Independent and Combined Effects of Telomere Shortening and mtDNA4977 Deletion on Long-term Outcomes of Patients with Coronary Artery Disease

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Baseline Characteristics and Correlations with LTL and mtDNA4977 Deletion
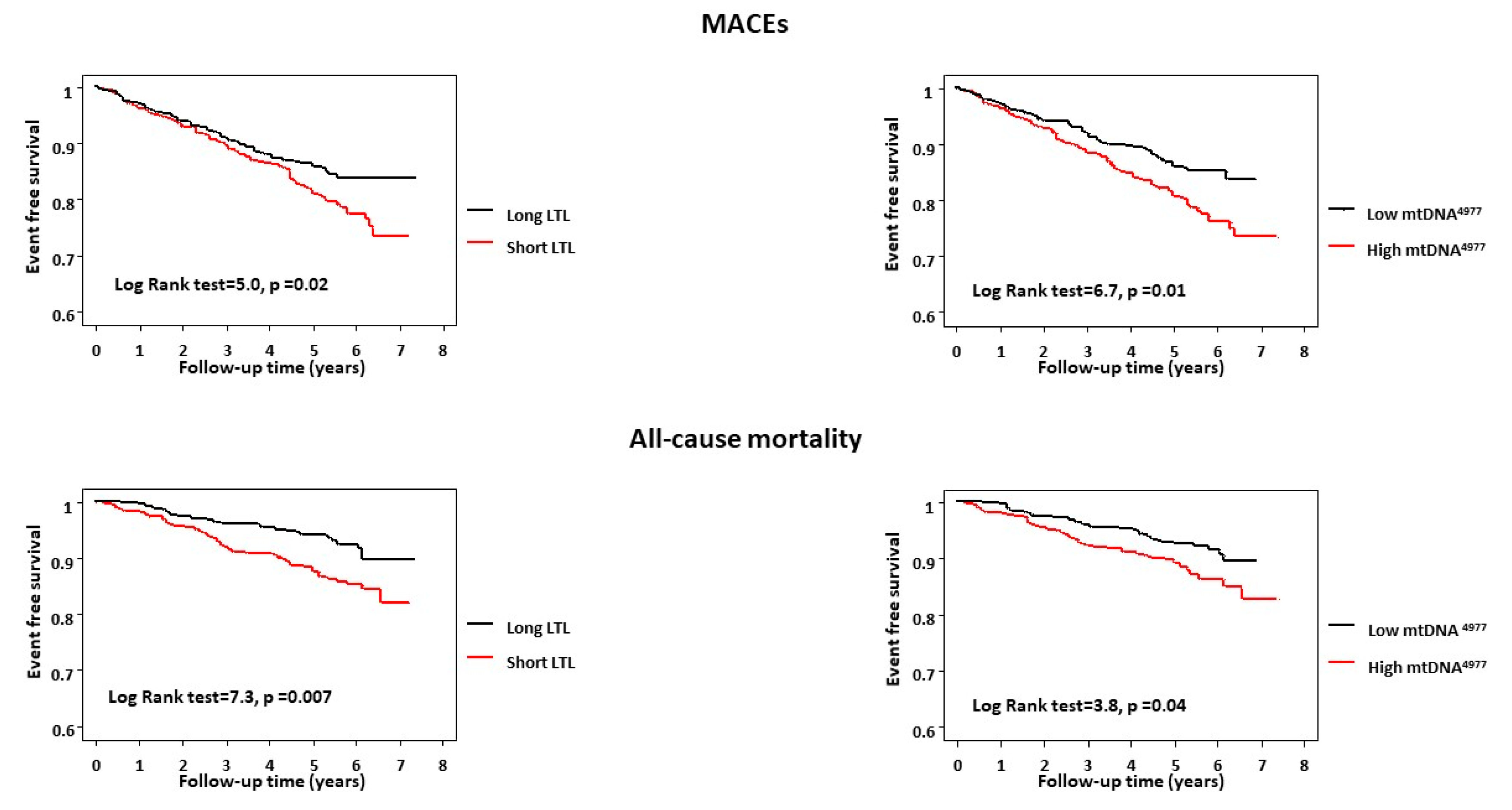
2.2. LTL, mtDNA4977 Deletion, and Outcome
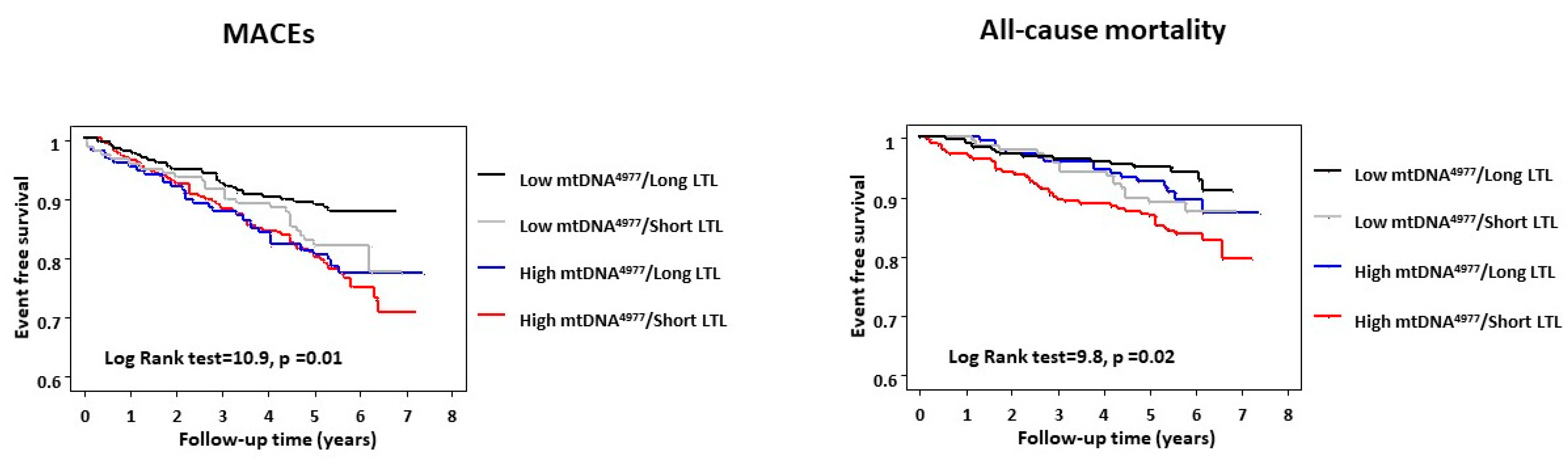
2.3. Regression Analyses and Combined Prognostic Value of LTL and mtDNA4977
3. Discussion
3.1. Comparison with Other Studies
3.2. Study Limitations
4. Materials and Methods
4.1. Study Population and Follow-Up
4.2. Leukocyte Telomere Length and mtDNA4977 Deletion Measurement
4.3. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Paneni, F.; Diaz Cañestro, C.; Libby, P.; Lüscher, T.F.; Camici, G.G. The Aging Cardiovascular System: Understanding It at the Cellular and Clinical Levels. J. Am. Coll. Cardiol. 2017, 69, 1952–1967. [Google Scholar] [CrossRef] [PubMed]
- Lowsky, D.J.; Olshansky, S.J.; Bhattacharya, J.; Goldman, D.P. Heterogeneity in healthy aging. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Pallis, A.G.; Hatse, S.; Brouwers, B.; Pawelec, G.; Falandry, C.; Wedding, U.; Dal Lago, L.; Repetto, L.; Ring, A.; Wildiers, H. Evaluating the physiological reserves of older patients with cancer: The value of potential biomarkers of aging? J. Geriatr. Oncol. 2014, 5, 204–218. [Google Scholar] [CrossRef] [PubMed]
- Jylhävä, J.; Pedersen, N.L.; Hägg, S. Biological Age Predictors. EBioMedicine 2017, 21, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Fajemiroye, J.O.; Cunha, L.C.; Saavedra-Rodríguez, R.; Rodrigues, K.L.; Naves, L.M.; Mourão, A.A.; Silva, E.F.; Williams, N.E.; Martins, J.L.; Sousa, R.B.; et al. Aging-Induced Biological Changes and Cardiovascular Diseases. Biomed. Res. Int. 2018, 7156435. [Google Scholar] [CrossRef]
- O’Sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181. [Google Scholar] [CrossRef]
- Andreassi, M.G.; Piccaluga, E.; Gargani, L.; Sabatino, L.; Borghini, A.; Faita, F.; Bruno, R.M.; Padovani, R.; Guagliumi, G.; Picano, E. Subclinical carotid atherosclerosis and early vascular aging from long-term low-dose ionizing radiation exposure: A genetic, telomere, and vascular ultrasound study in cardiac catheterization laboratory staff. JACC Cardiovasc. Interv. 2015, 8, 616–627. [Google Scholar] [CrossRef]
