ATRX Contributes to MeCP2-Mediated Pericentric Heterochromatin Organization during Neural Differentiation

 ,
,  , and
, and
Abstract
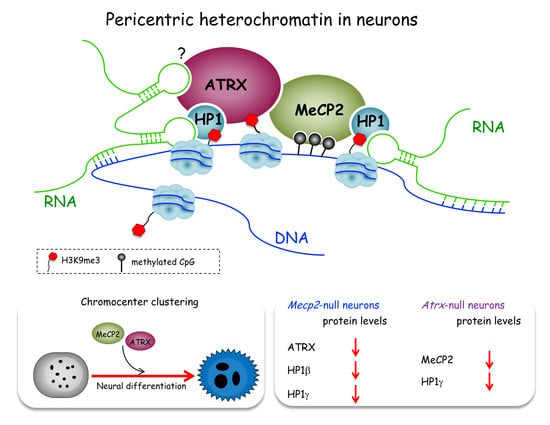
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
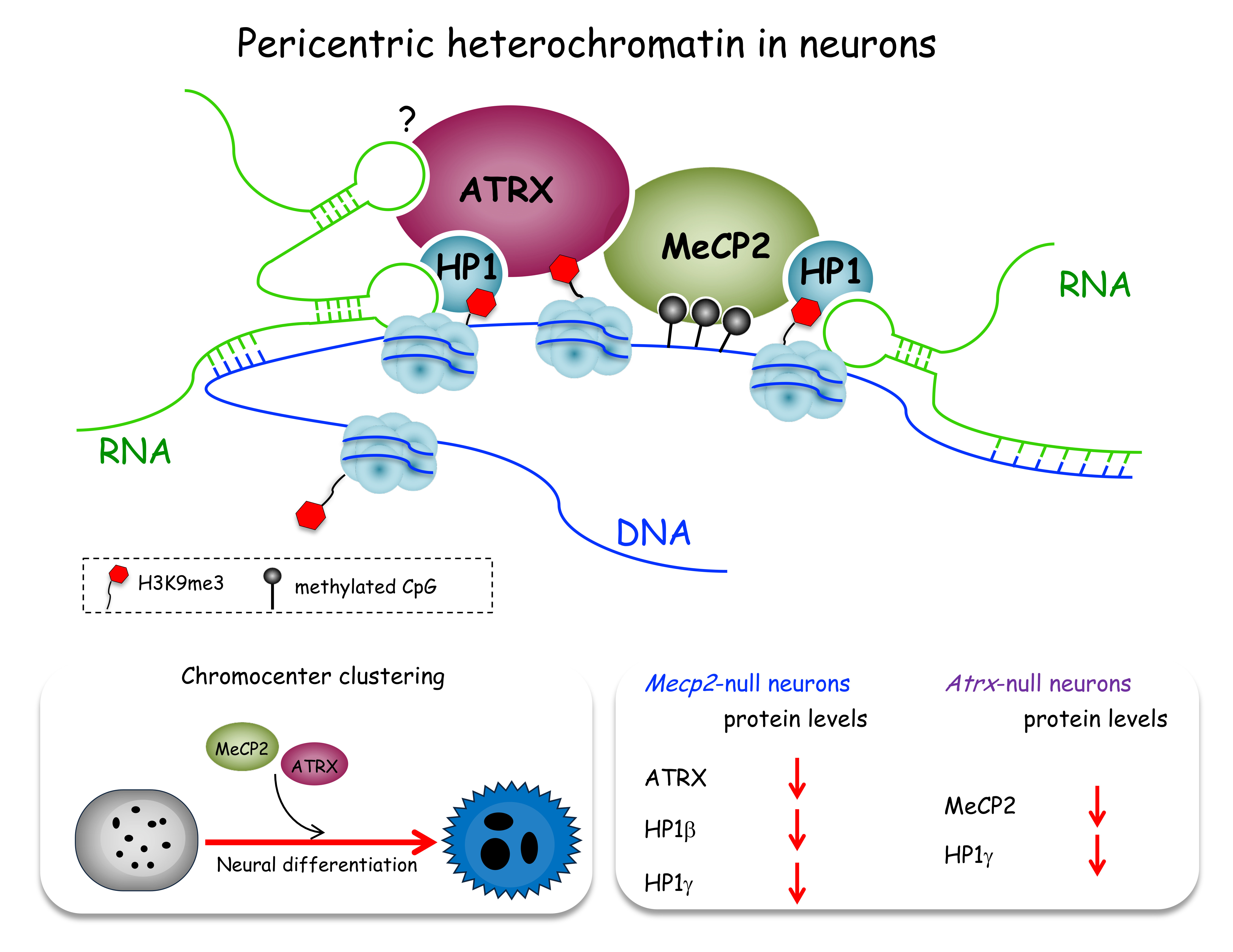
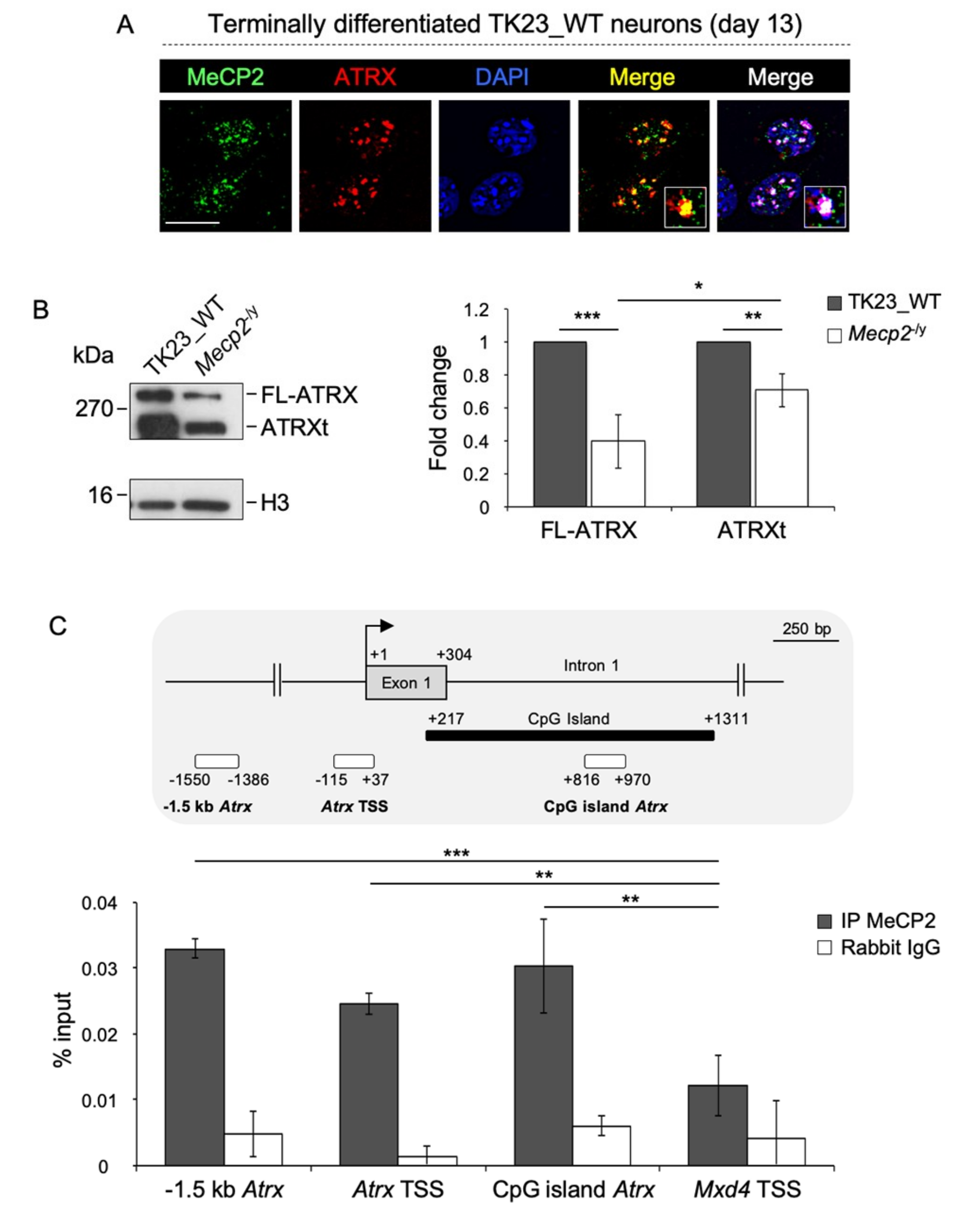
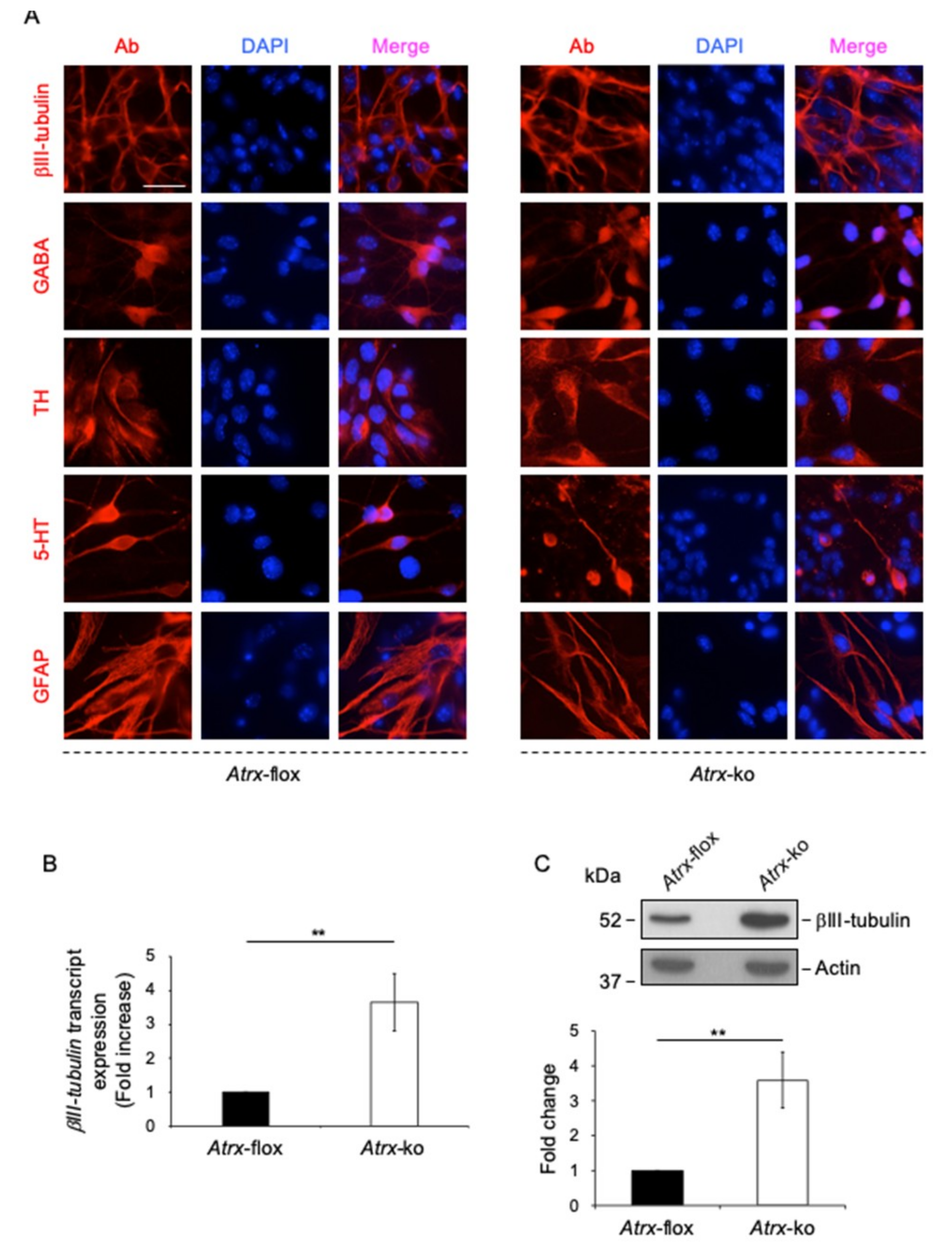
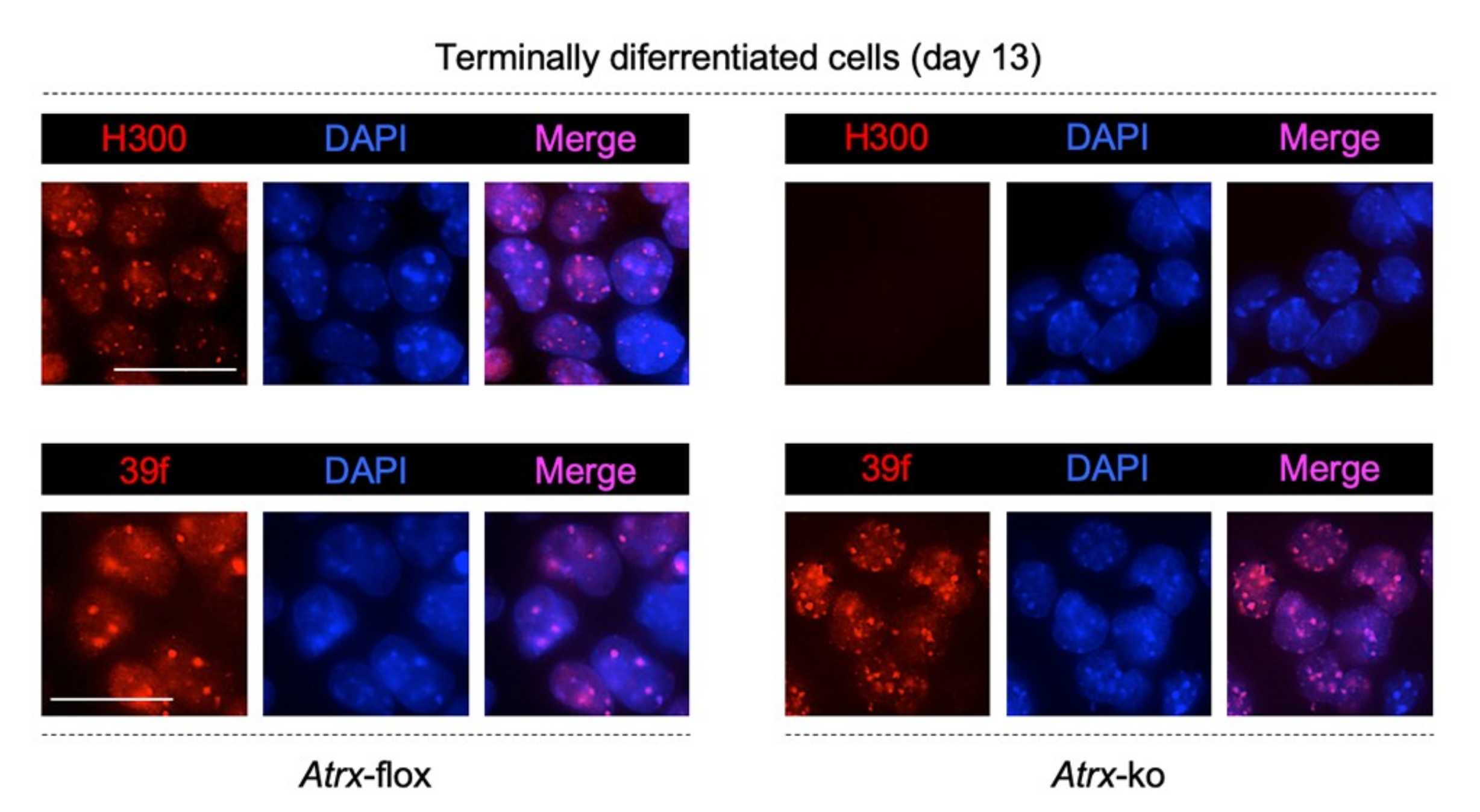
2.1. MeCP2 Co-Localizes with ATRX at Pericentric Heterochromatin and Regulates its Expression in Terminally Differentiated Neurons
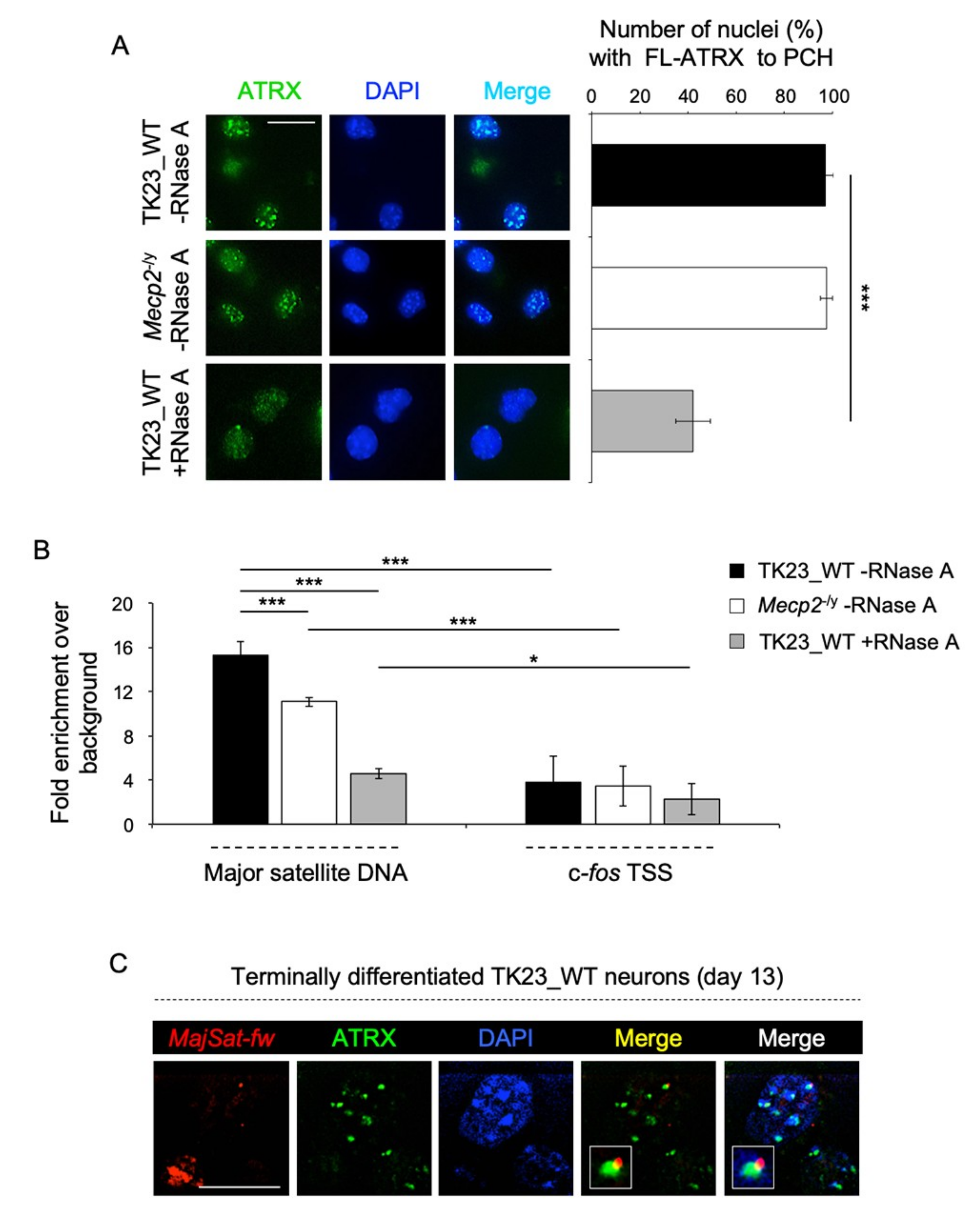
2.2. MeCP2 and an RNA Component Are Involved in ATRX Targeting to Pericentric Heterochromatin
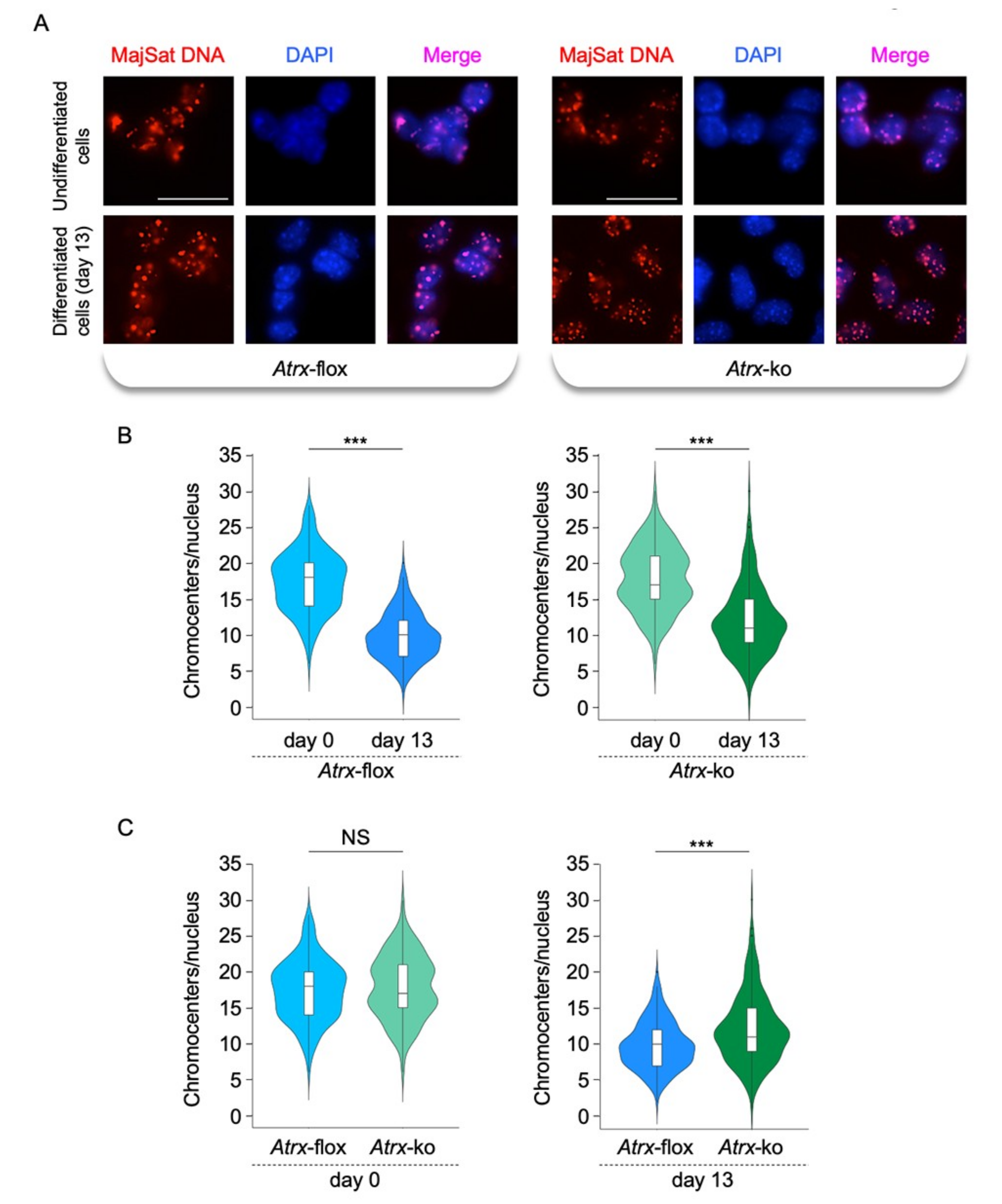
2.3. FL-ATRX Plays a Role in the Chromocenter Clustering during Neural Differentiation
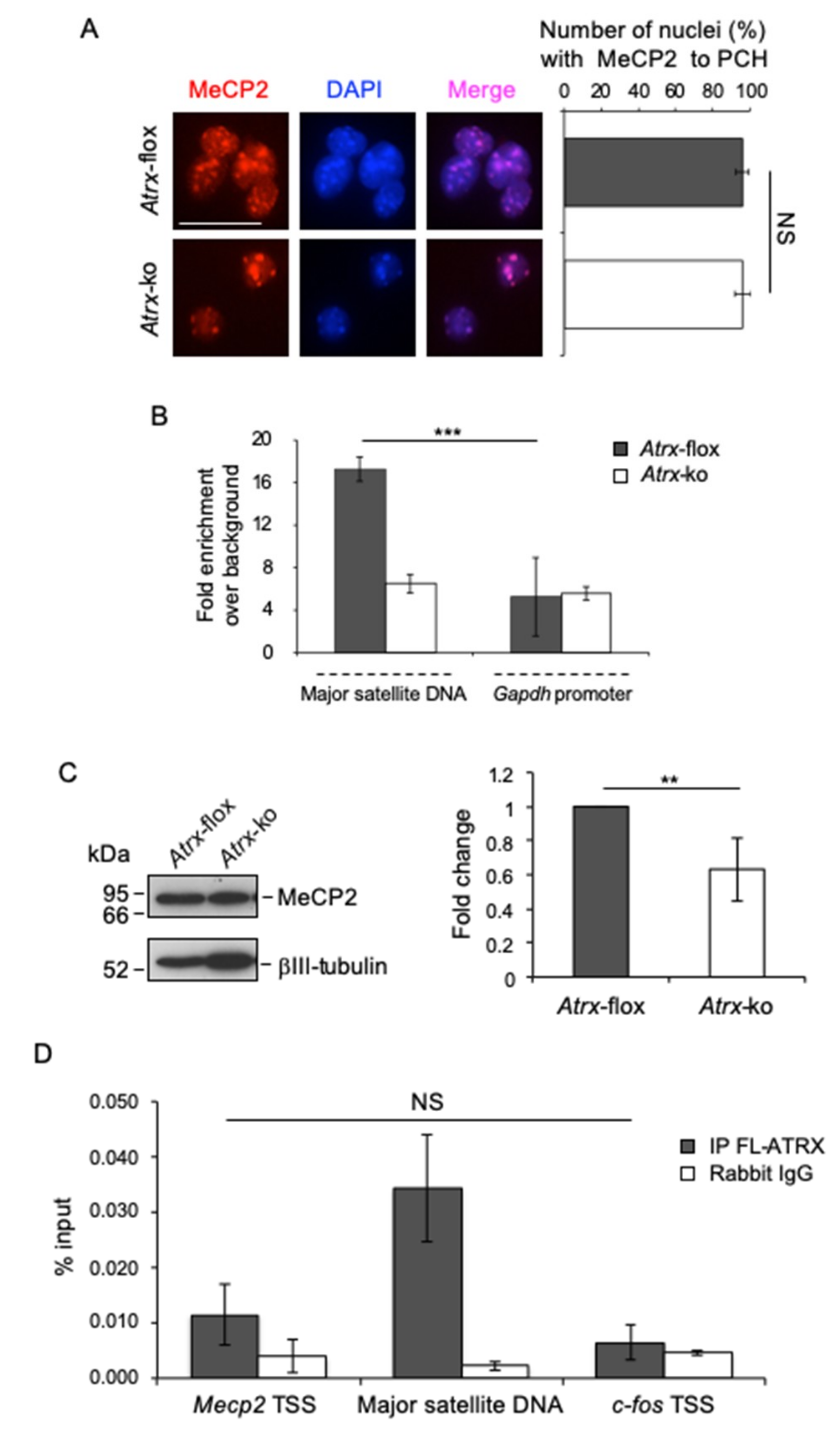
2.4. FL-ATRX Contributes to MeCP2 Enrichment at Pericentric Heterochromatin Foci and to its Transcriptional Regulation
2.5. FL-ATRX Promotes the Targeting of HP1α and HP1γ to Pericentric Heterochromatin and Regulates HP1γ Expression
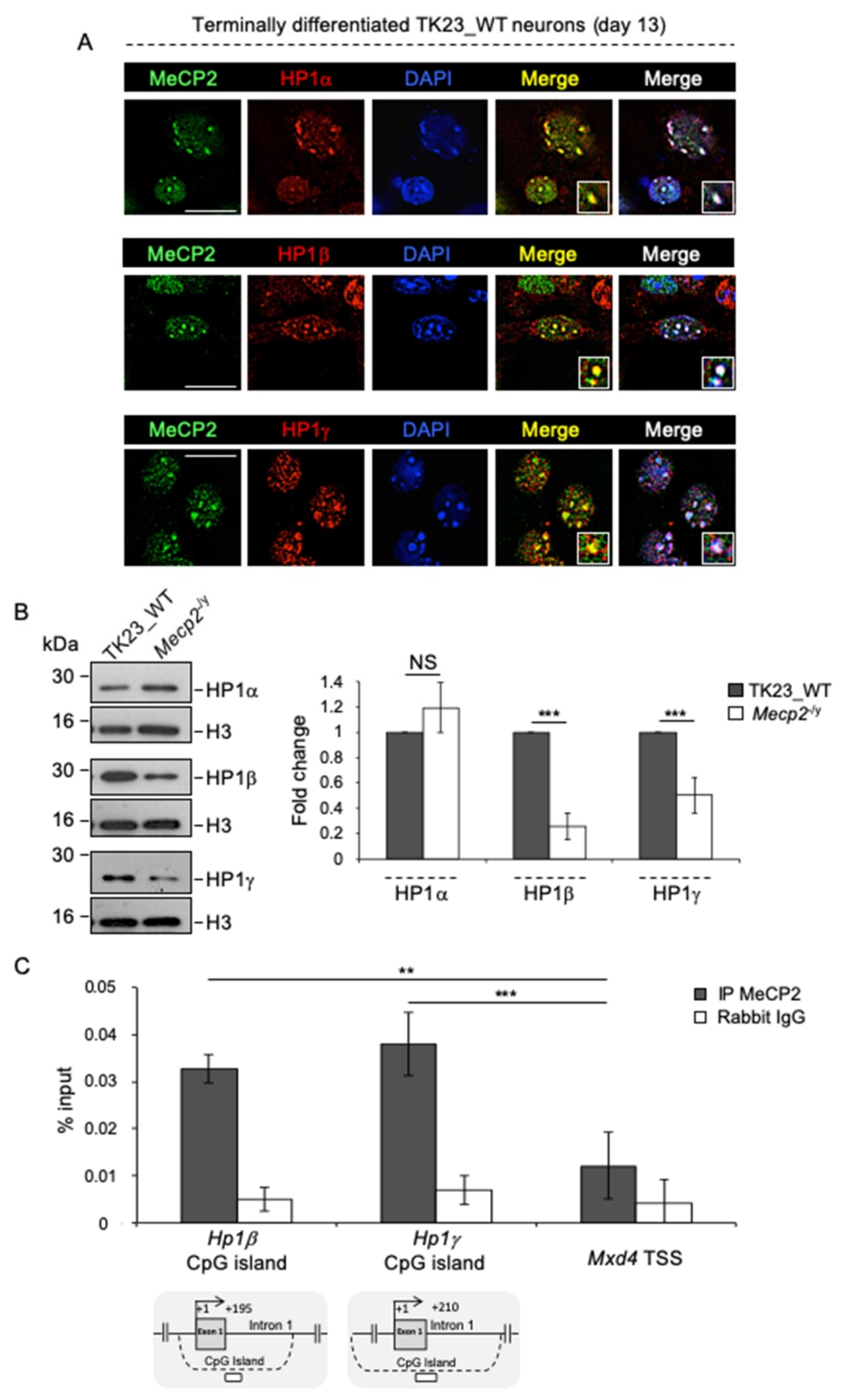
2.6. MeCP2 Promotes the Expression of HP1β and HP1γ in Terminally Differentiated Neurons
2.7. The Targeting of HP1α, HP1β and HP1γ to Pericentric Heterochromatin in Terminally Differentiated Neurons Depends on an RNA Component
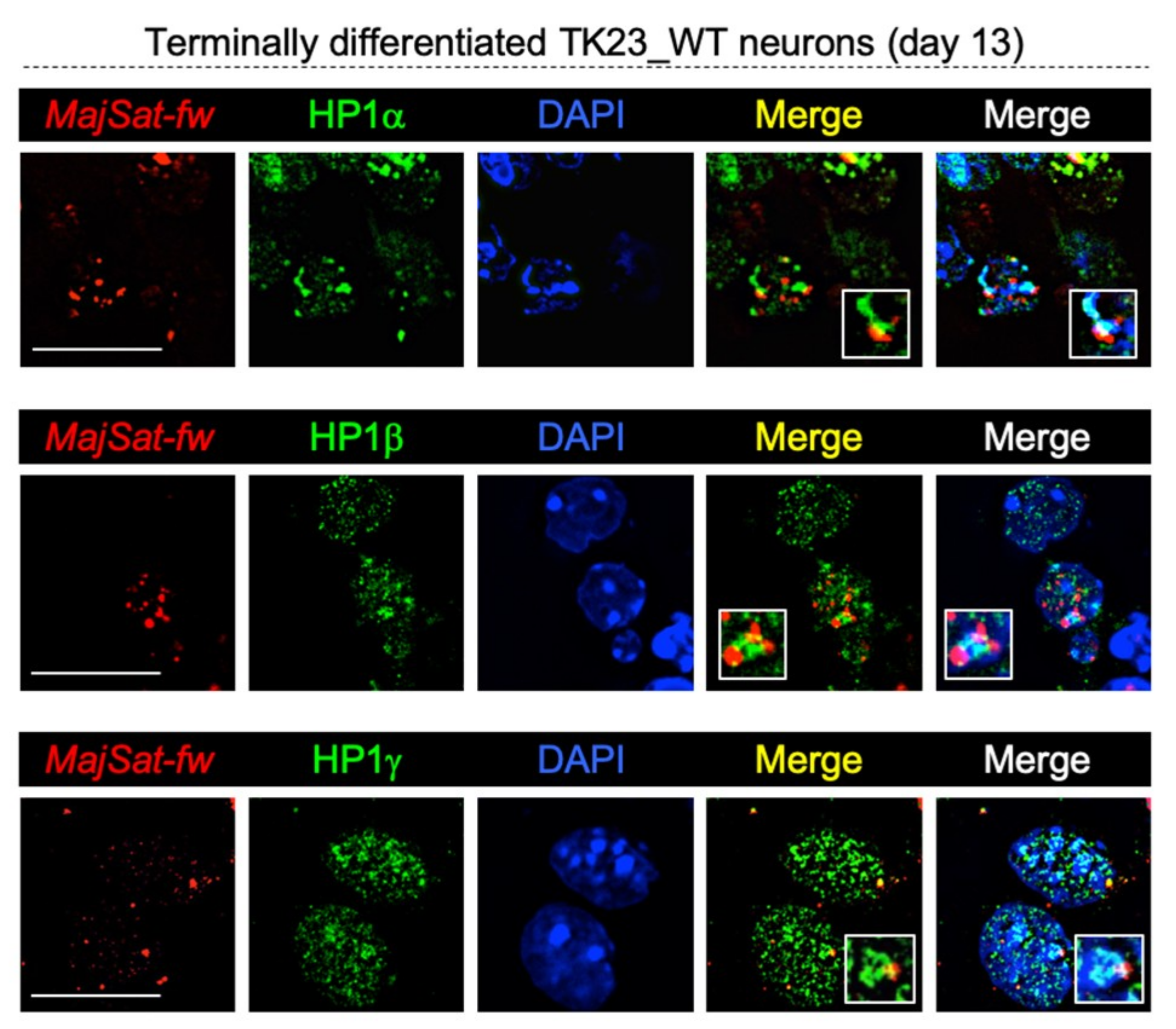
2.8. HP1 Proteins Co-Localize with Major Satellite Forward Transcript to Chromocenters in Terminally Differentiated Neurons
3. Discussion
4. Materials and Methods
4.1. Stem Cell Culture and Differentiation
4.2. RNA Extraction and Reverse Transcription qPCR
4.3. Western Blot
4.4. Immunofluorescences
4.5. DNA FISH and Immuno-RNA FISH
4.6. Microscopy and Image Analysis
4.7. Chromatin Immunoprecipitation (ChIP)
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-HT | 5-hydroxytryptamine |
| ATRX | Alpha-thalassemia/mental retardation syndrome X-linked protein |
| ATRXt | Truncated ATRX |
| c-fos | c-FBJ osteosarcoma oncogene |
| c-kit | c-KIT proto-oncogene receptor tyrosine kinase |
| ChIP | Chromatin immunoprecipitation |
| ChRO1 | Chromatin reorganization 1 |
| CSK | Cytoskeletal |
| DAPI | 4′,6-diamidino-2-phenylindole |
| EGS | Ethylene glycol bis(succinimidyl succinate) |
| FISH | Fluorescence In Situ Hybridization |
| FL-ATRX | Full-length ATRX |
| GABA | γ-aminobutyric acid |
| Gapdh | Glyceraldehyde 3 phosphate dehydrogenase |
