Synergy Between Low Dose Metronomic Chemotherapy and the pH-Centered Approach Against Cancer


1
Via Pier Capponi 6, 50132 Florence, Italy
2
Department of Biosciences, Biotechnology and Biopharmaceutics, University of Bari, 70126 Bari, Italy
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(21), 5438; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215438
Submission received: 11 September 2019
/
Revised: 25 October 2019
/
Accepted: 29 October 2019
/
Published: 31 October 2019
(This article belongs to the Special Issue The Role of Nonmainstream Approach in Science Discoveries)
Abstract
:Low dose metronomic chemotherapy (MC) is becoming a mainstream treatment for cancer in veterinary medicine. Its mechanism of action is anti-angiogenesis by lowering vascular endothelial growth factor (VEGF) and increasing trombospondin-1 (TSP1). It has also been adopted as a compassionate treatment in very advanced human cancer. However, one of the main limitations of this therapy is its short-term effectiveness: 6 to 12 months, after which resistance develops. pH-centered cancer treatment (pHT) has been proposed as a complementary therapy in cancer, but it has not been adopted or tested as a mainstream protocol, in spite of existing evidence of its advantages and benefits. Many of the factors directly or indirectly involved in MC and anti-angiogenic treatment resistance are appropriately antagonized by pHT. This led to the testing of an association between these two treatments. Preliminary evidence indicates that the association of MC and pHT has the ability to reduce anti-angiogenic treatment limitations and develop synergistic anti-cancer effects. This review will describe each of these treatments and will analyze the fundamentals of their synergy.
1. Introduction
1.1. Metronomic Chemotherapy (MC) vs. Maximum Tolerated Dose (MTD) Chemotherapy
Classical conventional chemotherapeutic regimens are designed to kill as many tumor cells as possible by treating them with “maximum tolerated doses” (MTDs) of cytotoxic agents. Side effects such as neurotoxicity, cardiotoxicity, and damage to proliferating cells in healthy tissues (hematopoietic system, intestinal mucosa, etc.) pose serious problems and limitations to this chemotherapy.
Metronomic chemotherapy (MC) is defined as frequent uninterrupted administration of low dose cytotoxic chemotherapy for long periods of time. The doses should be low enough to avoid myelo-suppression, but at the same time, high enough to kill endothelial cells.
Although many researchers before 1971 had described the association between growing malignant tumors and new vessels [1], it was Judah Folkman who reported for the first time that solid tumors were angiogenesis-dependent [2]. Angiogenesis means the development of new blood vessels from preexisting ones [3]. Folkman’s seminal discovery was that tumors could not grow without angiogenesis. Early in tumor development an angiogenic switch occurs activating endothelial cells in the malignant tissue. Activated endothelial cells degrade the extracellular matrix, proliferate and migrate generating new vascular sprouts [4].
He also hypothesized that there might be an unknown factor produced by the tumor that increased its vascular supply [5]: “the tumor angiogenesis factor”. Three different research groups independently and almost simultaneously (1983–1989) isolated and purified the tumor angiogenesis factor which was named Vascular Endothelial Growth Factor (VEGF) [6,7,8].
1.2. Anti-Angiogenic Treatment
Fumagillin, a toxic fungal product was the first anti-angiogenic drug discovered by the Folkman group, and a few years later a non-toxic derivative, TNP470, was synthesized [9]. α interferon was also successfully used for anti-angiogenic treatment of hemangiomas. Thrombospondin was identified as a natural inhibitor of angiogenesis via its inhibition of the synthesis of both VEGF [10,11,12] and FGF (fibrinogen growth factor) [13,14,15]. Angiogenesis can also be inhibited by blocking VEGF’s receptor: VEGFR [16,17,18]. From the beginning of the 1990s full-fledged research for anti-angiogenic compounds developed many new molecules like monoclonal antibodies against VEGF, 2-methoxyestradiol [19], the re-discovery of the anti-angiogenic properties of old drugs such as thalidomide [20], and low-dose chemotherapeutic agents when used metronomically, were also found to prevent angiogenesis [21].
At the beginning of the twenty-first century two different types of anti-angiogenic drugs have represented the proof of concept of anti-angiogenesis (Figure 1):
- (1)
- Bevacizumab: a monoclonal antibody that binds with VEGF.
- (2)
- Sunitinib and sorafenib: small protein inhibitors of VEGF receptor tyrosine-kinases.
While the anti-angiogenic compounds have had impressive success in preclinical models, and many new agents are entering into clinical trials, this success proved short lived in bedside medicine [22,23,24]. This lack of better clinical results is the consequence of the development of resistance to anti-angiogenic treatments [25,26,27]. The resistance can be [28]:
- (A)
- Primary: no response at all and supposedly due to intrinsic factors.
- (B)
- Secondary or evasive: initial response and then resistance.
At the beginning of the “anti-angiogenic era” it was a widespread belief that resistance would not appear [29], because the treatment is directed against endothelial cells that are not mutated, have no genetic instability and are not supposed to over-express pro-mitotic pathways. Now we have evidence that this is not so. Tumor-associated endothelial cells (TAECs) are different from their normal tissue counterparts. Genes expressed in TAECs differ from normal endothelial cells. Croix et al. identified 79 genes that were differently expressed in TAECs [30], and Lu et al. [31] found more than 400 genes differentially expressed in ovarian cancer TAECs. Akino et al. [32] also found frequent cytogenetic changes in renal cell carcinoma TAECs. Endothelial cells of glioma are resistant to apoptosis by over-expressing GRP78 (a stress chaperone protein) [33] and survivin [34]. This creates resistance to conventional chemotherapies. Therefore, TAECs can develop resistance to treatment and this is one of the reasons for the failure of the anti-angiogenic approach.
However, we believe that the lack of the real success of anti-angiogenesis rests on a biased concept of angiogenesis, because it is much more than the VEGF-VEGFR interaction. Some of this “much more” concept is known, but there remains a large gap of unknown factors. The “much more” participants in the angiogenic process has been named the “angiome” [35,36,37] in which 1233 proteins have been identified as playing a role in the angiome. Therefore, addressing angiogenesis by downregulating or inhibiting VEGF alone seems sort of naïve. Among the known factors are other growth factors that induce angiogenesis besides VEGF:
This list of growth factors inducing angiogenesis explains clearly why the inhibition of VEGF or VEGFR is insufficient to fully stop angiogenesis or its recurrence. Placental growth factor is the only one that binds VEGFR, while the rest bind their own receptors. This was called “the growth factor redundancy [59]”. This angiogenic growth factor redundancy explains the limited results of targeting only one of them. Furthermore, the added hypoxia produced by anti-angiogenic treatments is an inducer for the expression of other growth factors [60] and explains the increased rate of metastasis found after anti-angiogenic treatments [61].
There are cytokines that act in a similar way to growth factors, e.g., interleukin 8 (IL 8) [62,63] ephrin A1, ephrin A2, etc. These proteins are usually over-expressed in the escape from anti-angiogenesis. Some chemokines like SDF1/CXCR4 are also angiogenesis inducers [64,65,66]. Chemokines of the CXC family containing an ELR motif are strong angiogenesis inducers while those that lack this motif show an antagonistic action [67]. Other angiogenic proteins frequently found in tumor stroma include the heparin-binding protein CYR61 that promotes migration and adhesion of endothelial cells inducing neovascularization [68], MCP-1 (monocyte chemotactic protein 1) [69], CEA related cell adhesion molecule-1 [70], tumor necrosis factor α [71], brain-derived neurotrophic factor [72], platelet-derived endothelial cell growth factor [73], BMP2 (bone morphogenetic protein 2) [74] are among other angiogenic proteins frequently found in tumor stroma. Endothelial cells over-expressing the fatty acid-binding proteins FABP4 and FABP5 (fatty acid-binding proteins) are prone to migration, proliferation and angiogenic response. While FABP4 is VEGF-dependent, FABP-5 is VEGF-independent [75]. Metabotropic glutamate receptor-1 (mGluR1) is another player in angiogenesis [76]. Its inhibition (by riluzole) significantly handicaps the process of new vessel formation [77]. Endothelin-3, a vasoactive peptide released by vascular smooth muscle cells and endothelin converting enzyme-1, a protease that activates endothelin-3 also play a role in angiogenesis [78,79,80] and especially in tumor angiogenesis [81,82,83,84,85]. See Scheme 1.
Some hormones have also been identified as pro-angiogenic. This is the case of prolactin. Prolactin is the best example of the complexity of the angiogenesis problem. While the intact prolactin molecule is angiogenic, the N-terminal portion of this hormone is anti-angiogenic [86]. Furthermore, when this N-terminal portion of prolactin binds PAI-1 (plasminogen activator inhibitor-1), another pro-angiogenic molecule, it becomes fully anti-angiogenic [87].
Galectin-1 [88] and galectin-3 [89] are other essential proteins for angiogenesis [90,91]. Their downregulation inhibits angiogenesis and decreases tumor growth [92]. Anginex [93], a designed anti-angiogenic, seems to act through the inhibition of galectin-1 [94] without relation to the VEGF-VEGFR axis. Endoglin, an endothelial co-receptor for TGF-β, also plays a role in resistance to anti-angiogenesis [95].
Using a database search to look for a complementary drug to improve and prolong the effects of anti-angiogenic treatments, we found fenofibrate (FF), a powerful anti-angiogenic drug [96] that at the same time has the ability to act on many of the angiogenic escape factors. Fenofibrate is an agonist of PPARα (peroxisome proliferator-activated receptor alpha), which is a nuclear transcription factor and is actively being used as a lipid-lowering drug. FF decreases VEGF production and increases TSP1 expression thus inhibiting angiogenesis. Added to these fundamental effects (Figure 2), FF also:
Treating a small number of dogs with natural cancers with the metronomic chemotherapy scheme based on cyclophosphamide, celecoxib and cimetidine a rate of response (complete response, partial response and stable disease) of 50% was achieved that increased to 75% when fenofibrate was added to the scheme (unpublished data). However, resistance to treatment appeared within twelve months.
A similar response rate and time to develop resistance was found when another group of dogs was treated with pH-centered therapies.
1.3. The pH Centered Therapies
Cancer cells need a very particular pH homeostasis in order to sustain growth and invasion in a very different environment from that found in normal tissues.
Normal cells have a slightly alkaline intracellular pH (pHi), around 7.1, and a much more alkaline extracellular pH (pHe), around 7.35. The difference seems very small, but pH is a logarithmic expression of H+ (proton) concentration. Therefore, a difference of 0.25 in pH means a great difference in proton concentration inside and outside the cell. These pH values are strictly controlled and maintained. Cells need a higher pHi in order to proliferate [114,115,116,117,118,119] and this is concordant with the fact that microtubules assembly and motility also require a higher pHi [120].
A very early step of the malignant transformation is an increase of pHi [121]. Furthermore, cancer cells require a lower extracellular pH (pHe) for motility, degradation of extracellular matrix [122], invasion [123,124] and metastasis [125,126]. Hypoxia, a major contributor to tumor development and progression [127] is a strong player in the alterations of pH in cancer. The first step in the pH modification is induced by the genetic mutation(s) that leads or lead to cancer by alkalinizing intracellular pH. Increased intracellular alkalinity, the resultant metabolic switch and hypoxia work together to drive extracellular acidification, achieving pHe values well below those of normal tissues. The metabolic switch is probably a consequence of hypoxia coupled with intracellular alkalinization. This metabolic switch consists of two features:
- (a)
- oxidative mitochondrial metabolism is reduced while glycolytic cytoplasmic metabolism is increased;
- (b)
- this creates a decreased production of energy which is compensated by increasing glucose uptake and metabolism.
The increased but energetically inefficient cancer cells’ metabolism increases the production of acidic molecules: lactic acid and CO2. The acidic load thus produced must be swiftly extruded from the malignant cells in order to maintain an alkaline cytoplasm that favors proliferation. The consequence of this acid extrusion is microenvironmental acidification. At this stage, the intracellular pH is higher than the pH of the microenvironment: inversion of the pH gradient.
Now, the pH frame is perfect for increased proliferation (increased pHi) and mobility, migration, invasion, degradation of the extracellular matrix, angiogenesis, and metastasis (decreased pHe). As an added value, extracellular acidity also permits the escape from the immune system and resistance to drug therapy. The group headed by Barber called this dysregulated pH situation “the perfect storm” [128].
There are multiple cellular mechanisms involved in creating the inversion of the pH gradient and keeping it that way throughout the cancer’s life. The main mechanisms are represented by a wide range of different membrane-associated proteins working as channels, exchangers, transporters or enzymes. We have called all the elements participating in the creation of the perfect storm the “pHtome”, namely:
- (1)
- Na+/H+ exchangers (NHEs) and specifically the NHE1 isoform: promote the reversible electroneutral exchange of Na+/H+. In cancer, these exchangers extrude H+ from the cytoplasm to the extracellular space.
- (2)
- Monocarboxylate transporters (MCTs) and specifically the MCT1 and 4 isoforms. These MCTs extrude lactate and proton associated with lactate. The metabolic switch that increases glycolytic metabolism and decreases oxidative phosphorylation overloads the cell with lactate which is removed from the cell through MCTs.
- (3)
- Membrane carbonic anhydrases (CAs) isoforms 9 and 12 (CAIX and CAXII) mediate the reversible hydration of CO2 (which is produced in excess in malignant cells) producing carbonic acid.
- (4)
- V-ATPase proton pumps extrude protons across membranes while consuming energy.
