Evidence for Nanoparticle-Induced Lysosomal Dysfunction in Lung Adenocarcinoma (A549) Cells

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
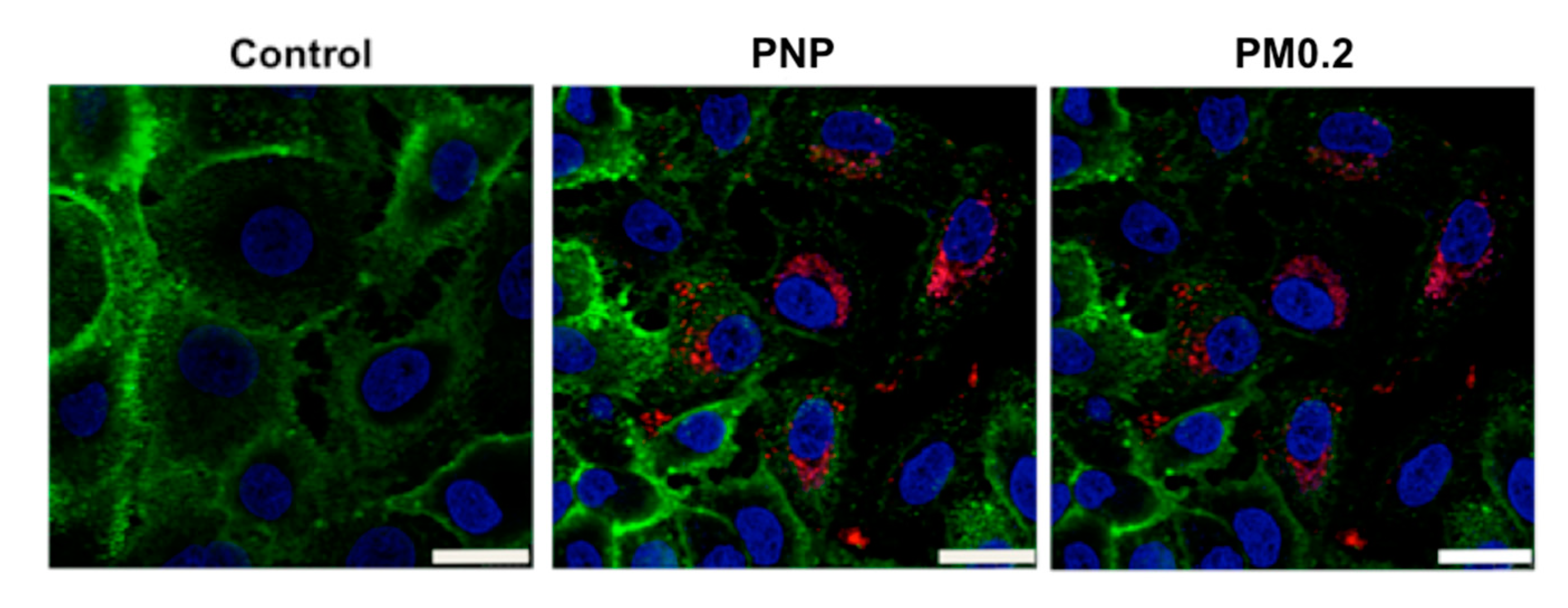
2.1. Live Cell Imaging of Intracellular PNP in A549 Cells
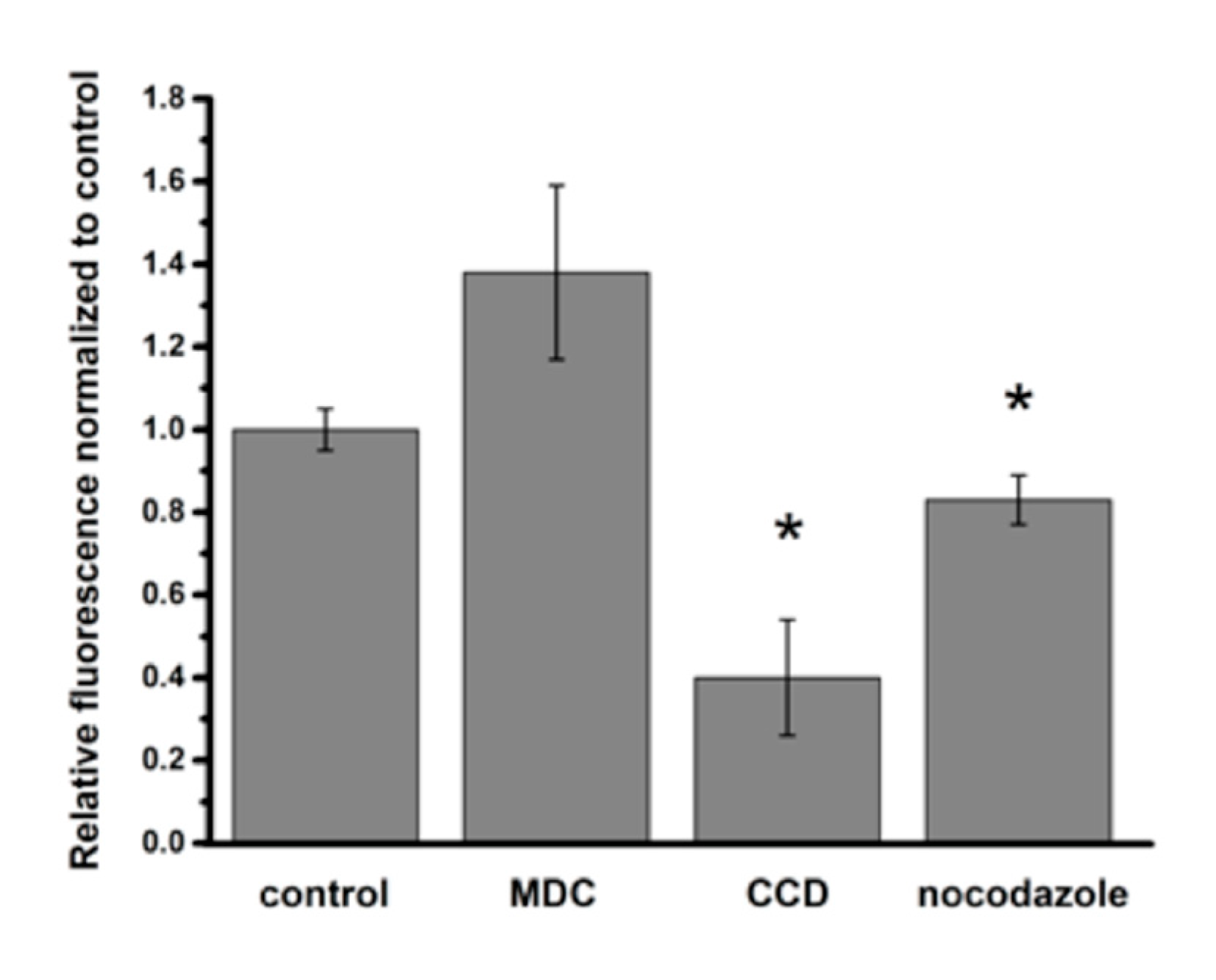
2.2. Mechanism(s) of PNP Entry into A549 Cells
2.3. PNP Egress from A549 Cells
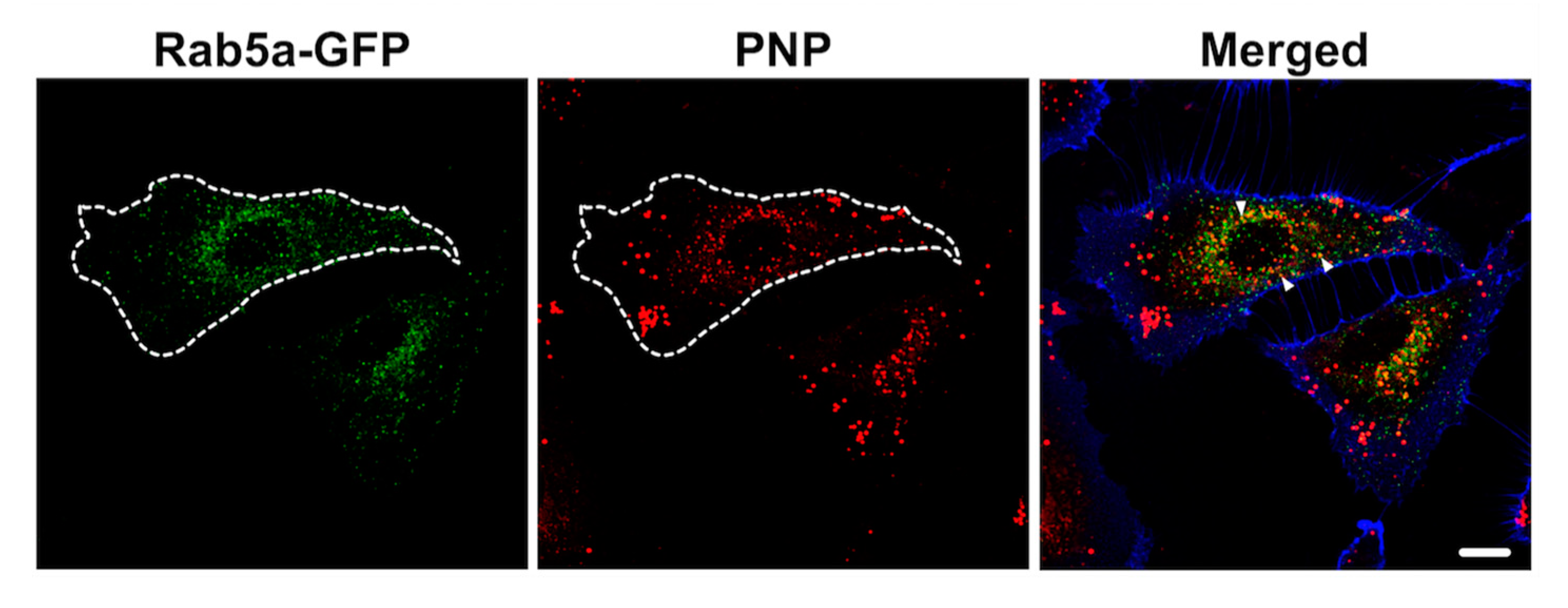
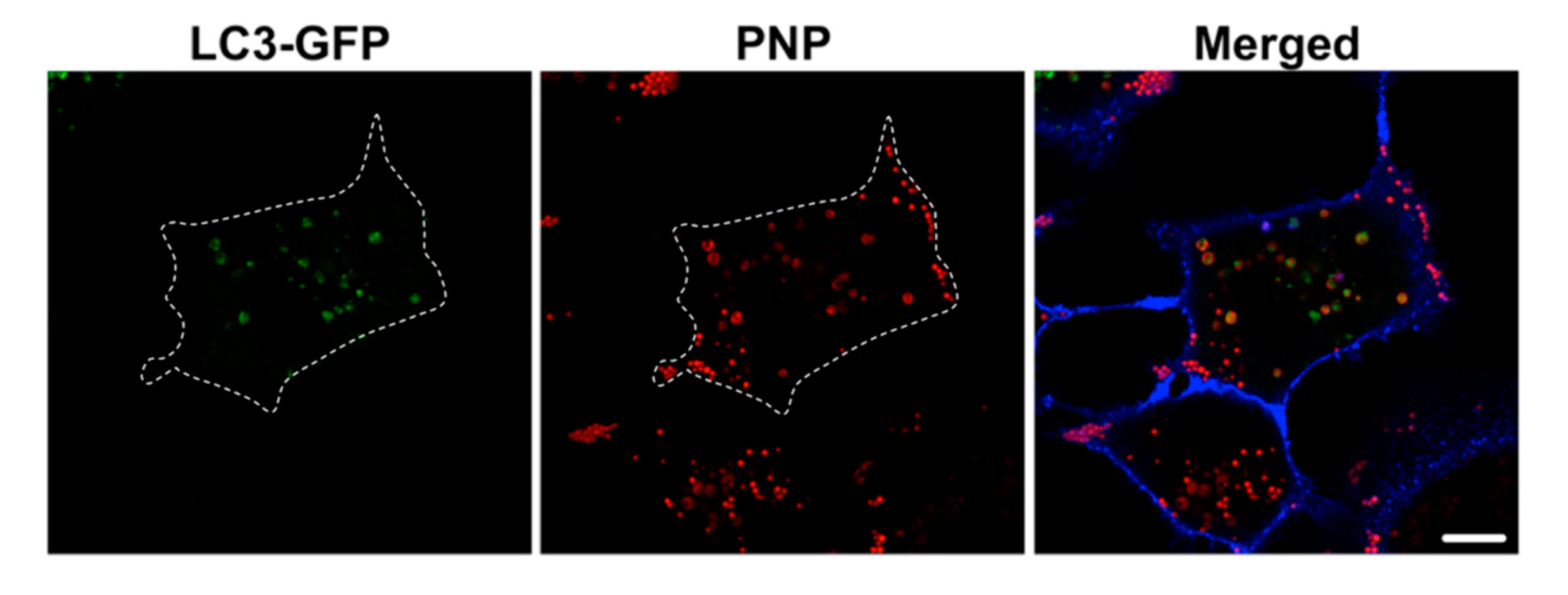
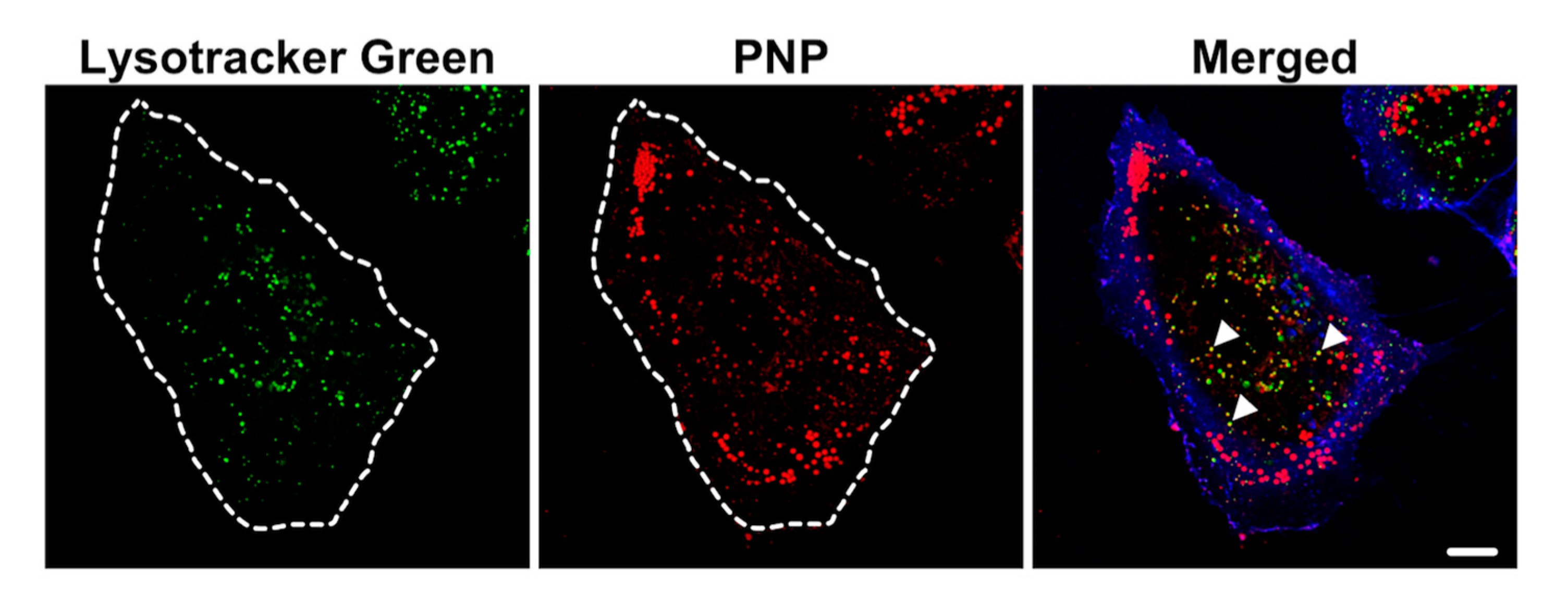
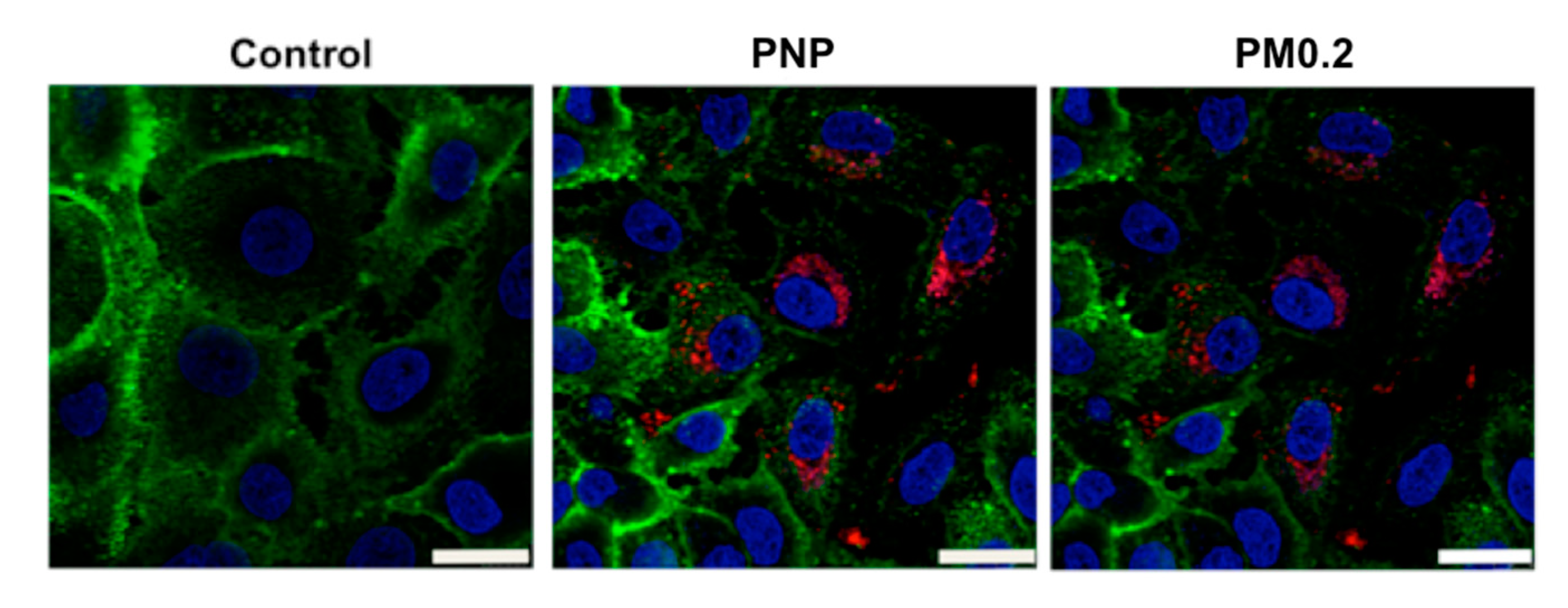
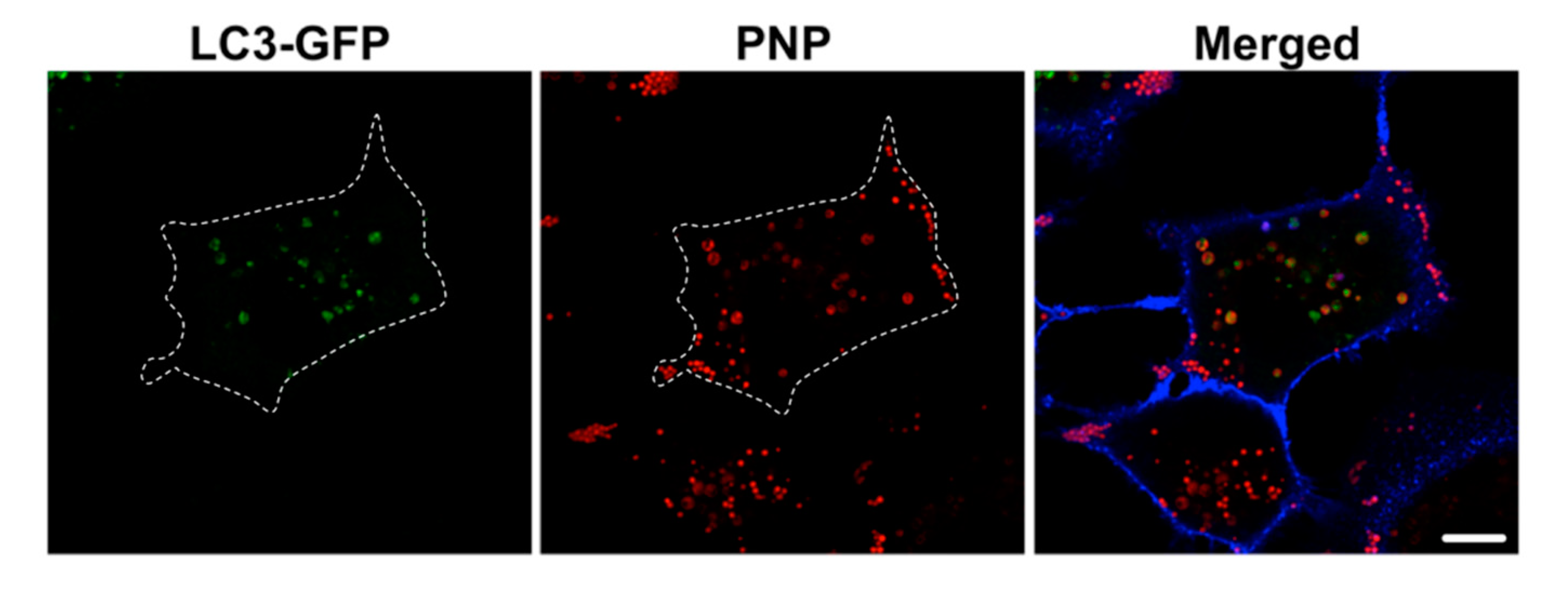
2.4. Intracellular NP Processing in A549 Cells
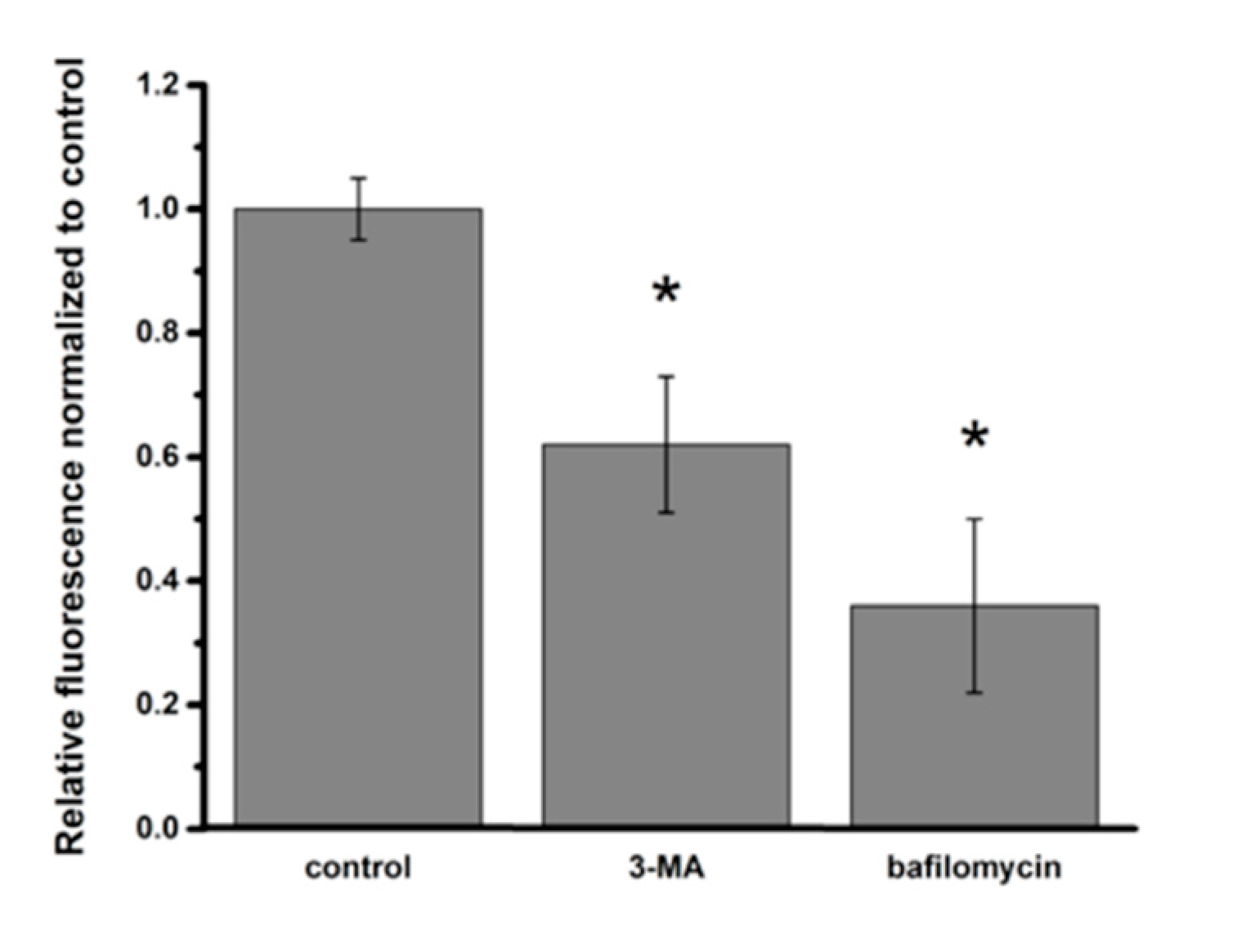
2.5. Assessment of NP Exposure-Induced Lysosomal Dysfunction
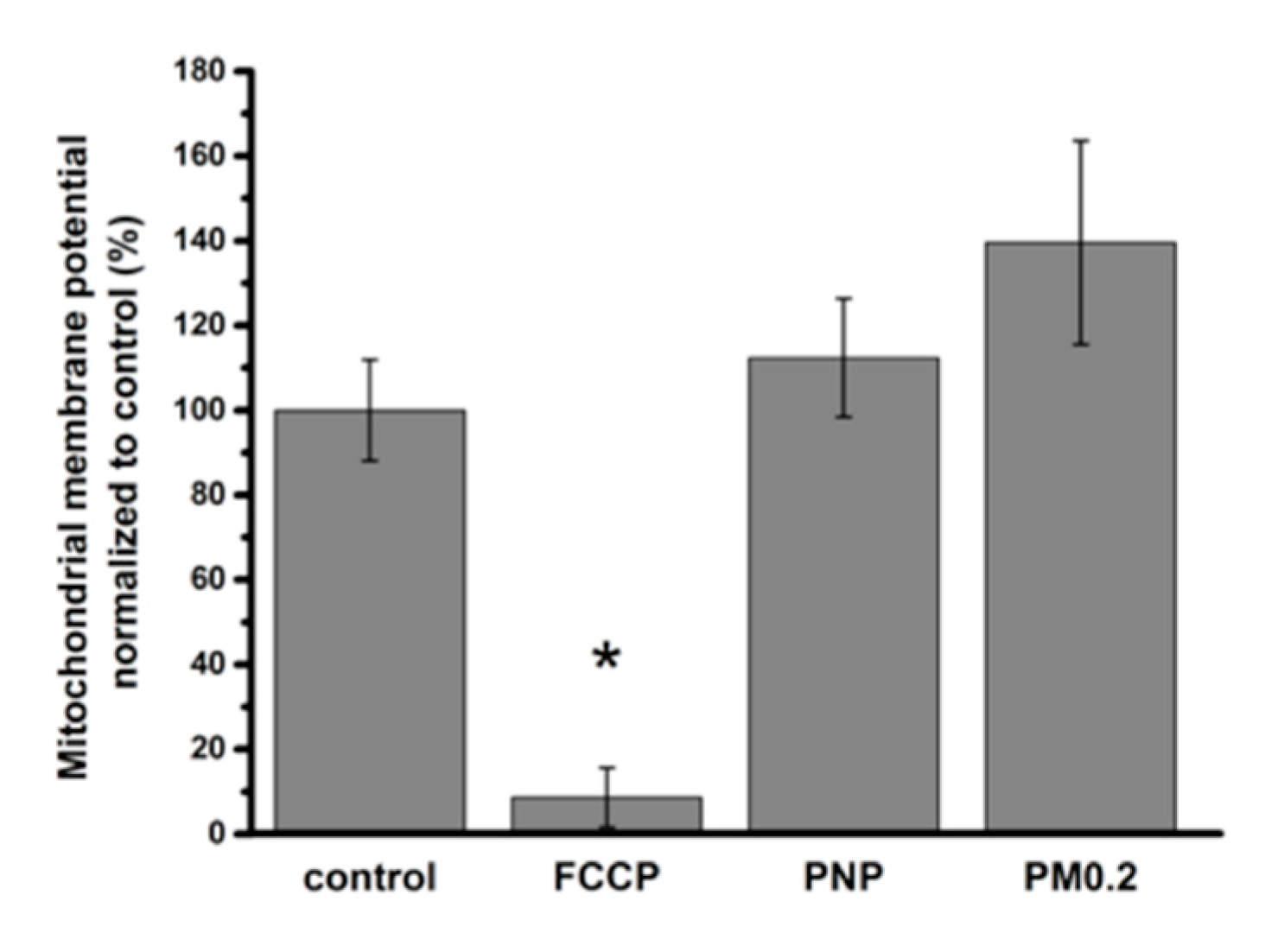
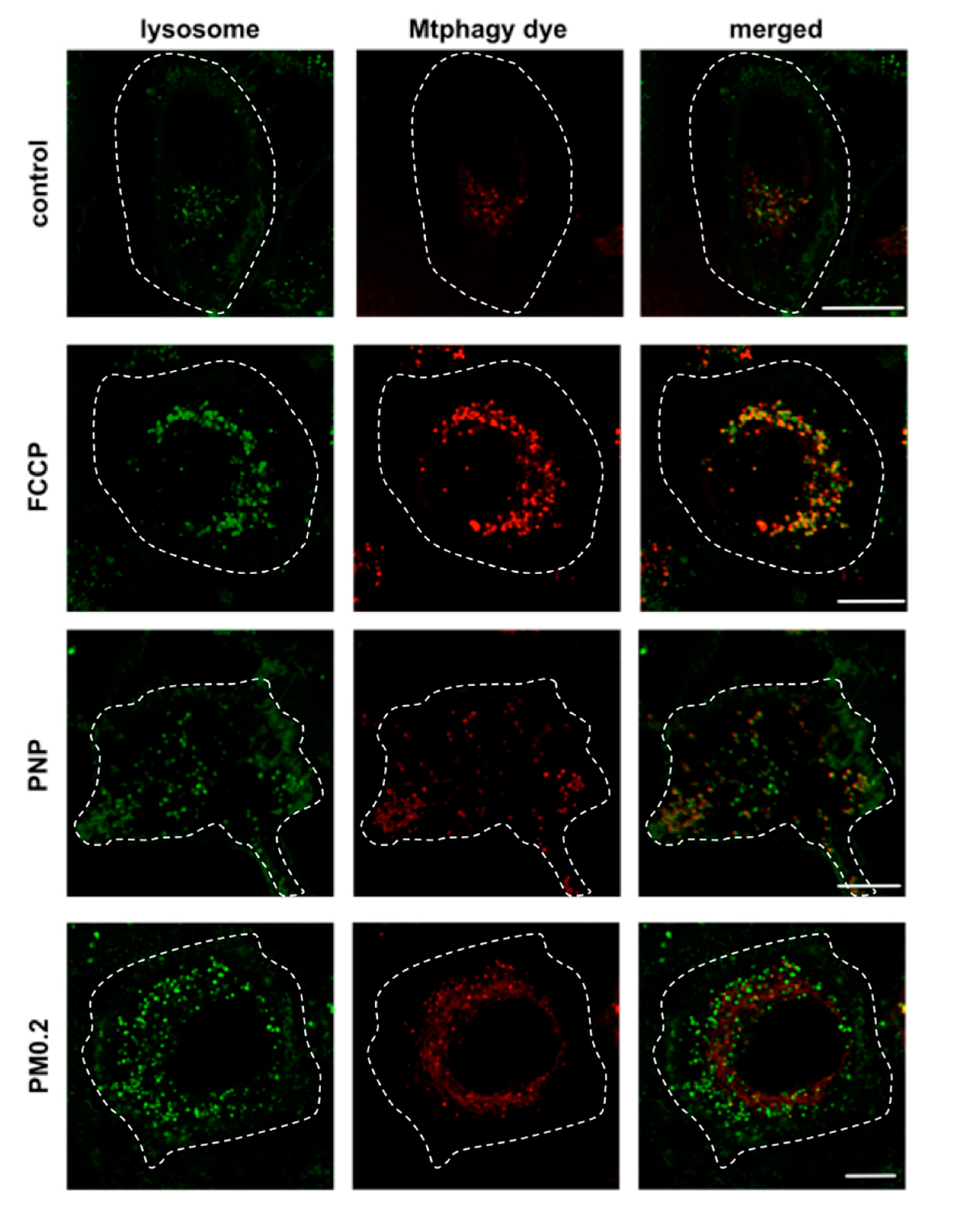
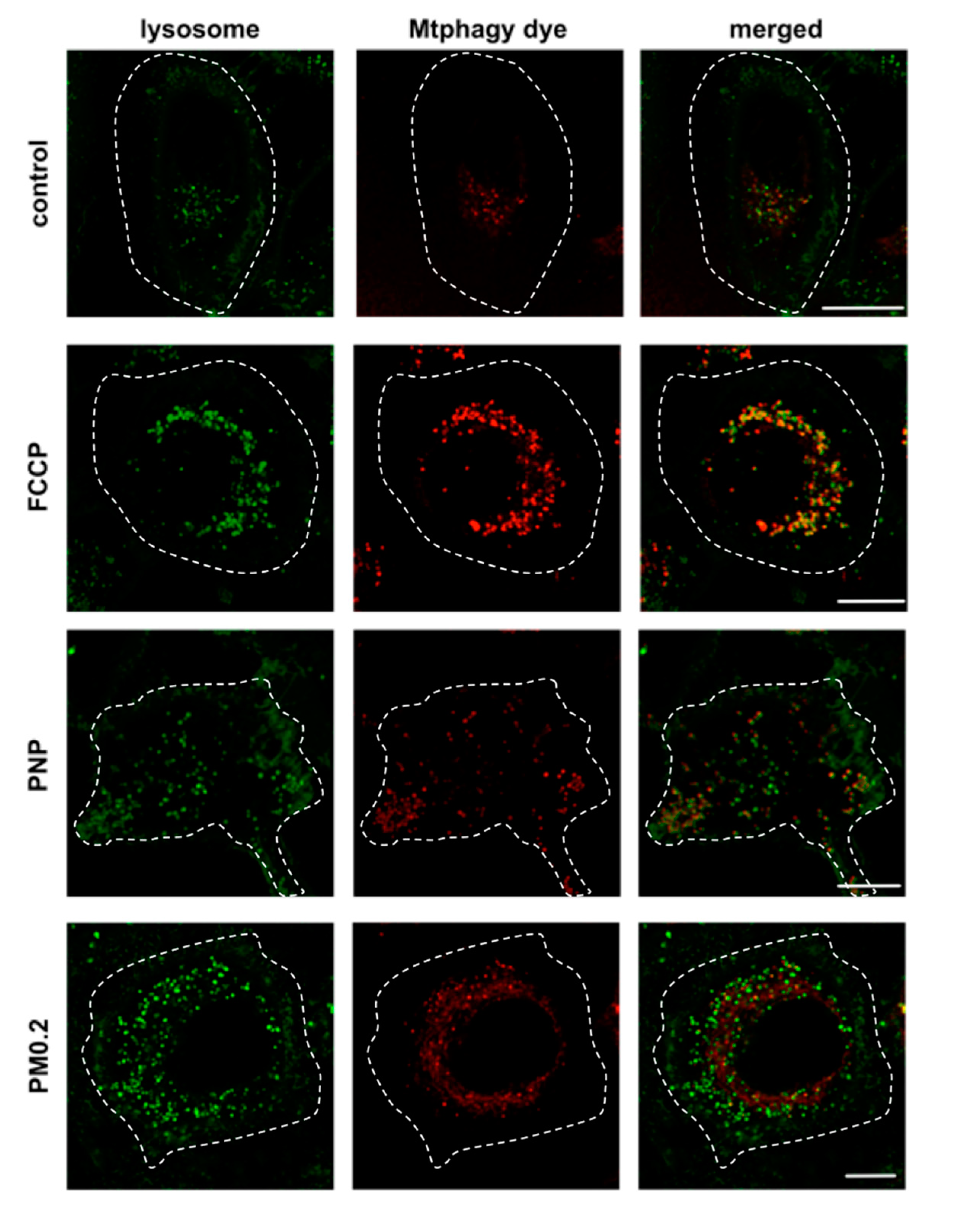
2.6. NP Exposure and Mitochondrial Function in A549 Cells
3. Discussion
3.1. Uptake of NP into A549 Cells
3.2. Egress of NP from A549 Cells
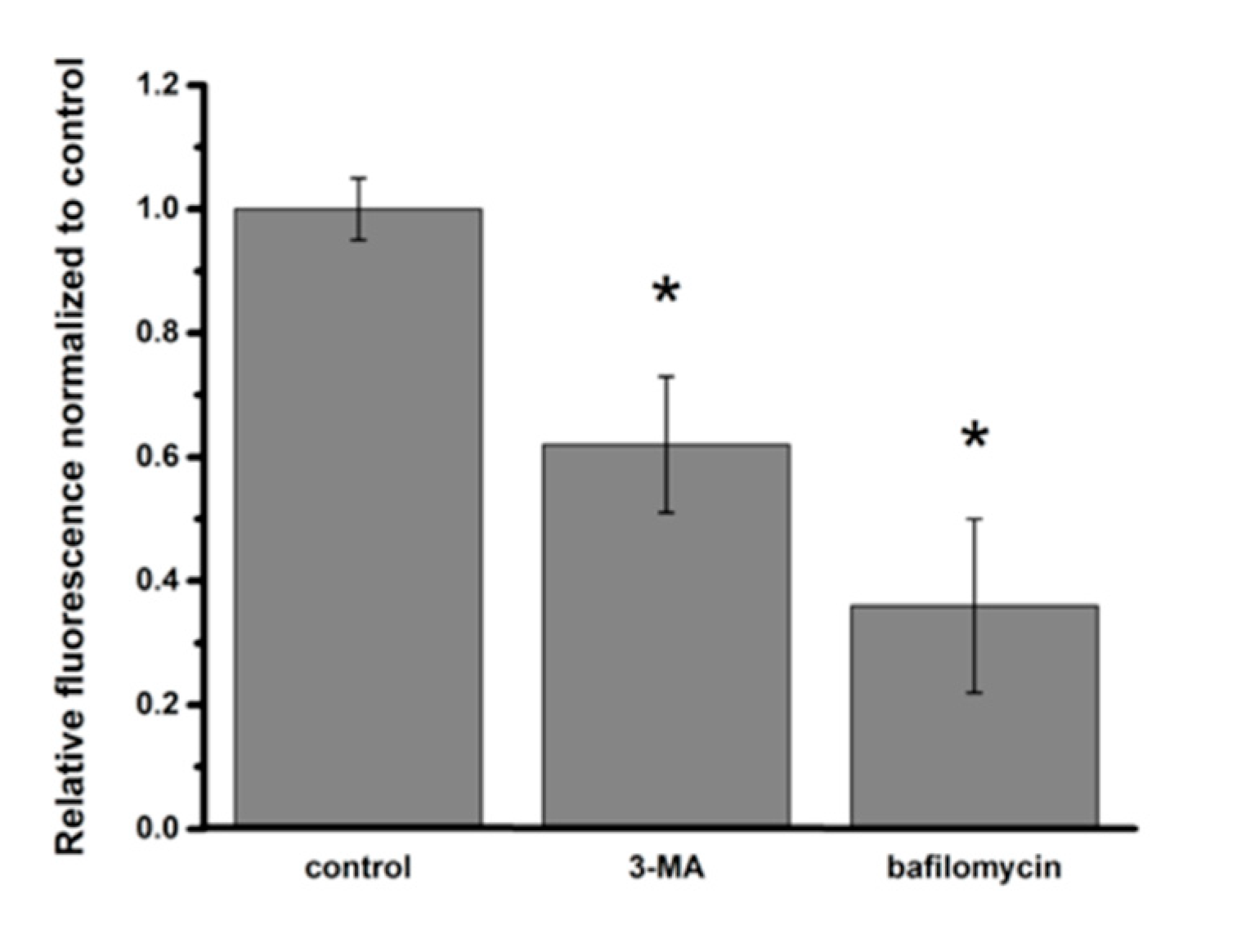
3.3. Activation of Autophagy in Intracellular Processing of NP in A549 Cells
3.4. Lysosomal Dysfunction in NP-Exposed A549 Cells
3.5. Absence of Mitochondrial Dysfunction in NP-Exposed A549 Cells
3.6. Summary
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Live Cell Imaging
4.4. Nanoparticle (NP) Exposure of A549 Cells
4.5. Intracellular PNP Content Assessed after 24 h of Apical PNP Exposure
