Celastrol Induces Necroptosis and Ameliorates Inflammation via Targeting Biglycan in Human Gastric Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Celastrol Induces Gastric Cancer Cell Death, Possibly via Necroptosis
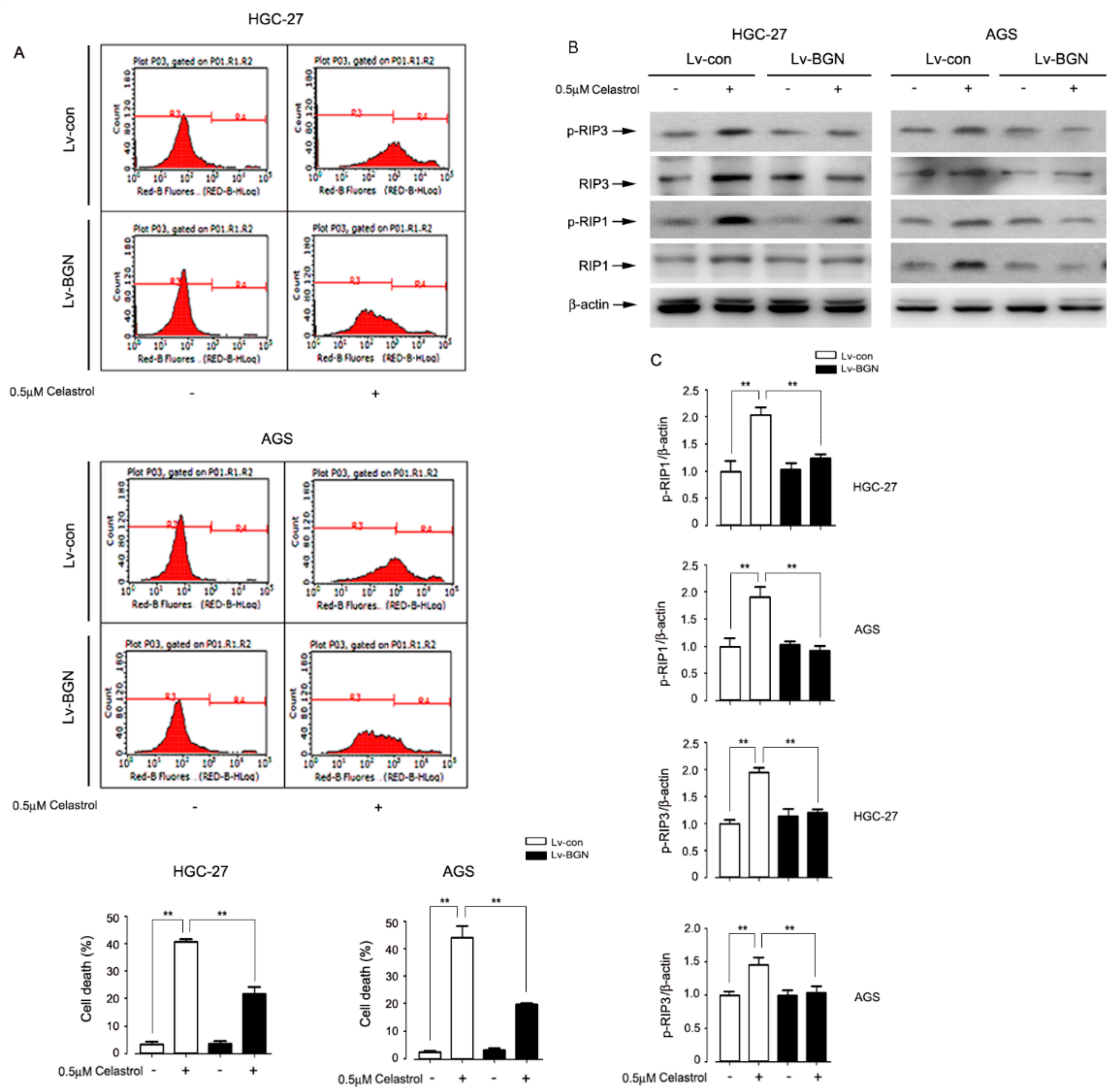
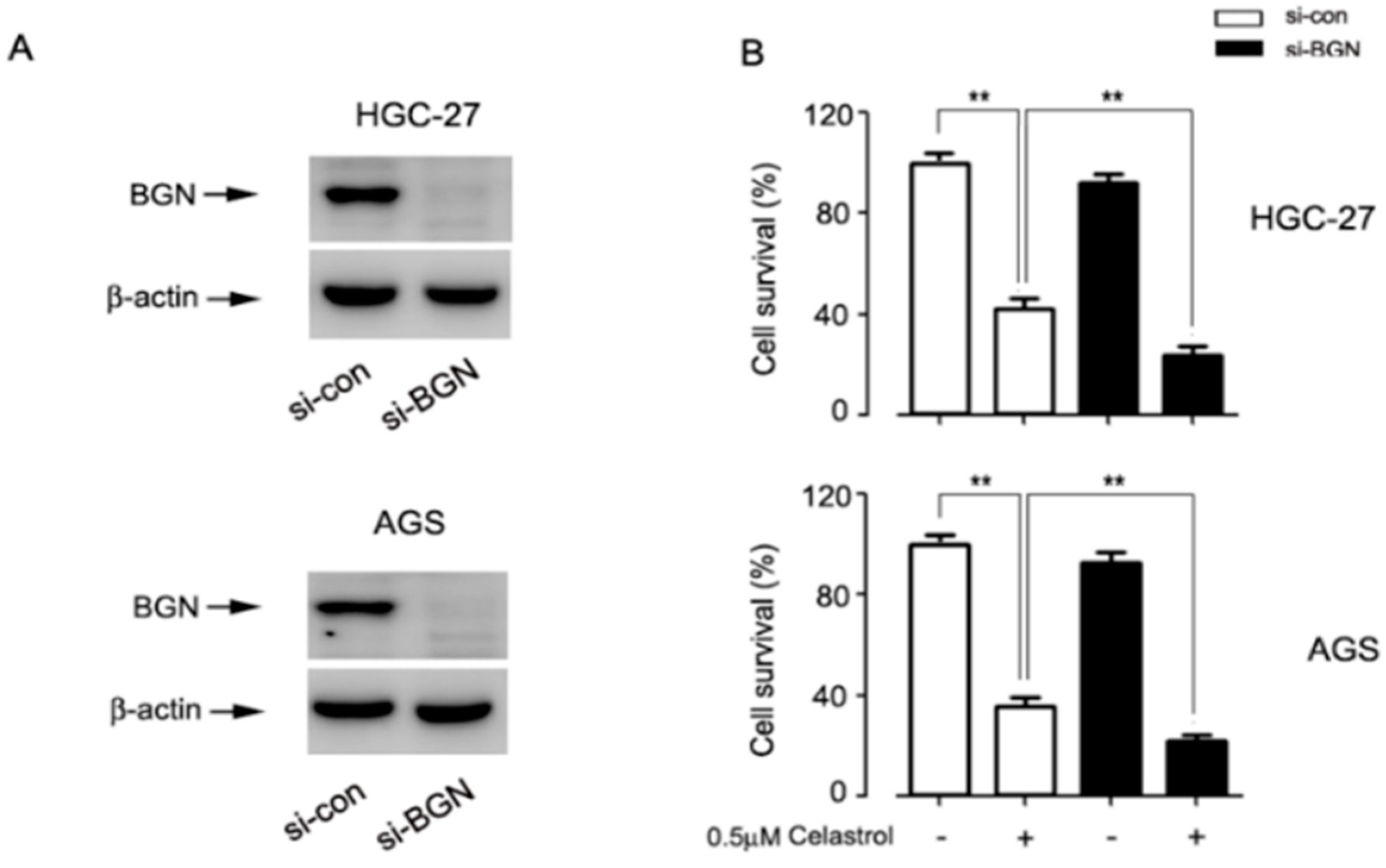
2.2. Celastrol Down-Regulated the Expression of BGN Leading to HGC-27 and AGS Cell Death
2.3. Celastrol Down-Regulated the Expression of BGN to Activate RIP1/RIP3 Necroptosis Signaling in HGC-27 and AGS Cells
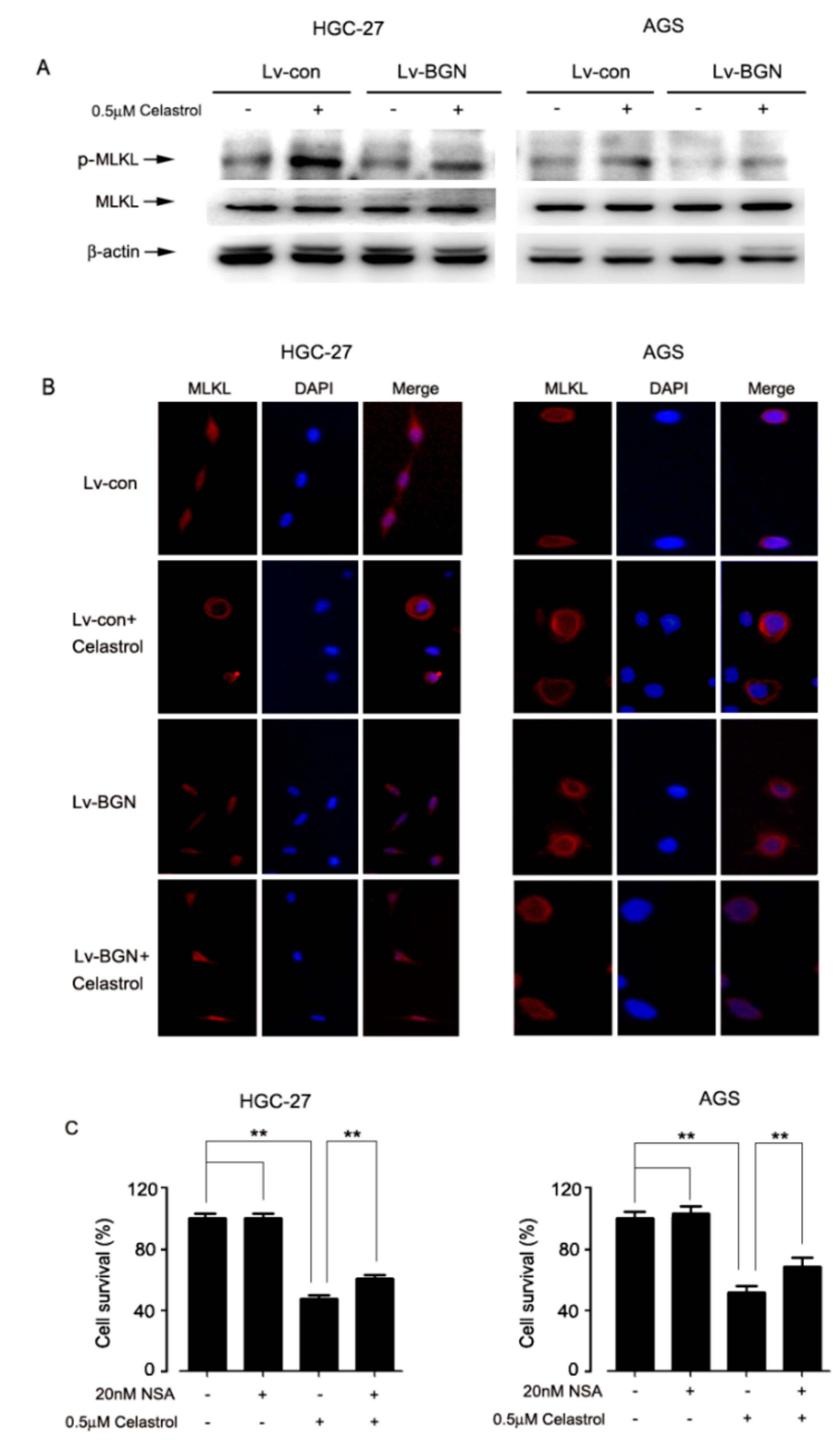
2.4. Celastrol Promoted the Translation of MLKL, Leading to Necroptosis in HGC-27 and AGS Cells
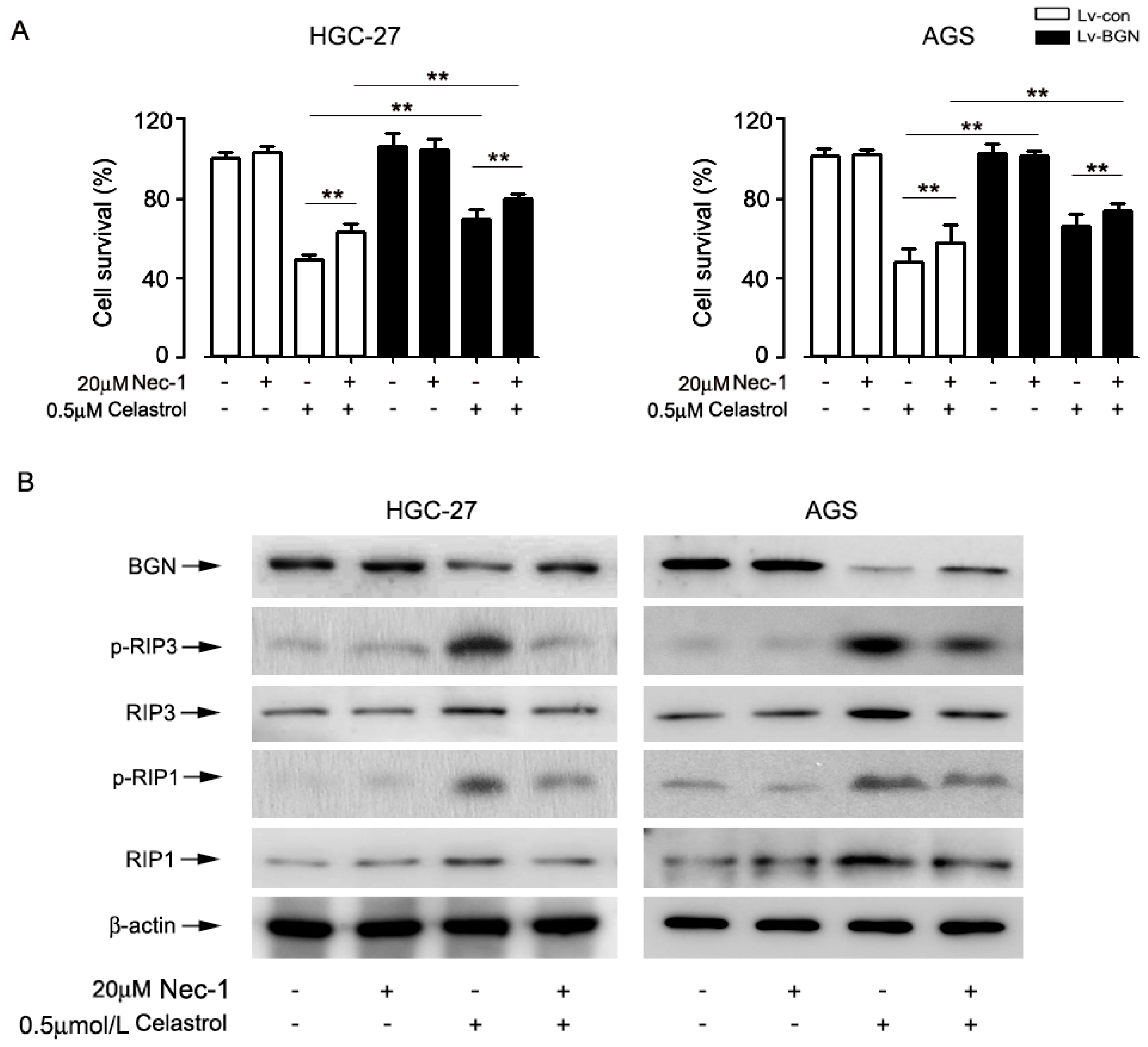
2.5. Inhibition of Necroptosis by Nec-1 Attenuated Celastrol-Induced Cell Death in Part by Up-Regulating the Expression of BGN in HGC-27 and AGS cells
2.6. Celastrol Blocking BGN Was Accompanied by Suppressing Pro-Inflammatory Cytokine Production in HGC-27 and AGS Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Western Blot Analysis
4.3. Cell Viability Assay
4.4. Cell Morphological Analysis
4.5. Analysis of Cell Death by Flow Cytometry
4.6. Immunofluorescence Assay
4.7. Cytokine Evaluation
4.8. Lentivirus Production and Transfection
4.9. siRNA Transfection
4.10. Statistical Analysis
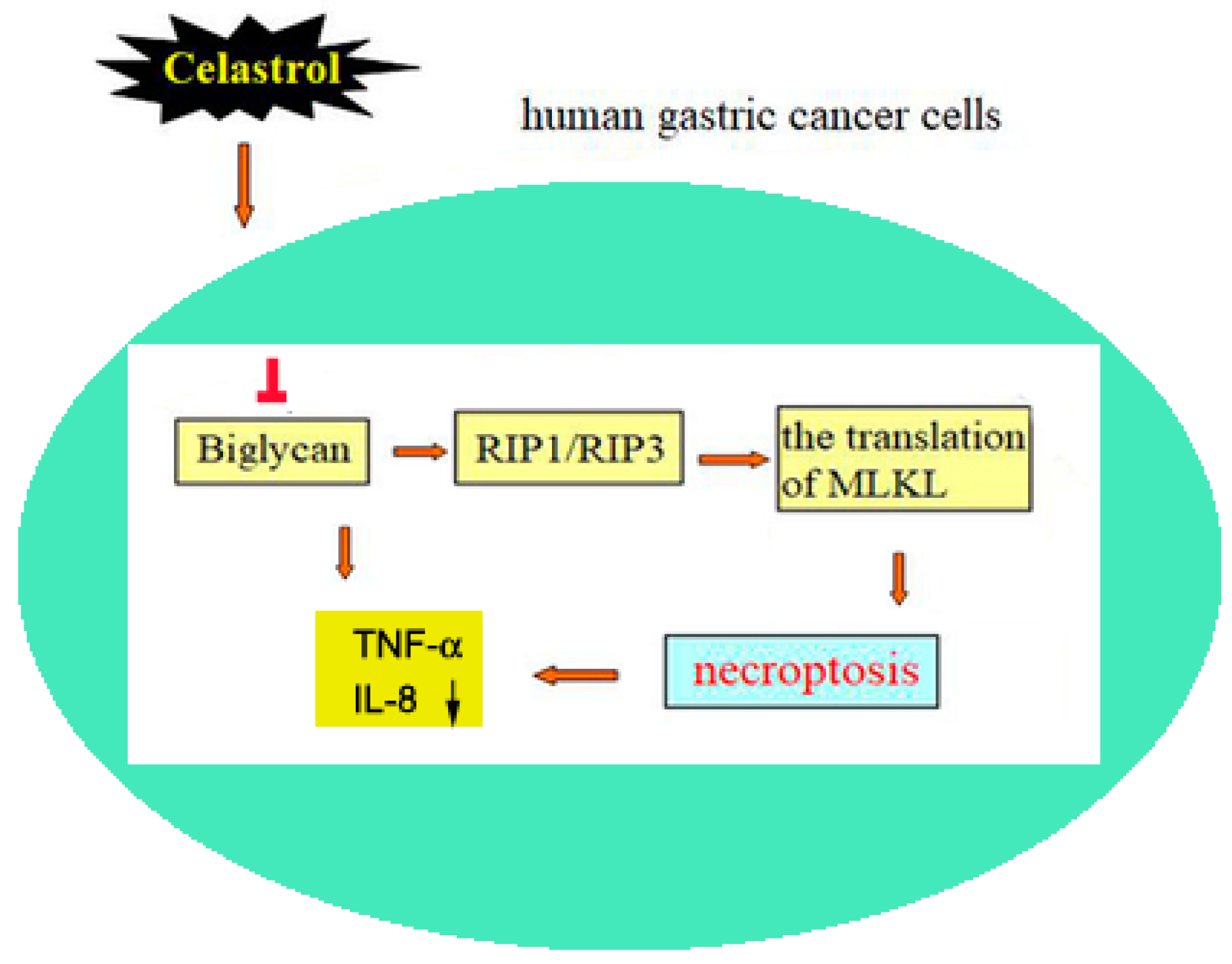
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BGN | biglycan |
| FADD | Fas-associated death domain protein |
| MLKL | mixed-lineage kinase domain-like |
| Nec-1 | necrostatin-1 |
| NSA | necrosulphonamide |
| SLRP | small leucine repeat proteoglycan family |
| RIP1 and RIP3 | receptor-interacting protein 1 and 3 |
Appendix A

References