- Calado, R.T.; Young, N.S. Telomere diseases. N. Engl. J. Med. 2009, 361, 2353–2365. [Google Scholar] [CrossRef]
- Fyhrquist, F.; Saijonmaa, O.; Strandberg, T. The roles of senescence and telomere shortening in cardiovascular disease. Nat. Rev. Cardiol. 2013, 10, 274–283. [Google Scholar] [CrossRef]
- Mons, U.; Müezzinler, A.; Schöttker, B.; Dieffenbach, A.K.; Butterbach, K.; Schick, M.; Peasey, A.; De Vivo, I.; Trichopoulou, A.; Boffetta, P.; et al. Leukocyte Telomere Length and All-Cause, Cardiovascular Disease, and Cancer Mortality: Results From Individual-Participant-Data Meta-Analysis of 2 Large Prospective Cohort Studies. Am. J. Epidemiol. 2017, 185, 1317–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammadah, M.; Al Mheid, I.; Wilmot, K.; Ramadan, R.; Abdelhadi, N.; Alkhoder, A.; Obideen, M.; Pimple, P.M.; Levantsevych, O.; Kelli, H.M.; et al. Telomere Shortening, Regenerative Capacity, and Cardiovascular Outcomes. Circ. Res. 2017, 120, 1130–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, C.; Bruse, P.; Mohamed, S.A.; Schulz, A.; Warnk, H.; Storm, T.; Oehmichen, M. The 4977 bp deletion of mitochondrial DNA in human skeletal muscle, heart and different areas of the brain: A useful biomarker or more? Exp. Gerontol. 2008, 43, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug. Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corral-Debrinski, M.; Shoffner, J.M.; Lott, M.T.; Wallace, D.C. Association of mitochondrial DNA damage with aging and coronary atherosclerotic heart disease. Mutat. Res. 1992, 275, 169–180. [Google Scholar] [CrossRef]
- Botto, N.; Berti, S.; Manfredi, S.; Al-Jabri, A.; Federici, C.; Clerico, A.; Ciofini, E.; Biagini, A.; Andreassi, M.G. Detection of mtDNA with 4977 bp deletion in blood cells and atherosclerotic lesions of patients with coronary artery disease. Mutat. Res. 2005, 570, 81–88. [Google Scholar] [CrossRef]
- Vecoli, C.; Borghini, A.; Pulignani, S.; Mercuri, A.; Turchi, S.; Carpeggiani, C.; Picano, E.; Andreassi, M.G. Prognostic value of mitochondrial DNA4977 deletion and mitochondrial DNA copy number in patients with stable coronary artery disease. Atherosclerosis 2018, 276, 91–97. [Google Scholar] [CrossRef]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Fang, E.F.; Scheibye-Knudsen, M.; Chua, K.F.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016, 17, 308–321. [Google Scholar] [CrossRef] [Green Version]