4.6. Effect of Endocytosis Inhibitors on Intracellular PNP Content
4.7. Egress of Intracellular PNP from A549 Cells
4.8. Cytosolic Ca2+ Mobilization and PNP Exocytosis
4.9. Nanoparticle Exposure-Induced Activation of Autophagy in A549 Cells
4.10. Effects of Inhibitors of Autophagosome or Autolysosome Formation on Intracellular PNP Content
4.11. Assessment of Lysosomal Dysfunction
4.12. Assessment of Mitochondrial Function
4.13. Data Analysis
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-MA | 3-methyladenine |
| AEC | Alveolar epithelial cell |
| AM | Acetoxymethyl ester |
| ATP | Adenosine triphosphate |
| CCD | Cytochalasin D |
| EDTA | Ethylenediaminetetraacetic acid |
| LMP | Lysosomal membrane permeability |
| MDC | Monodansylcadaverine |
| NP | Nanoparticle |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3B |
| PM0.2 | Ambient air pollution particles with a diameter <0.2 μm |
| PNP | Polystyrene nanoparticle |
| RAECM | Rat alveolar epithelial cell monolayer |
References
- Buzea, C.; Pacheco, I.I.; Robbie, K. Nanomaterials and nanoparticles: Sources and toxicity. Biointerphases 2007, 2, 17–71. [Google Scholar] [CrossRef]
- Khan, M.I.; Mohammad, A.; Patil, G.; Naqvi, S.A.H.; Chauhan, L.K.S.; Ahmad, I. Induction of ROS, mitochondrial damage and autophagy in lung epithelial cancer cells by iron oxide nanoparticles. Biomaterials 2012, 33, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Panariti, A.; Miserocchi, G.; Rivolta, I. The effect of nanoparticle uptake on cellular behavior: Disrupting or enabling functions? Nanotechnol. Sci. Appl. 2012, 5, 87–100. [Google Scholar] [PubMed]