- Cervantes, A.; Rosello, S.; Roda, D.; Rodriguez-Braun, E. The treatment of advanced gastric cancer: Current strategies and future perspectives. Ann. Oncol. 2008, 19, v103–v107. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Ajani, J.A. Multidisciplinary management of gastric cancer. Curr. Opin. Gastroenterol. 2010, 26, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Rivera, F.; Vega-Villegas, M.E.; Lopez-Brea, M.F. Chemotherapy of advanced gastric cancer. Cancer Treat. Rev. 2007, 33, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Chun, K.H.; Suh, P.G.; Kim, I.H. Alterations in cell proliferation related gene expressions in gastric cancer. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 237–254. [Google Scholar] [CrossRef] [PubMed]
- Feoktistova, M.; Leverkus, M. Programmed necrosis and necroptosis signalling. FEBS J. 2015, 282, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Duan, Y.T.; Li, J.F.; Su, L.P.; Yan, M.; Zhu, Z.G.; Liu, B.; Yang, Q. Biglycan enhances gastric cancer invasion by activating FAK signaling pathway. Oncotarget 2014, 5, 1885–1896. [Google Scholar] [CrossRef]
- Karl, I.; Jossberger-Werner, M.; Schmidt, N.; Horn, S.; Goebeler, M.; Leverkus, M.; Wajant, H.; Giner, T. TRAF2 inhibits TRAIL- and CD95L-induced apoptosis and necroptosis. Cell Death Dis. 2014, 5, e1444. [Google Scholar] [CrossRef]
- Li, J.; McQuade, T.; Siemer, A.B.; Napetschnig, J.; Moriwaki, K.; Hsiao, Y.S.; Damko, E.; Moquin, D.; Walz, T.; McDermott, A.; et al. The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 2012, 150, 339–350. [Google Scholar] [CrossRef]
- Wilson, N.S.; Dixit, V.; Ashkenazi, A. Death receptor signal transducers: Nodes of coordination in immune signaling networks. Nat. Immunol. 2009, 10, 348–355. [Google Scholar] [CrossRef]
- Iozzo, R.V. The biology of the small leucine-rich proteoglycans. Functional network of interactive proteins. J. Biol. Chem. 1999, 274, 18843–18846. [Google Scholar] [CrossRef]
- Fisher, L.W.; Heegaard, A.M.; Vetter, U.; Vogel, W.; Just, W.; Termine, J.D.; Young, M.F. Human biglycan gene. Putative promoter, intron-exon junctions, and chromosomal localization. J. Biol. Chem. 1991, 266, 14371–14377. [Google Scholar] [PubMed]