- (5)
- Anion Exchangers (AEs) mediate the electroneutral transmembrane exchange of HCO3- by Cl−.
- (6)
- Specificity protein 1 (Sp1) is a transcription factor and enhancer that increases the transcription of HIF-1α, CAIX, NHE1, and some protein domains that form the V-ATPase proton pump.
- (7)
- Voltage-gated sodium channels (VGSCs) are channels that upon a stimulus incorporate Na+ into the cell. While intracellular Na+ has a role in increasing pHi, the main action of VGSCs in the pH inversion scheme are related to NHE1 activation.
The inversion of the pH gradient in cancer is a constant finding and there is compelling evidence that more than one of these pH regulators are the engines driving the inversion.
The fundamentals behind the pH-centered treatment of cancer are based on the following concepts:
- (1)
- The inversion of the pH gradient (the perfect storm) is not merely an “innocent” consequence of cancer progression, but an important etiopathogenic and determinant factor in the origin and development of cancer and its progression.
- (2)
- The inverted pH gradient is a constant finding in all types of malignant tumors.
- (3)
- The proteins involved in this process are the components of the pHtome.
- (4)
- Reverting the inverted pH gradient creates an inadequate environment for cancer growth and progression that leads to apoptosis or at least to a slowing down of proliferation and invasion.
- (5)
- This means that the proteins of the pHtome must be downregulated, removed, blocked or inhibited.
- (6)
- It is useless to inhibit only one of the soldiers, because the others would take up the functions of the lost comrade.
- (7)
- It is not possible to fully systemically attack the proteins, because most of them also perform other functions that are beneficial and necessary for normal cells (housekeeping proteins).
- (8)
- However, it is possible to downregulate or decrease the activity of many of them without affecting normal cells.
- (9)
- The simultaneous and partial inhibition of many of the participants of the pHtome will decrease tumor progression.
- (10)
- The partial inhibition of the pHtome does not only go along with other chemotherapeutical approaches, but also improves their results.
A treatment based on these concepts uses:
- (a)
- an NHE1 inhibitor such as amiloride, a diuretic that has been used for almost fifty years;
- (b)
- an inhibitor of carbonic anhydrases such as acetazolamide, another diuretic which has been in clinical practice since the 1940s;
- (c)
- (d)
- a MCT inhibitor like the nutraceutical quercetin wrongly considered to be a food supplement that is sold over-the-counter, but which has clear pharmacological effects;
- (e)
- a VGSC inhibitor like topiramate, used as an anticonvulsant in the treatment of epilepsy; but which is also a CA inhibitor.
Unfortunately, there are no known AE inhibitors that can be used in clinical practice; those that are available are exclusively suitable for experimental purposes and are toxic enough to preclude them for in vivo experiments.
The five types of drugs listed above represent the core of the pH-centered treatment.
When a small group of dogs with natural cancers was treated with both schemes (MT and pHT) the overall survival increased from 9–11 months to an average of 22 months in those who responded to treatment. (See Supplementary Table). It is premature to draw conclusions from this finding because the different groups of dogs considered in this review were quite different regarding breed, age, previous treatments, and types of cancers. However, the difference in overall survival is significant enough to encourage testing the MT-pHT association in a standardized larger trial.
On a theoretical basis there are many issues that can explain the apparent synergy between both treatments:
- ❖
- ❖
- ❖
- Proton pump inhibitors induce endothelial cell senescence [138]: Chronic use of PPIs impaired endothelial function through telomere length reduction.
- ❖
- ❖
- ❖
From the extensive evidence showed above, it is clear that the pHT has an anti-angiogenic effect. However, this does not explain why the MC-pHT association effects extend for a longer period than when they are used separately.
The first reason is probably based on the shared characteristic of the pHT drugs: they all increase extracellular pH (and at the same time decrease intracellular pH).
The next question is: what is the relationship between extracellular acidity and anti-angiogenic treatment?
Faes et al. [151] found that sorafenib (a VEGFR tyrosin-kinase inhibitor) combined with sodium bicarbonate had a stronger anti-tumoral effect than each drug used independently. They also found that the reason behind this was a higher number of endothelial cells expressing VEGFR as pHe increased and suggested that strategies that increase pHe improve anti-angiogenic treatment outcomes. This would also explain the better results found with the MC-pHT association.
Shi et al. [152] found that acidosis increased VEGF expression. Both publications, (Faes et al., and Shi et al.), concurred that the alkalinization of the extracellular space was adequate to decrease angiogenesis and improve the results of anti-angiogenic treatments.
1.4. Many Ion And Water Channels/Exchangers Downregulated by pHT Are Angiogenic
- Aquaporin 1, a water channel that is inhibited by acetazolamide and topiramate, is strongly expressed in endothelial cells [156]. Aquaporin 1 plays an important role in endothelial cell migration and favors angiogenesis. In aquaporin 1 null mice the migration of endothelial cells is compromised [157]. Downregulation of aquaporin 1 decreases angiogenesis [158].
- VGSCs are pro-angiogenic. NaV1.5 and NaV1.7 are the predominant isoforms found in endothelial cells. VGSCs showed a modulatory effect on the pro-angiogenic properties of VEGF [159].
- CAIX seems to be pro-angiogenic [160]. We use the term “seems” because CAIX is a hallmark of hypoxia and increased HIF-1α activity. Therefore, it is difficult to establish whether CAIX is angiogenic per se or actually the hypoxia-HIF-1α-VEGF pathway is the reason for this pro-angiogenic effect. What we do know is that inhibition of CAIX enhances anti-angiogenic treatment results [161].
- V-ATPase proton pump inhibitors handicap endothelial cell proliferation and migration with inhibition of VEGFR2 signaling and decreasing the amount of VEGFR2 at the cell surface [162]. Therefore, V-ATPase proton pump inhibitors are clearly anti-angiogenic. There is laboratory and clinical evidence on using proton pump inhibitors in cancer treatment [163,164,165,166,167]. Furthermore, there is evidence showing synergy between MC and proton pump inhibition [168].
2. Discussion
The short benefit of clinical anti-angiogenesis is an important limiting factor of these types of therapies. Acting only on the VEGF-VEGFR axis is insufficient for achieving lasting results because many escape doors remain open [169,170,171,172]. Initial successful results are lost as soon as these escape doors open. As Quesada et al. [173] clearly stated; playing only one instrument may not be enough in anti-angiogenesis. Resistance to anti-angiogenesis is not related to mutations or to multidrug resistance mechanisms. It is related to alternative angiogenic pathways [174]. These alternative pathways are targeted by the pHT approach.
There are a few pharmaceuticals that can be added to mainstream anti-angiogenesis, such as fenofibrate or riluzole. These drugs could impede the opening of some escape routes. However, we believe that the main cooperation for lasting anti-angiogenesis would come from alkalinizing the extracellular space, which is highly acidic in all malignancies. The best way to achieve extracellular alkalinization is through intracellular acidification. The benefits of extracellular buffers like sodium bicarbonate are short lived, while the partial inhibition of many participants of the pHtome, like proton extruders, is well tolerated by patients and has more lasting effects.
The incubation of human pancreatic cancer cells in an acidic medium increased the expression of IL-8. When cells with low IL-8 expression were implanted in the pancreases of mice they showed lower angiogenesis than those with higher IL-8 expression [175]. IL-8 is a pro-angiogenic cytokine and serves as an example of low pHe promoting angiogenesis.
Solving the acidity problem of the extracellular space with a pH-centered approach should have two different but converging effects on angiogenesis.
2.1. It Would Decrease the VEGF-VEGFR Axis Activity
However, this might seem a controversial issue because VEGF expression does not show a linear correlation with extracellular acidity. When a malignant cell was placed in a low pH medium, VEGF expression was increased during the first 6 h of incubation. Further incubation showed increased VEGF expression at pH 6.9 to 7.1 but a decrease at longer incubation times [146]. This, at prima facie, seems to be the opposite of what would be expected in cancer. In a second step of their investigation, the authors found that “the VEGF mRNA half-life of cells at a pH of 6.9 was much longer than that of cells at pH 7.4”. Thus, the determination of VEGF expression does not reflect what happens with VEGF’s activity that is increased at low extracellular pH. In other words, after the first six hours in which low pHe increased the synthesis of VGEF at transcriptional level, the low pHe increased the activity of VEGF at a post-transcriptional level. Raising extracellular pH level decreases VEGF activity at the transcriptional level first and at the post-transcriptional level afterwards (Figure 4).
2.2. It Would Decrease the Activity/Expression of Other Molecules Involved in Angiogenesis And in Anti-Angiogenic Escape
We have already mentioned the increased IL-8 expression with low pHe. The TRPV4 (transient receptor potential vanilloid 4; also known as vanilloid receptor) ion channel is another player in angiogenesis [176,177,178,179] that is activated by low pHe [180,181]. Acidosis also induces the expression and modulates the activity of SDF-1α in both normal [182,183] and in malignant cells [184]. Acidic preconditioning improves endothelial cells colony formation and angiogenesis [185]. Furthermore, Galectin-3 is up-regulated by acidosis [186].
The pHT approach decreases cellular migration by multiple mechanisms:
- (a)
- It increases extracellular pH, which decreases the activity of proteolytic cathepsins, necessary for matrix degradation.
- (b)
- It decreases in the same way metalloproteases maturation also necessary for matrix degradation.
- (c)
- It decreases invadopodia formation and activity.
- (d)
- It inhibits aquaporin 1 which is essential for migration.
These activities are not restricted to malignant cells but also affect tumor-associated endothelial cells, therefore pHT has a clear anti-angiogenic effect that acts in a different way than the VEGF-VEGFR pathway inhibition.
At high doses amiloride derivatives induce endothelial cell apoptosis in extracellular alkalosis [187]. Amiloride and its derivatives also inhibit endothelial production of MCP1 (monocyte chemoattractant protein 1) [188] that is a pro-angiogenic protein [189,190]
From the above evidence, we can see that pHT addresses many issues that are “left out” from classical anti-angiogenic treatments. The synergy between the two approaches is explained by the fact that pHT complements the anti-angiogenic treatments.
A third pillar has been proposed for this combined MC-pHT approach: targeting the immune system dysfunction. McDonald et al. [191] suggest the use of immune checkpoint inhibitors. These modern drugs that have been shown to be very useful in the treatment of many types of cancer are however very expensive and produce many side effects that are not tolerated by all patients.
The advantage of metronomic chemotherapy based on cyclophosphamide and cimetidine is that it is quite efficient in targeting immune dysfunction without all the side effects of immune checkpoint inhibitors and its exorbitant cost.
In this respect, Loges et al. [192] wrote about the need to develop third generation anti-angiogenic drugs taking into account the resistance problem. We consider that the pHT-MC association somehow, represents a third generation scheme of anti-angiogenesis.
Conclusions from Table 1: Low dose cyclophosphamide and low dose metronomic cyclophosphamide (50 mg per os, daily, with no interruptions) not only have anti-angiogenic effects but also enhance immunologic modulation against cancer by decreasing T regulatory cells and increasing T-helper lymphocytes and NK cells.
For detailed recent reviews on cyclophosphamide as an immune regulator in cancer, read Hughes et al. [202] and the phase I clinical trial of metronomic cyclophosphamide and everolimus by Huijts et al. [203].
Conclusions from Table 2: Cimetidine antagonizes histamine’s immuno-suppressive effects and has powerful stimulatory effects on blood cells such as neutrophils and monocytes, and also macrophages, dendritic cells, NK cells, T-helpers, and CD8+ cytotoxic T cells [214]. For a review of cimetidine’s immunological actions see Pantziarka et al. [215]. The usual dose of cimetidine is 400 mg three times a day.
While this association in a metronomic scheme restores immunological defenses and has anti-angiogenic activity, bevacizumab has not shown immuno-modulatory abilities. Therefore, the metronomic low dose cyclophosphamide plus cimetidine restores immunologic functions that makes immune checkpoint inhibitors unnecessary in a scheme of MC plus pHT.
3. Conclusions
Tumor angiogenesis is much more complex than originally thought. Although VEGF and VEGFR seem to be the main players, the limited bedside success achieved with their inhibition shows that there are many other players that must be targeted. Present day treatments do not address all the other participants in the process. The direct result of this biased view of anti-angiogenesis is resistance to treatment. On the other hand, resolving the pH paradigm of cancer seems to delay this phenomenon substantially. pH-centered treatment targeting channels, transporters, exchangers, and enzymes that form part of the pHtome seems to have an impact on anti-angiogenesis. Evidence, that still needs further confirmation, shows that pH centered treatments would be a good companion to classic anti-angiogenesis therapies. Riluzole, a potent inhibitor of the pro-angiogenic protein kinase C and antagonist of the also pro-angiogenic metabotronic glutamate receptor-1 on one side and voltage gated sodium channel inhibitors on the other, should also be incorporated into anti-angiogenic and pH centered treatments because this adds other three targets to the anti-angiogenic scheme. Alkalinization of the extracellular space that is achievable through pHT is a powerful enhancer of anti-angiogenic treatments and delays resistance.