- Cao, Y.; Long, J.; Liu, L.; He, T.; Jiang, L.; Zhao, C.; Li, Z. A review of endoplasmic reticulum (ER) stress and nanoparticle (NP) exposure. Life Sci. 2017, 186, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Andersson, P.O.; Lejon, C.; Ekstrand-Hammarström, B.; Akfur, C.; Ahlinder, L.; Bucht, A.; Österlund, L. Polymorph- and size-dependent uptake and toxicity of TiO2 nanoparticles in living lung epithelial cells. Small 2011, 7, 514–523. [Google Scholar] [CrossRef]
- Bussy, C.; Pinault, M.; Cambedouzou, J.; Landry, M.J.; Jegou, P.; Mayne-L’hermite, M.; Launois, P.; Boczkowski, J.; Lanone, S. Critical role of surface chemical modifications induced by length shortening on multi-walled carbon nanotubes-induced toxicity. Part. Fibre Toxicol. 2012, 9, 46. [Google Scholar] [CrossRef]
- Gazzano, E.; Ghiazza, M.; Polimeni, M.; Bolis, V.; Fenoglio, I.; Attanasio, A.; Mazzucco, G.; Fubini, B.; Ghigo, D. Physicochemical determinants in the cellular responses to nanostructured amorphous silicas. Toxicol. Sci. 2012, 128, 158–170. [Google Scholar] [CrossRef]
- Chusuei, C.C.; Wu, C.-H.; Mallavarapu, S.; Hou, F.Y.S.; Hsu, C.-M.; Winiarz, J.G.; Aronstam, R.S.; Huang, Y.-W. Cytotoxicity in the age of nano: The role of fourth period transition metal oxide nanoparticle physicochemical properties. Chem. Biol. Interact. 