- Hasenohrl, R.U.; Frisch, C.; Junghans, U.; Muller, H.W.; Huston, J.P. Facilitation of learning following injection of the chondroitin sulfate proteoglycan biglycan into the vicinity of the nucleus basalis magnocellularis. Behav. Brain Res. 1995, 70, 59–67. [Google Scholar] [CrossRef]
- Huston, J.P.; Weth, K.; De Souza Silva, A.; Junghans, U.; Muller, H.W.; Hasenohrl, R.U. Facilitation of learning and long-term ventral pallidal-cortical cholinergic activation by proteoglycan biglycan and chondroitin sulfate C. Neuroscience 2000, 100, 355–361. [Google Scholar] [CrossRef]
- Aprile, G.; Avellini, C.; Reni, M.; Mazzer, M.; Foltran, L.; Rossi, D.; Cereda, S.; Iaiza, E.; Fasola, G.; Piga, A. Biglycan expression and clinical outcome in patients with pancreatic adenocarcinoma. Tumour. Biol. 2013, 34, 131–137. [Google Scholar] [CrossRef]
- Williams, K.J. Arterial wall chondroitin sulfate proteoglycans: Diverse molecules with distinct roles in lipoprotein retention and atherogenesis. Curr. Opin. Lipidol. 2001, 12, 477–487. [Google Scholar] [CrossRef]
- Mikula, M.; Rubel, T.; Karczmarski, J.; Goryca, K.; Dadlez, M.; Ostrowski, J. Integrating proteomic and transcriptomic high-throughput surveys for search of new biomarkers of colon tumors. Funct. Integr. Genomics 2011, 11, 215–224. [Google Scholar] [CrossRef]
- Ameye, L.; Aria, D.; Jepsen, K.; Oldberg, A.; Xu, T.; Young, M.F. Abnormal collagen fibrils in tendons of biglycan/fibromodulin-deficient mice lead to gait impairment, ectopic ossification, and osteoarthritis. FASEB J. 2002, 16, 673–680. [Google Scholar] [CrossRef]
- Nishino, R.; Honda, M.; Yamashita, T.; Takatori, H.; Minato, H.; Zen, Y.; Sasaki, M.; Takamura, H.; Horimoto, K.; Ohta, T.; et al. Identification of novel candidate tumour marker genes for intrahepatic cholangiocarcinoma. J. Hepatol. 2008, 49, 207–216. [Google Scholar] [CrossRef]