| GFAP | Glial fibrillary acidic protein |
| H3K9me3 | Trimethylated H3-Lys9 |
| H4K20me4 | Trimethylated H4-Lys20 |
| HDAC1 | Histone deacetylase 1 |
| HP1 | Heterochromatin protein 1 |
| ko | Knockout |
| LNA | Locked nucleic acids |
| MajSat | Major satellite |
| MajSat-fw | Major satellite forward |
| MeCP2 | Methyl-CpG binding protein 2 |
| mESC | Murine embryonic stem cell |
| Mxd4 | Max dimerization protein 4 |
| ncRNAs | Non-coding RNAs |
| NGS | Normal goat serum |
| PCH | Pericentric heterochromatin |
| PHD | Plant homeodomain |
| PRC2 | Polycomb-repressive complex 2 |
| qPCR | Quantitative PCR |
| RT-qPCR | qPCR after reverse transcription |
| RTT | Rett syndrome |
| SDK | Suv3(9)h1 and h2 double knock-out |
| Sin3A | Switch-independent 3A |
| SNF2 | Sucrose Non-Fermentable 2 |
| SUMO | Small ubiquitin-like modifier |
| Suv39H1-H2 | Suppressor of variegation 3-9 homolog 1-2 |
| SWI/SNF | SWItch/Sucrose Non-Fermentable |
| TH | Tyrosine hydroxylase |
| TSS | Transcriptional start site |
| VRC | Vanadyl ribonucleoside complex |
| WT | Wild-type |
| Xist | X-inactive specific transcript |
References
- Lewis, J.D.; Meehan, R.R.; Henzel, W.J.; Maurer-Fogy, I.; Jeppesen, P.; Klein, F.; Bird, A. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 1992, 69, 905–914. [Google Scholar] [CrossRef]
- Ben-Shachar, S.; Chahrour, M.; Thaller, C.; Shaw, C.A.; Zoghbi, H.Y. Mouse models of MeCP2 disorders share gene expression changes in the cerebellum and hypothalamus. Hum. Mol. Genet. 2009, 18, 2431–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Nan, X.; Ng, H.H.; Johnson, C.A.; Laherty, C.D.; Turner, B.M.; Eisenman, R.N.; Bird, A. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 1998, 393, 386–389. [Google Scholar] [CrossRef]
- Klose, R.; Bird, A. Molecular biology. MeCP2 repression goes nonglobal. Science 2003, 302, 793–795. [Google Scholar] [CrossRef]
- Della Ragione, F.; Filosa, S.; Scalabri, F.; D’Esposito, M. MeCP2 as a genome-wide modulator: The renewal of an old story. Front. Genet. 2012, 3, 181. [Google Scholar] [CrossRef]
- Bienvenu, T.; Chelly, J. Molecular genetics of Rett syndrome: When DNA methylation goes unrecognized. Nat. Rev. Genet. 2006, 7, 415–426. [Google Scholar] [CrossRef]
