I.; Rubio-Gozalbo, M.E. Impaired fertility and motor function in a zebrafish model for classic galactosaemia. J. Inherit. Metab. Dis. 2018, 41, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Gubbels, C.S.; Welt, C.K.; Dumoulin, J.C.; Robben, S.G.; Gordon, C.M.; Dunselman, G.A.; Rubio-Gozalbo, M.E.; Berry, G.T. The male reproductive system in classic galactosaemia: Cyptorchidism and low semen volume. J. Inherit. Metab. Dis. 2013, 36, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Siddiqi, A.; Witt, B.; Yuzyuk, T.; Johnson, B.; Fraser, N.; Chen, W.; Rascon, R.; Yin, X.; Goli, H.; et al. Subfertility and growth restriction in a new galactose-1-phosphate uridylytransferase (GALT)-deficient mouse model. Eur. J. Hum. Genet. 2014, 22, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.L. Reproductive effects of maternal metabolic disorders: Implications for pediatrics and obstetrics. Turk. J. Pediatr. 1996, 38, 335–344. [Google Scholar]
- Forges, T.; Monnier-Barbarino, P.; Leheup, B.; Jouvet, P. Pathophysiology of impaired ovarian function in galactosaemia. Hum. Reprod. Update 2006, 12, 537–584. [Google Scholar] [CrossRef]
- Menezo, Y.J.; Lescaille, M.; Nicollet, B.; Servy, E.J. Pregnancy and delivery after stimulation with rFSH of a galatosemia patient suffering hypergonadotropic hypogonadism: Case report. J. Assist. Reprod. Genet. 2004, 21, 89–90. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, K. Cellular and molecular regulation of the activation of mammalian primordial follicles: Somatic cells initiate follicle activation in adulthood. Hum. Reprod. Update 2015, 21, 779–786. [Google Scholar] [CrossRef]
- Kim, S.Y.; Ebbert, K.; Cordeiro, M.H.; Romero, M.; Zhu, J.; Serna, V.A.; Whelan, K.A.; Woodruff, T.K.; Kurita, T. Cell autonomous phosphoinositide 3-kinase activation on oocytes disrupts normal ovarian function through promoting survival and overgrowth of ovarian follicles. Endocrinology 2015, 156, 1464–1476. [Google Scholar] [CrossRef]
- Li, J.; Kawamura, K.; Cheung, Y.; Liu, S.; Klein, C.; Liu, S.; Duan, E.K.; Hsueh, A.J. Activation of dormant ovarian follicles to generate mature eggs. Proc. Natl. Acad. Sci. 2010, 107, 10280–10284. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Kim, J.; Li, X.X.; Hsueh, A.J. Promotion of ovarian follicle growth following mTOR activation: Synergistic effects of AKT stimulators. PLoS ONE 2015, 10, e0117769. [Google Scholar] [CrossRef]
- Sun, X.; Su, Y.; He, Y.; Zhang, J.; Liu, W.; Zhang, H.; Hou, Z.; Liu, J.; Li, J. New strategy for in vitro activation of primordial follicles with mTOR and PI3K stimulators. Cell Cycle 2015, 14, 721–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welling, L.; Bernstein, L.E.; Berry, G.T.; Burlina, A.B.; Eyskens, F.; Gautschi, M.; Grünewald, S.; Gubbels, C.S.; Knerr, I.; Labrune, P.; et al. Galactosemia Network (GalNet). International clinical guideline for the management of classical galactosaemia: Diagnosis, treatment and follow up. J. Inherit. Metab. Dis. 2017, 40, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Roness, H.; Meirow, D. Follicle reserve loss in ovarian tissue transplantation. Reproduction 2019. [Google Scholar] [CrossRef] [PubMed]
- Shea, L.D.; Woodruff, T.K.; Shikanov, A. Bioengineering the ovarian follicle microenvironment. Annu. Rev. Biomed. Eng. 2014, 16, 29–52. [Google Scholar] [CrossRef] [PubMed]