- Pan, S.; Cheng, L.; White, J.T.; Lu, W.; Utleg, A.G.; Yan, X.; Urban, N.D.; Drescher, C.W.; Hood, L.; Lin, B. Quantitative proteomics analysis integrated with microarray data reveals that extracellular matrix proteins, catenins, and p53 binding protein 1 are important for chemotherapy response in ovarian cancers. OMICS 2009, 13, 345–354. [Google Scholar] [CrossRef]
- Shimizu-Hirota, R.; Sasamura, H.; Kuroda, M.; Kobayashi, E.; Hayashi, M.; Saruta, T. Extracellular matrix glycoprotein biglycan enhances vascular smooth muscle cell proliferation and migration. Circ. Res. 2004, 94, 1067–1074. [Google Scholar] [CrossRef]
- Xu, T.; Bianco, P.; Fisher, L.W.; Longenecker, G.; Smith, E.; Goldstein, S.; Bonadio, J.; Boskey, A.; Heegaard, A.M.; Sommer, B.; et al. Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice. Nat. Genet. 1998, 20, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Lechner, B.E.; Lim, J.H.; Mercado, M.L.; Fallon, J.R. Developmental regulation of biglycan expression in muscle and tendon. Muscle Nerve 2006, 34, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Mercado, M.L.; Amenta, A.R.; Hagiwara, H.; Rafii, M.S.; Lechner, B.E.; Owens, R.T.; McQuillan, D.J.; Froehner, S.C.; Fallon, J.R. Biglycan regulates the expression and sarcolemmal localization of dystrobrevin, syntrophin, and nNOS. FASEB J. 2006, 20, 1724–1726. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L.; Tredup, C.; Gubbiotti, M.A.; Iozzo, R.V. Proteolycan neofunctions: Regulation of inflammation and autophagy in cancer biology. FEBS J. 2017, 284, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Pesic, M.; Greten, F.R. Inflammation and cancer: Tissue regeneration gone awry. Curr. Opin. Cell Biol. 2016, 43, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Van Gorp, H.; Lamkanfi, M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019, 20, e47575. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.M.; Silva, G.A.; Monteiro, M.R.; Albuquerque, S. Trypanocidal activity of quinonemethide triterpenoids from Cheiloclinium cognatum (Hippocrateaceae). Z. Naturforsch. C. 2008, 63, 207–210. [Google Scholar] [CrossRef]
