Multiple Sclerosis: Melatonin, Orexin, and Ceramide Interact with Platelet Activation Coagulation Factors and Gut-Microbiome-Derived Butyrate in the Circadian Dysregulation of Mitochondria in Glia and Immune Cells




{kind=link}
{kind=link}
Abstract
:1. Introduction
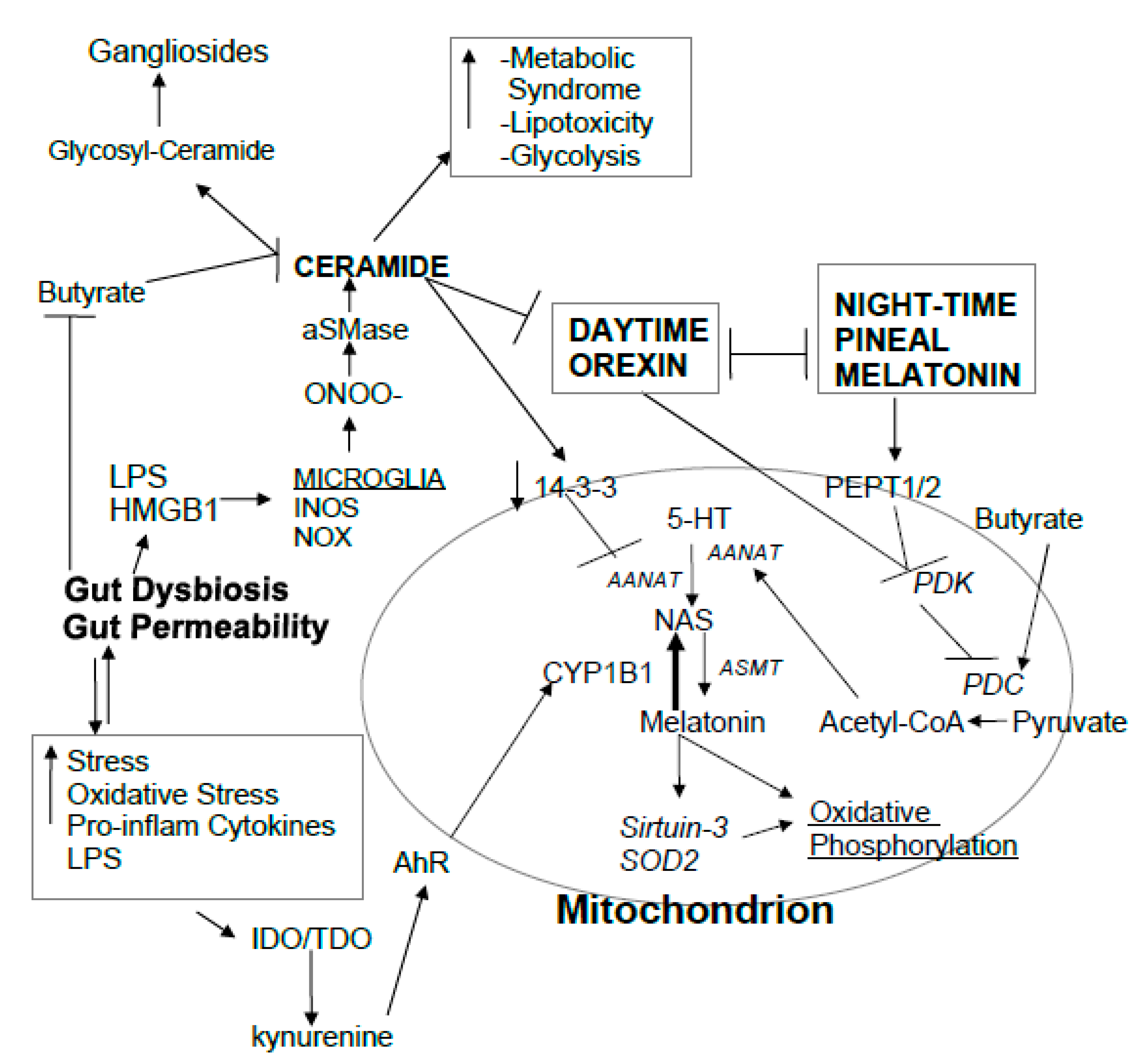
2. MS Pathophysiology
2.1. Circadian Rhythms, the Gut Microbiome, and Melatonin
2.1.1. Circadian Rhythms
2.1.2. Gut Microbiome-Derived Butyrate and Mitochondria
2.1.3. Pineal Gland and Mitochondria Melatonin
2.1.4. Mitochondria at the Heart of Circadian Rhythm
2.2. Ceramide, Mitochondria, and MS
2.2.1. Ceramide, Oxidants, and Acid Sphingomyelinase
2.2.2. Astrocytes and Ceramide
2.2.3. Oligodendrocytes and Ceramide
2.2.4. Butyrate and Ceramide
2.3. Orexin, Melatonin, and Mitochondria Circadian Regulation
2.3.1. Orexin: Wake Promotion and Mitochondria
2.3.2. Orexin and Obesity in MS
2.3.3. Orexin and Dynorphin
2.3.4. Orexin, Stress, and the Kynurenine Pathway
2.3.5. Orexin, Melatonin, and Glutamate in MS
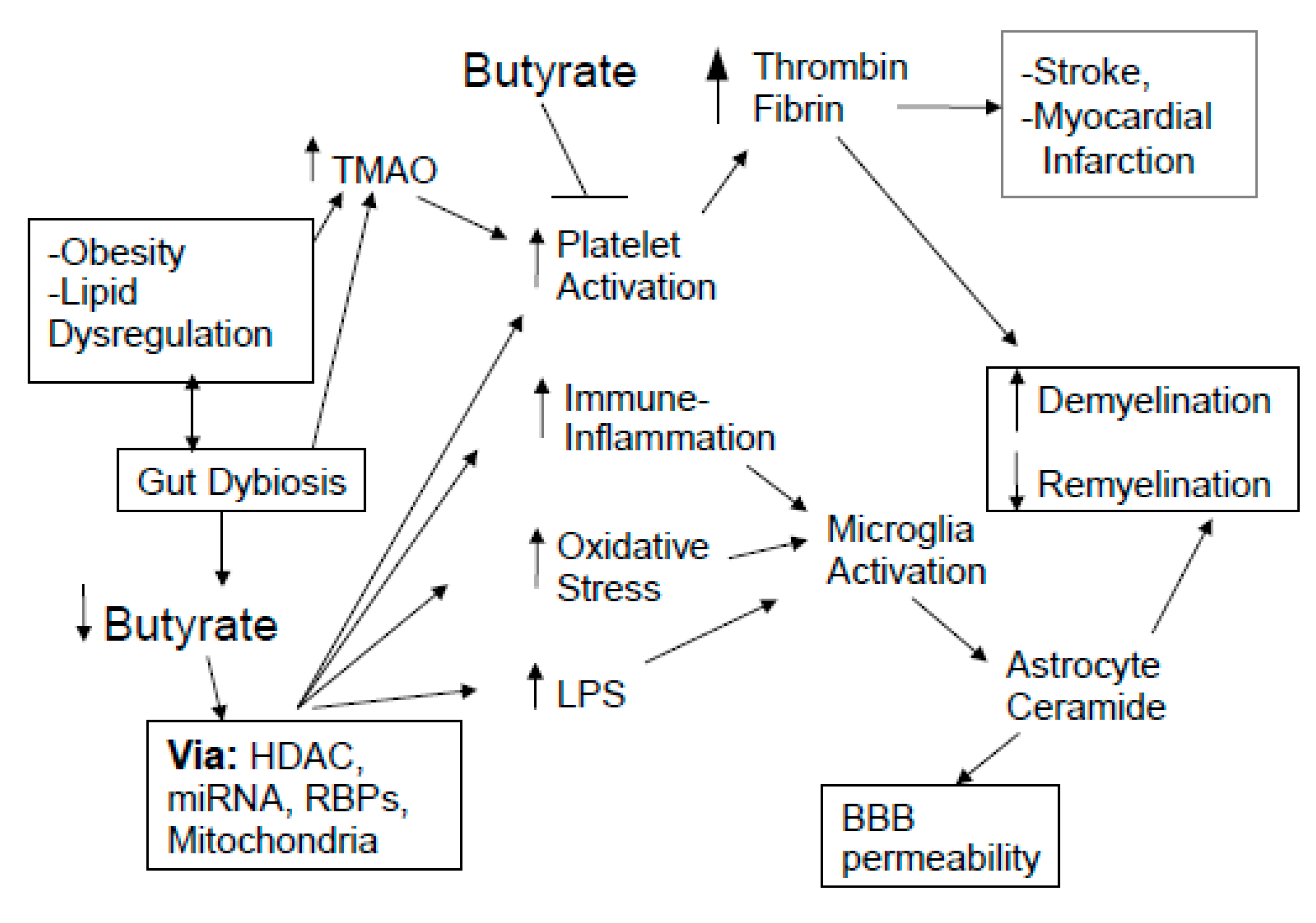
3. Integrating MS Pathophysiology
3.1. Platelets, Coagulation Factors, and Glia
3.2. microRNAs and mRNA Binding Proteins
4. Future Research
- (1)
- Does the modulation of OPCs’ mitochondria by butyrate also act to regulate their remyelinating capacity as well as the putative OPC association with, and disruption of, the BBB?
- (2)
- Caffeine increases levels of orexin neuron activity [186], and may decrease the risk, and attenuate the course, of MS [187], including in the EAE model [188]. Are caffeine benefits via orexin neuronal activity, and thereby on daytime mitochondrial function? As caffeine increases peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) and mitochondrial biogenesis in isolated cells, it is likely to also have direct effects on wider mitochondrial functioning [189]. PGC-1α is widely regarded as the mitochondria master regulator;
- (3)
- What is the role of the mitochondrial melatonergic pathways within orexin neurons of the lateral hypothalamus/perifornical area?
- (4)
- (5)
- Given preclinical data indicating greater effects of stress in the inhibition of male orexin neurons [126], is this relevant to sex differences in MS, including increased rates of males shifting to SPMS?
- (6)
- As pineal gland-derived melatonin is a significant modulator of the effects of chronic stress in rodents [191], how do variations in pineal melatonin modulate the effects of chronic stress on orexin neuronal structure and function, including across genders?