- Gonzales-Ebsen, A.C.; Gregersen, N.; Olsen, R.K. Linking telomere loss and mitochondrial dysfunction in chronic disease. Front Biosci. (Landmark Ed.) 2017, 22, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Moslehi, J.; DePinho, R.A.; Sahin, E. Telomeres and Mitochondria in the Aging Heart. Circ. Res. 2012, 110, 1226–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haycock, P.C.; Heydon, E.E.; Kaptoge, S.; Butterworth, A.S.; Thompson, A.; Willeit, P. Leucocyte telomere length and risk of cardiovascular disease: Systematic review and meta-analysis. BMJ 2014, 349, g4227. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, M.J.; Heydon, E.E.; Kaptoge, S.; Butterworth, A.S.; Thompson, A.; Willeit, P. Association between shortened leukocyte telomere length and cardiometabolic outcomes: Systematic review and meta-analysis. Circ. Cardiovasc. Genet. 2015, 8, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Codd, V.; Nelson, C.P.; Albrecht, E.; Mangino, M.; Deelen, J.; Buxton, J.L.; Hottenga, J.J.; Fischer, K.; Esko, T.; Surakka, I.; et al. Identification of seven loci affecting mean telomere length and their association with disease. Nat. Genet. 2013, 45, 422–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farzaneh-Far, R.; Cawthon, R.M.; Na, B.; Browner, W.S.; Schiller, N.B.; Whooley, M.A. Prognostic value of leukocyte telomere length in patients with stable coronary artery disease: Data from the Heart and Soul Study. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1379–1384. [Google Scholar] [CrossRef]
- Goglin, S.E.; Farzaneh-Far, R.; Epel, E.S.; Lin, J.; Blackburn, E.H.; Whooley, M.A. Change in Leukocyte Telomere Length Predicts Mortality in Patients with Stable Coronary Heart Disease from the Heart and Soul Study. PLoS ONE 2016, 11, e0160748. [Google Scholar]
- Perez-Rivera, J.A.; Pabon-Osuna, P.; Cieza-Borrella, C.; Duran-Bobin, O.; Martin-Herrero, F.; Gonzalez-Porras, J.R.; Gonzalez-Sarmiento, R. Effect of telomere length on prognosis in men with acute coronary syndrome. Am. J. Cardiol. 2014, 113, 418–421. [Google Scholar] [CrossRef]