- Satoh, H. Pharmacological effectiveness of the active phytochemicals contained in foods and herbs. J. Intercult. Ethnopharmacol. 2014, 3, 196–200. [Google Scholar] [CrossRef]
- Aragones, G.; Ardid-Ruiz, A.; Ibars, M.; Suarez, M.; Blade, C. Modulation of leptin resistance by food compounds. Mol. Nutr. Food Res. 2016, 60, 1789–1803. [Google Scholar] [CrossRef]
- Liu, J.; Lee, J.; Salazar Hernandez, M.A.; Mazitschek, R.; Ozcan, U. Treatment of obesity with celastrol. Cell 2015, 161, 999–1011. [Google Scholar] [CrossRef]
- Kannaiyan, R.; Manu, K.A.; Chen, L.; Li, F.; Rajendran, P.; Subramaniam, A.; Lam, P.; Kumar, A.P.; Sethi, G. Celastrol inhibits tumor cell proliferation and promotes apoptosis through the activation of c-Jun N-terminal kinase and suppression of PI3 K/Akt signaling pathways. Apoptosis 2011, 16, 1028–1041. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Yoon, I.S.; Lee, M.S.; Cha, E.Y.; Thuong, P.T.; Diep, T.T.; Kim, J.R. Anticancer activity of pristimerin in epidermal growth factor receptor 2-positive SKBR3 human breast cancer cells. Biol. Pharm. Bull. 2013, 36, 316–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Lehtonen, M.; Paimela, T.; Kaarniranta, K. Celastrol: Molecular targets of Thunder God Vine. Biochem. Biophys. Res. Commun. 2010, 394, 439–442. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Jeengar, M.K.; Reddy, V.S.; Reddy, G.B.; Naidu, V.G. Anticancer effect of celastrol on human triple negative breast cancer: Possible involvement of oxidative stress, mitochondrial dysfunction, apoptosis and PI3K/Akt pathways. Exp. Mol. Pathol. 2015, 98, 313–327. [Google Scholar] [CrossRef]
