Gender Related Changes in Gene Expression Induced by Valproic Acid in A Mouse Model of Autism and the Correction by S-adenosyl Methionine. Does It Explain the Gender Differences in Autistic Like Behavior?


Abstract
:1. Introduction
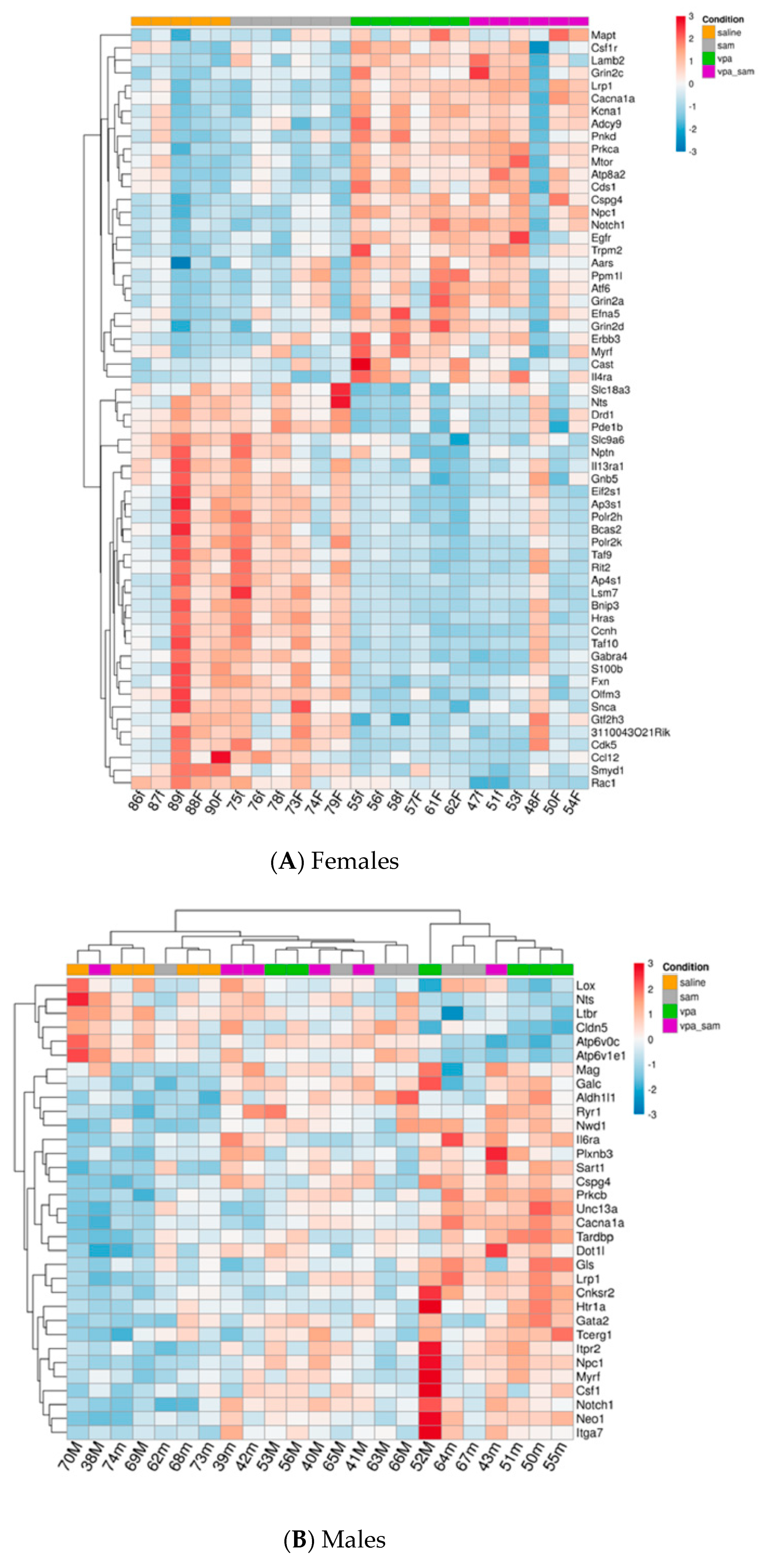
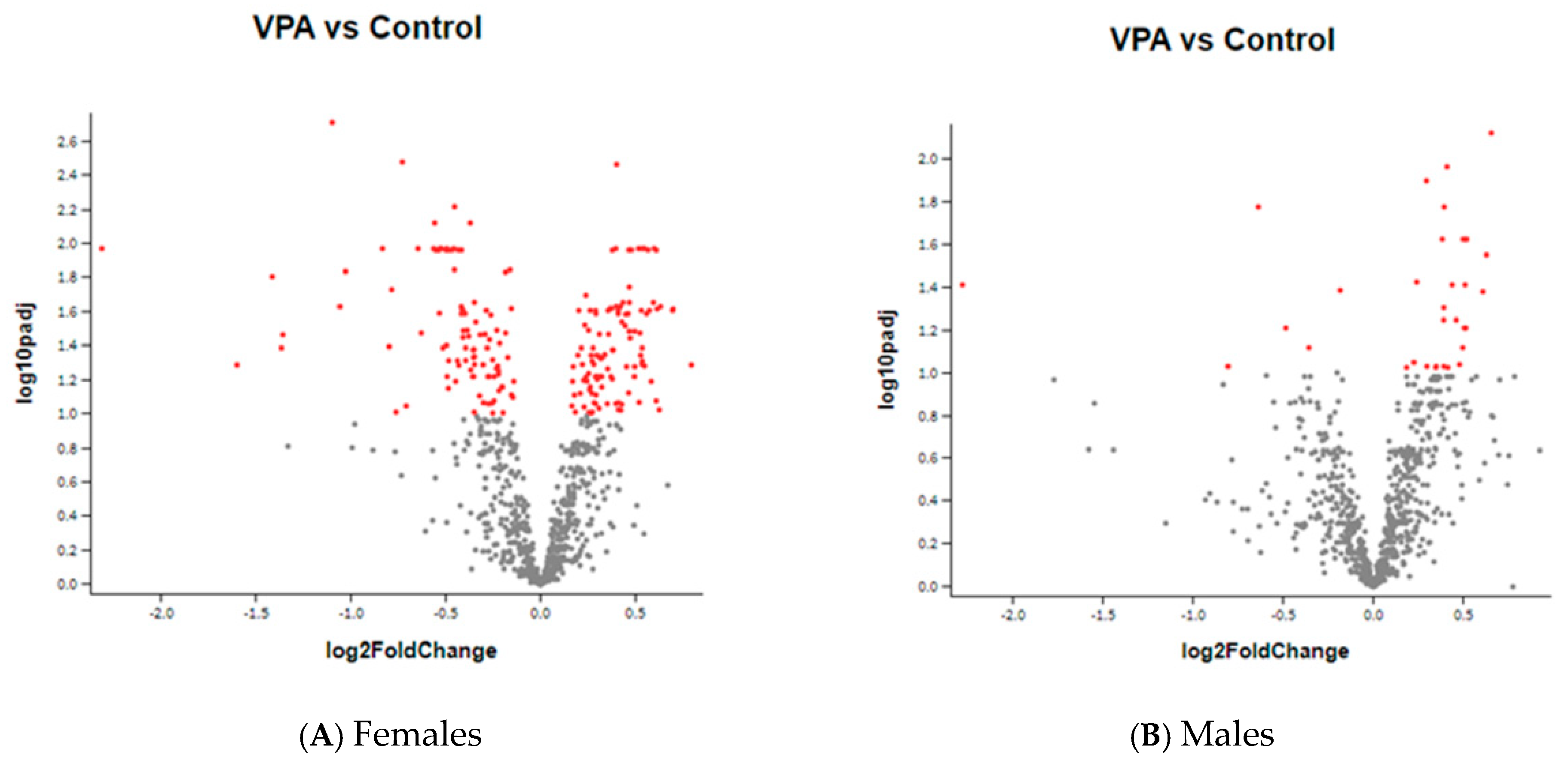
2. Results
SAM Administration to Controls
3. Discussion
3.1. Genes Whose Expression Was Significantly Changed in Females
3.2. Genes Whose Expression Was Significantly Changed Only in Males
4. Materials and Methods
4.1. Animals
4.2. RNA Extraction and Gene Expression Analysis
4.3. Gene Functional Enrichment
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ASD | Autism Spectrum disorder |
| VPA | Valproic Acid |
| SAM | S- Adenosyl Methionine |
| SFARI | Simons Foundation Autism Research Initiative |
| ADHD | Attention Deficit Hyperactivity Disorder |
| CAT | Catalase |
| SOD | Superoxide Dismutase |
| HDAC | Histone deacetylase |
| NMDA | N-methyl-D-aspartate |
| VEGF | Vascular endothelial growth factor |
| CHAT | Choline acetyltransferase |
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; Association, A.P., Ed.; American Psychiatric Association: Washington DC, USA, 2013; pp. 50–59. [Google Scholar]
- Ornoy, A.; Weinstein-Fudim, L.; Ergaz, Z. Prenatal factors associated with autism spectrum disorder (ASD). Reprod. Toxicol. 2015, 56, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Developmental Disabilities Monitoring Network Surveillance Year Principal Investigators, Centers for Disease, and Prevention. Prevalence of autism spectrum disorder among children aged 8 years-autism and developmental disabilities monitoring network, 11 sites, United States, 2010. MMWR Surveill. Summ. 2014, 63, 1–21. [Google Scholar]
- Baio, J.; Wiggins, L.; Christensen, D.L.; Maenner, M.J.; Daniels, J.; Warren, Z.; Kurzius-Spencer, M.; Zahorodny, W.; Robinson, C.; Rosenberg, C.R.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years—Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill. Summ. 2018, 67, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Lai, M.C.; Lombardo, M.V.; Auyeung, B.; Chakrabarti, B.; Baron-Cohen, S. Sex/gender differences and autism: Setting the scene for future research. J. Am. Acad. Child Adolesc. Psychiatry 2015, 54, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Beggiato, A.; Peyre, H.; Maruani, A.; Scheid, I.; Rastam, M.; Amsellem, F.; Gillberg, C.I.; Leboyer, M.; Bourgeron, T.; Gillberg, C.; et al. Gender differences in autism spectrum disorders: Divergence among specific core symptoms. Autism Res. 2017, 10, 680–689. [Google Scholar] [CrossRef] [PubMed]
- Szatmari, P.; Liu, X.Q.; Goldberg, J.; Zwaigenbaum, L.; Paterson, A.D.; Woodbury-Smith, M.; Georgiades, S.; Duku, E.; Thompson, A. Sex differences in repetitive stereotyped behaviors in autism: Implications for genetic liability. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2012, 159, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Van Wijngaarden-Cremers, P.J.; van Eeten, E.; Groen, W.B.; Van Deurzen, P.A.; Oosterling, I.J.; Van der Gaag, R.J. Gender and age differences in the core triad of impairments in autism spectrum disorders: A systematic review and meta-analysis. J. Autism Dev. Disord. 2014, 44, 627–635. [Google Scholar] [CrossRef]
- Loomes, R.; Hull, L.; Mandy, W.P.L. What Is the Male-to-Female Ratio in Autism Spectrum Disorder? A Systematic Review and Meta-Analysis. J. Am. Acad. Child Adolesc. Psychiatry 2017, 56, 466–474. [Google Scholar] [CrossRef]
- Rodier, P.M.; Ingram, J.L.; Tisdale, B.; Nelson, S.; Romano, J. Embryological origin for autism: Developmental anomalies of the cranial nerve motor nuclei. J. Comp. Neurol. 1996, 370, 247–261. [Google Scholar] [CrossRef]
- Ornoy, A. Valproic acid in pregnancy: How much are we endangering the embryo and fetus? Reprod. Toxicol. 2009, 28, 1–10. [Google Scholar] [CrossRef]
- Silberstein, S.D. Preventive migraine treatment. Neurol. Clin. 2009, 27, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Post, M.R.; Weiss, S.R. Tolerance to the prophylactic effects of carbamazepine and related mood stabilizers in the treatment of bipolar disorders. CNS Neurosci. 2011, 17, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.; King, J.; Cunningham, M.; Stephan, M.; Kerr, B.; Hersh, J.H. Fetal valproate syndrome and autism: Additional evidence of an association. Dev. Med. Child Neurol. 2001, 43, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.C.; Reuhl, K.R.; Cheh, M.; McRae, P.; Halladay, A.K. A New Neurobehavioral Model of Autism in Mice: Pre- and Postnatal Exposure to Sodium Valproate. J. Autism Dev. Disord. 2006, 36, 779–793. [Google Scholar] [CrossRef]
- Bambini-Junior, V.; Rodrigues, L.; Behr, G.A.; Moreira, J.C.F.; Riesgo, R.; Gottfried, C. Animal model of autism induced by prenatal exposure to valproate: Behavioral changes and liver parameters. Brain Res. 2011, 1408, 8–16. [Google Scholar] [CrossRef]
- Kolozsi, E.; MacKenzie, R.; Roullet, F.; Decatanzaro, D.; Foster, J. Prenatal exposure to valproic acid leads to reduced expression of synaptic adhesion molecule neuroligin 3 in mice. Neuroscience 2009, 163, 1201–1210. [Google Scholar] [CrossRef]