2013, 206, 319–326. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Kim, J.-E.; Kim, D.; Woo, C.G.; Pikhitsa, P.V.; Cho, M.-H.; Choi, M. Comparison of cellular toxicity between multi-walled carbon nanotubes and onion-like shell-shaped carbon nanoparticles. J. Nanopart. Res. 2015, 17, 378. [Google Scholar] [CrossRef]
- Yu, K.-N.; Yoon, T.-J.; Minai-Tehrani, A.; Kim, J.-E.; Park, S.J.; Jeong, M.S.; Ha, S.-W.; Lee, J.-K.; Kim, J.S.; Cho, M.-H. Zinc oxide nanoparticle induced autophagic cell death and mitochondrial damage via reactive oxygen species generation. Toxicol. In Vitro 2013, 27, 1187–1195. [Google Scholar] [CrossRef]
- Guo, C.; Wang, J.; Jing, L.; Ma, R.; Liu, X.; Gao, L.; Cao, L.; Duan, J.; Zhou, X.; Li, Y.; et al. Mitochondrial dysfunction, perturbations of mitochondrial dynamics and biogenesis involved in endothelial injury induced by silica nanoparticles. Environ. Pollut. 2018, 236, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.-N.; Chang, S.-H.; Park, S.J.; Lim, J.; Lee, J.; Yoon, T.-J.; Kim, J.-S.; Cho, M.-H. Titanium dioxide nanoparticles induce endoplasmic reticulum stress-mediated autophagic cell death via mitochondria-associated endoplasmic reticulum membrane disruption in normal lung cells. PLoS ONE 2015, 10, e0131208. [Google Scholar] [CrossRef] [PubMed]