- (7)
- (8)
- Is the ceramide inhibition of cellular 14–3-3 levels relevant to mitochondrial 14-3-3 levels and thereby to mitochondrial AANAT stabilization and melatonergic pathway activity?
- (9)
- Are ceramide effects via miRNAs associated with 14-3-3 suppression, including by miR-451, miR-375, and miR-7, reviewed in [192]?
- (10)
- Is pineal gland NAS/melatonin ratio relevant to MS pathophysiology?
- (11)
- Is the increased NAS in SPMS associated with p75NTR activation, leading to increased ceramide synthesis?
- (12)
- Are the benefits of exogenous orexin in MS mediated via orexin’s suppression of gut dysbiosis/permeability, with orexin thereby lowering circulating LPS, exosomal HMGB1, and associated brain microglia activation [193]? This would suggest gut, rather than central, effects of exogenous orexin;
- (13)
- How relevant are the circadian rhythms of pineal gland-derived melatonin and hypothalamic orexin to the circadian regulation of ceramide [155]?
- (14)
- Astrocyte network connectivity, via connexin-43 gap junctions, is important to circadian regulation by orexin [194]. In the absence of a functional lateral hypothalamic astrocyte network and energy provision, animals show excessive sleepiness and difficulty in maintaining alert states [194]. Because reactive astrocytes usually disengage from the astrocyte network, does this indicate that ONOO- or LPS/HMGB1 activation in the lateral hypothalamus may contribute to a decreased synchronization of the astrocyte network and thereby to the suppression of orexin?
- (15)
- As well as AhR-induced CYP1b1, a number of other factors can drive the ‘backward’ conversion of melatonin to NAS, including CYP2C19, high ATP levels, mGluR5 activation, and O-demethylation, see [31]. What are the role of these factors in cellular changes in MS?
5. Treatment Implications
- (1)
- From the role of the gut microbiome in MS, a number of proposed treatments emerge, including the use of probiotics, fecal microbiota transplantation, bile acid supplementation, and gut barrier enhancers. Sodium butyrate may have more immediate utility. It not only immediately increases the butyrate availability, but also encourages the growth of butyrate-producing gut bacteria. This would have a number of impacts on MS pathophysiology, including the inhibition of ceramide and optimization of mitochondrial function;
- (2)
- A number of studies have highlighted the beneficial effects of melatonin in MS patients, with melatonin significantly decreasing the levels of pro-inflammatory cytokines and NO products [195]. However, the effects of melatonin may be enhanced by the adjunctive use of sodium butyrate, which, via the processes outlined above, such as ceramide inhibition and decreasing CYP1b1, would better optimize the effects of melatonin;
- (3)
- As noted throughout, fingolimod has many effects on MS pathophysiology that are replicated by the actions of butyrate. Preclinical data show that sodium butyrate facilitates recovery of the BBB following trauma-induced increases in BBB permeability [196]. As BBB permeability has long been associated with MS etiology and relapse, including by permitting the effects of thrombin and fibrin on MS pathophysiology, it requires clinical investigation as to the utility of sodium butyrate as an adjunctive to fingolimod treatment, including as to whether there are additive or synergistic effects in their overlapping modes of efficacy;
- (4)
- A combination of daytime orexin and night-time melatonin administration would target the putative loss of the circadian regulation of mitochondrial function and the resetting of mitochondrial oxidative phosphorylation. This will be important to investigate in MS preclinical models, including as to the impact of both treatments on the mitochondrial melatonergic pathway;
- (5)
- As urban air quality is a risk factor for pediatric MS [197], with many air pollutants acting via the AhR, and therefore increasing CYP1b1 and the NAS/melatonin ratio, targeting air pollution may be an important preventative strategy. This may be a particular target in SPMS, given its association with increased NAS [156].
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AANAT | aralkylamine N-acetyltransferase |
| AhR | aryl hydrocarbon receptor |
| ARNTL | aryl hydrocarbon receptor nuclear translocator like |
| aSMase | acid sphingomyelinase |
| ASMT | N-acetylserotonin o-methyltransferase |
| BBB | blood-brain barrier |
| BDNF | brain-derived neurotrophic factor |
| BMI | body mass index |
| CLOCK | circadian locomotor uutput cycles kaput |
| CRH | corticotrophin releasing hormone |
| CSF | cerebrospinal fluid |
| CYP | cytochrome P450 |
| EAE | experimental autoimmune encephalomyelitis |
| ER | endoplasmic reticulum |
| HDAC | histone deacetylase |
| HMGB | high-mobility group box |
| HuR | human antigen R |
| IDO | indoleamine 2,3-dioxygenase |
| Ig | immunoglobulin |
| IL | interleukin |
| iNOS | inducible nitric oxide synthase |
| LPS | lipopolysaccharide |
| MMP | matrix metalloproteinase |
| MS | multiple sclerosis |
| NAS | N-acetylserotonin |
| O&NS | oxidative and nitrosative stress |
| ONOO | peroxynitrite |
| OPC | oligodendrocyte precursor cell |
| PDC | pyruvate dehydrogenase complex |
| PDK | pyruvate dehydrogenase kinase |
| PGC-1α | peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| RBP | mRNA binding protein |
| RRMS | relapsing-remitting multiple sclerosis |
| SNP | single nucleotide polymorphism |
| SOD2 | manganese superoxide dismutase |
| SPMS | secondary progressive multiple sclerosis |
| TDO | tryptophan 2,3-dioxygenase |
| TLR | toll-like receptor |
| TMAO | trimethylamine N-oxide |
| TNF | tumor necrosis factor |
| TTP | tristetraprolin |
| VMH | ventromedial hypothalamus |
References
- Golalipour, M.; Maleki, Z.; Farazmandfar, T.; Shahbazi, M. PER3 VNTR polymorphism in Multiple Sclerosis: A new insight to impact of sleep disturbances in MS. Mult. Scler. Relat. Disord. 2017, 17, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Lavtar, P.; Rudolf, G.; Maver, A.; Hodžić, A.; Starčević Čizmarević, N.; Živković, M.; Šega Jazbec, S.; Klemenc Ketiš, Z.; Kapović, M.; Dinčić, E.; et al. Association of circadian rhythm genes ARNTL/BMAL1 and CLOCK with multiple sclerosis. PLoS ONE 2018, 13, e0190601. [Google Scholar] [CrossRef] [PubMed]
- Goischke, H.K. Vitamin D Supplementation as Add-on Therapy in Multiple Sclerosis-Balance between Benefit and Risk? A Commentary on Vitamin D Supplementation in Central Nervous System Demyelinating Disease-Enough Is Enough. Int. J. Mol. Sci. 2019, 20, 1513. [Google Scholar] [CrossRef] [PubMed]
- Ghareghani, M.; Reiter, R.J.; Zibara, K.; Farhadi, N. Latitude, Vitamin D, Melatonin, and Gut Microbiota Act in Concert to Initiate Multiple Sclerosis: A New Mechanistic Pathway. Front. Immunol. 2018, 9, 2484. [Google Scholar] [CrossRef] [PubMed]
- Miclea, A.; Miclea, M.; Pistor, M.; Hoepner, A.; Chan, A.; Hoepner, R. Vitamin D supplementation differentially affects seasonal multiple sclerosis disease activity. Brain Behav. 2017, 7, e00761. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G. Prenatal corticosteroids: Pretermer outcomes, stress, schizophrenia, multiple sclerosis and the developmental role of melatonin and vitamin D3. J. Pediatr. Adolesc. Gynecol. 2010, 23, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Åkerstedt, T.; Olsson, T.; Alfredsson, L. Shift work influences multiple sclerosis risk. Mult. Scler. 2015, 21, 1195–1199. [Google Scholar] [CrossRef]
- Neufeld-Cohen, A.; Robles, M.S.; Aviram, R.; Manella, G.; Adamovich, Y.; Ladeuix, B.; Nir, D.; Rousso-Noori, L.; Kuperman, Y.; Golik, M.; et al. Circadian control of oscillations in mitochondrial rate-limiting enzymes and nutrient utilization by PERIOD proteins. Proc. Natl. Acad. Sci. USA 2016, 113, E1673–E1682. [Google Scholar] [CrossRef]
- Anderson, G. Gut Dysbiosis Dysregulates Central and Systemic Homeostasis via Decreased Melatonin and Suboptimal Mitochondria Functioning: Pathoetiological and Pathophysiological Implications. Melatonin. Res. 2019, 2, 70–85. [Google Scholar] [CrossRef]
- Anderson, G.; Seo, M.; Berk, M.; Carvalho, A.F.; Maes, M. Gut Permeability and Microbiota in Parkinson’s Disease: Role of Depression, Tryptophan Catabolites, Oxidative and Nitrosative Stress and Melatonergic Pathways. Curr. Pharm. Des. 2016, 22, 6142–6151. [Google Scholar] [CrossRef]