- Rodier, P.M.; Ingram, J.L.; Tisdale, B.; Croog, V.J. Linking etiologies in humans and animal models: Studies of autism. Reprod. Toxicol. 1997, 11, 417–422. [Google Scholar] [CrossRef]
- Ingram, J.L.; Peckham, S.M.; Tisdale, B.; Rodier, P.M. Prenatal exposure of rats to valproic acid reproduces the cerebellar anomalies associated with autism. Neurotoxicol. Teratol. 2000, 22, 319–324. [Google Scholar] [CrossRef]
- Nicolini, C.; Fahnestock, M. The valproic acid-induced rodent model of autism. Exp. Neurol. 2018, 299, 217–227. [Google Scholar] [CrossRef]
- Rasalam, A.; Hailey, H.; Williams, J.; Moore, S.; Turnpenny, P.; Lloyd, D.; Dean, J. Characteristics of fetal anticonvulsant syndrome associated autistic disorder. Dev. Med. Child Neurol. 2005, 47, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.J.; Gonzales, E.L.; Mabunga, D.F.N.; Valencia, S.T.; Kim, D.G.; Kim, Y.; Adil, K.J.L.; Shin, D.; Park, D.; Shin, C.Y. Sex-specific Behavioral Features of Rodent Models of Autism Spectrum Disorder. Exp. Neurobiol. 2018, 27, 321–343. [Google Scholar] [CrossRef] [PubMed]
- Ornoy, A.; Weinstein-Fudim, L.; Tfilin, M.; Ergaz, Z.; Yanai, J.; Szyf, M.; Turgeman, G. S-adenosyl methionine prevents ASD like behaviors triggered by early postnatal valproic acid exposure in very young mice. Neurotoxicol. Teratol. 2019, 71, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Schneider, T.; Roman, A.; Basta-Kaim, A.; Kubera, M.; Budziszewska, B.; Schneider, K.; Przewłocki, R. Gender-specific behavioral and immunological alterations in an animal model of autism induced by prenatal exposure to valproic acid. Psychoneuroendocrinology 2008, 33, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, N.; Seiffe, A.; Campolongo, M.; Zappala, C.; Depino, A.M. Sex-specific effects of prenatal valproic acid exposure on sociability and neuroinflammation: Relevance for susceptibility and resilience in autism. Psychoneuroendocrinology 2019, 110, 104441. [Google Scholar] [CrossRef]
- Sailer, L.; Duclot, F.; Wang, Z.; Kabbaj, M. Consequences of prenatal exposure to valproic acid in the socially monogamous prairie voles. Sci. Rep. 2019, 9, 2453. [Google Scholar] [CrossRef]
- Mabunga, D.F.N.; Gonzales, E.L.T.; Kim, J.-W.; Kim, K.C.; Shin, C.Y. Exploring the Validity of Valproic Acid Animal Model of Autism. Exp. Neurobiol. 2015, 24, 285–300. [Google Scholar] [CrossRef] [Green Version]
- Kataoka, S.; Takuma, K.; Hara, Y.; Maeda, Y.; Ago, Y.; Matsuda, T. Autism-like behaviours with transient histone hyperacetylation in mice treated prenatally with valproic acid. Int. J. Neuropsychopharmacol. 2013, 16, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Choi, C.S.; Gonzales, E.L.; Kim, K.C.; Yang, S.M.; Kim, J.-W.; Mabunga, D.F.; Cheong, J.H.; Han, S.-H.; Bahn, G.H.; Shin, C.Y. The transgenerational inheritance of autism-like phenotypes in mice exposed to valproic acid during pregnancy. Sci. Rep. 2016, 6, 36250. [Google Scholar] [CrossRef]
- Wiśniowiecka-Kowalnik, B.; Nowakowska, B.A. Genetics and epigenetics of autism spectrum disorder-current evidence in the field. J. Appl. Genet. 2019, 60, 37–47. [Google Scholar] [CrossRef]
- Lauber, E.; Filice, F.; Schwaller, B. Prenatal Valproate Exposure Differentially Affects Parvalbumin-Expressing Neurons and Related Circuits in the Cortex and Striatum of Mice. Front. Mol. Neurosci. 2016, 9, 150. [Google Scholar] [CrossRef] [Green Version]
- Hou, Q.; Wang, Y.; Li, Y.; Chen, D.; Yang, F.; Wang, S. A Developmental Study of Abnormal Behaviors and Altered GABAergic Signaling in the VPA-Treated Rat Model of Autism. Front. Behav. Neurosci. 2018, 12, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, L.; Dai, X.; Yin, Y.; Yin, Y. Valproic acid exposure sequentially activates Wnt and mTOR pathways in rats. Mol. Cell. Neurosci. 2016, 75, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhou, J.; Ren, J.; Sun, S.; Di, Y.; Wang, H.; An, X.; Zhang, K.; Zhang, J.; Qian, Z.; et al. Transcriptional and splicing dysregulation in the prefrontal cortex in valproic acid rat model of autism. Reprod. Toxicol. 2018, 77, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Kotajima-Murakami, H.; Kobayashi, T.; Kashii, H.; Sato, A.; Hagino, Y.; Tanaka, M.; Nishito, Y.; Takamatsu, Y.; Uchino, S.; Ikeda, K. Effects of rapamycin on social interaction deficits and gene expression in mice exposed to valproic acid in utero. Mol. Brain 2019, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.S.; Cabrera, R.; Schultz, D.W.; Zhu, H.; Lu, W.; Finnell, R.H.; Wlodarczyk, B.J. Autism-Like Behavior and Epigenetic Changes Associated with Autism as Consequences of in UteroExposure to Environmental Pollutants in a Mouse Model. Behav. Neurol. 2015, 2015, 426263. [Google Scholar] [CrossRef]
- Bezgin, G.; Lewis, J.D.; Evans, A.C. Developmental changes of cortical white-gray contrast as predictors of autism diagnosis and severity. Transl. Psychiatry 2018, 8, 249. [Google Scholar] [CrossRef]
- Wang, X.; Guo, J.; Song, Y.; Wang, Q.; Hu, S.; Gou, L.; Gao, Y. Decreased Number and Expression of nNOS-Positive Interneurons in Basolateral Amygdala in Two Mouse Models of Autism. Front. Cell. Neurosci. 2018, 12, 251. [Google Scholar] [CrossRef] [Green Version]