- Kishi, N.; Macklis, J.D. MECP2 is progressively expressed in post-migratory neurons and is involved in neuronal maturation rather than cell fate decisions. Mol. Cell. Neurosci. 2004, 27, 306–321. [Google Scholar] [CrossRef]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Guy, J.; Cheval, H.; Selfridge, J.; Bird, A. The role of MeCP2 in the brain. Annu. Rev. Cell Dev. Biol. 2011, 27, 631–652. [Google Scholar] [CrossRef]
- Della Ragione, F.; Vacca, M.; Fioriniello, S.; Pepe, G.; D’Esposito, M. MECP2, a multi-talented modulator of chromatin architecture. Brief Funct. Genom. 2016, 15, 420–431. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, B. Clinical manifestations and stages of Rett syndrome. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, B.; Aicardi, J.; Dias, K.; Ramos, O. A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: Report of 35 cases. Ann. Neurol. 1983, 14, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Chahrour, M.; Zoghbi, H.Y. The story of Rett syndrome: From clinic to neurobiology. Neuron 2007, 56, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Leonard, H.; Cobb, S.; Downs, J. Clinical and biological progress over 50 years in Rett syndrome. Nat. Rev. Neurol. 2017, 13, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.Z.; Akbarian, S.; Tudor, M.; Jaenisch, R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat. Genet. 2001, 27, 327–331. [Google Scholar] [CrossRef]
- Guy, J.; Hendrich, B.; Holmes, M.; Martin, J.E.; Bird, A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat. Genet. 2001, 27, 322–326. [Google Scholar] [CrossRef]
- Nan, X.; Tate, P.; Li, E.; Bird, A. DNA methylation specifies chromosomal localization of MeCP2. Mol. Cell. Biol. 1996, 16, 414–421. [Google Scholar] [CrossRef] [Green Version]
- Probst, A.V.; Almouzni, G. Pericentric heterochromatin: Dynamic organization during early development in mammals. Differentiation 2008, 76, 15–23. [Google Scholar] [CrossRef]
- Wijchers, P.J.; Geeven, G.; Eyres, M.; Bergsma, A.J.; Janssen, M.; Verstegen, M.; Zhu, Y.; Schell, Y.; Vermeulen, C.; de Wit, E.; et al. Characterization and dynamics of pericentromere-associated domains in mice. Genome Res. 2015, 25, 958–969. [Google Scholar] [CrossRef] [Green Version]