Supplementary Materials
Supplementary materials can be found at https://0-www-mdpi-com.brum.beds.ac.uk/1422-0067/20/21/5438/s1.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest
References
- Burk, D.; Algire, G.H.; Hesselbach, M.L.; Fischer, C.E.; Legallais, F.Y. Characterization of tissue metabolism of transplanted mouse melanomas by high oxidative response to paraphenylenediamine. J. Natl. Cancer Inst. 1947, 7, 425–429. [Google Scholar] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Larrivée, B.; Freitas, C.; Suchting, S.; Brunet, I.; Eichmann, A. Guidance of vascular development: Lessons from the nervous system. Circ. Res. 2009, 104, 428–441. [Google Scholar] [CrossRef] [PubMed]
- Van Beijnum, J.R.; Nowak-Sliwinska, P.; Huijbers, E.J.; Thijssen, V.L.; Griffioen, A.W. The great escape; the hallmarks of resistance to antiangiogenic therapy. Pharmacol. Rev. 2015, 67, 441–461. [Google Scholar] [CrossRef]
- Donahoe, P.K. Judah Folkman: 1933–2008. A Biographical Memoir Nacional Academy of Sciences. 2014. Available online: http://www.nasonline.org/publications/biographical-memoirs/memoir-pdfs/folkman-judah.pdf (accessed on 30 October 2019).
- Ferrara, N.; Henzel, W.J. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989, 161, 851–858. [Google Scholar] [CrossRef]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef]
- Strydom, D.J.; Fett, J.W.; Lobb, R.R.; Alderman, E.M.; Bethune, J.L.; Riordan, J.F.; Vallee, B.L. Amino acid sequence of human tumor derived angiogenin. Biochemistry 1985, 24, 5486–5494. [Google Scholar] [CrossRef]
- Ingber, D.; Fujita, T.; Kishimoto, S.; Sudo, K.; Kanamaru, T.; Brem, H.; Folkman, J. Synthetic analogues of fumagillin that inhibit angiogenesis and suppress tumour growth. Nature 1990, 348, 555–557. [Google Scholar] [CrossRef]
- Kaur, S.; Martin-Manso, G.; Pendrak, M.L.; Garfield, S.H.; Isenberg, J.S.; Roberts, D.D. Thrombospondin-1 inhibits VEGF receptor-2 signaling by disrupting its association with CD47. J. Biol. Chem. 2010, 285, 38923–38932. [Google Scholar] [CrossRef]
- Greenaway, J.; Lawler, J.; Moorehead, R.; Bornstein, P.; Lamarre, J.; Petrik, J. Thrombospondin-1 inhibits VEGF levels in the ovary directly by binding and internalization via the low density lipoprotein receptor-related protein-1 (LRP-1). J. Cell. Physiol. 2007, 210, 807–818. [Google Scholar] [CrossRef]
- Chu, L.Y.; Ramakrishnan, D.P.; Silverstein, R.L. Thrombospondin-1 modulates VEGF signaling via CD36 by recruiting SHP-1 to VEGFR2 complex in microvascular endothelial cells. Blood 2013, 122, 1822–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanda, S.; Shono, T.; Tomasini-Johansson, B.; Klint, P.; Saito, Y. Role of thrombospondin-1-derived peptide, 4N1K, in FGF-2-induced angiogenesis. Exp. Cell Res. 1999, 252, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Bagavandoss, P.; Wilks, J.W. Specific inhibition of endothelial cell proliferation by thrombospondin. Biochem. Biophys. Res. Commun. 1990, 170, 867–872. [Google Scholar] [CrossRef]
- Colombo, G.; Margosio, B.; Ragona, L.; Neves, M.; Bonifacio, S.; Annis, D.S.; Presta, M. Non-peptidic thrombospondin-1 mimics as fibroblast growth factor-2 inhibitors an integrated strategy for the development of new antiangiogenic compounds. J. Biol. Chem. 2010, 285, 8733–8742. [Google Scholar] [CrossRef]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Orf, J. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef] [PubMed]
- Heffelfinger, S.C.; Yan, M.; Gear, R.B.; Schneider, J.; LaDow, K.; Warshawsky, D. Inhibition of VEGFR2 prevents DMBA-induced mammary tumor formation. Lab. Investig. 2004, 84, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Dias, S.; Hattori, K.; Heissig, B.; Zhu, Z.; Wu, Y.; Witte, L.; Rafii, S. Inhibition of both paracrine and autocrine VEGF/VEGFR-2 signaling pathways is essential to induce long-term remission of xenotransplanted human leukemias. Proc. Natl. Acad. Sci. USA 2001, 98, 10857–10862. [Google Scholar] [CrossRef]
- Yue, T.L.; Wang, X.; Louden, C.S.; Gupta, S.; Pillarisetti, K.; Gu, J.L.; Feuerstein, G.Z. 2-Methoxyestradiol, an endogenous estrogen metabolite, induces apoptosis in endothelial cells and inhibits angiogenesis: Possible role for stress-activated protein kinase signaling pathway and Fas expression. Mol. Pharmacol. 1997, 51, 951–962. [Google Scholar] [CrossRef]
- Masiero, L.; Figg, W.D.; Kohn, E.C. New anti-angiogenesis agents: Review of the clinical experience with carboxyamido-triazole (CAI), thalidomide, TNP-470 and interleukin-12. Angiogenesis 1997, 1, 23–35. [Google Scholar] [CrossRef]
- Kerbel, R.S.; Kamen, B.A. The anti-angiogenic basis of metronomic chemotherapy. Nat. Rev. Cancer 2004, 4, 423–436. [Google Scholar] [CrossRef]
- Ribatti, D.; Nico, B. Limits of anti-angiogenic therapy. Res. Clin. Med. 2016, 1, 2. [Google Scholar]
- Mancuso, M.R.; Davis, R.; Norberg, S.M.; O’Brien, S.; Sennino, B.; Nakahara, T.; Shalinsky, D.R. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J. Clin. Investig. 2006, 116, 2610–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gacche, R.N.; Meshram, R.J. Angiogenic factors as potential drug target: Efficacy and limitations of anti-angiogenic therapy. Biochim. Biophys. Acta (BBA) Rev. Cancer 2014, 1846, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Casanovas, O.; Hicklin, D.J.; Bergers, G.; Hanahan, D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 2005, 8, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Kindler, H.L.; Niedzwiecki, D.; Hollis, D.; Oraefo, E.; Schrag, D.; Hurwitz, H.; Goldberg, R.M. A double-blind, placebo-controlled, randomized phase III trial of gemcitabine (G) plus bevacizumab (B) versus gemcitabine plus placebo (P) in patients (pts) with advanced pancreatic cancer (PC): A preliminary analysis of Cancer and Leukemia Group B CALGB. J. Clin. Oncol. 2007, 25 (Suppl. 18), 4508. [Google Scholar]
- Azam, F.; Mehta, S.; Harris, A.L. Mechanisms of resistance to antiangiogenesis therapy. Eur. J. Cancer 2010, 46, 1323–1332. [Google Scholar] [CrossRef]
- Bergers, G.; Hanahan, D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Kerbel, R.S. A cancer therapy resistant to resistance. Nature 1997, 390, 335–336. [Google Scholar] [CrossRef]
- Croix, B.S.; Rago, C.; Velculescu, V.; Traverso, G.; Romans, K.E.; Montgomery, E.; Lal, A.; Riggins, G.J.; Lengauer, C.; Kinzler, K.W.; et al. Genes expressed in human tumor endothelium. Science 2000, 289, 1197–1202. [Google Scholar] [CrossRef]
- Lu, C.; Bonome, T.; Li, Y.; Kamat, A.A.; Han, L.Y.; Schmandt, R.; Coleman, R.L.; Gershenson, D.M.; Jaffe, R.B.; Sood, A.K.; et al. Gene alterations identified by expression profiling in tumor-associated endothelial cells from invasive ovarian carcinoma. Cancer Res. 2007, 67, 1757–1768. [Google Scholar] [CrossRef]
- Akino, T.; Hida, K.; Hida, Y.; Tsuchiya, K.; Freedman, D.; Muraki, C.; Ohga, N.; Matsuda, K.; Akiyama, K.; Shinohara, N.; et al. Cytogenetic abnormalities of tumor-associated endothelial cells in human malignant tumors. Am. J. Pathol. 2009, 175, 2657–2667. [Google Scholar] [CrossRef] [PubMed]
- Virrey, J.J.; Dong, D.; Stiles, C.; Patterson, J.B.; Pen, L. Stress chaperone GRP78/BiP confers chemoresistance to tumor-associated endothelial cells. Mol. Cancer 2008, 6, 1268–1275. [Google Scholar] [CrossRef] [PubMed]
- Virrey, J.J.; Guan, S.; Li, W.; Schönthal, A.H.; Chen, T.C.; Hofman, F.M. Increased survivin expression confers chemoresistance to tumor-associated endothelial cells. Am. J. Pathol. 2008, 173, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.H.; Rivera, C.G.; Popel, A.S.; Bader, J.S. Constructing the angiome: A global angiogenesis protein interaction network. Physiol. Genom. 2012, 44, 915–924. [Google Scholar] [CrossRef]
- Chu, L.H.; Lee, E.; Bader, J.S.; Popel, A.S. Angiogenesis interactome and time course microarray data reveal the distinct activation patterns in endothelial cells. PLoS ONE 2014, 9, e110871. [Google Scholar] [CrossRef]
- Rivera, C.G.; Chu, L.H.; Bader, J.S.; Popel, A.S. Applications of network bioinformatics to cancer angiogenesis. In Systems Biology in Cancer Research and Drug Discovery; Springer: Dordrecht, The Netherlands, 2012; pp. 229–244. [Google Scholar]
- Folkman, J.; Klagsbrun, M. Angiogenic factors. Science 1987, 235, 442–447. [Google Scholar] [CrossRef]
- Pepper, M.S.; Ferrara, N.; Orci, L.; Montesano, R. Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochem. Biophys. Res. Commun. 1992, 189, 824–831. [Google Scholar] [CrossRef]
- Moroianu, J.; Riordan, J.F. Nuclear translocation of angiogenin in proliferating endothelial cells is essential to its angiogenic activity. Proc. Natl. Acad. Sci. USA 1994, 91, 1677–1681. [Google Scholar] [CrossRef]
- Schreiber, A.B.; Winkler, M.E.; Derynck, R. Transforming growth factor-alpha: A more potent angiogenic mediator than epidermal growth factor. Science 1986, 232, 1250–1253. [Google Scholar] [CrossRef]
- Roberts, A.B.; Sporn, M.B.; Assoian, R.K.; Smith, J.M.; Roche, N.S.; Wakefield, L.M.; Kehrl, J.H. Transforming growth factor type beta: Rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 4167–4171. [Google Scholar] [CrossRef]