- Tooze, S.A.; Abada, A.; Elazar, Z. Endocytosis and autophagy: Exploitation or cooperation? Cold Spring Harb. Perspect. Biol. 2014, 6, a018358. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Codogno, P.; Meijer, A.J. Autophagy and signaling: Their role in cell survival and cell death. Cell Death Differ. 2005, 12, 1509–1518. [Google Scholar] [CrossRef]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef]
- Denton, D.; Kumar, S. Autophagy-dependent cell death. Cell Death Differ. 2019, 26, 605–616. [Google Scholar] [CrossRef]
- Klionsky, D.J. Autophagy: From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Eskelinen, E.-L.; Deretic, V. Autophagosomes, phagosomes, autolysosomes, phagolysosomes, autophagolysosomes... wait, I’m confused. Autophagy 2014, 10, 549–551. [Google Scholar] [CrossRef]
- Manshian, B.B.; Pokhrel, S.; Mädler, L.; Soenen, S.J. The impact of nanoparticle-driven lysosomal alkalinization on cellular functionality. J. Nanobiotechnol. 2018, 16, 85. [Google Scholar] [CrossRef] [PubMed]
- Conti, S.; Harari, S.; Caminati, A.; Zanobetti, A.; Schwartz, J.D.; Bertazzi, P.A.; Cesana, G.; Madotto, F. The association between air pollution and the incidence of idiopathic pulmonary fibrosis in Northern Italy. Eur. Respir. J. 2018, 51, 1700397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurt, O.K.; Zhang, J.; Pinkerton, K.E. Pulmonary health effects of air pollution. Curr. Opin. Pulm. Med. 2016, 22, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Chen, Z.; Zhou, L.-F.; Huang, S.-X. Air pollutants and early origins of respiratory diseases. Chronic Dis. Transl. Med. 2018, 4, 75–94. [Google Scholar] [CrossRef]