- McDowell, T.L.; Gibbons, R.J.; Sutherland, H.; O’Rourke, D.M.; Bickmore, W.A.; Pombo, A.; Turley, H.; Gatter, K.; Picketts, D.J.; Buckle, V.J.; et al. Localization of a putative transcriptional regulator (ATRX) at pericentromeric heterochromatin and the short arms of acrocentric chromosomes. Proc. Natl. Acad. Sci. USA 1999, 96, 13983–13988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almouzni, G.; Probst, A.V. Heterochromatin maintenance and establishment: Lessons from the mouse pericentromere. Nucleus 2011, 2, 332–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenatri, M.; Bailly, D.; Maison, C.; Almouzni, G. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J. Cell Biol. 2004, 166, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Bertulat, B.; De Bonis, M.L.; Della Ragione, F.; Lehmkuhl, A.; Milden, M.; Storm, C.; Jost, K.L.; Scala, S.; Hendrich, B.; D’Esposito, M.; et al. MeCP2 dependent heterochromatin reorganization during neural differentiation of a novel Mecp2-deficient embryonic stem cell reporter line. PLoS ONE 2012, 7, e47848. [Google Scholar] [CrossRef]
- Brero, A.; Easwaran, H.P.; Nowak, D.; Grunewald, I.; Cremer, T.; Leonhardt, H.; Cardoso, M.C. Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J. Cell Biol. 2005, 169, 733–743. [Google Scholar] [CrossRef]
- Baker, S.A.; Chen, L.; Wilkins, A.D.; Yu, P.; Lichtarge, O.; Zoghbi, H.Y. An AT-hook domain in MeCP2 determines the clinical course of Rett syndrome and related disorders. Cell 2013, 152, 984–996. [Google Scholar] [CrossRef]
- Nan, X.; Hou, J.; Maclean, A.; Nasir, J.; Lafuente, M.J.; Shu, X.; Kriaucionis, S.; Bird, A. Interaction between chromatin proteins MECP2 and ATRX is disrupted by mutations that cause inherited mental retardation. Proc. Natl. Acad. Sci. USA 2007, 104, 2709–2714. [Google Scholar] [CrossRef] [Green Version]
- Kernohan, K.D.; Jiang, Y.; Tremblay, D.C.; Bonvissuto, A.C.; Eubanks, J.H.; Mann, M.R.; Berube, N.G. ATRX partners with cohesin and MeCP2 and contributes to developmental silencing of imprinted genes in the brain. Dev. Cell 2010, 18, 191–202. [Google Scholar] [CrossRef]
- Gibbons, R.J.; Picketts, D.J.; Higgs, D.R. Syndromal mental retardation due to mutations in a regulator of gene expression. Hum. Mol. Genet. 1995, 4, 1705–1709. [Google Scholar] [CrossRef]
- Gibbons, R.J.; Picketts, D.J.; Villard, L.; Higgs, D.R. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome). Cell 1995, 80, 837–845. [Google Scholar] [CrossRef]