- Wiseman, D.M.; Polverini, P.J.; Kamp, D.W.; Leibovich, S.J. Transforming growth factor-beta (TGFβ) is chemotactic for human monocytes and induces their expression of angiogenic activity. Biochem. Biophys. Res. Commun. 1988, 157, 793–800. [Google Scholar] [CrossRef]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Semenza, G.L. Modulation of hypoxia-inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 60, 1541–1545. [Google Scholar] [PubMed]
- Petit, A.M.; Rak, J.; Hung, M.C.; Rockwell, P.; Goldstein, N.; Fendly, B.; Kerbel, R.S. Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases downregulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: Angiogenic implications for signal transduction therapy of solid tumors. Am. J. Pathol. 1997, 151, 1523–1530. [Google Scholar] [PubMed]
- Perrotte, P.; Matsumoto, T.; Inoue, K.; Kuniyasu, H.; Eve, B.Y.; Hicklin, D.J.; Dinney, C.P. Anti-epidermal growth factor receptor antibody C225 inhibits angiogenesis in human transitional cell carcinoma growing orthotopically in nude mice. Clin. Cancer Res. 1999, 5, 257–264. [Google Scholar] [PubMed]
- Huang, S.M.; Harari, P.M. Modulation of radiation response after epidermal growth factor receptor blockade in squamous cell carcinomas: Inhibition of damage repair, cell cycle kinetics, and tumor angiogenesis. Clin. Cancer Res. 2000, 6, 2166–2174. [Google Scholar]
- Viloria-Petit, A.; Crombet, T.; Jothy, S.; Hicklin, D.; Bohlen, P.; Schlaeppi, J.M.; Kerbel, R.S. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: A role for altered tumor angiogenesis. Cancer Res. 2001, 61, 5090–5101. [Google Scholar]
- Gospodarowicz, D.; Bialecki, H.; Thakral, T.K. The angiogenic activity of the fibroblast and epidermal growth factor. Exp. Eye Res. 1979, 28, 501–514. [Google Scholar] [CrossRef]
- Relf, M.; LeJeune, S.; Scott, P.A.; Fox, S.; Smith, K.; Leek, R.; Harris, A.L. Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor β-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res. 1997, 57, 963–969. [Google Scholar]
- Risau, W.; Drexler, H.; Mironov, V.; Smits, A.; Siegbahn, A.; Funa, K.; Heldin, C.H. Platelet-derived growth factor is angiogenic in vivo. Growth Factors 1992, 7, 261–266. [Google Scholar] [CrossRef]
- Sato, N.; Beitz, J.G.; Kato, J.; Yamamoto, M.; Clark, J.W.; Calabresi, P.; Raymond, A.; Frackelton, A.R., Jr. Platelet-derived growth factor indirectly stimulates angiogenesis in vitro. Am. J. Pathol. 1993, 142, 1119–1130. [Google Scholar]
- Ziche, M.; Maglione, D.; Ribatti, D.; Morbidelli, L.; Lago, C.T.; Battisti, M.; Vincenti, V. Placenta growth factor-1 is chemotactic, mitogenic, and angiogenic. Lab. Investig. A J. Tech. Methods Pathol. 1997, 76, 517–531. [Google Scholar]
- Zhang, Y.W.; Su, Y.; Volpert, O.V.; Woude, G.F.V. Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 12718–12723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussolino, F.; di Renzo, M.F.; Ziche, M.; Bocchietto, E.; Olivero, M.; Naldini, L.; Gaudino, G.; Tamagnone, L.; Coffer, A.; Comoglio, P.M. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J. Cell Biol. 1992, 119, 629–641. [Google Scholar] [CrossRef]
- Grant, M.B.; Mames, R.N.; Fitzgerald, C.; Ellis, E.A.; Aboufriekha, M.; Guy, J. Insulin-like growth factor I acts as an angiogenic agent in rabbit cornea and retina: Comparative studies with basic fibroblast growth factor. Diabetologia 1993, 36, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Calza, L.; Giardino, L.; Giuliani, A.; Aloe, L.; Levi-Montalcini, R. Nerve growth factor control of neuronal expression of angiogenetic and vasoactive factors. Proc. Natl. Acad. Sci. USA 2001, 98, 4160–4165. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Hui, A.M.; Su, Q.; Vortmeyer, A.; Kotliarov, Y.; Pastorino, S.; Rosenblum, M. Neuronal and glioma-derived stem cell factor induces angiogenesis within the brain. Cancer Cell 2006, 9, 287–300. [Google Scholar] [CrossRef] [Green Version]
- Kerbel, R.S. Therapeutic implications of intrinsic or induced angiogenic growth factor redundancy in tumors revealed. Cancer Cell 2005, 8, 269–271. [Google Scholar] [CrossRef] [Green Version]
- Ebos, J.M.; Lee, C.R.; Christensen, J.G.; Mutsaers, A.J.; Kerbel, R.S. Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy. Proc. Natl. Acad. Sci. USA 2007, 104, 17069–17074. [Google Scholar] [CrossRef] [Green Version]
- Ebos, J.M.; Lee, C.R.; Cruz-Munoz, W.; Bjarnason, G.A.; Christensen, J.G.; Kerbel, R.S. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009, 15, 232–239. [Google Scholar] [CrossRef]
- Mizukami, Y.; Jo, W.S.; Duerr, E.M.; Gala, M.; Li, J.; Zhang, X.; Lynch, M.P. Induction of interleukin-8 preserves the angiogenic response in HIF-1α–deficient colon cancer cells. Nat. Med. 2005, 11, 992–997. [Google Scholar] [CrossRef]
- Koch, A.E.; Polverini, P.J.; Kunkel, S.L.; Harlow, L.A.; DiPietro, L.A.; Elner, V.M.; Strieter, R.M. Interleukin-8 as a macrophage-derived mediator of angiogenesis. Science 1992, 258, 1798–1801. [Google Scholar] [CrossRef] [PubMed]
- Deshane, J.; Chen, S.; Caballero, S.; Grochot-Przeczek, A.; Was, H.; Calzi, S.L.; Siegal, G.P. Stromal cell–derived factor 1 promotes angiogenesis via a heme oxygenase 1–dependent mechanism. J. Exp. Med. 2007, 204, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Laiva, A.L.; Raftery, R.M.; Keogh, M.B.; O’brien, F.J. Pro-angiogenic impact of SDF-1α gene-activated collagen-based scaffolds in stem cell driven angiogenesis. Int. J. Pharm. 2018, 544, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Xue, F.; Guan, J.; Zhang, Z.; Yin, J.; Kang, Q. Stromal-cell-derived factor (SDF) 1-alpha overexpression promotes bone regeneration by osteogenesis and angiogenesis in osteonecrosis of the femoral head. Cell. Physiol. Biochem. 2018, 46, 2561–2575. [Google Scholar] [CrossRef]
- Strieter, R.M.; Polverini, P.J.; Kunkel, S.L.; Arenberg, D.A.; Burdick, M.D.; Kasper, J.; Chan, S.Y. The functional role of the ELR motif in CXC chemokine-mediated angiogenesis. J. Biol. Chem. 1995, 270, 27348–27357. [Google Scholar] [CrossRef]
- Babic, A.M.; Kireeva, M.L.; Kolesnikova, T.V.; Lau, L.F. CYR61, a product of a growth factor-inducible immediate early gene, promotes angiogenesis and tumor growth. Proc. Natl. Acad. Sci. USA 1998, 95, 6355–6360. [Google Scholar] [CrossRef] [Green Version]
- Niu, J.; Azfer, A.; Zhelyabovska, O.; Fatma, S.; Kolattukudy, P.E. Monocyte chemotactic protein (MCP)-1 promotes angiogenesis via a novel transcription factor, MCP-1-induced protein (MCPIP). J. Biol. Chem. 2008, 283, 14542–14551. [Google Scholar] [CrossRef]
- Ergün, S.; Kilic, N.; Ziegeler, G.; Hansen, A.; Nollau, P.; Götze, J.; Wagener, C. CEA-related cell adhesion molecule 1: A potent angiogenic factor and a major effector of vascular endothelial growth factor. Mol. Cell 2000, 5, 311–320. [Google Scholar] [CrossRef]
- Leibovich, S.J.; Polverini, P.J.; Shepard, H.M.; Wiseman, D.M.; Shively, V.; Nuseir, N. Macrophage-induced angiogenesis is mediated by tumour necrosis factor-α. Nature 1987, 329, 630–632. [Google Scholar] [CrossRef]
- Kermani, P.; Hempstead, B. Brain-derived neurotrophic factor: A newly described mediator of angiogenesis. Trends Cardiovasc. Med. 2007, 17, 140–143. [Google Scholar] [CrossRef]
- Ishikawa, F.; Miyazono, K.; Hellman, U.; Drexler, H.; Wernstedt, C.; Hagiwara, K.; Heldin, C.H. Identification of angiogenic activity and the cloning and expression of platelet-derived endothelial cell growth factor. Nature 1989, 338, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Raida, M.; Clement, J.H.; Leek, R.D.; Ameri, K.; Bicknell, R.; Niederwieser, D.; Harris, A.L. Bone morphogenetic protein 2 (BMP-2) and induction of tumor angiogenesis. J. Cancer Res. Clin. Oncol. 2005, 131, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.W.; Liang, X.; Lipsky, S.; Karaaslan, C.; Kozakewich, H.; Hotamisligil, G.S.; Bischoff, J.; Cataltepe, S. Dual role of fatty acid-binding protein 5 on endothelial cell fate: A potential link between lipid metabolism and angiogenic responses. Angiogenesis 2016, 19, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Grabell, D.A.; Wen, Y.; Semlani, N.; Li, J.; Goydos, J. Pre-clinical study targeting metabotropic glutamate receptor (GRM1) angiogenesis pathway. Cancer Res. 2013. [Google Scholar] [CrossRef]
- Speyer, C.L.; Hachem, A.H.; Assi, A.A.; Johnson, J.S.; DeVries, J.A.; Gorski, D.H. Metabotropic glutamate receptor-1 as a novel target for the antiangiogenic treatment of breast cancer. PLoS ONE 2014, 9, e88830. [Google Scholar] [CrossRef]
- Makita, T.; Sucov, H.M.; Gariepy, C.E.; Yanagisawa, M.; Ginty, D.D. Endothelins are vascular-derived axonal guidance cues for developing sympathetic neurons. Nature 2008, 452, 759–763. [Google Scholar] [CrossRef] [Green Version]
- Salani, D.; Taraboletti, G.; Rosano, L.; Di Castro, V.; Borsotti, P.; Giavazzi, R.; Bagnato, A. Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am. J. Pathol. 2000, 157, 1703–1711. [Google Scholar] [CrossRef]
- Bek, E.L.; McMillen, M.A. Endothelins are angiogenic. J. Cardiovasc. Pharmacol. 2000, 36, S135–S139. [Google Scholar] [CrossRef]
- Bagnato, A.; Spinella, F. Emerging role of endothelin-1 in tumor angiogenesis. Trends Endocrinol. Metab. 2003, 14, 44–50. [Google Scholar] [CrossRef]
- Wülfing, P.; Kersting, C.; Tio, J.; Fischer, R.J.; Wülfing, C.; Poremba, C.; Kiesel, L. Endothelin-1-, endothelin-A-, and endothelin-B-receptor expression is correlated with vascular endothelial growth factor expression and angiogenesis in breast cancer. Clin. Cancer Res. 2004, 10, 2393–2400. [Google Scholar] [CrossRef]