- Rinaldi, T.; Perrodin, C.; Markram, H. Hyper-Connectivity and Hyper-Plasticity in the Medial Prefrontal Cortex in the Valproic Acid Animal Model of Autism. Front. Neural Circuits 2008, 2, 4. [Google Scholar] [CrossRef]
- Otero-Losada, M.E.; Rubio, M.C. Acute changes in 5-HT metabolism after S-adenosyl-L-methionine administration. Gen. Pharmacol. Vasc. Syst. 1989, 20, 403–406. [Google Scholar] [CrossRef]
- Villalobos, M.; De La Cruz, J.P.; Cuerda, M.; Ortiz, P.; Smith-Agreda, J.; De La Cuesta, F.S. Effect of S-adenosyl-l-methionine on rat brain oxidative stress damage in a combined model of permanent focal ischemia and global ischemia-reperfusion. Brain Res. 2000, 883, 31–40. [Google Scholar] [CrossRef]
- Gonzalez-Correa, J.A.; De La Cruz, J.P.; Martin-Aurioles, E.; Lopez-Egea, M.A.; Ortiz, P.; De La Cuesta, F.S.; Gonzalez-Correa, J.A.; Martin-Aurioles, E.; Lopez-Egea, M.A. Effects ofS-adenosyl-L-methionine on hepatic and renal oxidative stress in an experimental model of acute biliary obstruction in rats. Hepatology 1997, 26, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Cui, J.; Fang, C.; Liu, M.; Min, G.; Li, L. S-Adenosylmethionine Attenuates Oxidative Stress and Neuroinflammation Induced by Amyloid-beta Through Modulation of Glutathione Metabolism. J. Alzheimers Dis. 2017, 58, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-Y.; Hong, G.H.; Kwon, H.-S.; Park, S.; Park, S.Y.; Shin, B.; Kim, T.-B.; Moon, H.-B.; Cho, Y.S. S-adenosylmethionine reduces airway inflammation and fibrosis in a murine model of chronic severe asthma via suppression of oxidative stress. Exp. Mol. Med. 2016, 48, e236. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.D.; Smith, D.D.; Kish, S.J. Brain S-adenosylmethionine levels are severely decreased in Alzheimer’s disease. J. Neurochem. 1996, 67, 1328–1331. [Google Scholar] [CrossRef]
- Mischoulon, D.; Price, L.H.; Carpenter, L.L.; Tyrka, A.R.; Papakostas, G.I.; Baer, L.; Dording, C.M.; Clain, A.J.; Durham, K.; Walker, R.; et al. A double-blind, randomized, placebo-controlled clinical trial of S-adenosyl-L-methionine (SAMe) versus escitalopram in major depressive disorder. J. Clin. Psychiatry 2014, 75, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Sarris, J.; Murphy, J.; Mischoulon, D.; Papakostas, G.I.; Fava, M.; Berk, M.; Ng, C.H. Adjunctive Nutraceuticals for Depression: A Systematic Review and Meta-Analyses. Am. J. Psychiatry 2016, 173, 575–587. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.-L.; Girard, C.; Jui, D.; Sabina, A.; Katz, D.L. S-adenosylmethionine (SAMe) as treatment for depression: A systematic review. Clin. Investig. Med. 2005, 28, 132–139. [Google Scholar]
- Sharma, A.; Gerbarg, P.; Bottiglieri, T.; Massoumi, L.; Carpenter, L.L.; Lavretsky, H.; Muskin, P.R.; Brown, R.P.; Mischoulon, D. S-Adenosylmethionine (SAMe) for Neuropsychiatric Disorders: A Clinician-Oriented Review of Research. J. Clin. Psychiatry 2017, 78, e656–e667. [Google Scholar] [CrossRef]
- Sarris, J.; Price, L.H.; Carpenter, L.L.; Tyrka, A.R.; Ng, C.H.; Papakostas, G.I.; Jaeger, A.; Fava, M.; Mischoulon, D. Is S-Adenosyl Methionine (SAMe) for Depression Only Effective in Males? A Re-Analysis of Data from a Randomized Clinical Trial. Pharmacopsychiatry 2015, 48, 141–144. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.; Higgins, D.P.; Yadav, D.K.; Godbole, A.A.; Pukkila-Worley, R.; Walker, A.K. Stress-responsive and metabolic gene regulation are altered in low S-adenosylmethionine. PLoS Genet. 2018, 14, e1007812. [Google Scholar] [CrossRef]
- Christensen, D.L.; Baio, J.; Van Naarden Braun, K.; Bilder, D.; Charles, J.; Constantino, J.N.; Daniels, J.; Durkin, M.S.; Fitzgerald, R.T.; Kurzius-Spencer, M.; et al. Prevalence and Characteristics of Autism Spectrum Disorder Among Children Aged 8 Years-Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2012. MMWR Surveill. Summ. 2016, 65, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Kim, C.H.; Knight, E.Q.; Oh, H.W.; Park, B.; Kim, D.G.; Park, H.-J. Changes in brain metabolic connectivity underlie autistic-like social deficits in a rat model of autism spectrum disorder. Sci. Rep. 2017, 7, 13213. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.C.; Cho, K.S.; Yang, S.M.; Gonzales, E.L.; Valencia, S.; Eun, P.H.; Choi, C.S.; Mabunga, D.F.; Kim, J.-W.; Noh, J.K.; et al. Sex Differences in Autism-Like Behavioral Phenotypes and Postsynaptic Receptors Expression in the Prefrontal Cortex of TERT Transgenic Mice. Biomol. Ther. 2017, 25, 374–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Win-Shwe, T.-T.; Nway, N.C.; Imai, M.; Lwin, T.-T.; Mar, O.; Watanabe, H. Social behavior, neuroimmune markers and glutamic acid decarboxylase levels in a rat model of valproic acid-induced autism. J. Toxicol. Sci. 2018, 43, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.E.; Boules, M.; Williams, K.; Richelson, E. NTS1 and NTS2 mediate analgesia following neurotensin analog treatment in a mouse model for visceral pain. Behav. Brain Res. 2012, 232, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Merullo, D.P.; Cordes, M.A.; Devries, M.S.; Stevenson, S.A.; Riters, L.V. Neurotensin neural mRNA expression correlates with vocal communication and other highly-motivated social behaviors in male European starlings. Physiol. Behav. 2015, 151, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Gammie, S.C.; D'Anna, K.L.; Gerstein, H.; Stevenson, S.A. Neurotensin inversely modulates maternal aggression. Neuroscience 2009, 158, 1215–1223. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, K.; Winrow, C.J.; Gotter, A.L.; Millstein, J.; Arbuzova, J.; Brunner, J.; Kasarskis, A.; Vitaterna, M.H.; Renger, J.J.; Turek, F.W. Altered Sleep and Affect in the Neurotensin Receptor 1 Knockout Mouse. Sleep 2012, 35, 949–956. [Google Scholar] [CrossRef] [Green Version]