- Garrick, D.; Samara, V.; McDowell, T.L.; Smith, A.J.; Dobbie, L.; Higgs, D.R.; Gibbons, R.J. A conserved truncated isoform of the ATR-X syndrome protein lacking the SWI/SNF-homology domain. Gene 2004, 326, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Ratnakumar, K.; Duarte, L.F.; LeRoy, G.; Hasson, D.; Smeets, D.; Vardabasso, C.; Bonisch, C.; Zeng, T.; Xiang, B.; Zhang, D.Y.; et al. ATRX-mediated chromatin association of histone variant macroH2A1 regulates alpha-globin expression. Genes Dev. 2012, 26, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Kernohan, K.D.; Vernimmen, D.; Gloor, G.B.; Berube, N.G. Analysis of neonatal brain lacking ATRX or MeCP2 reveals changes in nucleosome density, CTCF binding and chromatin looping. Nucleic Acids Res. 2014, 42, 8356–8368. [Google Scholar] [CrossRef] [PubMed]
- Drane, P.; Ouararhni, K.; Depaux, A.; Shuaib, M.; Hamiche, A. The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 2010, 24, 1253–1265. [Google Scholar] [CrossRef] [Green Version]
- Wong, L.H.; McGhie, J.D.; Sim, M.; Anderson, M.A.; Ahn, S.; Hannan, R.D.; George, A.J.; Morgan, K.A.; Mann, J.R.; Choo, K.H. ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res. 2010, 20, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Voon, H.P.; Hughes, J.R.; Rode, C.; De La Rosa-Velazquez, I.A.; Jenuwein, T.; Feil, R.; Higgs, D.R.; Gibbons, R.J. ATRX Plays a Key Role in Maintaining Silencing at Interstitial Heterochromatic Loci and Imprinted Genes. Cell Rep. 2015, 11, 405–418. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, H.; Han, N.; Kwak, S.; Lee, H.T.; Kim, J.H.; Kang, K.; Youn, B.H.; Yang, J.H.; Jeong, H.J.; et al. Long non-coding RNA ChRO1 facilitates ATRX/DAXX-dependent H3.3 deposition for transcription-associated heterochromatin reorganization. Nucleic Acids Res. 2018, 46, 11759–11775. [Google Scholar] [CrossRef] [Green Version]
- Wallrath, L.L.; Elgin, S.C. Position effect variegation in Drosophila is associated with an altered chromatin structure. Genes Dev. 1995, 9, 1263–1277. [Google Scholar] [CrossRef]
- Maison, C.; Almouzni, G. HP1 and the dynamics of heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 2004, 5, 296–304. [Google Scholar] [CrossRef]
- Yamamoto, K.; Sonoda, M. Self-interaction of heterochromatin protein 1 is required for direct binding to histone methyltransferase, SUV39H1. Biochem. Biophys. Res. Commun. 2003, 301, 287–292. [Google Scholar] [CrossRef]