- Knowles, J.; Loizidou, M.; Taylor, I. Endothelin-1 and angiogenesis in cancer. Curr. Vasc. Pharmacol. 2005, 3, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Ratna, A.; Das, S.K. Endothelin: Ominous player in breast cancer. J. Cancer Clin. Trials 2016, 1, e102. [Google Scholar] [PubMed]
- Zhang, Z.Y.; Chen, L.L.; Xu, W.; Sigdel, K.; Jiang, X.T. Effects of silencing endothelin-1 on invasion and vascular formation in lung cancer. Oncol. Lett. 2017, 13, 4390–4396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Struman, I.; Bentzien, F.; Lee, H.; Mainfroid, V.; D’Angelo, G.; Goffin, V.; Martial, J.A. Opposing actions of intact and N-terminal fragments of the human prolactin/growth hormone family members on angiogenesis: An efficient mechanism for the regulation of angiogenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 1246–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajou, K.; Herkenne, S.; Thijssen, V.L.; D’amico, S.; Bouché, A.; Tabruyn, S.; Cornelissen, I. PAI-1 mediates the antiangiogenic and profibrinolytic effects of 16K prolactin. Nat. Med. 2014, 20, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, V.L.; Postel, R.; Brandwijk, R.J.; Dings, R.P.; Nesmelova, I.; Satijn, S.; Mayo, K.H. Galectin-1 is essential in tumor angiogenesis and is a target for antiangiogenesis therapy. Proc. Natl. Acad. Sci. USA 2006, 103, 15975–15980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nangia-Makker, P.; Honjo, Y.; Sarvis, R.; Akahani, S.; Hogan, V.; Pienta, K.J.; Raz, A. Galectin-3 induces endothelial cell morphogenesis and angiogenesis. Am. J. Pathol. 2000, 156, 899–909. [Google Scholar] [CrossRef]
- Newlaczyl, A.U.; Yu, L.G. Galectin-3–a jack-of-all-trades in cancer. Cancer Lett. 2011, 313, 123–128. [Google Scholar] [CrossRef]
- Markowska, A.I.; Liu, F.T.; Panjwani, N. Galectin-3 is an important mediator of VEGF-and bFGF-mediated angiogenic response. J. Exp. Med. 2010, 207, 1981–1993. [Google Scholar] [CrossRef]
- Thijssen, V.L.; Barkan, B.; Shoji, H.; Aries, I.M.; Mathieu, V.; Deltour, L.; Hackeng, T.M.; Kiss, R.; Kloog, Y.; Griffioen, A.W.; et al. Tumor cells secrete galectin-1 to enhance endothelial cell activity. Cancer Res. 2010, 70, 6216–6224. [Google Scholar] [CrossRef]
- Griffioen, A.W.; van der Schaft, D.W.; Barendsz-Janson, A.F.; Andrew, C.O.X.; Boudier, H.A.S.; Hillen, H.F. Anginex, a designed peptide that inhibits angiogenesis. Biochem. J. 2001, 354, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Salomonsson, E.; Thijssen, V.L.; Griffioen, A.W.; Nilsson, U.J.; Leffler, H. The anti-angiogenic peptide anginex greatly enhances galectin-1 binding affinity for glycoproteins. J. Biol. Chem. 2011, 286, 13801–13804. [Google Scholar] [CrossRef] [PubMed]
- Duff, S.E.; Li, C.; Garland, J.M.; Kumar, S. CD105 is important for angiogenesis: Evidence and potential applications. FASEB J. 2003, 17, 984–992. [Google Scholar] [CrossRef] [PubMed]
- Panigrahy, D.; Kaipainen, A.; Huang, S.; Butterfield, C.E.; Barnés, C.M.; Fannon, M.; Kieran, M.W. PPARα agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc. Natl. Acad. Sci. USA 2008, 105, 985–990. [Google Scholar] [CrossRef]
- Drukala, J.; Urbanska, K.; Wilk, A.; Grabacka, M.; Wybieralska, E.; del Valle, L.; Reiss, K. ROS accumulation and IGF-IR inhibition contribute to fenofibrate/PPARα-mediated inhibition of glioma cell motility in vitro. Mol. Cancer 2010, 9, 159. [Google Scholar] [CrossRef]
- Lian, X.; Gu, J.; Gao, B.; Li, Y.; Damodaran, C.; Wei, W.; Cai, L. Fenofibrate inhibits mTOR-p70S6K signaling and simultaneously induces cell death in human prostate cancer cells. Biochem. Biophys. Res. Commun. 2018, 496, 70–75. [Google Scholar] [CrossRef]
- Han, D.; Wei, W.; Chen, X.; Zhang, Y.; Wang, Y.; Zhang, J.; You, Y. NF-κB/RelA-PKM2 mediates inhibition of glycolysis by fenofibrate in glioblastoma cells. Oncotarget 2015, 6, 26119–26128. [Google Scholar] [CrossRef]
- Madej, A.; Okopien, B.; Kowalski, J.; Zielinski, M.; Wysocki, J.; Szygula, B.; Herman, Z.S. Effects of fenofibrate on plasma cytokine concentrations in patients with atherosclerosis and hyperlipoproteinemia IIb. Int. J. Clin. Pharmacol. Ther. 1998, 36, 345–349. [Google Scholar]
- Zak, Z.; Gelebart, P.; Lai, R. Fenofibrate induces effective apoptosis in mantle cell lymphoma by inhibiting the TNFα/NF-κB signaling axis. Leukemia 2010, 24, 1476–1486. [Google Scholar] [CrossRef]
- Ge, Y.; Liu, J.; Yang, X.; Zhu, H.; Yang, B.; Zhao, K.; Ge, Q. Fenofibrate enhances radiosensitivity of esophageal squamous cell carcinoma by suppressing hypoxia-inducible factor-1α expression. Tumor Biol. 2014, 35, 10765–10771. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, S.; Xue, J.; Avery, J.; Wu, J.; Lind, S.E.; Ding, W.Q. Activation of peroxisome proliferator-activated receptor α (PPARα) suppresses hypoxia-inducible factor-1α (HIF-1α) signaling in cancer cells. J. Biol. Chem. 2012, 287, 35161–35169. [Google Scholar] [CrossRef] [PubMed]
- Avis, I.; Hong, S.H.; Martínez, A.; Moody, T.; Choi, Y.H.; Trepel, J.; Mulshine, J.L. Five-lipoxygenase inhibitors can mediate apoptosis in human breast cancer cell lines through complex eicosanoid interactions. FASEB J. 2001, 15, 2007–2009. [Google Scholar] [CrossRef] [PubMed]
- Tsimihodimos, V.; Kakafika, A.; Tambaki, A.P.; Bairaktari, E.; Chapman, M.J.; Elisaf, M.; Tselepis, A.D. Fenofibrate induces HDL-associated PAF-AH but attenuates enzyme activity associated with apoB-containing lipoproteins. J. Lipid Res. 2003, 44, 927–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delerive, P.; de Bosscher, K.; Besnard, S.; Berghe, W.V.; Peters, J.M.; Gonzalez, F.J.; Staels, B. Peroxisome proliferator-activated receptor α negatively regulates the vascular inflammatory gene response by negative cross-talk with transcription factors NF-κB and AP-1. J. Biol. Chem. 1999, 274, 32048–32054. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, L.; Takemura, T.; Eriksson, P.; Hamsten, A. Effects of fibrate compounds on expression of plasminogen activator inhibitor-1 by cultured endothelial cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Look, M.P.; van Putten, W.L.; Duffy, M.J.; Harbeck, N.; Christensen, I.J.; Thomssen, C.; Sweep, C.F. Pooled analysis of prognostic impact of urokinase-type plasminogen activator and its inhibitor PAI-1 in 8377 breast cancer patients. J. Natl. Cancer Inst. 2002, 94, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.; Grøndahl-Hansen, J.; Francis, D.; Østerlind, K.; Hansen, H.H.; Danø, K.; Brünner, N. Urokinase and plasminogen activator inhibitor type 1 in pulmonary adenocarcinoma. Cancer Res. 1994, 54, 120–123. [Google Scholar]
- Zhang, K.L.; Han, L.; Chen, L.Y.; Shi, Z.D.; Yang, M.; Ren, Y.; Kang, C.S. Blockage of a miR-21/EGFR regulatory feedback loop augments anti-EGFR therapy in glioblastomas. Cancer Lett. 2014, 342, 139–149. [Google Scholar] [CrossRef]
- Chang, N.W.; Tsai, M.H.; Lin, C.; Hsu, H.T.; Chu, P.Y.; Yeh, C.M.; Yeh, K.T. Fenofibrate exhibits a high potential to suppress the formation of squamous cell carcinoma in an oral-specific 4-nitroquinoline 1-oxide/arecoline mouse model. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2011, 1812, 558–564. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Su, C.; Jiang, M.; Shen, Y.; Shi, A.; Zhao, F.; Tang, W. Fenofibrate inhibited pancreatic cancer cells proliferation via activation of p53 mediated by upregulation of LncRNA MEG3. Biochem. Biophys. Res. Commun. 2016, 471, 290–295. [Google Scholar] [CrossRef]
- Koltai, T. Fenofibrate in cancer: Mechanisms involved in anticancer activity. F1000Research 2015, 4, 55. [Google Scholar] [CrossRef]
- Gerson, D.F.; Kiefer, H. High intracellular pH accompanies mitotic activity in murine lymphocytes. J. Cell. Physiol. 1982, 112, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Gerson, D.F.; Burton, A.C. The relation of cycling of intracellular pH to mitosis in the acellular slime mould Physarum polycephalum. J. Cell. Physiol. 1977, 91, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Madshus, I.H. Regulation of intracellular pH in eukaryotic cells. Biochem. J. 1988, 250, 1. [Google Scholar] [CrossRef] [PubMed]
- Busa, W.B.; Nuccitelli, R. Metabolic regulation via intracellular pH. Am. J. Physiol. Regul. Integr. Comp. Physiol. 1984, 246, R409–R438. [Google Scholar] [CrossRef] [Green Version]
- Putney, L.K.; Barber, D.L. Na-H exchange-dependent increase in intracellular pH times G2/M entry and transition. J. Biol. Chem. 2003, 278, 44645–44649. [Google Scholar] [CrossRef]
- Gagliardi, L.J.; Shain, D.H. Is intracellular pH a clock for mitosis? Theor. Biol. Med Model. 2013, 10, 8. [Google Scholar] [CrossRef]
- Schatten, G.; Bestor, T.; Balczon, R.; Henson, J.; Schatten, H. Intracellular pH shift leads to microtubule assembly and microtubule-mediated motility during sea urchin fertilization: Correlations between elevated intracellular pH and microtubule activity and depressed intracellular pH and microtubule disassembly. Eur. J. Cell Biol. 1985, 36, 116–127. [Google Scholar]
- Reshkin, S.J.; Bellizzi, A.; Caldeira, S.; Albarani, V.; Malanchi, I.; Poignee, M.; Tommasino, M. Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypes. FASEB J. 2000, 14, 2185–2197. [Google Scholar] [CrossRef]