- Rodriguez, M.; Wootla, B.; Anderson, G. Multiple Sclerosis, Gut Microbiota and Permeability: Role of Tryptophan Catabolites, Depression and the Driving Down of Local Melatonin. Curr. Pharm. Des. 2016, 22, 6134–6141. [Google Scholar] [CrossRef] [PubMed]
- Rapaport, B.; Karceski, S. Multiple sclerosis and stress. Neurology 2012, 79, e47–e49. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Junli, G.; Liu, X.; Chen, C.; Sun, X.; Li, H.; Zhou, Y.; Cui, C.; Wang, Y.; Yang, Y.; et al. Gut dysbiosis and lack of short chain fatty acids in a Chinese cohort of patients with multiple sclerosis. Neurochem. Int. 2019, 129, 104468. [Google Scholar] [CrossRef] [PubMed]
- Gianfrancesco, M.A.; Stridh, P.; Shao, X.; Rhead, B.; Graves, J.S.; Chitnis, T.; Waldman, A.; Lotze, T.; Schreiner, T.; Belman, A.; et al. Genetic risk factors for pediatric-onset multiple sclerosis. Mult. Scler. 2018, 24, 1825–1834. [Google Scholar] [CrossRef]
- Riccio, P.; Rossano, R. Diet, Gut Microbiota, and Vitamins D+ A in Multiple Sclerosis. Neurotherapeutics 2018, 15, 75–91. [Google Scholar] [CrossRef]
- Thaiss, C.A.; Levy, M.; Korem, T.; Dohnalová, L.; Shapiro, H.; Jaitin, D.A.; David, E.; Winter, D.R.; Gury-BenAri, M.; Tatirovsky, E.; et al. Microbiota Diurnal Rhythmicity Programs Host Transcriptome Oscillations. Cell 2016, 167, 1495–1510. [Google Scholar] [CrossRef]
- Berezowska, M.; Coe, S.; Dawes, H. Effectiveness of Vitamin D Supplementation in the Management of Multiple Sclerosis: A Systematic Review. Int. J. Mol. Sci. 2019, 20, 1301. [Google Scholar] [CrossRef]
- Engkasan, J.P. Does Vitamin D reduce disease activity in people with multiple sclerosis? A Cochrane Review summary with commentary. NeuroRehabilitation 2019, 1–2. [Google Scholar] [CrossRef]
- O’Neil, D.S.; Stewart, C.J.; Chu, D.M.; Goodspeed, D.M.; Gonzalez-Rodriguez, P.J.; Shope, C.D.; Aagaard, K.M. Conditional postnatal deletion of the neonatal murine hepatic circadian gene, Npas2, alters the gut microbiome following restricted feeding. Am. J. Obstet. Gynecol. 2017, 217, e1–e218. [Google Scholar] [CrossRef]
- Reynolds, A.C.; Paterson, J.L.; Ferguson, S.A.; Stanley, D.; Wright, K.P., Jr.; Dawson, D. The shift work and health research agenda: Considering changes in gut microbiota as a pathway linking shift work, sleep loss and circadian misalignment, and metabolic disease. Sleep. Med. Rev. 2017, 34, 3–9. [Google Scholar] [CrossRef]
- Khalyfa, A.; Poroyko, V.A.; Qiao, Z.; Gileles-Hillel, A.; Khalyfa, A.A.; Akbarpour, M.; Almendros, I.; Farré, R.; Gozal, D. Exosomes and Metabolic Function in Mice Exposed to Alternating Dark-Light Cycles Mimicking Night Shift Work Schedules. Front. Physiol. 2017, 8, 882. [Google Scholar] [CrossRef] [PubMed]
- Matheus, V.A.; Monteiro, L.; Oliveira, R.B.; Maschio, D.A.; Collares-Buzato, C.B. Butyrate reduces high-fat diet-induced metabolic alterations, hepatic steatosis and pancreatic beta cell and intestinal barrier dysfunctions in prediabetic mice. Exp. Biol. Med. (Maywood) 2017, 242, 1214–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fetisova, E.K.; Muntyan, M.S.; Lyamzaev, K.G.; Chernyak, B.V. Therapeutic Effect of the Mitochondria-Targeted Antioxidant SkQ1 on the Culture Model of Multiple Sclerosis. Oxid. Med. Cell. Longev. 2019, 2019, 2082561. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wu, Y.; Sun, Y.; Dong, X.; Wang, Z.; Zhang, Z.; Xiao, Y.; Dong, G. Bacterial endotoxin decreased histone H3 acetylation of bovine mammary epithelial cells and the adverse effect was suppressed by sodium butyrate. BMC Vet. Res. 2019, 15, 267. [Google Scholar] [CrossRef]
- Roy, A.C.; Chang, G.; Ma, N.; Wang, Y.; Roy, S.; Liu, J.; Aabdin, Z.U.; Shen, X. Sodium butyrate suppresses NOD1-mediated inflammatory molecules expressed in bovine hepatocytes during iE-DAP and LPS treatment. J. Cell. Physiol. 2019, 234, 19602–19620. [Google Scholar] [CrossRef]
- Liu, J.; Chang, G.; Huang, J.; Wang, Y.; Ma, N.; Roy, A.C.; Shen, X. Sodium Butyrate Inhibits the Inflammation of Lipopolysaccharide-Induced Acute Lung Injury in Mice by Regulating the Toll-Like Receptor 4/Nuclear Factor κB Signaling Pathway. J. Agric. Food. Chem. 2019, 67, 1674–1682. [Google Scholar] [CrossRef]
- Yamawaki, Y.; Yoshioka, N.; Nozaki, K.; Ito, H.; Oda, K.; Harada, K.; Shirawachi, S.; Asano, S.; Aizawa, H.; Yamawaki, S.; et al. Sodium butyrate abolishes lipopolysaccharide-induced depression-like behaviors and hippocampal microglial activation in mice. Brain. Res. 2018, 1680, 13–38. [Google Scholar] [CrossRef]
- Jin, C.J.; Engstler, A.J.; Sellmann, C.; Ziegenhardt, D.; Landmann, M.; Kanuri, G.; Lounis, H.; Schröder, M.; Vetter, W.; Bergheim, I. Sodium butyrate protects mice from the development of the early signs of non-alcoholic fatty liver disease: Role of melatonin and lipid peroxidation. Br. J. Nutr. 2016, 23, 1–12. [Google Scholar] [CrossRef]
- Rodriguez, M. Virus-induced demyelination in mice: “dying back” of oligodendrocytes. Mayo. Clin. Proc. 1985, 60, 433–438. [Google Scholar] [CrossRef]
- Park, J.; Wang, Q.; Wu, Q.; Mao-Draayer, Y.; Kim, C.H. Bidirectional regulatory potentials of short-chain fatty acids and their G-protein-coupled receptors in autoimmune neuroinflammation. Sci. Rep. 2019, 9, 8837. [Google Scholar] [CrossRef]
- Anderson, G.; Reiter, R.J. Glioblastoma:Role of Mitochondria N-acetylserotonin/Melatonin Ratio in Mediating Effects of miR-451, Aryl Hydrocarbon Receptor and in Co-ordinating Wider Biochemical Changes. Int. J. Tryptophan. Res. 2019, 2, 44–66. [Google Scholar] [CrossRef]
- Anderson, G.; Mazzoccoli, G. Left Ventricular Hypertrophy: Roles of Mitochondria CYP1B1 and Melatonergic Pathways in Co-Ordinating Wider Pathophysiology. Int. J. Mol. Sci. 2019, 20, 4068. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Breast Cancer: Occluded Role of Mitochondria N-acetylserotonin/Melatonin Ratio in Co-ordinating Pathophysiology. Biochem. Pharmacol. 2019, 168, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G. Endometriosis Pathoetiology and Pathophysiology: Roles of Vitamin A, Estrogen, Immunity, Adipocytes, Gut Microbiome and Melatonergic Pathway on Mitochondria Regulation. Biomol. Concepts. 2019, 10, 133–149. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Noto, D.; Hoshino, Y.; Mizuno, M.; Miyake, S. Butyrate suppresses demyelination and enhances remyelination. J. Neuroinflammation 2019, 16, 165. [Google Scholar] [CrossRef]
- Niu, J.; Tsai, H.H.; Hoi, K.K.; Huang, N.; Yu, G.; Kim, K.; Baranzini, S.E.; Xiao, L.; Chan, J.R.; Fancy, S.P.J. Aberrant oligodendroglial-vascular interactions disrupt the blood-brain barrier, triggering CNS inflammation. Nat. Neurosci. 2019, 22, 709–718. [Google Scholar] [CrossRef]
- Galloway, D.A.; Gowing, E.; Setayeshgar, S.; Kothary, R. Inhibitory milieu at the multiple sclerosis lesion site and the challenges for remyelination. Glia 2019. [Google Scholar] [CrossRef]
- Chamberlain, K.A.; Chapey, K.S.; Nanescu, S.E.; Huang, J.K. Creatine Enhances Mitochondrial-Mediated Oligodendrocyte Survival After Demyelinating Injury. J. Neurosci. 2017, 37, 1479–1492. [Google Scholar] [CrossRef]
- De Biasi, S.; Simone, A.M.; Bianchini, E.; Lo Tartaro, D.; Pecorini, S.; Nasi, M.; Patergnani, S.; Carnevale, G.; Gibellini, L.; Ferraro, D.; et al. Mitochondrial functionality and metabolism in T cells from progressive multiple sclerosis patients. Eur. J. Immunol. 2019. [Google Scholar] [CrossRef]
- Reiter, R.J.; Tan, D.X.; Rosales-Corral, S.; Galano, A.; Jou, M.J.; Acuna-Castroviejo, D. Melatonin Mitigates Mitochondrial Meltdown: Interactions with SIRT3. Int. J. Mol. Sci. 2018, 19, 2439. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Escames, G. Inhibition of Mitochondrial Pyruvate Dehydrogenase Kinase: A Proposed Mechanism by Which Melatonin Causes Cancer Cells to Overcome Aerobic Glycolysis, Limit Tumor Growth and Reverse Insensitivity to Chemotherapy. Melatonin. Res. 2019, 2, 105–119. [Google Scholar] [CrossRef]
- Anderson, G. Circadian and Local Melatonin: Role in Warburg Effect vs Oxidative Phosphorylation in Breast Cancer. Melatonin Res. 2019, in press. [Google Scholar]
- Wentling, M.; Lopez-Gomez, C.; Park, H.J.; Amatruda, M.; Ntranos, A.; Aramini, J.; Petracca, M.; Rusielewicz, T.; Chen, E.; Tolstikov, V.; et al. A metabolic perspective on CSF-mediated neurodegeneration in multiple sclerosis. Brain 2019, 142, 2756–2774. [Google Scholar] [CrossRef] [PubMed]