- Agarwal, N.; Hardt, T.; Brero, A.; Nowak, D.; Rothbauer, U.; Becker, A.; Leonhardt, H.; Cardoso, M.C. MeCP2 interacts with HP1 and modulates its heterochromatin association during myogenic differentiation. Nucleic Acids Res. 2007, 35, 5402–5408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnertz, B.; Ueda, Y.; Derijck, A.A.; Braunschweig, U.; Perez-Burgos, L.; Kubicek, S.; Chen, T.; Li, E.; Jenuwein, T.; Peters, A.H. Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr. Biol. 2003, 13, 1192–1200. [Google Scholar] [CrossRef]
- Berube, N.G.; Smeenk, C.A.; Picketts, D.J. Cell cycle-dependent phosphorylation of the ATRX protein correlates with changes in nuclear matrix and chromatin association. Hum. Mol. Genet. 2000, 9, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Eustermann, S.; Yang, J.C.; Law, M.J.; Amos, R.; Chapman, L.M.; Jelinska, C.; Garrick, D.; Clynes, D.; Gibbons, R.J.; Rhodes, D.; et al. Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat. Struct. Mol. Biol. 2011, 18, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.S.; Schultz, D.C.; Negorev, D.; Maul, G.G.; Rauscher, F.J. The mammalian heterochromatin protein 1 binds diverse nuclear proteins through a common motif that targets the chromoshadow domain. Biochem. Biophys. Res. Commun. 2005, 331, 929–937. [Google Scholar] [CrossRef]
- Johnson, W.L.; Yewdell, W.T.; Bell, J.C.; McNulty, S.M.; Duda, Z.; O’Neill, R.J.; Sullivan, B.A.; Straight, A.F. RNA-dependent stabilization of SUV39H1 at constitutive heterochromatin. Elife 2017, 6, e25299. [Google Scholar] [CrossRef]
- Maison, C.; Bailly, D.; Roche, D.; Montes de Oca, R.; Probst, A.V.; Vassias, I.; Dingli, F.; Lombard, B.; Loew, D.; Quivy, J.P.; et al. SUMOylation promotes de novo targeting of HP1alpha to pericentric heterochromatin. Nat. Genet. 2011, 43, 220–227. [Google Scholar] [CrossRef]
- Velazquez Camacho, O.; Galan, C.; Swist-Rosowska, K.; Ching, R.; Gamalinda, M.; Karabiber, F.; De La Rosa-Velazquez, I.; Engist, B.; Koschorz, B.; Shukeir, N.; et al. Major satellite repeat RNA stabilize heterochromatin retention of Suv39h enzymes by RNA-nucleosome association and RNA:DNA hybrid formation. Elife 2017, 6, e25293. [Google Scholar] [CrossRef]
- Maison, C.; Bailly, D.; Peters, A.H.; Quivy, J.P.; Roche, D.; Taddei, A.; Lachner, M.; Jenuwein, T.; Almouzni, G. Higher-order structure in pericentric heterochromatin involves a distinct pattern of histone modification and an RNA component. Nat. Genet. 2002, 30, 329–334. [Google Scholar] [CrossRef]
- Ying, Q.L.; Stavridis, M.; Griffiths, D.; Li, M.; Smith, A. Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nat. Biotechnol. 2003, 21, 183–186. [Google Scholar] [CrossRef]