- Dhup, S.; Kumar Dadhich, R.; Ettore Porporato, P.; Sonveaux, P. Multiple biological activities of lactic acid in cancer: Influences on tumor growth, angiogenesis and metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef]
- Brisson, L.; Reshkin, S.J.; Gore, J.; Roger, S. pH regulators in invadosomal functioning: Proton delivery for matrix tasting. Eur. J. Cell Biol. 2012, 91, 847–860. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gawlinski, E.T.; Gmitro, A.F.; Kaylor, B.; Gillies, R.J. Acid-mediated tumor invasion: A multidisciplinary study. Cancer Res. 2006, 66, 5216–5223. [Google Scholar] [CrossRef] [PubMed]
- Cardone, R.A.; Casavola, V.; Reshkin, S.J. The role of disturbed pH dynamics and the Na+/H+ exchanger in metastasis. Nat. Rev. Cancer 2005, 5, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Harguindey, S.; Arranz, J.L.; Orozco, J.D.P.; Rauch, C.; Fais, S.; Cardone, R.A.; Reshkin, S.J. Cariporide and other new and powerful NHE1 inhibitors as potentially selective anticancer drugs—An integral molecular/biochemical/metabolic/clinical approach after one hundred years of cancer research. J. Transl. Med. 2013, 11, 282. [Google Scholar] [CrossRef]
- Vaupel, P.; Mayer, A. Hypoxia in cancer: Significance and impact on clinical outcome. Cancer Metastasis Rev. 2007, 26, 225–239. [Google Scholar] [CrossRef]
- Webb, B.A.; Chimenti, M.; Jacobson, M.P.; Barber, D.L. Dysregulated pH: A perfect storm for cancer progression. Nat. Rev. Cancer 2011, 11, 671–677. [Google Scholar] [CrossRef]
- Fais, S.; de Milito, A.; You, H.; Qin, W. Targeting vacuolar H+-ATPases as a new strategy against cancer. Cancer Res. 2007, 67, 10627–10630. [Google Scholar] [CrossRef]
- Spugnini, E.P.; Sonveaux, P.; Stock, C.; Perez-Sayans, M.; de Milito, A.; Avnet, S.; Fais, S. Proton channels and exchangers in cancer. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1848, 2715–2726. [Google Scholar] [CrossRef] [Green Version]
- Spugnini, E.P.; Citro, G.; Fais, S. Proton pump inhibitors as anti-vacuolar-ATPases drugs: A novel anticancer strategy. J. Exp. Clin. Cancer Res. 2010, 29, 44. [Google Scholar] [CrossRef]
- Xiang, Y.; Ma, B.; Li, T.; Gao, J.W.; Yu, H.M.; Li, X.J. Acetazolamide inhibits aquaporin-1 protein expression and angiogenesis. Acta Pharmacol. Sin. 2004, 25, 812–816. [Google Scholar]
- Ran, X.; Wang, H.; Chen, Y.; Zeng, Z.; Zhou, Q.; Zheng, R.; Sun, J.; Wang, B.; Lv, X.; Zhang, K. Aquaporin-1 expression and angiogenesis in rabbit chronic myocardial ischemia is decreased by acetazolamide. Heart Vessel. 2010, 25, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Nico, B.; Ribatti, D. Aquaporins in tumor growth and angiogenesis. Cancer Lett. 2010, 294, 135–138. [Google Scholar] [CrossRef]
- Ma, B.; Pan, Y.; Song, Q.; Tie, L.; Zhang, Y.; Xiao, Y.; Yu, H.M. The effect of topiramate on tumor-related angiogenesis and on the serum proteome of mice bearing Lewis lung carcinoma. Eur. J. Pharmacol. 2011, 663, 9–16. [Google Scholar] [CrossRef]
- Bing, M.A.; Yang, X.; Li, T.; Yu, H.M.; Li, X.J. Inhibitory effect of topiramate on Lewis lung carcinoma metastasis and its relation with AQP1 water channel. Acta Pharmacol. Sin. 2004, 25, 54–60. [Google Scholar]
- Xu, G.; Fang, Z.; Clark, L.H.; Sun, W.; Yin, Y.; Zhang, R.; Zhou, C. Topiramate exhibits anti-tumorigenic and metastatic effects in ovarian cancer cells. Am. J. Transl. Res. 2018, 10, 1663–1676. [Google Scholar] [PubMed]
- Yepuri, G.; Sukhovershin, R.; Nazari-Shafti, T.Z.; Petrascheck, M.; Ghebre, Y.T.; Cooke, J.P. Proton pump inhibitors accelerate endothelial senescence. Circ. Res. 2016, 118, e36–e42. [Google Scholar] [CrossRef] [PubMed]
- Alliegro, M.C.; Alliegro, M.A.; Cragoe, E.J.; Glaser, B.M. Amiloride inhibition of angiogenesis in vitro. J. Exp. Zool. 1993, 267, 245–252. [Google Scholar] [CrossRef]
- Avery, R.L.; Connor, T.B.; Farazdaghi, M. Systemic amiloride inhibits experimentally induced neovascularization. Arch. Ophthalmol. 1990, 108, 1474–1476. [Google Scholar] [CrossRef] [PubMed]
- Ignjatović, Z.; Nikolić, L. Inhibition of angiogenesis in the cornea with amiloride. Srp. Arh. Celok. Lek. 1996, 124, 120–123. [Google Scholar] [PubMed]
- Evans, D.M.; Sloan-Stakleff, K.; Arvan, M.; Guyton, D.P. Time and dose dependency of the suppression of pulmonary metastases of rat mammary cancer by amiloride. Clin. Exp. Metastasis 1998, 16, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Borentain, P.; Carmona, S.; Mathieu, S.; Jouve, E.; El-Battari, A.; Gérolami, R. Inhibition of E-selectin expression on the surface of endothelial cells inhibits hepatocellular carcinoma growth by preventing tumor angiogenesis. Cancer Chemother. Pharmacol. 2016, 77, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Igura, K.; Ohta, T.; Kuroda, Y.; Kaji, K. Resveratrol and quercetin inhibit angiogenesis in vitro. Cancer Lett. 2001, 171, 11–16. [Google Scholar] [CrossRef]
- Pratheeshkumar, P.; Budhraja, A.; Son, Y.O.; Wang, X.; Zhang, Z.; Ding, S.; Chen, G. Quercetin inhibits angiogenesis mediated human prostate tumor growth by targeting VEGFR-2 regulated AKT/mTOR/P70S6K signaling pathways. PLoS ONE 2012, 7, e47516. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Shi, D.; Liu, L.; Wang, J.; Xie, X.; Kang, T.; Deng, W. Quercetin suppresses cyclooxygenase-2 expression and angiogenesis through inactivation of P300 signaling. PLoS ONE 2011, 6, e22934. [Google Scholar] [CrossRef]
- Oh, S.J.; Kim, O.; Lee, J.S.; Kim, J.A.; Kim, M.R.; Choi, H.S.; Kim, Y.C. Inhibition of angiogenesis by quercetin in tamoxifen-resistant breast cancer cells. Food Chem. Toxicol. 2010, 48, 3227–3234. [Google Scholar] [CrossRef]
- Ma, Z.S.; Huynh, T.H.; Ng, C.P.; Do, P.T.; Nguyen, T.H.; Huynh, H. Reduction of CWR22 prostate tumor xenograft growth by combined tamoxifen-quercetin treatment is associated with inhibition of angiogenesis and cellular proliferation. Int. J. Oncol. 2004, 24, 1297–1304. [Google Scholar] [CrossRef]
- Zhao, D.; Qin, C.; Fan, X.; Li, Y.; Gu, B. Inhibitory effects of quercetin on angiogenesis in larval zebrafish and human umbilical vein endothelial cells. Eur. J. Pharmacol. 2014, 723, 360–367. [Google Scholar] [CrossRef]
- Plate, K.H.; Breier, G.; Millauer, B.; Ullrich, A.; Risau, W. Up-regulation of vascular endothelial growth factor and its cognate receptors in a rat glioma model of tumor angiogenesis. Cancer Res. 1993, 53, 5822–5827. [Google Scholar]
- Faes, S.; Uldry, E.; Planche, A.; Santoro, T.; Pythoud, C.; Demartines, N.; Dormond, O. Acidic pH reduces VEGF-mediated endothelial cell responses by downregulation of VEGFR-2; relevance for anti-angiogenic therapies. Oncotarget 2016, 7, 86026–86038. [Google Scholar] [CrossRef] [Green Version]
- Shi, Q.; Le, X.; Wang, B.; Abbruzzese, J.L.; Xiong, Q.; He, Y.; Xie, K. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene 2001, 20, 3751–3756. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Chang, G.; Wang, J.; Jin, W.; Wang, L.; Lin, Y.; Pang, T. Inhibition of K562 leukemia angiogenesis and growth by selective Na+/H+ exchanger inhibitor cariporide through down-regulation of pro-angiogenesis factor VEGF. Leuk. Res. 2011, 35, 1506–1511. [Google Scholar] [CrossRef] [PubMed]
- Mo, X.G.; Chen, Q.W.; Li, X.S.; Zheng, M.M.; Ke, D.Z.; Deng, W.; Wang, P. Suppression of NHE1 by small interfering RNA inhibits HIF-1α-induced angiogenesis in vitro via modulation of calpain activity. Microvasc. Res. 2011, 81, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Orive, G.; Reshkin, S.J.; Harguindey, S.; Pedraz, J.L. Hydrogen ion dynamics and the Na+/H+ exchanger in cancer angiogenesis and antiangiogenesis. Br. J. Cancer 2003, 89, 1395–1399. [Google Scholar] [CrossRef] [PubMed]
- Endo, M.; Jain, R.K.; Witwer, B.; Brown, D. Water channel (aquaporin 1) expression and distribution in mammary carcinomas and glioblastomas. Microvasc. Res. 1999, 58, 89–98. [Google Scholar] [CrossRef]
- Monzani, E.; Bazzotti, R.; Perego, C.; la Porta, C.A. AQP1 is not only a water channel: It contributes to cell migration through Lin7/beta-catenin. PLoS ONE 2009, 4, e6167. [Google Scholar] [CrossRef]
- Saadoun, S.; Papadopoulos, M.C.; Hara-Chikuma, M.; Verkman, A.S. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature 2005, 434, 786–792. [Google Scholar] [CrossRef]
- Andrikopoulos, P.; Fraser, S.P.; Patterson, L.; Ahmad, Z.; Burcu, H.; Ottaviani, D.; Djamgoz, M.B. Angiogenic Functions of Voltage-gated Na+ Channels in Human Endothelial Cells Modulation of Vascular Endothelial Growth Factor (VEGF) Signaling. J. Biol. Chem. 2011, 286, 16846–16860. [Google Scholar] [CrossRef]
- Giatromanolaki, A.; Koukourakis, M.I.; Sivridis, E.; Pastorek, J.; Wykoff, C.C.; Gatter, K.C.; Harris, A.L. Expression of hypoxia-inducible carbonic anhydrase-9 relates to angiogenic pathways and independently to poor outcome in non-small cell lung cancer. Cancer Res. 2001, 61, 7992–7998. [Google Scholar]
- McIntyre, A.; Patiar, S.; Wigfield, S.; Li, J.L.; Ledaki, I.; Turley, H.; Vaughan-Jones, R.D. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clin. Cancer Res. 2012, 18, 3100–3111. [Google Scholar] [CrossRef]
- Rath, S.; Liebl, J.; Fürst, R.; Vollmar, A.M.; Zahler, S. Regulation of endothelial signaling and migration by v-ATPase. Angiogenesis 2014, 17, 587–601. [Google Scholar] [CrossRef]