- Levitas-Djerbi, T.; Sagi, D.; Lebenthal-Loinger, I.; Lerer-Goldshtein, T.; Appelbaum, L. Neurotensin Enhances Locomotor Activity and Arousal, and Inhibits Melanin-Concentrating Hormone Signalings. Neuroendocrinology 2019, in press. [Google Scholar] [CrossRef]
- Cáceda, R.; Kinkead, B.; Nemeroff, C.B. Neurotensin: Role in psychiatric and neurological diseases. Peptides 2006, 27, 2385–2404. [Google Scholar] [CrossRef]
- Shilling, P.D.; Feifel, D. The neurotensin-1 receptor agonist PD149163 blocks fear-potentiated startle. Pharmacol. Biochem. Behav. 2008, 90, 748–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervo, L.; Rossi, C.; Tatarczyńska, E.; Samanin, R. Antidepressant-like effect of neurotensin administered in the ventral tegmental area in the forced swimming test. Psychopharmacology 1992, 109, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Binder, E.B.; Kinkead, B.; Owens, M.J.; Nemeroff, C.B. Neurotensin and dopamine interactions. Pharmacol. Rev. 2001, 53, 453–486. [Google Scholar] [PubMed]
- Dobner, P.R. Multitasking with neurotensin in the central nervous system. Cell. Mol. Life Sci. 2005, 62, 1946–1963. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.H.; Adermark, L.; Lovinger, D.M. Neurotensin reduces glutamatergic transmission in the dorsolateral striatum via retrograde endocannabinoid signaling. Neuropharmacology 2008, 54, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Kadiri, N.; Rodeau, J.L.; Schlichter, R.; Hugel, S. Neurotensin inhibits background K+ channels and facilitates glutamatergic transmission in rat spinal cord dorsal horn. Eur. J. Neurosci. 2011, 34, 1230–1240. [Google Scholar] [CrossRef]
- Ferraro, L.; Tomasini, M.C.; Mazza, R.; Fuxe, K.; Fournier, J.; Tanganelli, S.; Antonelli, T. Neurotensin receptors as modulators of glutamatergic transmission. Brain Res. Rev. 2008, 58, 365–373. [Google Scholar] [CrossRef]
- Rakovska, A.; Giovannini, M.; Corte, L.; Kalfin, R.; Bianchi, L.; Pepeu, G. Neurotensin modulation of acetylcholine and gaba release from the rat hippocampus: An in vivo microdialysis study. Neurochem. Int. 1998, 33, 335–340. [Google Scholar] [CrossRef]
- Petkova-Kirova, P.; Rakovska, A.; Della Corte, L.; Zaekova, G.; Radomirov, R.; Mayer, A. Neurotensin modulation of acetylcholine, GABA, and aspartate release from rat prefrontal cortex studied in vivo with microdialysis. Brain Res. Bull. 2008, 77, 129–135. [Google Scholar] [CrossRef]
- Petrie, K.A.; Schmidt, D.; Bubser, M.; Fadel, J.; Carraway, R.E.; Deutch, A.Y. Neurotensin Activates GABAergic Interneurons in the Prefrontal Cortex. J. Neurosci. 2005, 25, 1629–1636. [Google Scholar] [CrossRef] [Green Version]
- Vincent, J.-P.; Mazella, J.; Kitabgi, P. Neurotensin and neurotensin receptors. Trends Pharmacol. Sci. 1999, 20, 302–309. [Google Scholar] [CrossRef]
- Martin, S.; Vincent, J.-P.; Mazella, J. Involvement of the Neurotensin Receptor-3 in the Neurotensin-Induced Migration of Human Microglia. J. Neurosci. 2003, 23, 1198–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vadnie, C.A.; Hinton, D.J.; Choi, S.; Choi, Y.; Ruby, C.L.; Oliveros, A.; Prieto, M.L.; Park, J.H.; Choi, D.-S. Activation of neurotensin receptor type 1 attenuates locomotor activity. Neuropharmacology 2014, 85, 482–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keiser, A.A.; Matazel, K.S.; Esser, M.K.; Feifel, D.; Prus, A.J. Systemic administration of the neurotensin NTS-receptor agonist PD149163 improves performance on a memory task in naturally deficient male brown Norway rats. Exp. Clin. Psychopharmacol. 2014, 22, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Angelidou, A.; Francis, K.; Vasiadi, M.; Alysandratos, K.-D.; Zhang, B.; Theoharides, A.; Lykouras, L.; Sideri, K.; Kalogeromitros, D.; Theoharides, T.C. Neurotensin is increased in serum of young children with autistic disorder. J. Neuroinflamm. 2010, 7, 48. [Google Scholar] [CrossRef]
- Tsilioni, I.; Dodman, N.; Petra, A.I.; Taliou, A.; Francis, K.; Moon-Fanelli, A.; Shuster, L.; Theoharides, T.C. Elevated serum neurotensin and CRH levels in children with autistic spectrum disorders and tail-chasing Bull Terriers with a phenotype similar to autism. Transl. Psychiatry 2014, 4, e466. [Google Scholar] [CrossRef]
- Patel, A.B.; Tsilioni, I.; Leeman, S.E.; Theoharides, T.C. Neurotensin stimulates sortilin and mTOR in human microglia inhibitable by methoxyluteolin, a potential therapeutic target for autism. Proc. Natl. Acad. Sci. USA 2016, 113, E7049–E7058. [Google Scholar] [CrossRef] [Green Version]
- Ghanizadeh, A. Targeting neurotensin as a potential novel approach for the treatment of autism. J. Neuroinflamm. 2010, 7, 58. [Google Scholar] [CrossRef]