- Regenold, W.T.; Phatak, P.; Makley, M.J.; Stone, R.D.; Kling, M.A. Cerebrospinal fluid evidence of increased extra-mitochondrial glucose metabolism implicates mitochondrial dysfunction in multiple sclerosis disease progression. J. Neurol. Sci. 2008, 275, 106–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurz, J.; Brunkhorst, R.; Foerch, C.; Blum, L.; Henke, M.; Gabriel, L.; Ulshöfer, T.; Ferreirós, N.; Parnham, M.J.; Geisslinger, G.; et al. The relevance of ceramides and their synthesizing enzymes for multiple sclerosis. Clin. Sci. (Lond) 2018, 132, 1963–1976. [Google Scholar] [CrossRef] [Green Version]
- Albanese, M.; Zagaglia, S.; Landi, D.; Boffa, L.; Nicoletti, C.G.; Marciani, M.G.; Mandolesi, G.; Marfia, G.A.; Buttari, F.; Mori, F.; et al. Cerebrospinal fluid lactate is associated with multiple sclerosis disease progression. J. Neuroinflammation 2016, 13, 36. [Google Scholar] [CrossRef]
- La Rocca, C.; Carbone, F.; De Rosa, V.; Colamatteo, A.; Galgani, M.; Perna, F.; Lanzillo, R.; Brescia Morra, V.; Orefice, G.; Cerillo, I.; et al. Immunometabolic profiling of T cells from patients with relapsing-remitting multiple sclerosis reveals an impairment in glycolysis and mitochondrial respiration. Metabolism 2017, 77, 39–46. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Sun, H.; Bai, Y.; Zhi, F. Gut dysbiosis-derived exosomes trigger hepatic steatosis by transiting HMGB1 from intestinal to liver in mice. Biochem. Biophys. Res. Commun. 2019, 509, 767–772. [Google Scholar] [CrossRef]
- Zhen, C.; Wang, Y.; Li, D.; Zhang, W.; Zhang, H.; Yu, X.; Wang, X. Relationship of High-mobility group box 1 levels and multiple sclerosis: A systematic review and meta-analysis. Mult. Scler. Relat. Disord. 2019, 31, 87–92. [Google Scholar] [CrossRef]
- Doğan, H.O.; Yildiz, Ö.K. Serum NADPH oxidase concentrations and the associations with iron metabolism in relapsing remitting multiple sclerosis. J. Trace. Elem. Med. Biol. 2019, 55, 39–43. [Google Scholar] [CrossRef]
- Sonar, S.A.; Lal, G. The iNOS Activity during an Immune Response Controls the CNS Pathology in Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2019, 10, 710. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.; Antel, J.; Brück, W.; Kuhlmann, T. Contrasting potential of nitric oxide and peroxynitrite to mediate oligodendrocyte injury in multiple sclerosis. Glia 2007, 55, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Wilbanks, B.; Maher, L.J.; Rodriguez, M. Glial cells as therapeutic targets in progressive multiple sclerosis. Expert. Rev. Neurother. 2019, 19, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.J.; Woo, D.H.; Lee, P.R.; Pajevic, S.; Bukalo, O.; Huffman, W.C.; Wake, H.; Basser, P.J.; SheikhBahaei, S.; Lazarevic, V.; et al. Regulation of myelin structure and conduction velocity by perinodal astrocytes. Proc. Natl. Acad. Sci. USA 2018, 115, 11832–11837. [Google Scholar] [CrossRef] [Green Version]
- Petersen, M.A.; Ryu, J.K.; Chang, K.J.; Etxeberria, A.; Bardehle, S.; Mendiola, A.S.; Kamau-Devers, W.; Fancy, S.P.J.; Thor, A.; Bushong, E.A.; et al. Fibrinogen Activates BMP Signaling in Oligodendrocyte Progenitor Cells and Inhibits Remyelination after Vascular Damage. Neuron 2017, 96, 1003–1017. [Google Scholar] [CrossRef]
- Wang, G.; Dinkins, M.; He, Q.; Zhu, G.; Poirier, C.; Campbell, A.; Mayer-Proschel, M.; Bieberich, E. Astrocytes secrete exosomes enriched with proapoptotic ceramide and prostate apoptosis response 4 (PAR-4): Potential mechanism of apoptosis induction in Alzheimer disease (AD). J. Biol. Chem. 2012, 287, 21384–21395. [Google Scholar] [CrossRef]
- De Wit, N.M.; den Hoedt, S.; Martinez-Martinez, P.; Rozemuller, A.J.; Mulder, M.T.; de Vries, H.E. Astrocytic ceramide as possible indicator of neuroinflammation. J. Neuroinflammation 2019, 16, 48. [Google Scholar] [CrossRef]
- Becker, K.A.; Halmer, R.; Davies, L.; Henry, B.D.; Ziobro-Henry, R.; Decker, Y.; Liu, Y.; Gulbins, E.; Fassbender, K.; Walter, S. Blockade of Experimental Multiple Sclerosis by Inhibition of the Acid Sphingomyelinase/Ceramide System. Neurosignals 2017, 25, 88–97. [Google Scholar] [CrossRef]
- Van Doorn, R.; Nijland, P.G.; Dekker, N.; Witte, M.E.; Lopes-Pinheiro, M.A.; van het Hof, B.; Kooij, G.; Reijerkerk, A.; Dijkstra, C.; van van der Valk, P.; et al. Fingolimod attenuates ceramide-induced blood-brain barrier dysfunction in multiple sclerosis by targeting reactive astrocytes. Acta Neuropathol. 2012, 124, 397–410. [Google Scholar] [CrossRef]
- Scheiblich, H.; Schlütter, A.; Golenbock, D.T.; Latz, E.; Martinez-Martinez, P.; Heneka, M.T. Activation of the NLRP3 inflammasome in microglia: The role of ceramide. J. Neurochem. 2017, 143, 534–550. [Google Scholar] [CrossRef]
- Serbulea, V.; Upchurch, C.M.; Ahern, K.W.; Bories, G.; Voigt, P.; DeWeese, D.E.; Meher, A.K.; Harris, T.E.; Leitinger, N. Macrophages sensing oxidized DAMPs reprogram their metabolism to support redox homeostasis and inflammation through a TLR2-Syk-ceramide dependent mechanism. Mol. Metab. 2018, 7, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Jha, A.K.; Huang, S.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.N.; Zhu, Z.; Itokazu, Y.; Wang, G.; Dinkins, M.B.; Zhong, L.; Lin, H.P.; Elsherbini, A.; Leanhart, S.; Jiang, X.; et al. Novel function of ceramide for regulation of mitochondrial ATP release in astrocytes. J. Lipid. Res. 2018, 59, 488–506. [Google Scholar] [CrossRef] [Green Version]
- Warrington, A.E.; Asakura, K.; Bieber, A.; Ciric, B.; Van Keulen, V.; Kaveri, S.V.; Kyle, R.; Weinshenker, B.; Pease, L.; Rodriguez, M. Human Monoclonal Antibodies Promote Remyelination of Spinal Cord Lesions in a Model of Multiple Sclerosis. PNAS 2000, 97, 6820–6825. [Google Scholar] [CrossRef]
- Grassi, S.; Giussani, P.; Prioni, S.; Button, D.; Cao, J.; Hakimi, I.; Sarmiere, P.; Srinivas, M.; Cabitta, L.; Sonnino, S.; et al. Human Remyelination Promoting Antibody Stimulates Astrocytes Proliferation Through Modulation of the Sphingolipid Rheostat in Primary Rat Mixed Glial Cultures. Neurochem. Res. 2019, 44, 1460–1474. [Google Scholar] [CrossRef]
- Eisen, A.; Greenberg, B.M.; Bowen, J.D.; Arnold, D.L.; Caggiano, A.O. A double-blind, placebo-controlled, single ascending-dose study of remyelinating antibody rHIgM22 in people with multiple sclerosis. Mult. Scler. J. Exp. Transl. Clin. 2017, 3, 2055217317743097. [Google Scholar] [CrossRef]
- Podbielska, M.; Szulc, Z.M.; Kurowska, E.; Hogan, E.L.; Bielawski, J.; Bielawska, A.; Bhat, N.R. Cytokine-induced release of ceramide-enriched exosomes as a mediator of cell death signaling in an oligodendroglioma cell line. J. Lipid. Res. 2016, 57, 2028–2039. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, S.; Ray, S.K. Diverse Biological Functions of Sphingolipids in the CNS: Ceramide and Sphingosine Regulate Myelination in Developing Brain but Stimulate Demyelination during Pathogenesis of Multiple Sclerosis. J. Neurol. Psychol. 2017, 5. [Google Scholar] [CrossRef]
- Chami, M.; Halmer, R.; Schnoeder, L.; Anne Becker, K.; Meier, C.; Fassbender, K.; Gulbins, E.; Walter, S. Acid sphingomyelinase deficiency enhances myelin repair after acute and chronic demyelination. PLoS ONE 2017, 12, e0178622. [Google Scholar] [CrossRef]
- Novgorodov, S.A.; Voltin, J.R.; Gooz, M.A.; Li, L.; Lemasters, J.J.; Gudz, T.I. Acid sphingomyelinase promotes mitochondrial dysfunction due to glutamate-induced regulated necrosis. J. Lipid. Res. 2018, 59, 312–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granzotto, A.; Bomba, M.; Castelli, V.; Navarra, R.; Massetti, N.; d’Aurora, M.; Onofrj, M.; Cicalini, I.; Del Boccio, P.; Gatta, V.; et al. Inhibition of de novo ceramide biosynthesis affects aging phenotype in an in vitro model of neuronal senescence. Aging (Albany NY) 2019, 11, 6336–6357. [Google Scholar] [CrossRef] [PubMed]
- Kogot-Levin, A.; Saada, A. Ceramide and the mitochondrial respiratory chain. Biochimie 2014, 100, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.; Che, P.; Zhao, X.K.; Li, N.; Ding, Y.; Liu, J.; Li, S.; Ding, K.; Han, L.; Huang, Z.; et al. Molecular mechanism of tumour necrosis factor alpha regulates hypocretin (orexin) expression, sleep and behaviour. J. Cell. Mol. Med. 2019. [Google Scholar] [CrossRef]