- Ferrari, S.; Perut, F.; Fagioli, F.; del Prever, A.B.; Meazza, C.; Parafioriti, A.; Fais, S. Proton pump inhibitor chemosensitization in human osteosarcoma: From the bench to the patients’ bed. J. Transl. Med. 2013, 11, 268. [Google Scholar] [CrossRef] [PubMed]
- Azzarito, T.; Venturi, G.; Cesolini, A.; Fais, S. Lansoprazole induces sensitivity to suboptimal doses of paclitaxel in human melanoma. Cancer Lett. 2015, 356, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Spugnini, E.P.; Baldi, A.; Buglioni, S.; Carocci, F.; de Bazzichini, G.M.; Betti, G.; Fais, S. Lansoprazole as a rescue agent in chemoresistant tumors: A phase I/II study in companion animals with spontaneously occurring tumors. J. Transl. Med. 2011, 9, 221. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.Y.; Zhang, J.; Wang, J.L.; Sun, S.; Wang, Z.H.; Wang, L.P.; Zhang, Q.L.; Lv, F.F.; Cao, E.Y.; Fais, S.; et al. Intermittent high dose proton pump inhibitor enhances the antitumor effects of chemotherapy in metastatic breast cancer. J. Exp. Clin. Cancer Res. 2015, 34, 85. [Google Scholar] [CrossRef]
- Falcone, R.; Roberto, M.; D’Antonio, C.; Romiti, A.; Milano, A.; Onesti, C.E.; Marchetti, P.; Fais, S. High-doses of proton pump inhibitors in refractory gastro-intestinal cancer: A case series and the state of art. Dig. Liver Dis. 2016, 48, 1503–1505. [Google Scholar] [CrossRef]
- Spugnini, E.P.; Buglioni, S.; Carocci, F.; Francesco, M.; Vincenzi, B.; Fanciulli, M.; Fais, S. High dose lansoprazole combined with metronomic chemotherapy: A phase I/II study in companion animals with spontaneously occurring tumors. J. Transl. Med. 2014, 12, 225. [Google Scholar] [CrossRef]
- Abdollahi, A.; Folkman, J. Evading tumor evasion: Current concepts and perspectives of anti-angiogenic cancer therapy. Drug Resist. Updates 2010, 13, 16–28. [Google Scholar] [CrossRef]
- Sitohy, B.; Nagy, J.A.; Dvorak, H.F. Anti-VEGF/VEGFR therapy for cancer: Reassessing the target. Cancer Res. 2012, 72, 1909–1914. [Google Scholar] [CrossRef]
- Nagy, J.A.; Dvorak, H.F. Heterogeneity of the tumor vasculature: The need for new tumor blood vessel type-specific targets. Clin. Exp. Metástasis 2012, 29, 657–662. [Google Scholar] [CrossRef]
- Giuliano, S.; Pagès, G. Mechanisms of resistance to anti-angiogenesis therapies. Biochimie 2013, 95, 1110–1119. [Google Scholar] [CrossRef]
- Quesada, A.R.; Medina, M.Á.; Alba, E. Playing only one instrument may be not enough: Limitations and future of the antiangiogenic treatment of cancer. Bioessays 2007, 29, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Croci, D.O.; Mendez-Huergo, S.P.; Cerliani, J.P.; Rabinovich, G.A. Immune-mediated and hypoxia-regulated programs: Accomplices in resistance to anti-angiogenic therapies. In Mechanisms of Drug Resistance in Cancer Therapy; Mandalà, M., Romano, E., Eds.; Springer: Cham, Switzerland, 2017; Volume 249, pp. 31–61. [Google Scholar]
- Shi, Q.; Abbruzzese, J.L.; Huang, S.; Fidler, I.J.; Xiong, Q.; Xie, K. Constitutive and inducible interleukin 8 expression by hypoxia and acidosis renders human pancreatic cancer cells more tumorigenic and metastatic. Clin. Cancer Res. 1999, 5, 3711–3721. [Google Scholar] [PubMed]
- Thoppil, R.J.; Cappelli, H.C.; Adapala, R.K.; Kanugula, A.K.; Paruchuri, S.; Thodeti, C.K. TRPV4 channels regulate tumor angiogenesis via modulation of Rho/Rho kinase pathway. Oncotarget 2016, 7, 25849–25861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pla, A.F.; Ong, H.L.; Cheng, K.T.; Brossa, A.; Bussolati, B.; Lockwich, T.; Paria, B.; Munaron, L.; Ambudkar, I.S. TRPV4 mediates tumor-derived endothelial cell migration via arachidonic acid-activated actin remodeling. Oncogene 2012, 31, 200–212. [Google Scholar]
- Chen, C.K.; Hsu, P.Y.; Wang, T.M.; Miao, Z.F.; Lin, R.T.; Juo, S.H.H. TRPV4 activation contributes functional recovery from ischemic stroke via angiogenesis and neurogenesis. Mol. Neurobiol. 2018, 55, 4127–4135. [Google Scholar] [CrossRef]
- Pla, A.F.; Avanzato, D.; Munaron, L.; Ambudkar, I.S. Ion channels and transporters in cancer. 6. Vascularizing the tumor: TRP channels as molecular targets. Am. J. Physiol. Cell Physiol. 2011, 302, C9–C15. [Google Scholar] [CrossRef]
- Cortright, D.N.; Szallasi, A. Biochemical pharmacology of the vanilloid receptor TRPV1. An update. Eur. J. Biochem. 2004, 271, 1814–1819. [Google Scholar] [CrossRef]
- Szallasi, A.; Cortright, D.N.; Blum, C.A.; Eid, S.R. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat. Rev. Drug Discov. 2007, 6, 357–372. [Google Scholar] [CrossRef]
- Schwartz, G.J.; Gao, X.; Tsuruoka, S.; Purkerson, J.M.; Peng, H.; D’agati, V.; Picard, N.; Eladari, D.; Al-Awqati, Q. SDF1 induction by acidosis from principal cells regulates intercalated cell subtype distribution. J. Clin. Investig. 2015, 125, 4365–4374. [Google Scholar] [CrossRef] [Green Version]
- Veldkamp, C.T.; Peterson, F.C.; Pelzek, A.J.; Volkman, B.F. The monomer–dimer equilibrium of stromal cell-derived factor-1 (CXCL 12) is altered by pH, phosphate, sulfate, and heparin. Protein Sci. 2005, 14, 1071–1081. [Google Scholar] [CrossRef]
- Avnet, S.; Di Pompo, G.; Chano, T.; Errani, C.; Ibrahim-Hashim, A.; Gillies, R.J.; Baldini, N. Cancer-associated mesenchymal stroma fosters the stemness of osteosarcoma cells in response to intratumoral acidosis via NF-κB activation. Int. J. Cancer 2017, 140, 1331–1345. [Google Scholar] [CrossRef] [PubMed]
- Mena, H.A.; Lokajczyk, A.; Dizier, B.; Strier, S.E.; Voto, L.S.; Boisson-Vidal, C.; Negrotto, S. Acidic preconditioning improves the proangiogenic responses of endothelial colony forming cells. Angiogenesis 2014, 17, 867–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwaderer, A.L.; Vijayakumar, S.; Al-Awqati, Q.; Schwartz, G.J. Galectin-3 expression is induced in renal β-intercalated cells during metabolic acidosis. Am. J. Physiol. Ren. Physiol. 2006, 290, F148–F158. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Poburko, D.; Wollheim, C.B.; Demaurex, N. Amiloride derivatives induce apoptosis by depleting ER Ca2+ stores in vascular endothelial cells. Br. J. Pharmacol. 2009, 156, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Németh, Z.H.; Deitch, E.A.; Lu, Q.; Szabó, C.; Hasko, G. NHE blockade inhibits chemokine production and NF-κB activation in immunostimulated endothelial cells. Am. J. Physiol. Cell Physiol. 2002, 283, C396–C403. [Google Scholar] [CrossRef] [PubMed]
- Salcedo, R.; Ponce, M.L.; Young, H.A.; Wasserman, K.; Ward, J.M.; Kleinman, H.K.; Murphy, W.J. Human endothelial cells express CCR2 and respond to MCP-1: Direct role of MCP-1 in angiogenesis and tumor progression. Blood 2000, 96, 34–40. [Google Scholar] [CrossRef]
- Ma, J.; Wang, Q.; Fei, T.; Han, J.D.J.; Chen, Y.G. MCP-1 mediates TGF-β–induced angiogenesis by stimulating vascular smooth muscle cell migration. Blood 2007, 109, 987–994. [Google Scholar] [CrossRef]
- McDonald, P.C.; Chafe, S.C.; Dedhar, S. Overcoming Hypoxia-mediated tumor progression: Combinatorial approaches targeting pH regulation, angiogenesis and immune dysfunction. Front. Cell Dev. Biol. 2016, 4, 27. [Google Scholar] [CrossRef]
- Loges, S.; Schmidt, T.; Carmeliet, P. Mechanisms of resistance to anti-angiogenic therapy and development of third-generation anti-angiogenic drug candidates. Genes Cancer 2010, 1, 12–25. [Google Scholar] [CrossRef]
- Berd, D.; Mastrangelo, M.J. Effect of low dose cyclophosphamide on the immune system of cancer patients: Depletion of CD4+, 2H4+ suppressor-inducer T-cells. Cancer Res. 1988, 48, 1671–1675. [Google Scholar]
- Ghiringhelli, F.; Larmonier, N.; Schmitt, E.; Parcellier, A.; Cathelin, D.; Garrido, C.; Martin, F. CD4+ CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur. J. Immunol. 2004, 34, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Lutsiak, M.C.; Semnani, R.T.; de Pascalis, R.; Kashmiri, S.V.; Schlom, J.; Sabzevari, H. Inhibition of CD4+ 25+ T regulatory cell function implicated in enhanced immune response by low-dose cyclophosphamide. Blood 2005, 105, 2862–2868. [Google Scholar] [CrossRef] [PubMed]
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+ CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2007, 56, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Brode, S.; Cooke, A. Immune-potentiating effects of the chemotherapeutic drug cyclophosphamide. Crit. Rev. Immunol. 2008, 28, 109–126. [Google Scholar] [CrossRef]
- Zhao, J.; Cao, Y.; Lei, Z.; Yang, Z.; Zhang, B.; Huang, B. Selective depletion of CD4+ CD25+ Foxp3+ regulatory T cells by low-dose cyclophosphamide is explained by reduced intracellular ATP levels. Cancer Res. 2010, 70, 4850–4858. [Google Scholar] [CrossRef]
- Burton, J.H.; Mitchell, L.; Thamm, D.H.; Dow, S.W.; Biller, B.J. Low-dose cyclophosphamide selectively decreases regulatory T cells and inhibits angiogenesis in dogs with soft tissue sarcoma. J. Vet. Intern. Med. 2011, 25, 920–926. [Google Scholar] [CrossRef]
- Ge, Y.; Domschke, C.; Stoiber, N.; Schott, S.; Heil, J.; Rom, J.; Beckhove, P. Metronomic cyclophosphamide treatment in metastasized breast cancer patients: Immunological effects and clinical outcome. Cancer Immunol. Immunother. 2012, 61, 353–362. [Google Scholar] [CrossRef]
- Wu, J.; Waxman, D.J. Metronomic cyclophosphamide eradicates large implanted GL261 gliomas by activating antitumor Cd8+ T-cell responses and immune memory. Oncoimmunology 2015, 4, e1005521. [Google Scholar] [CrossRef]