- Kirsten, T.B.; Casarin, R.C.; Bernardi, M.M.; Felicio, L.F. Pioglitazone abolishes autistic-like behaviors via the IL-6 pathway. PLoS ONE 2018, 13, e0197060. [Google Scholar] [CrossRef]
- Kirsten, T.B.; Casarin, R.C.; Bernardi, M.M.; Felicio, L.F. Pioglitazone abolishes cognition impairments as well as BDNF and neurotensin disturbances in a rat model of autism. Biol. Open 2019, 8, bio041327. [Google Scholar] [CrossRef] [Green Version]
- Normandeau, C.P.; Ventura-Silva, A.P.; Hawken, E.R.; Angelis, S.; Sjaarda, C.; Liu, X.; Pego, J.M.; Dumont, E.C. A Key Role for Neurotensin in Chronic-Stress-Induced Anxiety-Like Behavior in Rats. Neuropsychopharmacology 2018, 43, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Lipstein, N.; Verhoeven-Duif, N.M.; Michelassi, F.E.; Calloway, N.; Van Hasselt, P.M.; Pienkowska, K.; Van Haaften, G.; Van Haelst, M.M.; Van Empelen, R.; Cuppen, I.; et al. Synaptic UNC13A protein variant causes increased neurotransmission and dyskinetic movement disorder. J. Clin. Investig. 2017, 127, 1005–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damaj, L.; Lupien-Meilleur, A.; Lortie, A.; Riou, E.; Ospina, L.H.; Gagnon, L.; Vanasse, C.; Rossignol, E. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur. J. Hum. Genet. 2015, 23, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.H.; Perlis, R.H.; Jung, J.-Y.; Byrne, E.M.; Rueckert, E.; Siburian, R.; Haddad, S.; Mayerfeld, C.E.; Heath, A.C.; Pergadia, M.L.; et al. Multi-locus genome-wide association analysis supports the role of glutamatergic synaptic transmission in the etiology of major depressive disorder. Transl. Psychiatry 2012, 2, e184. [Google Scholar] [CrossRef] [PubMed]
- Horder, J.; Lavender, T.; Mendez, M.A.; O'Gorman, R.; Daly, E.; Craig, M.C.; Lythgoe, D.J.; Barker, G.J.; Murphy, D.G. Reduced subcortical glutamate/glutamine in adults with autism spectrum disorders: A [H] MRS study. Transl. Psychiatry 2013, 3, e279. [Google Scholar] [CrossRef]
- Bernardi, S.; Anagnostou, E.; Shen, J.; Kolevzon, A.; Buxbaum, J.D.; Hollander, E.; Hof, P.R.; Fan, J. In vivo 1H-magnetic resonance spectroscopy study of the attentional networks in autism. Brain Res. 2011, 1380, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Page, L.A.; Daly, E.; Schmitz, N.; Simmons, A.; Toal, F.; Deeley, Q.; Ambery, F.; McAlonan, G.M.; Murphy, K.C.; Murphy, D.G.M. In Vivo 1 H-Magnetic Resonance Spectroscopy Study of Amygdala-Hippocampal and Parietal Regions in Autism. Am. J. Psychiatry 2006, 163, 2189–2192. [Google Scholar] [CrossRef]
- Wickens, M.M.; Bangasser, D.A.; Briand, L.A. Sex Differences in Psychiatric Disease: A Focus on the Glutamate System. Front. Mol. Neurosci. 2018, 11, 197. [Google Scholar] [CrossRef]
- Plavén-Sigray, P.; Gustavsson, P.; Farde, L.; Borg, J.; Stenkrona, P.; Nyberg, L.; Bäckman, L.; Červenka, S. Dopamine D1 receptor availability is related to social behavior: A positron emission tomography study. NeuroImage 2014, 102, 590–595. [Google Scholar] [CrossRef]
- Homberg, J.R.; Olivier, J.D.A.; VandenBroeke, M.; Youn, J.; Ellenbroek, A.K.; Karel, P.; Shan, L.; Van Boxtel, R.; Ooms, S.; Balemans, M.; et al. The role of the dopamine D1 receptor in social cognition: Studies using a novel genetic rat model. Dis. Model. Mech. 2016, 9, 1147–1158. [Google Scholar] [CrossRef]
- Canals, M.; Marcellino, D.; Fanelli, F.; Ciruela, F.; de Benedetti, P.; Goldberg, S.R.; Neve, K.; Fuxe, K.; Agnati, L.F.; Woods, A.S.; et al. Adenosine A2A-dopamine D2 receptor-receptor heteromerization: Qualitative and quantitative assessment by fluorescence and bioluminescence energy transfer. J. Biol. Chem. 2003, 278, 46741–46749. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.G.E.; Frank, M.J. Opponent actor learning (OpAL): Modeling interactive effects of striatal dopamine on reinforcement learning and choice incentive. Psychol. Rev. 2014, 121, 337–366. [Google Scholar] [CrossRef] [PubMed]
- Vorstman, J.A.; Morcus, M.E.; Duijff, S.N.; Klaassen, P.W.; Heineman-de Boer, J.A.; Beemer, F.A.; Swaab, H.; Kahn, R.S.; van Engeland, H. The 22q11.2 deletion in children: High rate of autistic disorders and early onset of psychotic symptoms. J. Am. Acad. Child Adolesc. Psychiatry 2006, 45, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Deckert, J.; Rietschel, M.; Wildenauer, D.; Bondy, B.; Ertl, M.A.; Knapp, M.; Schofield, P.R.; Albus, M.; Maier, W.; Propping, P. Human adenosine A2a receptor (A2aAR) gene: Systematic mutation screening in patients with schizophrenia. J. Neural Transm. 1996, 103, 1447–1455. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.-J.; Liu, H.-C.; Liu, T.-Y.; Liao, D.-L.; Tsai, S.-J. Association studies of the adenosine A2a receptor (1976T > C) genetic polymorphism in Parkinson’s disease and schizophrenia. J. Neural Transm. 2005, 112, 1503–1510. [Google Scholar] [CrossRef] [PubMed]
- Hohoff, C.; Mullings, E.L.; Heatherley, S.V.; Freitag, C.M.; Neumann, L.C.; Domschke, K.; Krakowitzky, P.; Rothermundt, M.; Keck, M.E.; Erhardt, A.; et al. Adenosine A2A receptor gene: Evidence for association of risk variants with panic disorder and anxious personality. J. Psychiatr. Res. 2010, 44, 930–937. [Google Scholar] [CrossRef]