- Li, X.; Ding, J.; Li, N.; Liu, W.; Ding, F.; Zheng, H.; Ning, Y.; Wang, H.; Liu, R.; Ren, S. Synthesis and biological evaluation of celastrol derivatives as anti-ovarian cancer stem cell agents. Eur. J. Med. Chem. 2019, 179, 667–679. [Google Scholar] [CrossRef]
- Xu, B.; Xu, M.; Tian, Y.; Yu, Q.; Zhao, Y.; Chen, X.; Mi, P.; Cao, H.; Zhang, B.; Song, G.; et al. Matrine induces RIP3-dependent necroptosis in cholangiocarcinoma cells. Cell Death Discov. 2017, 3, 16096. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.Y.; Chang, T.W.; Hsieh, W.H.; Hung, M.C.; Lin, I.H.; Lai, S.C.; Tzeng, Y.J. Simultaneous induction of apoptosis and necroptosis by Tanshinone IIA in human hepatocellular carcinoma HepG2 cells. Cell Death Discov. 2016, 2, 16065. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Deng, B.; Liao, Y.; Shan, L.; Yin, F.; Wang, Z.; Zeng, H.; Zuo, D.; Hua, Y.; Cai, Z. The anti-tumor effect of shikonin on osteosarcoma by inducing RIP1 and RIP3 dependent necroptosis. BMC Cancer 2013, 13, 580. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.W.; Jang, K.S.; Choi, H.J.; Jo, A.; Cheong, J.H.; Chun, K.H. Celastrol inhibits gastric cancer growth by induction of apoptosis and autophagy. BMB Rep. 2014, 47, 697–702. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Zhong, C.Q.; Zhang, D.W. Programmed necrosis: Backup to and competitor with apoptosis in the immune system. Nat. Immunol. 2011, 12, 1143–1149. [Google Scholar] [CrossRef]
- Wagner, A.D.; Moehler, M. Development of targeted therapies in advanced gastric cancer: Promising exploratory steps in a new era. Curr. Opin. Oncol. 2009, 21, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Ma, Y.; Chen, G.; Zhou, H.; Yamazaki, T.; Klein, C.; Pietrocola, F.; Vacchelli, E.; Souquere, S.; Sauvat, A.; et al. Contribution of RIP3 and MLKL to immunogenic cell death signaling in cancer chemotherapy. Oncoimmunology 2016, 