- Lagouge, M.; Larsson, N.G. The role of mitochondrial DNA mutations and free radicals in disease and ageing. J. Intern. Med. 2013, 273, 529–543. [Google Scholar] [CrossRef]
- Yu, E.; Mercer, J.; Bennett, M. Mitochondria in vascular disease. Cardiovasc. Res. 2012, 95, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Madamanchi, N.R.; Runge, M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007, 100, 460–473. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Wang, N.; Tall, A.R.; Tabas, I. Mitochondrial oxidative stress promotes atherosclerosis and neutrophil extracellular traps in aged mice. Arterioscler. Thromb. Vasc. Biol. 2017, 37, e99–e107. [Google Scholar] [CrossRef] [PubMed]
- Yu, E.; Calvert, P.A.; Mercer, J.R.; Harrison, J.; Baker, L.; Figg, N.L.; Kumar, S.; Wang, J.C.; Hurst, L.A.; Obaid, D.R.; et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation 2013, 128, 702–712. [Google Scholar] [CrossRef] [PubMed]
- Fetterman, J.L.; Holbrook, M.; Westbrook, D.G.; Brown, J.A.; Feeley, K.P.; Bretón-Romero, R.; Linder, E.A.; Berk, B.D.; Weisbrod, R.M.; Widlansky, M.E.; et al. Mitochondrial DNA damage and vascular function in patients with diabetes mellitus and atherosclerotic cardiovascular disease. Cardiovasc. Diabetol. 2016, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Sobenin, I.A.; Zhelankin, A.V.; Sinyov, V.V.; Bobryshev, Y.V.; Orekhov, A.N. Mitochondrial Aging: Focus on Mitochondrial DNA Damage in Atherosclerosis—A Mini-Review. Gerontology 2015, 61, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Trimarchi, J.R.; Smith, P.J.; Keefe, D.L. Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell 2002, 1, 40–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahin, E.; DePinho, R.A. Axis of ageing: Telomeres, p53 and mitochondria. Nat. Rev. Mol. Cell. Biol. 2012, 13, 397–404. [Google Scholar] [CrossRef]
- Ait-Aissa, K.; Ebben, J.D.; Kadlec, A.O.; Beyer, A.M. Friend or foe? Telomerase as a pharmacological target in cancer and cardiovascular disease. Pharmacol. Res. 2016, 111, 422–433. [Google Scholar] [CrossRef] [Green Version]