- Si, X.; Shang, W.; Zhou, Z.; Strappe, P.; Wang, B.; Bird, A.; Blanchard, C. Gut Microbiome-Induced Shift of Acetate to Butyrate Positively Manages Dysbiosis in High Fat Diet. Mol. Nutr. Food. Res. 2018, 62. [Google Scholar] [CrossRef]
- Drazba, M.A.; Holásková, I.; Sahyoun, N.R.; Ventura Marra, M. Associations of Adiposity and Diet Quality with Serum Ceramides in Middle-Aged Adults with Cardiovascular Risk Factors. J. Clin. Med. 2019, 8, 527. [Google Scholar] [CrossRef]
- Stampanoni Bassi, M.; Iezzi, E.; Buttari, F.; Gilio, L.; Simonelli, I.; Carbone, F.; Micillo, T.; De Rosa, V.; Sica, F.; Furlan, R.; et al. Obesity worsens central inflammation and disability in multiple sclerosis. Mult. Scler. 2019, 1352458519853473. [Google Scholar] [CrossRef]
- Castro, K.; Ntranos, A.; Amatruda, M.; Petracca, M.; Kosa, P.; Chen, E.Y.; Morstein, J.; Trauner, D.; Watson, C.T.; Kiebish, M.A.; et al. Body Mass Index in Multiple Sclerosis modulates ceramide-induced DNA methylation and disease course. EBioMedicine 2019, 43, 392–410. [Google Scholar] [CrossRef] [Green Version]
- De Melo, L.G.P.; Nunes, S.O.V.; Anderson, G.; Vargas, H.O.; Barbosa, D.S.; Galecki, P.; Carvalho, A.F.; Maes, M. Shared metabolic and immune-inflammatory, oxidative and nitrosative stress pathways in the metabolic syndrome and mood disorders. Prog. Neuropsychopharmacol Biol. Psychiatry 2017, 78, 34–50. [Google Scholar] [CrossRef]
- Rohrbach, T.D.; Asgharpour, A.; Maczis, M.A.; Montefusco, D.; Cowart, L.A.; Bedossa, P.; Sanyal, A.J.; Spiegel, S. FTY720/fingolimod decreases hepatic steatosis and expression of fatty acid synthase in diet-induced nonalcoholic fatty liver disease in mice. J. Lipid. Res. 2019, 60, 1311–1322. [Google Scholar] [CrossRef]
- Rumah, K.R.; Vartanian, T.K.; Fischetti, V.A. Oral Multiple Sclerosis Drugs Inhibit the In vitro Growth of Epsilon Toxin Producing Gut Bacterium, Clostridium perfringens. Front. Cell. Infect. Microbiol. 2017, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamane, M.; Yamane, S. The induction of colonocyte differentiation in CaCo-2 cells by sodium butyrate causes an increase in glucosylceramide synthesis in order to avoid apoptosis based on ceramide. Arch. Biochem. Biophys. 2007, 459, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Messner, M.C.; Cabot, M.C. Glucosylceramide in humans. Adv. Exp. Med. Biol. 2010, 688, 156–164. [Google Scholar] [PubMed]
- Qin, J.; Sikkema, A.H.; van der Bij, K.; de Jonge, J.C.; Klappe, K.; Nies, V.; Jonker, J.W.; Kok, J.W.; Hoekstra, D.; Baron, W. GD1a Overcomes Inhibition of Myelination by Fibronectin via Activation of Protein Kinase A: Implications for Multiple Sclerosis. J. Neurosci. 2017, 37, 9925–9938. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Hait, N.C.; Maceyka, M.; Colaco, A.; Maczis, M.; Wassif, C.A.; Cougnoux, A.; Porter, F.D.; Milstien, S.; Platt, N.; et al. FTY720/fingolimod increases NPC1 and NPC2 expression and reduces cholesterol and sphingolipid accumulation in Niemann-Pick type C mutant fibroblasts. FASEB J. 2017, 31, 1719–1730. [Google Scholar] [CrossRef]
- Parkar, S.G.; Kalsbeek, A.; Cheeseman, J.F. Potential Role for the Gut Microbiota in Modulating Host Circadian Rhythms and Metabolic Health. Microorganisms 2019, 7, 41. [Google Scholar] [CrossRef]
- Jang, Y.S.; Kang, Y.J.; Kim, T.J.; Bae, K. Temporal expression profiles of ceramide and ceramide-related genes in wild-type and mPer1/mPer2 double knockout mice. Mol. Biol. Rep. 2012, 39, 4215–4221. [Google Scholar] [CrossRef]
- Lenercept Multiple Sclerosis Study Group. TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. The Lenercept Multiple Sclerosis Study Group and the University of British Columbia MS/MRI Analysis Group. Neurology 1999, 53, 457–465. [Google Scholar] [CrossRef]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [Green Version]
- Nevárez, N.; de Lecea, L. Recent advances in understanding the roles of hypocretin/orexin in arousal, affect, and motivation. F1000Res 2018, 7. [Google Scholar] [CrossRef]
- Shariq, A.S.; Rosenblat, J.D.; Alageel, A.; Mansur, R.B.; Rong, C.; Ho, R.C.; Ragguett, R.M.; Pan, Z.; Brietzke, E.; McIntyre, R.S. Evaluating the role of orexins in the pathophysiology and treatment of depression: A comprehensive review. Prog. Neuropsychopharmacol Biol. Psychiatry 2019, 92, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gencer, M.; Akbayır, E.; Şen, M.; Arsoy, E.; Yılmaz, V.; Bulut, N.; Tüzün, E.; Türkoğlu, R. Serum orexin-A levels are associated with disease progression and motor impairment in multiple sclerosis. Neurol. Sci. 2019, 40, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Becquet, L.; Abad, C.; Leclercq, M.; Miel, C.; Jean, L.; Riou, G.; Couvineau, A.; Boyer, O.; Tan, Y.V. Systemic administration of orexin A ameliorates established experimental autoimmune encephalomyelitis by diminishing neuroinflammation. J. Neuroinflammation 2019, 16, 64. [Google Scholar] [CrossRef] [PubMed]
- Fatemi, I.; Shamsizadeh, A.; Ayoobi, F.; Taghipour, Z.; Sanati, M.H.; Roohbakhsh, A.; Motevalian, M. Role of orexin-A in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2016, 291, 101–109. [Google Scholar] [CrossRef]
- Qiu, W.; Raven, S.; Wu, J.S.; Bundell, C.; Hollingsworth, P.; Carroll, W.M.; Mastaglia, F.L.; Kermode, A.G. Hypothalamic lesions in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 819–822. [Google Scholar] [CrossRef]
- Omori, Y.; Kanbayashi, T.; Imanishi, A.; Tsutsui, K.; Sagawa, Y.; Kikuchi, Y.S.; Takeshima, M.; Yoshizawa, K.; Uemura, S.; Shimizu, T. Orexin/hypocretin levels in the cerebrospinal fluid and characteristics of patients with myotonic dystrophy type 1 with excessive daytime sleepiness. Neuropsychiatr. Dis. Treat. 2018, 14, 451–457. [Google Scholar] [CrossRef]
- Suzuki, K.; Miyamoto, M.; Miyamoto, T.; Matsubara, T.; Inoue, Y.; Iijima, M.; Mizuno, S.; Horie, J.; Hirata, K.; Shimizu, T.; et al. Cerebrospinal fluid orexin-A levels in systemic lupus erythematosus patients presenting with excessive daytime sleepiness. Lupus 2018, 27, 1847–1853. [Google Scholar] [CrossRef]
- Braga, D.M.; Prado, G.F.; Bichueti, D.B.; Oliveira, E.M. Positive correlation between functional disability, excessive daytime sleepiness, and fatigue in relapsing-remitting multiple sclerosis. Arq. Neuropsiquiatr. 2016, 74, 433–438. [Google Scholar] [CrossRef] [Green Version]
- Labuz-Roszak, B.; Kubicka-Bączyk, K.; Pierzchała, K.; Machowska-Majchrzak, A.; Skrzypek, M. Fatigue and its association with sleep disorders, depressive symptoms and anxiety in patients with multiple sclerosis. Neurol. Neurochir. Pol. 2012, 46, 309–317. [Google Scholar] [CrossRef]
- Fellows Maxwell, K.; Wahls, T.; Browne, R.W.; Rubenstein, L.; Bisht, B.; Chenard, C.A.; Snetselaar, L.; Weinstock-Guttman, B.; Ramanathan, M. Lipid profile is associated with decreased fatigue in individuals with progressive multiple sclerosis following a diet-based intervention: Results from a pilot study. PLoS ONE 2019, 14, e0218075. [Google Scholar] [CrossRef]
- Lassiter, K.; Greene, E.; Piekarski, A.; Faulkner, O.B.; Hargis, B.M.; Bottje, W.; Dridi, S. Orexin system is expressed in avian muscle cells and regulates mitochondrial dynamics. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, R173–R187. [Google Scholar] [CrossRef] [Green Version]
- Türkoğlu, R.; Benbir, G.; Özyurt, S.; Arsoy, E.; Akbayır, E.; Turan, S.; Karadeniz, D.; Yılmaz, V.; Gencer, M.; Tüzün, E. Sleep disturbance and cognitive decline in multiple sclerosis patients with isolated optic neuritis as the first demyelinating event. Int. Ophthalmol. 2019, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Duffy, C.M.; Hofmeister, J.J.; Nixon, J.P.; Butterick, T.A. High fat diet increases cognitive decline and neuroinflammation in a model of orexin loss. Neurobiol. Learn. Mem. 2019, 157, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Argüelles, A.; Méndez-Huerta, M.A.; Lozano, C.D.; Ruiz-Argüelles, G.J. Metabolomic profile of insulin resistance in patients with multiple sclerosis is associated to the severity of the disease. Mult. Scler. Relat. Disord. 2018, 316–321. [Google Scholar] [CrossRef]
- Liu, Y.; Zhao, Y.; Guo, L. Effects of orexin A on glucose metabolism in human hepatocellular carcinoma invvitro via PI3K/Akt/mTOR-dependent and -independent mechanism. Mol. Cell. Endocrinol. 2016, 420, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Tsuneki, H.; Tokai, E.; Sugawara, C.; Wada, T.; Sakurai, T.; Sasaoka, T. Hypothalamic orexin prevents hepatic insulin resistance induced by social defeat stress in mice. Neuropeptides 2013, 47, 213–219. [Google Scholar] [CrossRef]