5, e1149673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Wu, M.Y.; Jiang, M.; Zhi, Q.; Bian, X.; Xu, M.D.; Gong, F.R.; Hou, J.; Tao, M.; Shou, L.M.; et al. TNF-alpha sensitizes chemotherapy and radiotherapy against breast cancer cells. Cancer Cell Int. 2017, 17, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zagouri, F.; Papadimitriou, C.A.; Dimopoulos, M.A.; Pectasides, D. Molecularly targeted therapies in unresectable-metastatic gastric cancer: A systematic review. Cancer Treat. Rev. 2011, 37, 599–610. [Google Scholar] [CrossRef]
- Yu, X.; Deng, Q.; Li, W.; Xiao, L.; Luo, X.; Liu, X.; Yang, L.; Peng, S.; Ding, Z.; Feng, T.; et al. Neoalbaconol induces cell death through necroptosis by regulating RIPK-dependent autocrine TNF-a and ROS production. Oncotarget 2015, 6, 1995–2008. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, L.T.; Nastase, M.V.; Zeng-Brouwers, J.; Iozzo, R.V.; Schaefer, L. Soluble biglycan as a biomarker of inflammatory renal diseases. Int. J. Biochem. Cell Biol. 2014, 54, 223–235. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, L.; Babelova, A.; Kiss, E.; Hausser, H.-J.; Baliova, M.; Krzyzankova, M.; Marsche, G.; Young, M.F.; Mihalik, D.; Götte, M.; et al. The matrix component biglycan is proinflammatory and signals through toll-like receptors 4 and 2 in macrophages. J. Clin. Investig. 2005, 115, 2223–2233. [Google Scholar] [CrossRef]
- Moreth, K.; Frey, H.; Hubo, M.; Zeng-Brouwers, J.; Nastase, M.V.; Hsieh, L.T.; Haceni, R.; Pfeilschifter, J.; Iozzo, R.V.; Schaefer, L. Biglycan-triggered TLR-2- and TLR-4-signaling exacerbates the pathophysiology of ischemic acute kidney injury. Matrix Biol. 2014, 35, 143–151. [Google Scholar] [CrossRef]
- Moreth, K.; Brodbeck, R.; Babelova, A.; Gretz, N.; Spieker, T.; Zeng-Brouwers, J.; Pfeilschifter, J.; Young, M.F.; Schaefer, R.M.; Schaefer, L. The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis. J. Clin. Investig. 2010, 120, 4251–4272. [Google Scholar] [CrossRef] [Green Version]