- Nardone, L.L.; Andrews, S.B. Cell line A549 as a model of the type II pneumocyte. Biochim. Biophys. Acta 1979, 573, 276–295. [Google Scholar] [CrossRef]
- Sipos, A.; Kim, K.-J.; Chow, R.H.; Flodby, P.; Borok, Z.; Crandall, E.D. Alveolar epithelial cell processing of nanoparticles activates autophagy and lysosomal exocytosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 315, L286–L300. [Google Scholar] [CrossRef]
- Contini, C.; Schneemilch, M.; Gaisford, S.; Quirke, N. Nanoparticle—membrane interactions. J. Exp. Nanosci. 2018, 13, 62–81. [Google Scholar] [CrossRef]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.A.; Alkawareek, M.Y.; Dreaden, E.C.; Brown, D.; Alkilany, A.M.; Farokhzad, O.C.; Mahmoudi, M. Cellular uptake of nanoparticles: Journey inside the cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef]
- Treuel, L.; Jiang, X.; Nienhaus, G.U. New views on cellular uptake and trafficking of manufactured nanoparticles. J. R. Soc. Interface 2013, 10, 20120939. [Google Scholar] [CrossRef]
- Rodríguez, A.; Webster, P.; Ortego, J.; Andrews, N.W. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J. Cell Biol. 1997, 137, 93–104. [Google Scholar] [CrossRef]