- Hughes, E.; Scurr, M.; Campbell, E.; Jones, E.; Godkin, A.; Gallimore, A. T-cell modulation by cyclophosphamide for tumour therapy. Immunology 2018, 154, 62–68. [Google Scholar] [CrossRef]
- Huijts, C.M.; Lougheed, S.M.; Bodalal, Z.; van Herpen, C.M.; Hamberg, P.; Tascilar, M.; Haanen, J.B.; Verheul, H.M.; de Gruijl, T.D.; van der Vliet, H.J.; et al. The effect of everolimus and low-dose cyclophosphamide on immune cell subsets in patients with metastatic renal cell carcinoma: Results from a phase I clinical trial. Cancer Immunol. Immunother. 2019, 68, 503–515. [Google Scholar] [CrossRef]
- Osband, M.; Shen, Y.J.; Shlesinger, M.; Brown, A.; Hamilton, D.; Cohen, E.; Lavin, P.; Mccaffrey, R. Successful tumour immunotherapy with cimetidine in mice. Lancet 1981, 317, 636–638. [Google Scholar] [CrossRef]
- Ershler, W.B.; Hacker, M.P.; Burroughs, B.J.; Moore, L.; Myers, C.F. Cimetidine and the immune response: I. In vivo augmentation of nonspecific and specific immune response. Clin. Immunol. Immunopathol. 1983, 26, 10–17. [Google Scholar] [CrossRef]
- Jin, Z.; Kumar, A.; Cleveland, R.P.; Murray, D.L.; Kaufman, D.B. Inhibition of suppressor cell function by cimetidine in a murine model. Clin. Immunol. Immunopathol. 1986, 38, 350–356. [Google Scholar] [CrossRef]
- Hirai, N.; Hill, N.O.; Motoo, Y.; Osther, K. Cimetidine enhances interferon induced augmentation of NK cell activity and suppresses interferon production. Acta Pathol. Microbiol. Scand. Ser. C Immunol. 1985, 93, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Sahasrabudhe, D.M.; McCune, C.S.; O’donnell, R.W.; Henshaw, E.C. Inhibition of suppressor T lymphocytes (Ts) by cimetidine. J. Immunol. 1987, 138, 2760–2763. [Google Scholar]
- Dejiao, B.; Guoliang, Y.; Hongying, Y.; Yan, L.; Kun, W.; Hua, S. Perioperative cimetidine application modulates natural killer cells in patients with colorectal cancer: A randomized clinical study. J. Tongji Med Univ. 1999, 19, 300–303. [Google Scholar] [CrossRef]
- Kubota, T.; Fujiwara, H.; Ueda, Y.; Itoh, T.; Yamashita, T.; Yoshimura, T.; Yamagishi, H. Cimetidine modulates the antigen presenting capacity of dendritic cells from colorectal cancer patients. Br. J. Cancer 2002, 86, 1257–1261. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; Xu, M.; Li, X.; Jia, J.; Fan, K.; Lai, G. Cimetidine suppresses lung tumor growth in mice through proapoptosis of myeloid-derived suppressor cells. Mol. Immunol. 2013, 54, 74–83. [Google Scholar] [CrossRef]
- Pan, W.; Sun, Q.; Wang, Y.; Wang, J.; Cao, S.; Ren, X. Highlights on mechanisms of drugs targeting MDSCs: Providing a novel perspective on cancer treatment. Tumor Biol. 2015, 36, 3159–3169. [Google Scholar] [CrossRef]
- Vila-Leahey, A.; Oldford, S.A.; Marignani, P.A.; Wang, J.; Haidl, I.D.; Marshall, J.S. Ranitidine modifies myeloid cell populations and inhibits breast tumor development and spread in mice. Oncoimmunology 2016, 5, e1151591. [Google Scholar] [CrossRef]
- Jafarzadeh, A.; Nemati, M.; Khorramdelazad, H.; Hassan, Z.M. Immunomodulatory properties of cimetidine: Its therapeutic potentials for treatment of immune-related diseases. Int. Immunopharmacol. 2019, 70, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Pantziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P. Repurposing drugs in oncology (ReDO)—Cimetidine as an anti-cancer agent. Ecancermedicalscience 2014, 8, 485. [Google Scholar] [CrossRef] [PubMed]
- Dorr, R.T.; Alberts, D.S. Cimetidine enhancement of cyclophosphamide antitumour activity. Br. J. Cancer 1982, 45, 35–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorr, R.T.; Soble, M.J.; Alberts, D.S. Interaction of cimetidine but not ranitidine with cyclophosphamide in mice. Cancer Res. 1986, 46, 1795–1799. [Google Scholar]
Figure 1.
Two different mechanisms of anti-angiogenic therapy.

Scheme 1.
Mechanisms of resistance to anti-angiogenesis.

Figure 2.
Anti-angiogenic mechanisms of Fenofibrate (FF) [113].
Figure 2.
Anti-angiogenic mechanisms of Fenofibrate (FF) [113].

Figure 3.
The combination of different proton-extruder inhibitors (“a cocktail”) are the base of the pH centered treatment of cancer with two goals: Acidifying the cytoplasm and alkalinizing the extracellular pH.
Figure 3.
The combination of different proton-extruder inhibitors (“a cocktail”) are the base of the pH centered treatment of cancer with two goals: Acidifying the cytoplasm and alkalinizing the extracellular pH.

Figure 4.
Explanation in the text.


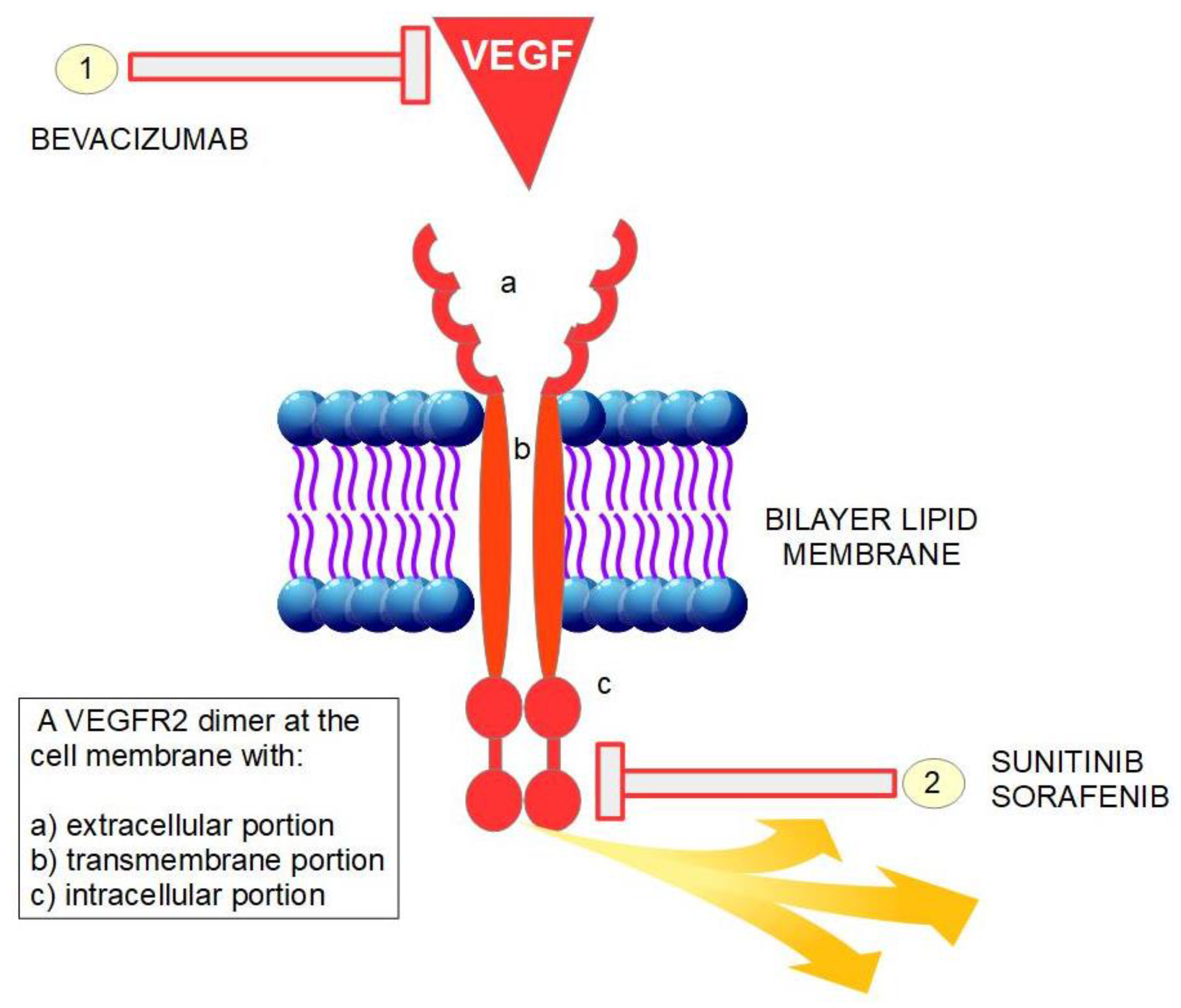{kind=link}
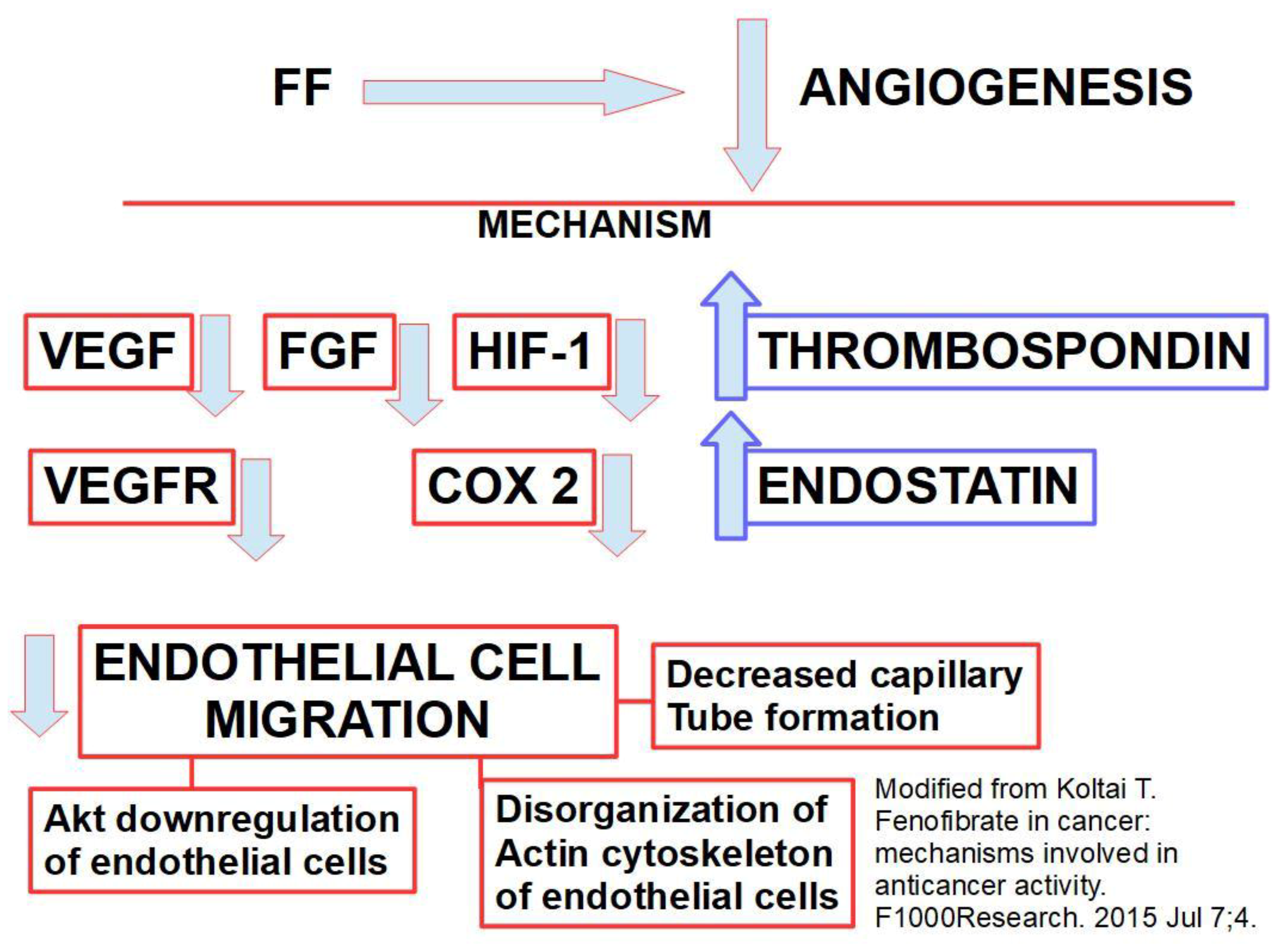{kind=link}
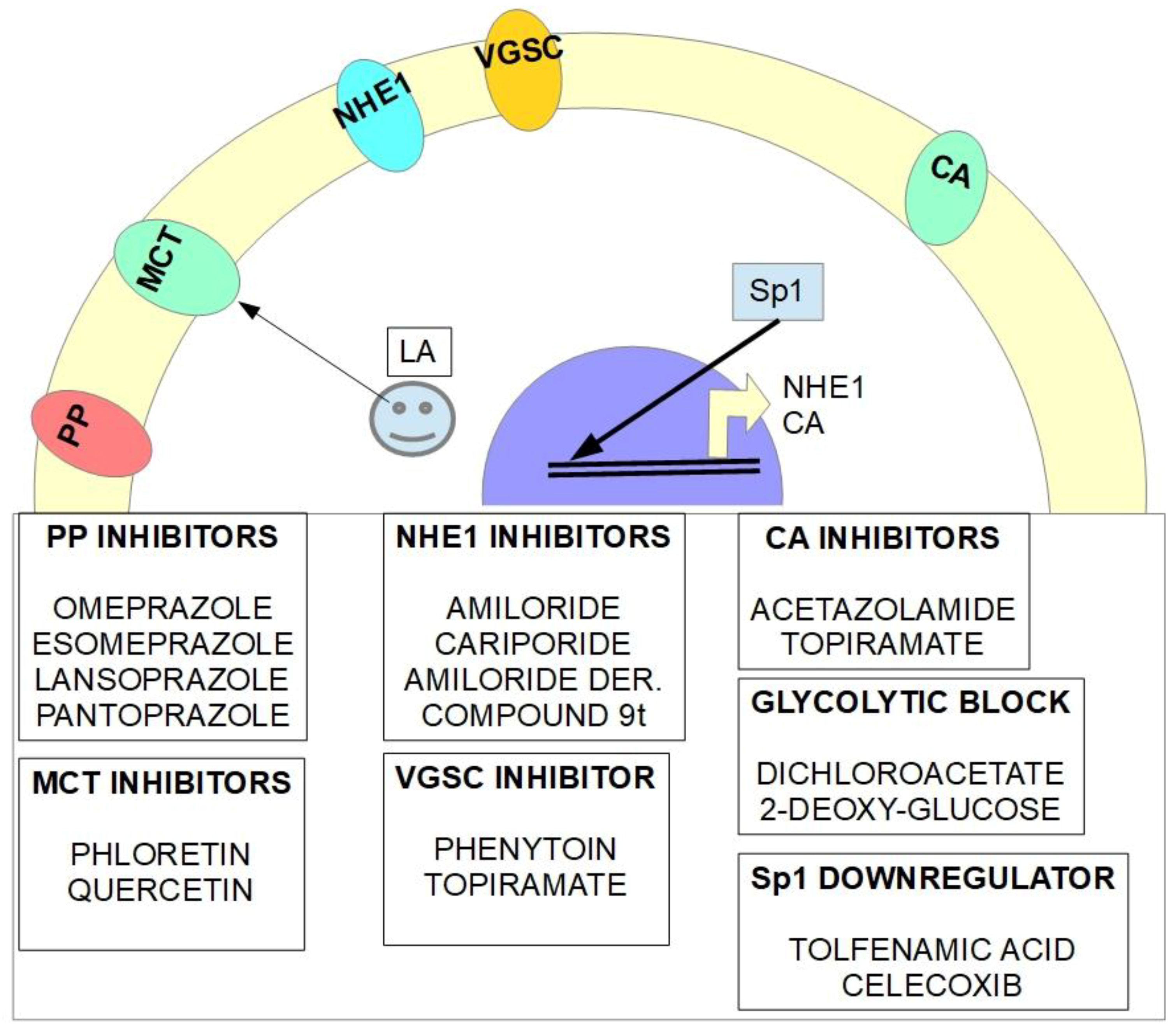{kind=link}
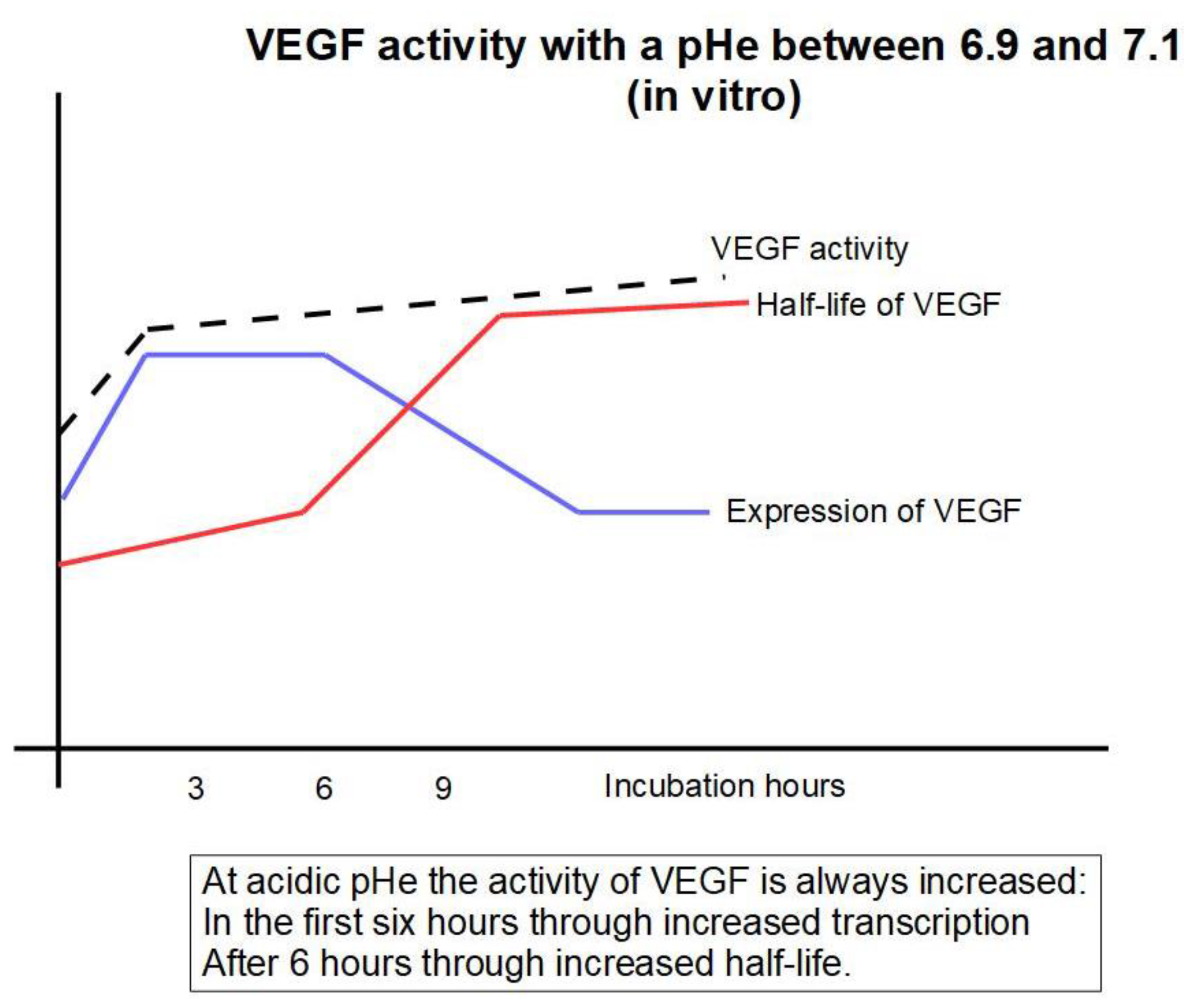{kind=link}
{kind=link}
Table 1.
Immune effects of low dose cyclophosphamide (LDC) and low dose metronomic cyclophosphamide (LDMC).
Table 1.
Immune effects of low dose cyclophosphamide (LDC) and low dose metronomic cyclophosphamide (LDMC).
| Reference | Findings |
|---|---|
| Berd et al. 1988 [193] | LDC showed progressive decrease of T suppressor CD4+ expressing 2H4+ cells. True helper cells were not reduced. |
| Ghiringhelli et al. 2004 [194] | A single dose of LDC depleted CD4+CD25+ T regulatory cells in tumor bearing animals, and significantly increased the effects of subsequent immunotherapy. |
| Lutsiak et al. 2005 [195] | LDC inhibited T cell regulatory cells CD4+CD25+ not only decreasing their number but also their functionality. |
| Ghiringhelli et al. 2007 [196] | In very advanced cancer patients, LDMC downregulated CD4+CD25+ regulatory T cells and at the same time restored the functionality of T and NK cells. |
| Brode et al. 2008 [197] | LDC inhibited regulatory T cells through inhibition of Foxp3+. |
| Xhao et al. 2010 [198] | LDC produced selective depletion of CD4+CD25+ Foxp3 Tregs by depleting intracellular ATP. |
| Burton et al. 2011 [199] | 11 dogs with soft tissue sarcoma treated with LDMC showed decreased Tregs and decreased angiogenesis |
| Ge et al. 2012 [200] | For 3 months the authors studied the effects of LDMC in 12 patients with treatment-refractory breast cancer with metastases. Initially there was a significant decrease in circulating Tregs for a short period. Furthermore, there was an important increase in reactive T cells that remained at high levels during the 3 months of the study. This meant that the recuperated activity of reactive T cells outlasted the short lived Treg decrease. |
| Wu et al. 2015 [201] | Mice with implanted glioma treated with LDMC showed an increase in tumor associated-cytotoxic CD8+ lymphocytes, NK cells and macrophages achieving tumor regression. |
Table 2.
Cimetidine’s immunologic effects.
| Reference | Findings |
|---|---|
| Osband et al. 1981 [204] | Cimetidine produced inactivation of suppressor cells, slowed metastases development and prolonged survival in tumor bearing mice. |
| Ershler et al. 1983 [205] | Mice treated with daily injections of cimetidine produced twice as much specific antibody in response to immunization. |
| Jin et al. 1986 [206] | Cimetidine induced loss of suppressor cells function. |
| Hirai et al. 1987 [207] | Cimetidine increased the interferon α enhancer effect on NK cell activity. Cimetidine decreased interferon α production, therefore both drugs have to be administered together in order to increase NK’s activity. |
| Sahasrabudhe et al. 1987 [208] | Cimetidine decreased suppressor T lymphocytes and decreased their activity. |
| Dejiao et al. 1999 [209] | Peri-operative administration of cimetidine in patients with colon cancer significantly increased NK cells in the surgical specimen as compared with patients not receiving it. |
| Kubota et al. 2002 [210] | Cimetidine increased the antigen presenting capacity of dendritic cells in tumors obtained from patients with colorectal cancer. |
| Zheng et al. 2013 [211] | Cimetidine induced apoptosis in myeloid derived suppressor cells inhibiting lung tumor growth in mice in vivo. It also increased interferon γ production. |
| Pan et al. 2015 [212] | Cimetidine inhibited T-cell suppression by myeloid derived suppressor cells. |
| Vila-Leahey et al. 2016 [213] | Ranitidine, another histamine receptor 2 antagonist, also significantly decreased the population of myeloid derived suppressor cells in spleen and bone marrow and decreased lung metastasis in tumor xenografted mice. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Koltai, T.; Cardone, R.A.; Reshkin, S.J. Synergy Between Low Dose Metronomic Chemotherapy and the pH-Centered Approach Against Cancer. Int. J. Mol. Sci. 2019, 20, 5438. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215438
AMA Style
Koltai T, Cardone RA, Reshkin SJ. Synergy Between Low Dose Metronomic Chemotherapy and the pH-Centered Approach Against Cancer. International Journal of Molecular Sciences. 2019; 20(21):5438. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215438
Chicago/Turabian StyleKoltai, Tomas, Rosa A. Cardone, and Stephan J. Reshkin. 2019. "Synergy Between Low Dose Metronomic Chemotherapy and the pH-Centered Approach Against Cancer" International Journal of Molecular Sciences 20, no. 21: 5438. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215438
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.