- Freitag, C.M.; Agelopoulos, K.; Huy, E.; Rothermundt, M.; Krakowitzky, P.; Meyer, J.; Deckert, J.; von Gontard, A.; Hohoff, C. Adenosine A (2A) receptor gene (ADORA2A) variants may increase autistic symptoms and anxiety in autism spectrum disorder. Eur. Child Adolesc. Psychiatry 2010, 19, 67–74. [Google Scholar] [CrossRef]
- Squillace, M.; Dodero, L.; Federici, M.; Migliarini, S.; Errico, F.; Napolitano, F.; Krashia, P.; Di Maio, A.; Galbusera, A.; Bifone, A.; et al. Dysfunctional dopaminergic neurotransmission in asocial BTBR mice. Transl. Psychiatry 2014, 4, e427. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Chen, X.S.; Reader, R.H.; Hoischen, A.; Veltman, J.A.; Simpson, N.H.; Francks, C.; Newbury, D.F.; Fisher, S.E. Next-generation DNA sequencing identifies novel gene variants and pathways involved in specific language impairment. Sci. Rep. 2017, 7, 46105. [Google Scholar] [CrossRef]
- Gao, K.; Tankovic, A.; Zhang, Y.; Kusumoto, H.; Zhang, J.; Chen, W.; Xiangwei, W.; Shaulsky, G.H.; Hu, C.; Traynelis, S.F.; et al. A de novo loss-of-function GRIN2A mutation associated with childhood focal epilepsy and acquired epileptic aphasia. PLoS ONE 2017, 12, e0170818. [Google Scholar] [CrossRef] [PubMed]
- Goswami, D.B.; Jernigan, C.S.; Chandran, A.; Iyo, A.H.; May, W.L.; Austin, M.C.; Stockmeier, C.A.; Karolewicz, B. Gene expression analysis of novel genes in the prefrontal cortex of major depressive disorder subjects. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 43, 126–133. [Google Scholar] [CrossRef]
- Kaut, O.; Schmitt, I.; Hofmann, A.; Hoffmann, P.; Schlaepfer, T.E.; Wüllner, U.; Hurlemann, R. Aberrant NMDA receptor DNA methylation detected by epigenome-wide analysis of hippocampus and prefrontal cortex in major depression. Eur. Arch. Psychiatry Clin. Neurosci. 2015, 265, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Chauhan, S.K.; Kay, E.; Dana, R. Flt-1 regulates vascular endothelial cell migration via a protein tyrosine kinase-7–dependent pathway. Blood 2011, 117, 5762–5771. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Differential roles of vascular endothelial growth factor receptor-1 and receptor-2 in angiogenesis. J. Biochem. Mol. Biol. 2006, 39, 469–478. [Google Scholar] [CrossRef]
- Mahoney, E.R.; Dumitrescu, L.; Moore, A.M.; Cambronero, F.E.; De Jager, P.L.; Koran, M.E.I.; Petyuk, V.A.; Robinson, R.A.S.; Goyal, S.; Schneider, J.A.; et al. Brain expression of the vascular endothelial growth factor gene family in cognitive aging and alzheimer’s disease. Mol. Psychiatry 2019, in press. [Google Scholar] [CrossRef]
- Hu, V.W.; Frank, B.C.; Heine, S.; Lee, N.H.; Quackenbush, J. Gene expression profiling of lymphoblastoid cell lines from monozygotic twins discordant in severity of autism reveals differential regulation of neurologically relevant genes. BMC Genom. 2006, 7, 118. [Google Scholar]
- Maurer, S.V.; Williams, C.L. The Cholinergic System Modulates Memory and Hippocampal Plasticity via Its Interactions with Non-Neuronal Cells. Front. Immunol. 2017, 8, 1489. [Google Scholar] [CrossRef]
- Ballinger, E.C.; Ananth, M.; Talmage, D.A.; Role, L.W. Basal Forebrain Cholinergic Circuits and Signaling in Cognition and Cognitive Decline. Neuron 2016, 91, 1199–1218. [Google Scholar] [CrossRef] [Green Version]
- Drenan, R.M.; Grady, S.R.; Steele, A.D.; McKinney, S.; Patzlaff, N.E.; McIntosh, J.M.; Marks, M.J.; Miwa, J.M.; Lester, H.A. Cholinergic modulation of locomotion and striatal dopamine release is mediated by alpha6alpha4* nicotinic acetylcholine receptors. J. Neurosci. 2010, 30, 9877–9889. [Google Scholar] [CrossRef]
- Sarter, M.; Lustig, C.; Taylor, S.F. Cholinergic contributions to the cognitive symptoms of schizophrenia and the viability of cholinergic treatments. Neuropharmacology 2012, 62, 1544–1553. [Google Scholar] [CrossRef] [PubMed]
- Hardan, A.Y.; Handen, B.L. A Retrospective Open Trial of Adjunctive Donepezil in Children and Adolescents with Autistic Disorder. J. Child Adolesc. Psychopharmacol. 2002, 12, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Almeida, L.E.F.; Spornick, N.A.; Kenyon, N.; Kamimura, S.; Khaibullina, A.; Nouraie, M.; Quezado, Z.M.N. Modulation of social deficits and repetitive behaviors in a mouse model of autism: The role of the nicotinic cholinergic system. Psychopharmacology 2015, 232, 4303–4316. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Martin-Ruiz, C.; Graham, A.; Court, J.; Jaros, E.; Perry, R.; Iversen, P.; Bauman, M.; Perry, E. Nicotinic receptor abnormalities in the cerebellar cortex in autism. Brain 2002, 125, 1483–1495. [Google Scholar] [CrossRef] [Green Version]
- Perry, E.K.; Lee, M.L.; Martin-Ruiz, C.M.; Court, J.A.; Volsen, S.G.; Merrit, J.; Folly, E.; Iversen, P.E.; Bauman, M.L.; Perry, R.H.; et al. Cholinergic Activity in Autism: Abnormalities in the Cerebral Cortex and Basal Forebrain. Am. J. Psychiatry 2001, 158, 1058–1066. [Google Scholar] [CrossRef]
- Han, S.; Yang, S.H.; Kim, J.Y.; Mo, S.; Yang, E.; Song, K.M.; Ham, B.-J.; Mechawar, N.; Turecki, G.; Lee, H.W.; et al. Author Correction: Down-regulation of cholinergic signaling in the habenula induces anhedonia-like behavior. Sci. Rep. 2017, 7, 17090. [Google Scholar] [CrossRef] [Green Version]