- Keamey, C.J.; Cullen, S.P.; Tynan, G.A.; Henry, C.M.; Clancy, D.; Lavelle, E.C.; Martin, S.J. Necroptosis suppresses inflammation via termination of TNF- or LPS-induced cytokine and chemokine production. Cell Death Differ. 2015, 22, 1313–1327. [Google Scholar]
- Kearney, C.J.; Martin, S.J. An Inflammatory Perspective on Necroptosis. Mol. Cell 2017, 65, 965–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silke, J.; Rickard, J.A.; Gerlic, M. The diverse role of RIP kinases in necroptosis and inflammation. Nat. Immunol. 2015, 16, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.J.; Guo, D.D.; Zhang, W.; Xie, Y.F.; Yang, H.J.; Cheng, B.F.; Wang, L.; Yang, R.; Bi, J.; Feng, Z. Biglycan, a Nitric Oxide-Downregulated Proteoglycan, Prevents Nitric Oxide-Induced Neuronal Cell Apoptosis via Targeting Erk1/2 and p38 Signaling Pathways. J. Mol. Neurosci. 2018, 66, 68–76. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, D.; Zhang, W.; Yang, H.; Bi, J.; Xie, Y.; Cheng, B.; Wang, Y.; Chen, S. Celastrol Induces Necroptosis and Ameliorates Inflammation via Targeting Biglycan in Human Gastric Carcinoma. Int. J. Mol. Sci. 2019, 20, 5716. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225716
Guo D, Zhang W, Yang H, Bi J, Xie Y, Cheng B, Wang Y, Chen S. Celastrol Induces Necroptosis and Ameliorates Inflammation via Targeting Biglycan in Human Gastric Carcinoma. International Journal of Molecular Sciences. 2019; 20(22):5716. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225716
Chicago/Turabian StyleGuo, Dandan, Wei Zhang, Haijie Yang, Jiajia Bi, Yunfei Xie, Binfeng Cheng, Yan Wang, and Sujuan Chen. 2019. "Celastrol Induces Necroptosis and Ameliorates Inflammation via Targeting Biglycan in Human Gastric Carcinoma" International Journal of Molecular Sciences 20, no. 22: 5716. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225716