- Fico, A.; Manganelli, G.; Simeone, M.; Guido, S.; Minchiotti, G.; Filosa, S. High-throughput screening-compatible single-step protocol to differentiate embryonic stem cells in neurons. Stem Cells Dev. 2008, 17, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Sarma, K.; Levasseur, P.; Aristarkhov, A.; Lee, J.T. Locked nucleic acids (LNAs) reveal sequence requirements and kinetics of Xist RNA localization to the X chromosome. Proc. Natl. Acad. Sci. USA 2010, 107, 22196–22201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrick, D.; Sharpe, J.A.; Arkell, R.; Dobbie, L.; Smith, A.J.; Wood, W.G.; Higgs, D.R.; Gibbons, R.J. Loss of Atrx affects trophoblast development and the pattern of X-inactivation in extraembryonic tissues. PLoS Genet. 2006, 2, e58. [Google Scholar] [CrossRef]
- Gonzales, M.L.; Adams, S.; Dunaway, K.W.; LaSalle, J.M. Phosphorylation of distinct sites in MeCP2 modifies cofactor associations and the dynamics of transcriptional regulation. Mol. Cell. Biol. 2012, 32, 2894–2903. [Google Scholar] [CrossRef]
- Noh, K.M.; Maze, I.; Zhao, D.; Xiang, B.; Wenderski, W.; Lewis, P.W.; Shen, L.; Li, H.; Allis, C.D. ATRX tolerates activity-dependent histone H3 methyl/phos switching to maintain repetitive element silencing in neurons. Proc. Natl. Acad. Sci. USA 2015, 112, 6820–6827. [Google Scholar] [CrossRef]
- Sarma, K.; Cifuentes-Rojas, C.; Ergun, A.; Del Rosario, A.; Jeon, Y.; White, F.; Sadreyev, R.; Lee, J.T. ATRX Directs Binding of PRC2 to Xist RNA and Polycomb Targets. Cell 2014, 159, 1228. [Google Scholar] [CrossRef] [Green Version]
- Kourmouli, N.; Sun, Y.M.; van der Sar, S.; Singh, P.B.; Brown, J.P. Epigenetic regulation of mammalian pericentric heterochromatin in vivo by HP1. Biochem. Biophys. Res. Commun. 2005, 337, 901–907. [Google Scholar] [CrossRef]
- Berube, N.G.; Mangelsdorf, M.; Jagla, M.; Vanderluit, J.; Garrick, D.; Gibbons, R.J.; Higgs, D.R.; Slack, R.S.; Picketts, D.J. The chromatin-remodeling protein ATRX is critical for neuronal survival during corticogenesis. J. Clin. Investig. 2005, 115, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, K.; Shioda, N.; Yabuki, Y.; Zhang, C.; Han, F.; Fukunaga, K. SA4503, A Potent Sigma-1 Receptor Ligand, Ameliorates Synaptic Abnormalities and Cognitive Dysfunction in a Mouse Model of ATR-X Syndrome. Int. J. Mol. Sci. 2018, 19, 2811. [Google Scholar] [CrossRef]