- Andreassi, M.G.; Adlerstein, D.; Carpeggiani, C.; Shehi, E.; Fantinato, S.; Ghezzi, E.; Botto, N.; Coceani, M.; L’Abbate, A. Individual and summed effects of high-risk genetic polymorphisms on recurrent cardiovascular events following ischemic heart disease. Atherosclerosis 2012, 223, 409–415. [Google Scholar] [CrossRef]
- Vecoli, C.; Borghini, A.; Foffa, I.; Ait-Ali, L.; Picano, E.; Andreassi, M.G. Leukocyte telomere shortening in grown-up patients with congenital heart disease. Int. J. Cardiol. 2016, 204, 17–22. [Google Scholar] [CrossRef]
- Pencina, M.J.; D’Agostino, R.B.S.; D’Agostino, R.B., Jr.; Vasan, R.S. Evaluating the added predictive ability of a new marker: From area under the ROC curve to reclassification and beyond. Stat. Med. 2008, 27, 157–172. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Characteristic | Value |
|---|---|
| Mean age (years ± SD) | 64.8 ± 8.3 |
| Male, n (%) | 673 (87) |
| Current smoker, n (%) | 477 (62) |
| Obesity, n (%) | 225 (29) |
| Hypercholesterolemia, n (%) | 533 (69) |
| Hypertension, n (%) | 370 (48) |
| Diabetes, n (%) | 112 (15) |
| LVEF (%) | 50 (48–58) |
| Number of diseased coronaries | |
| 1, n (%) | 398 (52) |
| 2, n (%) | 239 (31) |
| 3, n (%) | 133 (17) |
| Previous revascularization, n (%) | 111 (14) |
| Previous MI, n (%) | 398 (52) |
| Creatinine (mg/dL), described as (mean ± SD) | 1.1 ± 0.3 |
| Medical treatment | |
| Aspirin, n (%) | 708 (92) |
| Statin, n (%) | 554 (72) |
| ACEI/ARB, n (%) | 209 (27) |
| Beta-blocker, n (%) | 339 (44) |
| Calcium channel blocker, n (%) | 285 (37) |
| LTL, described as a T/S ratio | 1.0 (0.69–1.40) |
| mtDNA4977 deletion (%) | 0.56 (0.23–1.0) |
| MACEs | All-Cause Mortality | |||
|---|---|---|---|---|
| HR (95% CI) | p-Value | HR (95% CI) | p-Value | |
| Age, 1-year increase | 1.0 (0.9–1.0) | 0.99 | 1.1 (1.1–1.2) | <0.0001 |
| Male | 0.9 (0.4–2.1) | 0.80 | 0.9 (0.3–2.9) | 0.80 |
| Smoking | 1.2 (0.7–2.1) | 0.56 | 2.6 (1.2–6.0) | 0.02 |
| Obesity | 1.3 (0.7–2.2) | 0.44 | 1.1 (0.5–2.3) | 0.71 |
| Hypercholesterolemia | 0.7 (0.4–1.3) | 0.33 | 0.7 (0.4–1.4) | 0.32 |
| Hypertension | 1.1 (0.6–1.8) | 0.85 | 1.2 (0.6–2.3) | 0.57 |
| Diabetes | 1.3 (0.7–2.6) | 0.40 | 1.4 (0.6–3.1) | 0.41 |
| Ejection fraction (%) | 1.0 (0.9–1.0) | 0.49 | 0.9 (0.9–1.0) | 0.09 |
| Multivessel disease | 1.8 (0.8–2.4) | 0.18 | 1.2 (0.7–4.5) | 0.54 |
| Previous revascularization | 1.6 (0.8–3.1) | 0.18 | 2.0 (0.8–4.8) | 0.11 |