- Bierer, L.M.; Haroutunian, V.; Gabriel, S.; Knott, P.J.; Carlin, L.S.; Purohit, D.P.; Perl, D.P.; Schmeidler, J.; Kanof, P.; Davis, K.L. Neurochemical correlates of dementia severity in Alzheimer’s disease: Relative importance of the cholinergic deficits. J. Neurochem. 1995, 64, 749–760. [Google Scholar] [CrossRef]
- Giannini, G.; Conti, A.; Mammarella, S.; Scrobogna, M.; Sorrentino, V. The ryanodine receptor/calcium channel genes are widely and differentially expressed in murine brain and peripheral tissues. J. Cell Boil. 1995, 128, 893–904. [Google Scholar] [CrossRef]
- Galeotti, N.; Quattrone, A.; Vivoli, E.; Norcini, M.; Bartolini, A.; Ghelardini, C. Different involvement of type 1, 2, and 3 ryanodine receptors in memory processes. Learn. Mem. 2008, 15, 315–323. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Ochoa, E.O.; Pratt, S.J.P.; Lovering, R.M.; Schneider, M.F. Critical Role of Intracellular RyR1 Calcium Release Channels in Skeletal Muscle Function and Disease. Front. Physiol. 2015, 6, 420. [Google Scholar] [CrossRef]
- Kulkarni, M.M. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr. Protoc. Mol. Biol. 2011. [Google Scholar] [CrossRef]
- da Huang, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Sheilabi, M.A.; Bhattacharyya, D.; Kitchen, P.; Conner, A.C.; Bill, R.M.; Woodroofe, M.N.; Conner, M.T.; Princivalle, A.P. Transcriptome analysis suggests a role for the differential expression of cerebral aquaporins and the MAPK signalling pathway in human temporal lobe epilepsy. Eur. J. Neurosci. 2017, 46, 2121–2132. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Kitchen, P.; Woodroofe, M.N.; Bill, R.M.; Conner, A.C.; Heath, P.R.; Conner, M.T. Transcriptome Analysis of Gene Expression Provides New Insights into the Effect of Mild Therapeutic Hypothermia on Primary Human Cortical Astrocytes Cultured under Hypoxia. Front. Cell. Neurosci. 2017, 11, 386. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Boil. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.T.; Do, I.G.; Lee, J.; Sohn, I.; Kim, K.M.; Kang, W.K. The NanoString-based multigene assay as a novel platform to screen EGFR, HER2, and MET in patients with advanced gastric cancer. Clin. Transl. Oncol. 2015, 17, 462–468. [Google Scholar] [CrossRef]
- Leal, L.F.; Evangelista, A.F.; De Paula, F.E.; Almeida, G.C.; Carloni, A.C.; Saggioro, F.; Stavale, J.N.; Malheiros, S.M.; Mançano, B.; De Oliveira, M.A.; et al. Reproducibility of the NanoString 22-gene molecular subgroup assay for improved prognostic prediction of medulloblastoma. Neuropathology 2018, 38, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Łastowska, M.; Trubicka, J.; Niemira, M.; Paczkowska-Abdulsalam, M.; Karkucińska-Więckowska, A.; Kaleta, M.; Drogosiewicz, M.; Perek-Polnik, M.; Kretowski, A.; Cukrowska, B.; et al. Medulloblastoma with transitional features between Group 3 and Group 4 is associated with good prognosis. J. Neuro-Oncol. 2018, 138, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Zapka, P.; Dorner, E.; Dreschmann, V.; Sakamato, N.; Kristiansen, G.; Calaminus, G.; Vokuhl, C.; Leuschner, I.; Pietsch, T. Type, Frequency, and Spatial Distribution of Immune Cell Infiltrates in CNS Germinomas: Evidence for Inflammatory and Immunosuppressive Mechanisms. J. Neuropathol. Exp. Neurol. 2018, 77, 119–127. [Google Scholar] [CrossRef]
- Solomon, I.H.; De Girolami, U.; Chettimada, S.; Misra, V.; Singer, E.J.; Gabuzda, D. Brain and liver pathology, amyloid deposition, and interferon responses among older HIV-positive patients in the late HAART era. BMC Infect. Dis. 2017, 17, 151. [Google Scholar] [CrossRef]
- Prokopec, S.D.; Watson, J.D.; Waggott, D.M.; Smith, A.B.; Wu, A.H.; Okey, A.B.; Pohjanvirta, R.; Boutros, P.C. Systematic evaluation of medium-throughput mRNA abundance platforms. RNA 2013, 19, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Oldridge, N.B.; Streiner, D.L. The health belief model: Predicting compliance and dropout in cardiac rehabilitation. Med. Sci. Sports Exerc. 1990, 22, 678–683. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Category | Term | Count | % | p Value | Genes | Fold Enrichment | |
|---|---|---|---|---|---|---|---|
| KEGG_PATHWAY | mmu04510: Focal adhesion | 22 | 6% | 0.011 | PRKCA, EGFR, COL4A2, HRAS, COL4A1, FLT1, MAP2K1, PIK3CB, IGF1, PRKCG, BAD, CTNNB1, IGF1R, MAPK1, LAMB2, TNR, ITGA7, RAC1, PDGFRB, PIK3CA, PAK1, FN1 | 1.630 | |
| KEGG_PATHWAY | mmu03022: Basal transcription factors | 9 | 3% | 0.025 | TAF10, MNAT1, CCNH, GTF2H3, TAF9, CDK7, TAF6L, GTF2B, TBPL1 | 2.222 | |
| KEGG_PATHWAY | mmu05016: Huntington’s disease | 18 | 5% | 0.025 | POLR2H, HTT, CREBBP, DNAH1, SOD1, PPARGC1A, TFAM, AP2B1, EP300, PLCB4, HDAC2, GNAQ, CASP9, SP1, GRIN2B, BAX, TBPL1, HAP1 | 1.626 | |
| Females | KEGG_PATHWAY | mmu05215: Prostate cancer | 17 | 5% | 0.026 | EGFR, HRAS, MAP2K1, PIK3CB, CREBBP, IGF1, BAD, CTNNB1, IGF1R, MAPK1, CDKN1A, ATF4, EP300, CASP9, PDGFRB, PIK3CA, MTOR | 1.657 |
| KEGG_PATHWAY | mmu04151: PI3K-Akt signaling pathway | 31 | 9% | 0.029 | HRAS, IL4RA, PPP2R5C, IGF1R, LAMB2, CASP9, TNR, RAC1, PIK3CA, PRKAA2, INSR, CSF1R, FN1, PRKCA, EGFR, COL4A2, FLT1, COL4A1, MAP2K1, PIK3CB, IGF1, BAD, MAPK1, CDKN1A, ATF4, ITGA7, PDGFRB, GNB5, EFNA5, PPP2R5E, MTOR | 1.383 | |