- Saxena, A.; Carninci, P. Long non-coding RNA modifies chromatin: Epigenetic silencing by long non-coding RNAs. Bioessays 2011, 33, 830–839. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Gan, J.; Selfridge, J.; Cobb, S.; Bird, A. Reversal of neurological defects in a mouse model of Rett syndrome. Science 2007, 315, 1143–1147. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.V.; Okamoto, I.; Casanova, M.; El Marjou, F.; Le Baccon, P.; Almouzni, G. A strand-specific burst in transcription of pericentric satellites is required for chromocenter formation and early mouse development. Dev. Cell 2010, 19, 625–638. [Google Scholar] [CrossRef]
- Chaumeil, J.; Okamoto, I.; Guggiari, M.; Heard, E. Integrated kinetics of X chromosome inactivation in differentiating embryonic stem cells. Cytogenet. Genome Res. 2002, 99, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Law, M.J.; Lower, K.M.; Voon, H.P.; Hughes, J.R.; Garrick, D.; Viprakasit, V.; Mitson, M.; De Gobbi, M.; Marra, M.; Morris, A.; et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell 2010, 143, 367–378. [Google Scholar] [CrossRef]
- Skene, P.J.; Illingworth, R.S.; Webb, S.; Kerr, A.R.; James, K.D.; Turner, D.J.; Andrews, R.; Bird, A.P. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol. Cell 2010, 37, 457–468. [Google Scholar] [CrossRef]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marano, D.; Fioriniello, S.; Fiorillo, F.; Gibbons, R.J.; D’Esposito, M.; Della Ragione, F. ATRX Contributes to MeCP2-Mediated Pericentric Heterochromatin Organization during Neural Differentiation. Int. J. Mol. Sci. 2019, 20, 5371. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215371
Marano D, Fioriniello S, Fiorillo F, Gibbons RJ, D’Esposito M, Della Ragione F. ATRX Contributes to MeCP2-Mediated Pericentric Heterochromatin Organization during Neural Differentiation. International Journal of Molecular Sciences. 2019; 20(21):5371. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215371
Chicago/Turabian StyleMarano, Domenico, Salvatore Fioriniello, Francesca Fiorillo, Richard J. Gibbons, Maurizio D’Esposito, and Floriana Della Ragione. 2019. "ATRX Contributes to MeCP2-Mediated Pericentric Heterochromatin Organization during Neural Differentiation" International Journal of Molecular Sciences 20, no. 21: 5371. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215371