- Skory, R.M.; Xu, Y.; Shea, L.D.; Woodruff, T.K. Engineering the ovarian cycle using in vitro follicle culture. Hum. Reprod. 2015, 30, 1386–1395. [Google Scholar] [CrossRef] [Green Version]
- van Erven, B.; Gubbels, C.S.; van Golde, R.J.; Dunselman, G.A.; Derhaag, J.G.; de Wert, G.; Geraedts, J.P.; Bosch, A.M.; Treacy, E.P.; Welt, C.K.; et al. Fertility preservation in female classic galactosemia patients. Orphanet, J. Rare Dis. 2013, 8, 107. [Google Scholar] [CrossRef]
- Zhai, J.; Yao, G.; Dong, F.; Bu, Z.; Cheng, Y.; Sato, Y.; Hu, L.; Zhang, Y.; Wang, J.; Dai, S.; et al. In vitro activation of follicles and fresh tissue autotransplantation in primary ovarian insufficiency patients. J. Clin. Endocrinol. Metab. 2016, 101, 4405–4412. [Google Scholar] [CrossRef]
- Kawamura, K.; Kawamura, N.; Hsueh, A.J. Activation of dormant follicles: A new treatment for premature ovarian failure? Curr. Opin. Obstet. Gynecol. 2016, 28, 217–222. [Google Scholar] [CrossRef]
- Tang, M.; Odejinmi, S.I.; Vankayalapati, H.; Wierenga, K.J.; Lai, K. Innovative therapy for classic galactosemia – tale of two HTS. Mol. Genet. Metab. 2012, 105, 44–55. [Google Scholar] [CrossRef]
- Martini, P.G.V.; Guey, L.T. A new era for rare genetic diseases: messenger RNA therapy. Hum. Gene Ther. 2019, 30, 1180–1189. [Google Scholar] [CrossRef]
- Truman, A.M.; Tilly, J.L.; Woods, D.C. Ovarian regeneration: The potential for stem cell contribution in the postnatal ovary to sustained endocrine function. Mol. Cell Endocrinol. 2017, 445, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, T.K. Oncofertility: A grand collaboration between reproductive medicine and oncology. Reproduction 2015, 150, S1–S10. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(a) Illustration of the pathways of galactose metabolism, with inhibition of the key enzyme: galactose-1-phosphate uridylytransferase. The key enzymes involved are shaded. (b) Illustration of the steps in oogenesis and folliculogenesis over the prenatal to menopause time period that may be influenced by GALT deficiency [16,17,18,19,20] with the proposed site of the PI3K/AKT regulation as cited by Sanchez and Smitz [20]. The abbreviations used are listed in the abbreviations list.
Figure 1.
(a) Illustration of the pathways of galactose metabolism, with inhibition of the key enzyme: galactose-1-phosphate uridylytransferase. The key enzymes involved are shaded. (b) Illustration of the steps in oogenesis and folliculogenesis over the prenatal to menopause time period that may be influenced by GALT deficiency [16,17,18,19,20] with the proposed site of the PI3K/AKT regulation as cited by Sanchez and Smitz [20]. The abbreviations used are listed in the abbreviations list.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Abidin, Z.; Treacy, E.P. Insights into the Pathophysiology of Infertility in Females with Classical Galactosaemia. Int. J. Mol. Sci. 2019, 20, 5236. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205236
AMA Style
Abidin Z, Treacy EP. Insights into the Pathophysiology of Infertility in Females with Classical Galactosaemia. International Journal of Molecular Sciences. 2019; 20(20):5236. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205236
Chicago/Turabian StyleAbidin, Zaza, and Eileen P. Treacy. 2019. "Insights into the Pathophysiology of Infertility in Females with Classical Galactosaemia" International Journal of Molecular Sciences 20, no. 20: 5236. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20205236
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.