- Tsuneki, H.; Tokai, E.; Nakamura, Y.; Takahashi, K.; Fujita, M.; Asaoka, T.; Kon, K.; Anzawa, Y.; Wada, T.; Takasaki, I.; et al. Hypothalamic orexin prevents hepatic insulin resistance via daily bidirectional regulation of autonomic nervous system in mice. Diabetes 2015, 64, 459–470. [Google Scholar] [CrossRef]
- Stacchiotti, A.; Favero, G.; Lavazza, A.; Garcia-Gomez, R.; Monsalve, M.; Rezzani, R. Perspective: Mitochondria-ER Contacts in Metabolic Cellular Stress Assessed by Microscopy. Cells 2018, 8, 5. [Google Scholar] [CrossRef]
- Huppke, B.; Ellenberger, D.; Hummel, H.; Stark, W.; Röbl, M.; Gärtner, J.; Huppke, P. Association of Obesity with Multiple Sclerosis Risk and Response to First-line Disease Modifying Drugs in Children. JAMA. Neurol. 2019, 76, 1157–1165. [Google Scholar] [CrossRef]
- Harroud, A.; Morris, J.A.; Forgetta, V.; Mitchell, R.; Smith, G.D.; Sawcer, S.J.; Richards, J.B. Effect of age at puberty on risk of multiple sclerosis: A mendelian randomization study. Neurology 2019, 92, e1803–e1810. [Google Scholar] [CrossRef]
- Contreras, C.; González-García, I.; Martínez-Sánchez, N.; Seoane-Collazo, P.; Jacas, J.; Morgan, D.A.; Serra, D.; Gallego, R.; Gonzalez, F.; Casals, N.; et al. Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell. Rep. 2014, 9, 366–377. [Google Scholar] [CrossRef]
- Cox, J.E.; Powley, T.L. Intragastric pair feeding fails to prevent VMH obesity or hyperinsulinemia. Am. J. Physiol. 1981, 240, E566–E572. [Google Scholar] [CrossRef] [PubMed]
- Hirschberg, P.R.; Sarkar, P.; Teegala, S.B.; Routh, V.H. Ventromedial hypothalamus glucose-inhibited neurones: A role in glucose and energy homeostasis? J. Neuroendocrinol. 2019, 22, e12773. [Google Scholar] [CrossRef] [PubMed]
- Tupone, D.; Madden, C.J.; Cano, G.; Morrison, S.F. An orexinergic projection from perifornical hypothalamus to raphe pallidus increases rat brown adipose tissue thermogenesis. J. Neurosci. 2011, 31, 15944–15955. [Google Scholar] [CrossRef] [PubMed]
- Messina, G.; Valenzano, A.; Moscatelli, F.; Salerno, M.; Lonigro, A.; Esposito, T.; Monda, V.; Corso, G.; Messina, A.; Viggiano, A.; et al. Role of Autonomic Nervous System and Orexinergic System on Adipose Tissue. Front. Physiol. 2017, 8, 137. [Google Scholar] [CrossRef] [Green Version]
- Korim, W.S.; Llewellyn-Smith, I.J.; Verberne, A.J. Activation of Medulla-Projecting Perifornical Neurons Modulates the Adrenal Sympathetic Response to Hypoglycemia: Involvement of Orexin Type 2 (OX2-R) Receptors. Endocrinology 2016, 157, 810–819. [Google Scholar] [CrossRef]
- Aston-Jones, G.; Smith, R.J.; Sartor, G.C.; Moorman, D.E.; Massi, L.; Tahsili-Fahadan, P.; Richardson, K.A. Lateral hypothalamic orexin/hypocretin neurons: A role in reward-seeking and addiction. Brain. Res. 2010, 1314, 74–90. [Google Scholar] [CrossRef] [Green Version]
- Bonci, A.; Borgland, S. Role of orexin/hypocretin and CRF in the formation of drug-dependent synaptic plasticity in the mesolimbic system. Neuropharmacology 2009, 56, 107–111. [Google Scholar] [CrossRef]
- Kantor, S.; Mochizuki, T.; Janisiewicz, A.M.; Clark, E.; Nishino, S.; Scammell, T.E. Orexin neurons are necessary for the circadian control of REM sleep. Sleep 2009, 32, 1127–1134. [Google Scholar] [CrossRef]
- Wang, F.; Mei, F. Kappa opioid receptor and oligodendrocyte remyelination. Vitam. Horm. 2019, 111, 281–297. [Google Scholar] [CrossRef]
- Anderson, G. Pathoetiology and Pathophysiology of Borderline Personality: Role of Prenatal Factors, Gut Microbiome, mu- and kappa-opioid receptors in Amygdala-PFC Interactions. Prog. NeuroPsychoPharm. Biol. Psychiatr 2019, in press. [Google Scholar] [CrossRef]
- Cao, Z.; Liu, L.; Van Winkle, D.M. Activation of delta- and kappa-opioid receptors by opioid peptides protects cardiomyocytes via KATP channels. Am. J. Physiol. Heart. Circ. Physiol. 2003, 285, H1032–H1039. [Google Scholar] [CrossRef] [PubMed]
- Virgili, N.; Mancera, P.; Chanvillard, C.; Wegner, A.; Wappenhans, B.; Rodríguez, M.J.; Infante-Duarte, C.; Espinosa-Parrilla, J.F.; Pugliese, M. Diazoxide attenuates autoimmune encephalomyelitis and modulates lymphocyte proliferation and dendritic cell functionality. J. Neuroimmune. Pharmacol. 2014, 9, 558–568. [Google Scholar] [CrossRef] [PubMed]
- Ardianto, C.; Yonemochi, N.; Yamamoto, S.; Yang, L.; Takenoya, F.; Shioda, S.; Nagase, H.; Ikeda, H.; Kamei, J. Opioid systems in the lateral hypothalamus regulate feeding behavior through orexin and GABA neurons. Neuroscience 2016, 320, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Staton, C.D.; Yaeger, J.D.W.; Khalid, D.; Haroun, F.; Fernandez, B.S.; Fernandez, J.S.; Summers, B.K.; Summers, T.R.; Sathyanesan, M.; Newton, S.S.; et al. Orexin 2 receptor stimulation enhances resilience, while orexin 2 inhibition promotes susceptibility, to social stress, anxiety and depression. Neuropharmacology 2018, 143, 79–94. [Google Scholar] [CrossRef]
- Grafe, L.A.; Geng, E.; Corbett, B.; Urban, K.; Bhatnagar, S. Sex- and Stress-Dependent Effects on Dendritic Morphology and Spine Densities in Putative Orexin Neurons. Neuroscience 2019. [Google Scholar] [CrossRef]
- AlZahrani, A.S.; Alshamrani, F.J.; Al-Khamis, F.A.; Al-Sulaiman, A.A.; Al Ghamdi, W.S.; Al Ghamdi, O.A.; Mohammad, M.Y.; Alshayea, M.S.; Alhazmi, R.A.; Alkhaja, M.A. Association of acute stress with multiple sclerosis onset and relapse in Saudi Arabia. Saudi. Med. J. 2019, 40, 372–378. [Google Scholar] [CrossRef]
- Anderson, G.; Rodriguez, M. Multiple sclerosis: The role of melatonin and N-acetylserotonin. Mult. Scler. Relat. Disord. 2015, 4, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G.; Rodriguez, M. Multiple sclerosis, seizures, and antiepileptics: Role of IL-18, IDO, and melatonin. Eur. J. Neurol. 2011, 18, 680–685. [Google Scholar] [CrossRef]
- Meknatkhah, S.; Sharif Dashti, P.; Mousavi, M.S.; Zeynali, A.; Ahmadian, S.; Karima, S.; Saboury, A.A.; Riazi, G.H. Psychological stress effects on myelin degradation in the cuprizone-induced model of demyelination. Neuropathology 2019, 39, 14–21. [Google Scholar] [CrossRef]
- Adidharma, W.; Deats, S.P.; Ikeno, T.; Lipton, J.W.; Lonstein, J.S.; Yan, L. Orexinergic modulation of serotonin neurons in the dorsal raphe of a diurnal rodent, Arvicanthis niloticus. Horm. Behav. 2019, 116, 104584. [Google Scholar] [CrossRef]
- Sharma, R.; Sahota, P.; Thakkar, M.M. Melatonin promotes sleep in mice by inhibiting orexin neurons in the perifornical lateral hypothalamus. J. Pineal. Res. 2018, 65, e12498. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, J.D.; Hauser, F.; deLecea, L.; Sutcliffe, J.G.; Kilduff, T.S.; Calgari, C.; Pévet, P.; Simonneaux, V. Hypocretin (orexin) in the rat pineal gland: A central transmitter with effects on noradrenaline-induced release of melatonin. Eur. J. Neurosci. 2001, 14, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Buonfiglio, D.; Parthimos, R.; Dantas, R.; Cerqueira Silva, R.; Gomes, G.; Andrade-Silva, J.; Ramos-Lobo, A.; Amaral, F.G.; Matos, R.; Sinésio, J., Jr.; et al. Melatonin Absence Leads to Long-Term Leptin Resistance and Overweight in Rats. Front. Endocrinol (Lausanne) 2018, 9, 122. [Google Scholar] [CrossRef] [PubMed]
- Mäkelä, K.A.; Karhu, T.; Jurado Acosta, A.; Vakkuri, O.; Leppäluoto, J.; Herzig, K.H. Plasma Orexin-A Levels Do Not Undergo Circadian Rhythm in Young Healthy Male Subjects. Front. Endocrinol (Lausanne) 2018, 9, 710. [Google Scholar] [CrossRef]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell. Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef]
- Yi, C.X.; Serlie, M.J.; Ackermans, M.T.; Foppen, E.; Buijs, R.M.; Sauerwein, H.P.; Fliers, E.; Kalsbeek, A. A major role for perifornical orexin neurons in the control of glucose metabolism in rats. Diabetes 2009, 58, 1998–2005. [Google Scholar] [CrossRef]
- Fetissov, S.O.; Huang, P.; Zhang, Q.; Mimura, J.; Fujii-Kuriyama, Y.; Rannug, A.; Hökfelt, T.; Ceccatelli, S. Expression of hypothalamic neuropeptides after acute TCDD treatment and distribution of Ah receptor repressor. Regul. Pept. 2004, 119, 113–124. [Google Scholar] [CrossRef]
- Neavin, D.R.; Liu, D.; Ray, B.; Weinshilboum, R.M. The Role of the Aryl Hydrocarbon Receptor (AHR) in Immune and Inflammatory Diseases. Int. J. Mol. Sci. 2018, 19, 3851. [Google Scholar] [CrossRef]