| Previous MI | 0.8 (0.4–1.3) | 0.32 | 0.7 (0.3–1.4) | 0.32 |
| Creatinine | 1.3 (0.6–2.8) | 0.56 | 1.8 (0.7–4.6) | 0.22 |
| Short LTL | 2.2 (1.2–3.9) | 0.01 | 2.1 (1.1–4.6) | 0.04 |
| High mtDNA4977 | 1.7 (1.1–2.9) | 0.04 | 2.3 (1.1–4.9) | 0.02 |
| MACEs | All-Cause Mortality | |||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p-Value | HR | 95% CI | p-Value | |
| long LTL/low mtDNA4977 (n = 126) | 1.00 | — | 1.00 | — | ||
| long LTL/high mtDNA4977 (n = 76) | 2.3 | 0.9–6.2 | 0.09 | 3.4 | 0.9–12.6 | 0.07 |
| short LTL/low mtDNA4977 (n = 84) | 2.7 | 1.1–7.5 | 0.04 | 2.8 | 0.7–10.4 | 0.12 |
| short LTL/high mtDNA4977 (n = 134) | 4.3 | 1.9–9.6 | 0.0006 | 6.0 | 2.0–18.4 | 0.001 |
| Direction of Reclassification | ||||
|---|---|---|---|---|
| Upward | Downward | NRI (SE) | p-Value | |
| mtDNA4977 | ||||
| Event | 53 (38) | 185 (27) | 0.18 (0.07) | 0.01 |
| Non-event | 28 (20) | 185 (27) | ||
| LTL | ||||
| Event | 51 (36) | 196 (31) | 0.09 (0.07) | 0.2 |
| Non-event | 37 (26) | 189 (30) | ||
| mtDNA4977 + LTL | ||||
| Event | 57 (41) | 203 (32) | 0.19 (0.07) | 0.01 |
| Non-event | 33 (24) | 211 (33) | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vecoli, C.; Borghini, A.; Pulignani, S.; Mercuri, A.; Turchi, S.; Picano, E.; Andreassi, M.G. Independent and Combined Effects of Telomere Shortening and mtDNA4977 Deletion on Long-term Outcomes of Patients with Coronary Artery Disease. Int. J. Mol. Sci. 2019, 20, 5508. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215508
Vecoli C, Borghini A, Pulignani S, Mercuri A, Turchi S, Picano E, Andreassi MG. Independent and Combined Effects of Telomere Shortening and mtDNA4977 Deletion on Long-term Outcomes of Patients with Coronary Artery Disease. International Journal of Molecular Sciences. 2019; 20(21):5508. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215508
Chicago/Turabian StyleVecoli, Cecilia, Andrea Borghini, Silvia Pulignani, Antonella Mercuri, Stefano Turchi, Eugenio Picano, and Maria Grazia Andreassi. 2019. "Independent and Combined Effects of Telomere Shortening and mtDNA4977 Deletion on Long-term Outcomes of Patients with Coronary Artery Disease" International Journal of Molecular Sciences 20, no. 21: 5508. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215508