- Xu, J.; Toops, K.A.; Diaz, F.; Carvajal-Gonzalez, J.M.; Gravotta, D.; Mazzoni, F.; Schreiner, R.; Rodriguez-Boulan, E.; Lakkaraju, A. Mechanism of polarized lysosome exocytosis in epithelial cells. J. Cell Sci. 2012, 125, 5937–5943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Noda, T.; Yoshimori, T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007, 3, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, M.; Di Rienzo, M.; Piacentini, M.; Fimia, G.M. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem. Sci. 2017, 42, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-T.; Tan, H.-L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.-N.; Codogno, P.; Shen, H.-M. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef] [Green Version]
- Folkerts, H.; Hilgendorf, S.; Vellenga, E.; Bremer, E.; Wiersma, V.R. The multifaceted role of autophagy in cancer and the microenvironment. Med. Res. Rev. 2019, 39, 517–560. [Google Scholar] [CrossRef]
- Zhao, Y.G.; Zhang, H. Autophagosome maturation: An epic journey from the ER to lysosomes. J. Cell Biol. 2019, 218, 757–770. [Google Scholar] [CrossRef]
- Pierzyńska-Mach, A.; Janowski, P.A.; Dobrucki, J.W. Evaluation of acridine orange, LysoTracker Red, and quinacrine as fluorescent probes for long-term tracking of acidic vesicles. Cytometry A 2014, 85, 729–737. [Google Scholar] [CrossRef]
- Repnik, U.; Hafner Česen, M.; Turk, B. Strategies for assaying lysosomal membrane permeabilization. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef]