- Kostic, M.; Zivkovic, N.; Cvetanovic, A.; Stojanovic, I.; Colic, M. IL-17 signalling in astrocytes promotes glutamate excitotoxicity: Indications for the link between inflammatory and neurodegenerative events in multiple sclerosis. Mult. Scler. Relat. Disord. 2017, 11, 12–17. [Google Scholar] [CrossRef]
- Wang, D.D.; Jin, M.F.; Zhao, D.J.; Ni, H. Reduction of Mitophagy-Related Oxidative Stress and Preservation of Mitochondria Function Using Melatonin Therapy in an HT22 Hippocampal Neuronal Cell Model of Glutamate-Induced Excitotoxicity. Front. Endocrinol (Lausanne) 2019, 10, 550. [Google Scholar] [CrossRef] [Green Version]
- Shu, Q.; Zhang, J.; Ma, W.; Lei, Y.; Zhou, D. Orexin-A promotes Glu uptake by OX1R/PKCα/ERK1/2/GLT-1 pathway in astrocytes and protects co-cultured astrocytes and neurons against apoptosis in anoxia/hypoglycemic injury in vitro. Mol. Cell. Biochem. 2017, 425, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Sathornsumetee, S.; McGavern, D.B.; Ure, D.R.; Rodriguez, M. Quantitative ultrastructural analysis of a single spinal cord demyelinated lesion predicts total lesion load, axonal loss, and neurological dysfunction in a murine model of multiple sclerosis. Am. J. Pathol. 2000, 157, 1365–1376. [Google Scholar] [CrossRef]
- López-González, A.; Álvarez-Sánchez, N.; Lardone, P.J.; Cruz-Chamorro, I.; Martínez-López, A.; Guerrero, J.M.; Reiter, R.J.; Carrillo-Vico, A. Melatonin treatment improves primary progressive multiple sclerosis: A case report. J. Pineal. Res. 2015, 58, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Idowu, A.J.; Kumar, S.L.; Yidong, B.; Russel, R. Melatonin modulates neuronal mitochondria function during normal ageing in mice. Niger. J. Physiol. Sci. 2017, 32, 145–152. [Google Scholar]
- Pozuelo-Rubio, M. Proteomic and biochemical analysis of 14-3-3-binding proteins during C2-ceramide-induced apoptosis. FEBS J. 2010, 277, 3321–3342. [Google Scholar] [CrossRef]
- Naia, L.; Cunha-Oliveira, T.; Rodrigues, J.; Rosenstock, T.R.; Oliveira, A.; Ribeiro, M.; Carmo, C.; Oliveira-Sousa, S.I.; Duarte, A.I.; Hayden, M.R.; et al. Histone Deacetylase Inhibitors Protect Against Pyruvate Dehydrogenase Dysfunction in Huntington’s Disease. J. Neurosci. 2017, 37, 2776–2794. [Google Scholar] [CrossRef]
- Oliveira, S.R.; Kallaur, A.P.; Lopes, J.; Colado Simão, A.N.; Reiche, E.M.; de Almeida, E.R.D.; Morimoto, H.K.; de Carvalho Jennings de Pereira, W.L.; Alfieri, D.F.; Flauzino, T.; et al. Insulin resistance, atherogenicity, and iron metabolism in multiple sclerosis with and without depression: Associations with inflammatory and oxidative stress biomarkers and uric acid. Psychiatry Res. 2017, 250, 113–120. [Google Scholar] [CrossRef]
- Jorissen, W.; Wouters, E.; Bogie, J.F.; Vanmierlo, T.; Noben, J.P.; Sviridov, D.; Hellings, N.; Somers, V.; Valcke, R.; Vanwijmeersch, B.; et al. Relapsing-remitting multiple sclerosis patients display an altered lipoprotein profile with dysfunctional HDL. Sci. Rep. 2017, 7, 43410. [Google Scholar] [CrossRef]
- Yuyama, K.; Mitsutake, S.; Igarashi, Y. Pathological roles of ceramide and its metabolites in metabolic syndrome and Alzheimer’s disease. Biochim. Biophys. Acta. 2014, 1841, 793–798. [Google Scholar] [CrossRef]
- An, H.; Cho, M.H.; Kim, D.H.; Chung, S.; Yoon, S.Y. Orexin Impairs the Phagocytosis and Degradation of Amyloid-β Fibrils by Microglial Cells. J. Alzheimers. Dis. 2017, 58, 253–261. [Google Scholar] [CrossRef]
- Hu, L.; Zhang, S.; Wen, H.; Liu, T.; Cai, J.; Du, D.; Zhu, D.; Chen, F.; Xia, C. Melatonin decreases M1 polarization via attenuating mitochondrial oxidative damage depending on UCP2 pathway in prorenin-treated microglia. PLoS ONE 2019, 14, e0212138. [Google Scholar] [CrossRef] [PubMed]
- Song, J. Pineal gland dysfunction in Alzheimer’s disease: Relationship with the immune-pineal axis, sleep disturbance, and neurogenesis. Mol. Neurodegener. 2019, 14, 28. [Google Scholar] [CrossRef] [PubMed]
- Milanova, I.V.; Kalsbeek, M.J.T.; Wang, X.L.; Korpel, N.L.; Stenvers, D.J.; Wolff, S.E.C.; de Goede, P.; Heijboer, A.C.; Fliers, E.; la Fleur, S.E.; et al. Diet-Induced Obesity Disturbs Microglial Immunometabolism in a Time-of-Day Manner. Front. Endocrinol (Lausanne) 2019, 10, 424. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.W.; Liu, X.; Pradoldej, S.; Tosini, G.; Chang, Q.; Iuvone, P.M.; Ye, K. N-acetylserotonin activates TrkB receptor in a circadian rhythm. Proc. Natl. Acad. Sci. USA 2010, 107, 3876–3881. [Google Scholar] [CrossRef]
- Herman, S.; Åkerfeldt, T.; Spjuth, O.; Burman, J.; Kultima, K. Biochemical Differences in Cerebrospinal Fluid between Secondary Progressive and Relapsing–Remitting Multiple Sclerosis. Cells 2019, 8, 84. [Google Scholar] [CrossRef]
- Zorlu, N.; Hoffjan, S.; Haghikia, A.; Deyneko, I.V.; Epplen, J.T. Evaluation of variation in genes of the arylhydrocarbon receptor pathway for an association with multiple sclerosis. J. Neuroimmunol. 2019, 334, 576979. [Google Scholar] [CrossRef]
- Dowling, P.; Ming, X.; Raval, S.; Husar, W.; Casaccia-Bonnefil, P.; Chao, M.; Cook, S.; Blumberg, B. Up-regulated p75NTR neurotrophin receptor on glial cells in MS plaques. Neurology 1999, 53, 1676–1682. [Google Scholar] [CrossRef]
- Piao, C.; Ralay Ranaivo, H.; Rusie, A.; Wadhwani, N.; Koh, S.; Wainwright, M.S. Thrombin decreases expression of the glutamate transporter GLAST and inhibits glutamate uptake in primary cortical astrocytes via the Rho kinase pathway. Exp. Neurol. 2015, 273, 288–300. [Google Scholar] [CrossRef]
- Yates, R.L.; Esiri, M.M.; Palace, J.; Jacobs, B.; Perera, R.; DeLuca, G.C. Fibrin(ogen) and neurodegeneration in the progressive multiple sclerosis cortex. Ann. Neurol. 2017, 82, 259–270. [Google Scholar] [CrossRef]
- Merlini, M.; Rafalski, V.A.; Rios Coronado, P.E.; Gill, T.M.; Ellisman, M.; Muthukumar, G.; Subramanian, K.S.; Ryu, J.K.; Syme, C.A.; Davalos, D.; et al. Fibrinogen Induces Microglia-Mediated Spine Elimination and Cognitive Impairment in an Alzheimer’s Disease Model. Neuron 2019, 101, 1099–1108. [Google Scholar] [CrossRef]
- Ziliotto, N.; Bernardi, F.; Jakimovski, D.; Zivadinov, R. Coagulation Pathways in Neurological Diseases: Multiple Sclerosis. Front. Neurol. 2019, 10, 409. [Google Scholar] [CrossRef]
- Plantone, D.; Inglese, M.; Salvetti, M.; Koudriavtseva, T. A Perspective of Coagulation Dysfunction in Multiple Sclerosis and in Experimental Allergic Encephalomyelitis. Front. Neurol. 2019, 9, 1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [Green Version]
- Saluk-Bijak, J.; Dziedzic, A.; Bijak, M. Pro-Thrombotic Activity of Blood Platelets in Multiple Sclerosis. Cells 2019, 8, 110. [Google Scholar] [CrossRef]
- Dziedzic, A.; Bijak, M. Interactions between platelets and leukocytes in pathogenesis of multiple sclerosis. Adv. Clin. Exp. Med. 2019, 28, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Jäckel, S.; Kiouptsi, K.; Lillich, M.; Hendrikx, T.; Khandagale, A.; Kollar, B.; Hörmann, N.; Reiss, C.; Subramaniam, S.; Wilms, E.; et al. Gut microbiota regulate hepatic von Willebrand factor synthesis and arterial thrombus formation via Toll-like receptor-2. Blood 2017, 130, 542–553. [Google Scholar] [CrossRef] [Green Version]
- Peer, C.J.; Hall, O.M.; Sissung, T.M.; Piekarz, R.; Balasubramaniam, S.; Bates, S.E.; Figg, W.D. A population pharmacokinetic/toxicity model for the reduction of platelets during a 48-h continuous intravenous infusion of the histone deacetylase inhibitor belinostat. Cancer. Chemother. Pharmacol. 2018, 82, 565–570. [Google Scholar] [CrossRef]
- Larsson, P.; Alwis, I.; Niego, B.; Sashindranath, M.; Fogelstrand, P.; Wu, M.C.; Glise, L.; Magnusson, M.; Daglas, M.; Bergh, N.; et al. Valproic acid selectively increases vascular endothelial tissue-type plasminogen activator production and reduces thrombus formation in the mouse. J. Thromb. Haemost. 2016, 14, 2496–2508. [Google Scholar] [CrossRef] [Green Version]
- Münzer, P.; Borst, O.; Walker, B.; Schmid, E.; Feijge, M.A.; Cosemans, J.M.; Chatterjee, M.; Schmidt, E.M.; Schmidt, S.; Towhid, S.T.; et al. Acid sphingomyelinase regulates platelet cell membrane scrambling, secretion, and thrombus formation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 61–71. [Google Scholar] [CrossRef]