| KEGG_PATHWAY | mmu05010: Alzheimer’s disease | 21 | 6% | 0.038 | CDK5R1, ADAM10, SNCA, GRIN2A, BAD, CDK5, ATF6, MAPK1, APP, PLCB4, LRP1, GNAQ, CASP9, GRIN2B, GRIN2C, MAPT, RYR3, GRIN2D, BACE1, PPP3CC, CACNA1C | 1.496 | |
| KEGG_PATHWAY | mmu04020: Calcium signaling pathway | 22 | 6% | 0.044 | PRKCA, EGFR, SLC8A1, DRD1, ADORA2A, ERBB3, GRIN2A, PRKCG, ATP2B3, PLCB4, GNAQ, ADCY9, PDE1B, GRIN2C, RYR3, GRIN2D, PDGFRB, PPP3CC, RYR2, CACNA1C, CACNA1A, CACNA1B | 1.455 |
| Gene. | Official Full Name | Up (+)/down(−) Regulated by VPA | % Change | Adjusted p Value | Neuroinflammation | Neuroplasticity, Development & Aging | Metabolism | Compartmentalization and Structural Integrity | Neurotransmission | Normalized by SAM Administration | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Smyd1 | SET and MYND domain containing 1 | (−) | −63% | 0.001951 | − | + | − | − | − | − | |
| Cspg4 | chondroitin sulfate proteoglycan 4 | (+) | 51% | 0.01074 | + | + | − | − | − | − | |
| Nts | neurotensin | (−) | −71% | 0.01074 | − | − | − | + | − | + | |
| Il4ra | interleukin 4 receptor, alpha | (+) | 63% | 0.01097 | + | − | − | − | − | − | |
| Ccl12 | chemokine (C-C motif) ligand 12 | (−) | −58% | 0.01461 | + | + | + | + | + | − | |
| Drd1 | dopamine receptor D1 | (−) | −57% | 0.01567 | − | − | − | + | + | Reported in human and animal SFARI data base. | + |
| Slc18a3 | solute carrier family 18 (vesicular monoamine), member 3 | (−) | −58% | 0.02347 | − | − | − | + | + | + | |
| Notch1 | notch 1 | (+) | 51% | 0.02426 | − | + | − | − | − | − | |
| Ppm1l | protein phosphatase 1 (formerly 2C)-like | (+) | 60% | 0.02426 | + | − | − | − | − | + | |
| Grin2a | glutamate receptor, ionotropic, NMDA2A (epsilon 1) | (+) | 61% | 0.02478 | − | − | − | + | + | Reported in human SFARI data base. | + |
| Chat | choline acetyltransferase | (−) | −56% | 0.03445 | − | − | + | + | + | + | |
| Cd40 | CD40 antigen | (−) | −55% | 0.04037 | + | − | − | + | − | − | |
| Cd4 | CD4 antigen | (−) | −55% | 0.0411 | − | − | − | + | − | − | |
| Adora2a | adenosine A2a receptor | (−) | −59% | 0.05169 | − | − | − | − | + | Reported in human and animal SFARI data base. | + |
| Flt1 | FMS-like tyrosine kinase 1 | (+) | 83% | 0.05169 | + | + | − | − | − | Reported in human SFARI data base. | + |
| Gene. | Official Full Name | Up(+)/down(-) Regulated by VPA | % Change | Adjusted p Value | Neuroinflammation | Compartmentalization and Structural Integrity | Neurotransmission | Normalized by SAM Administration |
|---|---|---|---|---|---|---|---|---|
| Ryr1 | ryanodine receptor 1, skeletal muscle | (+) | 61% | 0.007544 | − | − | + | + |
| Nts ** | neurotensin | (−) | −76% | 0.03878 | − | + | − | + |
| Itga7 | integrin alpha 7 | (+) | 53% | 0.04183 | − | + | − | + |
| Gene. | Official Full Name | % Change Males | Adjusted p Value- Males | % Change Females | Adjusted p Value- Females | Neuroplasticity, Development & Aging | Metabolism | Compartmentalization and Structural Integrity | Neuron-Glia interaction | Neurotransmission | Normalized by SAM Males | Normalized by SAM Females | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Npc1 | Niemann-Pick type C1 | 31% | 0.01089 | 26% | 0.003442 | − | + | − | − | − | + | − | |
| Plxnb3 | plexin B3 | 34% | 0.02376 | 40% | 0.0259 | + | − | − | − | − | − | + | |
| Unc13a | unc-13 homolog A (C. elegans) | 0.24 | 0.02376 | 0.23 | 0.04218 | − | − | + | − | + | + | − | Reported in human SFARI data base |
| Myrf | myelin regulatory factor | 48% | 0.0281 | 30% | 0.02347 | − | − | − | + | − | + | + | |
| Notch1 | notch 1 | 0.43 | 0.03878 | 0.51 | 0.02426 | + | − | − | − | − | − | − | |
| Nts | neurotensin | −76% | 0.03878 | −71% | 0.01074 | − | − | + | − | − | −− | + | |
| Itga7 | integrin alpha 7 | 0.53 | 0.04183 | 0.3 | 0.04599 | − | − | + | − | − | + | − | |
| Cacna1a | calcium channel, voltage-dependent, P/Q type, alpha 1A subunit | 25% | 0.0497 | 36% | 0.02478 | + | − | + | − | + | + | − | Reported in human SFARI data base |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weinstein-Fudim, L.; Ergaz, Z.; Turgeman, G.; Yanai, J.; Szyf, M.; Ornoy, A. Gender Related Changes in Gene Expression Induced by Valproic Acid in A Mouse Model of Autism and the Correction by S-adenosyl Methionine. Does It Explain the Gender Differences in Autistic Like Behavior? Int. J. Mol. Sci. 2019, 20, 5278. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215278
Weinstein-Fudim L, Ergaz Z, Turgeman G, Yanai J, Szyf M, Ornoy A. Gender Related Changes in Gene Expression Induced by Valproic Acid in A Mouse Model of Autism and the Correction by S-adenosyl Methionine. Does It Explain the Gender Differences in Autistic Like Behavior? International Journal of Molecular Sciences. 2019; 20(21):5278. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215278
Chicago/Turabian StyleWeinstein-Fudim, Liza, Zivanit Ergaz, Gadi Turgeman, Joseph Yanai, Moshe Szyf, and Asher Ornoy. 2019. "Gender Related Changes in Gene Expression Induced by Valproic Acid in A Mouse Model of Autism and the Correction by S-adenosyl Methionine. Does It Explain the Gender Differences in Autistic Like Behavior?" International Journal of Molecular Sciences 20, no. 21: 5278. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215278