- Song, W.; Popp, L.; Yang, J.; Kumar, A.; Gangoli, V.S.; Segatori, L. The autophagic response to polystyrene nanoparticles is mediated by transcription factor EB and depends on surface charge. J. Nanobiotechnol. 2015, 13, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, Y.-K.; Lee, W.-C.; Hu, C.-H.; Hammond, G.L. The mitochondria are recognition organelles of cell stress. J. Surg. Res. 1996, 62, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Shirmohammadi, F.; Hasheminassab, S.; Saffari, A.; Schauer, J.J.; Delfino, R.J.; Sioutas, C. Fine and ultrafine particulate organic carbon in the Los Angeles basin: Trends in sources and composition. Sci. Total Environ. 2016, 541, 1083–1096. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sipos, A.; Kim, K.-J.; Sioutas, C.; Crandall, E.D. Evidence for Nanoparticle-Induced Lysosomal Dysfunction in Lung Adenocarcinoma (A549) Cells. Int. J. Mol. Sci. 2019, 20, 5253. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215253
Sipos A, Kim K-J, Sioutas C, Crandall ED. Evidence for Nanoparticle-Induced Lysosomal Dysfunction in Lung Adenocarcinoma (A549) Cells. International Journal of Molecular Sciences. 2019; 20(21):5253. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215253
Chicago/Turabian StyleSipos, Arnold, Kwang-Jin Kim, Constantinos Sioutas, and Edward D. Crandall. 2019. "Evidence for Nanoparticle-Induced Lysosomal Dysfunction in Lung Adenocarcinoma (A549) Cells" International Journal of Molecular Sciences 20, no. 21: 5253. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215253