- Bijak, M.; Olejnik, A.; Rokita, B.; Morel, A.; Dziedzic, A.; Miller, E.; Saluk-Bijak, J. Increased level of fibrinogen chains in the proteome of blood platelets in secondary progressive multiple sclerosis patients. J. Cell. Mol. Med. 2019, 23, 3476–3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Li, D.; Zhu, P.; Hu, S.; Hu, N.; Ma, S.; Zhang, Y.; Han, T.; Ren, J.; Cao, F.; et al. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARγ/FUNDC1/mitophagy pathways. J. Pineal. Res. 2017, 63. [Google Scholar] [CrossRef] [PubMed]
- Girish, K.S.; Paul, M.; Thushara, R.M.; Hemshekhar, M.; Shanmuga Sundaram, M.; Rangappa, K.S.; Kemparaju, K. Melatonin elevates apoptosis in human platelets via ROS mediated mitochondrial damage. Biochem. Biophys. Res. Commun. 2013, 438, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.; Maes, M. How Immune-inflammatory Processes Link CNS and Psychiatric Disorders: Classification and Treatment Implications. CNS. Neurol. Disord. Drug. Targets 2017, 16, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Astakhova, A.A.; Chistyakov, D.V.; Sergeeva, M.G.; Reiser, G. Regulation of the ARE-binding proteins, TTP (tristetraprolin) and HuR (human antigen R), in inflammatory response in astrocytes. Neurochem. Int. 2018, 118, 82–90. [Google Scholar] [CrossRef]
- Guo, Y.; Hong, W.; Wang, X.; Zhang, P.; Körner, H.; Tu, J.; Wei, W. MicroRNAs in Microglia: How do MicroRNAs Affect Activation, Inflammation, Polarization of Microglia and Mediate the Interaction Between Microglia and Glioma? Front. Mol. Neurosci. 2019, 12, 125. [Google Scholar] [CrossRef]
- Bae, J.E.; Park, S.J.; Hong, Y.; Jo, D.S.; Lee, H.; Park, N.Y.; Kim, J.B.; Park, H.J.; Bunch, H.; Chang, J.H.; et al. Loss of RNA binding protein, human antigen R enhances mitochondrial elongation by regulating Drp1 expression in SH-SY5Y cells. Biochem. Biophys. Res. Commun. 2019, 516, 713–718. [Google Scholar] [CrossRef]
- Liu, Y.; Wei, W.; Wang, Y.; Wan, C.; Bai, Y.; Sun, X.; Ma, J.; Zheng, F. TNF-α/calreticulin dual signaling induced NLRP3 inflammasome activation associated with HuR nucleocytoplasmic shuttling in rheumatoid arthritis. Inflamm. Res. 2019, 68, 597–611. [Google Scholar] [CrossRef]
- Torun, A.; Enayat, S.; Sheraj, I.; Tunçer, S.; Ülgen, D.H.; Banerjee, S. Butyrate mediated regulation of RNA binding proteins in the post-transcriptional regulation of inflammatory gene expression. Cell Signal 2019, 64, 109410. [Google Scholar] [CrossRef]
- Rhead, B.; Shao, X.; Graves, J.S.; Chitnis, T.; Waldman, A.T.; Lotze, T.; Schreiner, T.; Belman, A.; Krupp, L.; Greenberg, B.M.; et al. US Network of Pediatric MS Centers.miRNA contributions to pediatric-onset multiple sclerosis inferred from GWAS. Ann. Clin. Transl. Neurol. 2019, 6, 1053–1061. [Google Scholar] [CrossRef]
- Lee, J.; Park, E.J.; Kiyono, H. MicroRNA-orchestrated pathophysiologic control in gut homeostasis and inflammation. BMB Rep. 2016, 49, 263–269. [Google Scholar] [CrossRef]
- Lim, S.M.; Park, S.H.; Lee, J.H.; Kim, S.H.; Kim, J.Y.; Min, J.K.; Lee, G.M.; Kim, Y.G. Differential expression of microRNAs in recombinant Chinese hamster ovary cells treated with sodium butyrate using digital RNA counting. J. Biotechnol. 2018, 283, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Teymoori-Rad, M.; Mozhgani, S.H.; Zarei-Ghobadi, M.; Sahraian, M.A.; Nejati, A.; Amiri, M.M.; Shokri, F.; Marashi, S.M. Integrational analysis of miRNAs data sets as a plausible missing linker between Epstein-Barr virus and vitamin D in relapsing remitting MS patients. Gene 2019, 689, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Zhang, J. Dietary Modulation of Intestinal Microbiota: Future Opportunities in Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Front. Microbiol. 2019, 10, 740. [Google Scholar] [CrossRef] [PubMed]
- Eftekharian, M.M.; Komaki, A.; Mazdeh, M.; Arsang-Jang, S.; Taheri, M.; Ghafouri-Fard, S. Expression Profile of Selected MicroRNAs in the Peripheral Blood of Multiple Sclerosis Patients: A Multivariate Statistical Analysis with ROC Curve to Find New Biomarkers for Fingolimod. J. Mol. Neurosci. 2019, 68, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Yamakawa, T.; Kurauchi, Y.; Hisatsune, A.; Seki, T.; Katsuki, H. Endogenous Nitric Oxide Inhibits, Whereas Awakening Stimuli Increase, the Activity of a Subset of Orexin Neurons. Biol. Pharm. Bull. 2018, 41, 1859–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herden, L.; Weissert, R. The Impact of Coffee and Caffeine on Multiple Sclerosis Compared to Other Neurodegenerative Diseases. Front. Nutr. 2018, 5, 133. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Xi, N.N.; Chen, Y.; Shang, X.F.; Hu, Q.; Chen, J.F.; Zheng, R.Y. Chronic caffeine treatment protects against experimental autoimmune encephalomyelitis in mice: Therapeutic window and receptor subtype mechanism. Neuropharmacology 2014, 86, 203–211. [Google Scholar] [CrossRef]
- Schnuck, J.K.; Gould, L.M.; Parry, H.A.; Johnson, M.A.; Gannon, N.P.; Sunderland, K.L.; Vaughan, R.A. Metabolic effects of physiological levels of caffeine in myotubes. J. Physiol. Biochem. 2018, 74, 35–45. [Google Scholar] [CrossRef]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; de Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dähling, S.; Kastenmüller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8+ T Cells. Immunity. 2019, 51, 285–297. [Google Scholar] [CrossRef]
- Otsuka, M.; Kato, K.; Murai, I.; Asai, S.; Iwasaki, A.; Arakawa, Y. Roles of nocturnal melatonin and the pineal gland in modulation of water-immersion restraint stress-induced gastric mucosal lesions in rats. J. Pineal. Res. 2001, 30, 82–86. [Google Scholar] [CrossRef]
- George, A. Integrating Pathophysiology in Migraine: Role of the Gut Microbiome and Melatonin. Curr. Pharm. Des. 2019. [Google Scholar]
- Tunisi, L.; Forte, N.; Fernández-Rilo, A.C.; Mavaro, I.; Capasso, R.; D’Angelo, L.; Milić, N.; Cristino, L.; Di Marzo, V.; Palomba, L. Orexin-A Prevents Lipopolysaccharide-Induced Neuroinflammation at the Level of the Intestinal Barrier. Front. Endocrinol (Lausanne) 2019, 10, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clasadonte, J.; Scemes, E.; Wang, Z.; Boison, D.; Haydon, P.G. Connexin 43-Mediated Astroglial Metabolic Networks Contribute to the Regulation of the Sleep-Wake Cycle. Neuron 2017, 95, 1365–1380. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-López, A.L.; Ortiz, G.G.; Pacheco-Moises, F.P.; Mireles-Ramírez, M.A.; Bitzer-Quintero, O.K.; Delgado-Lara, D.L.C.; Ramírez-Jirano, L.J.; Velázquez-Brizuela, I.E. Efficacy of Melatonin on Serum Pro-inflammatory Cytokines and Oxidative Stress Markers in Relapsing Remitting Multiple Sclerosis. Arch. Med. Res. 2018, 49, 391–398. [Google Scholar] [CrossRef]
- Li, H.; Sun, J.; Wang, F.; Ding, G.; Chen, W.; Fang, R.; Yao, Y.; Pang, M.; Lu, Z.Q.; Liu, J. Sodium butyrate exerts neuroprotective effects by restoring the blood-brain barrier in traumatic brain injury mice. Brain Res. 2016, 1642, 70–78. [Google Scholar] [CrossRef]
- Lavery, A.M.; Waubant, E.; Casper, T.C.; Roalstad, S.; Candee, M.; Rose, J.; Belman, A.; Weinstock-Guttman, B.; Aaen, G.; Tillema, J.M.; et al. Urban air quality and associations with pediatric multiple sclerosis. Ann. Clin. Transl. Neurol. 2018, 5, 1146–1153. [Google Scholar] [CrossRef] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anderson, G.; Rodriguez, M.; Reiter, R.J. Multiple Sclerosis: Melatonin, Orexin, and Ceramide Interact with Platelet Activation Coagulation Factors and Gut-Microbiome-Derived Butyrate in the Circadian Dysregulation of Mitochondria in Glia and Immune Cells. Int. J. Mol. Sci. 2019, 20, 5500. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215500
Anderson G, Rodriguez M, Reiter RJ. Multiple Sclerosis: Melatonin, Orexin, and Ceramide Interact with Platelet Activation Coagulation Factors and Gut-Microbiome-Derived Butyrate in the Circadian Dysregulation of Mitochondria in Glia and Immune Cells. International Journal of Molecular Sciences. 2019; 20(21):5500. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215500
Chicago/Turabian StyleAnderson, George, Moses Rodriguez, and Russel J. Reiter. 2019. "Multiple Sclerosis: Melatonin, Orexin, and Ceramide Interact with Platelet Activation Coagulation Factors and Gut-Microbiome-Derived Butyrate in the Circadian Dysregulation of Mitochondria in Glia and Immune Cells" International Journal of Molecular Sciences 20, no. 21: 5500. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215500