B Vitamins and Fatty Acids: What Do They Share with Small Vessel Disease-Related Dementia?


Neurology Clinic, Department of Medical, Surgical and Health Sciences, University of Trieste, 34149 Trieste, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(22), 5797; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225797
Submission received: 21 September 2019
/
Revised: 21 October 2019
/
Accepted: 12 November 2019
/
Published: 18 November 2019
(This article belongs to the Special Issue Functional Mechanism of B-Vitamins and Their Metabolites)
{kind=link}
Abstract
:Many studies have been written on vitamin supplementation, fatty acid, and dementia, but results are still under debate, and no definite conclusion has yet been drawn. Nevertheless, a significant amount of lab evidence confirms that vitamins of the B group are tightly related to gene control for endothelium protection, act as antioxidants, play a co-enzymatic role in the most critical biochemical reactions inside the brain, and cooperate with many other elements, such as choline, for the synthesis of polyunsaturated phosphatidylcholine, through S-adenosyl-methionine (SAM) methyl donation. B-vitamins have anti-inflammatory properties and act in protective roles against neurodegenerative mechanisms, for example, through modulation of the glutamate currents and a reduction of the calcium currents. In addition, they also have extraordinary antioxidant properties. However, laboratory data are far from clinical practice. Many studies have tried to apply these results in everyday clinical activity, but results have been discouraging and far from a possible resolution of the associated mysteries, like those represented by Alzheimer’s disease (AD) or small vessel disease dementia. Above all, two significant problems emerge from the research: No consensus exists on general diagnostic criteria—MCI or AD? Which diagnostic criteria should be applied for small vessel disease-related dementia? In addition, no general schema exists for determining a possible correct time of implementation to have effective results. Here we present an up-to-date review of the literature on such topics, shedding some light on the possible interaction of vitamins and phosphatidylcholine, and their role in brain metabolism and catabolism. Further studies should take into account all of these questions, with well-designed and world-homogeneous trials.
1. Introduction
Discussion of vitamins and vascular dementia is akin to opening Pandora’s box. Much has been written on vitamin supplementation and dementia, but results are still under debate, and no real conclusion has yet been drawn [1]. Above all, two of the most significant problems have emerged from the debate. The first is that, from the hundreds of relevant studies, no consensus on the application of standard diagnostic criteria has been reached between Alzheimer’s disease (AD) or MCI, nor on the best term to diagnose small vessel disease-related dementia. The second problem is that a possible correct time of implementation has yet to be determined to have the most effective clinical result. Nevertheless, as no specific therapeutic options have been discovered for the two most globally relevant forms of dementia (AD and small vessel disease-related dementia), different risk factors for cognitive impairment have been researched, and vitamin supply and fatty acids could be a potential therapy.
2. Vascular Dementia and Small Vessel Disease-Related Dementia
Vascular dementia should be one of the simplest diagnosed pathologies due to the apparent temporal correlation between an acute vascular brain lesion and its onset. Nonetheless, consensus criteria for vascular cognitive impairment remain under debate, which began in 1983 when NINDS-AIREN criteria were written [2]. These criteria have been partially modified by the ICD-0 [3]. Despite multiple attempts, the current clinical diagnostic criteria for vascular dementia are still being debated. They lack a definite morphological substrate for the different types of cognitive disruption due to vascular causes. In fact, three different subtypes have been recognized: Genetic type of vascular dementia (CADASIL or CARASIL), macrovascular type of dementia (multi-infarct dementia or strategic infarct dementia), and microvascular type of dementia (subcortical vascular dementia or, more appropriately, small vessel disease-related dementia) [4,5,6].
The most recent effort to categorize vascular impairment relies on DSM V [7,8]. In the same year, the Standards for Reporting Vascular changes on Neuroimaging (STRIVE) study provided the same guidelines for recommended standards for research on vascular dementia with MRI and CT [9]. For the first time, a panel completed a standard advisory about the terms and definitions for features visible on MRI and minimum standards for image acquisition. Signs of small vessel disease include, in a conventional MRI, recent subcortical infarcts, white matter hyperintensities, lacunes, prominent perivascular spaces, and cerebral microbleeds, with possible consequent atrophy (see below for a more accurate description).
Small vessel disease (SVD) results from damage to the small penetrating arteries and arterioles in the pial and lepto-meningeal circulation, along with penetrating and parenchymal arteries and arterioles, pericytes, capillaries, and venules [10]. The prevalence of SVD increases exponentially with aging. A review of pathologic studies shows enormous differences in the incidence of subcortical vascular dementia (sVaD) ranging from 0.03% to 85.2%, with means around 11%, while in a series of recent autopsies from Japanese geriatric hospitals it was 23.6% to 35%. In Europe, prevalence rates of SVD-related dementia estimated between ages 65–69 to 80+ years ranged from 2.2% to 16.3% [11,12,13,14]. Aging is the most critical risk factor in developing small vessel disease; it leads to the loss of arterial elasticity. This fact causes a reduction of arterial compliance due to the altered mechanical small vessel walls [15]. The effect is the loss of the autoregulatory capabilities of cerebral arteries; as a consequence, the brain suffers from a higher vulnerability to hypotension, with possible major ischemia, when the systemic blood pressure dips below a critical threshold [16,17,18]. Arteriosclerosis and cerebral amyloid angiopathy (CAA) [19,20,21,22] are the most crucial pathogenic expressions of small vessel disease. The mechanism here described is supported by the concomitant age-related impairment of vascular autoregulation; it has been described as the low-level functioning of the autonomic nervous system, with direct and endothelium-mediated altered baroreflex activity [23,24,25,26]. Small vessel disease, due to the primary localization of the white matter hyperintensities, may affect the integrity of the adjacent medial cholinergic pathway [27] or could develop from the deafferentation of the basal forebrain cholinergic system to the tubero-mamillary tracts [28,29], observing, therefore, perverse procrastination of the hypoperfusion. Neural activity is typically supplemented by increased blood flow, originated by a retrograde vascular dilatation of upstream arterioles outside the activated area [30]. The altered “retrograde vasodilatation system” (which occurs in small vessel disease-related dementia) could be another factor that worsens vessel dysregulation [30].
Cerebral small vessel disease includes a neuroimaging and a combined pathological description, which comprise different imaging changes in the white matter and subcortical grey matter, including small subcortical infarct, lacunes, white matter hyperintensities (WMHs), prominent perivascular spaces (PVS), cerebral microbleeds (CMBs), and atrophy. Lacunes derive from a complete occlusion of the small arteries, leading to an infarct in a specific area (basal ganglia, capsule, pons). Moreover, SVD is characterized by an associated hypoperfusion progression, causing incomplete ischemia of the deep white matter [31,32,33,34] accompanied by inflammation, diffuse rarefaction of myelin sheaths, axonal disruption, and astrocyte gliosis [26]. In small vessel disease, occlusion of the deep periventricular-draining veins is also evident [35], with the disruption of the blood-brain barrier (BBB) as a consequence. The BBB disruption leads to a consequent leakage of fluid and plasma cells, which eventually potentiates the perivascular inflammation, the demyelination process, and gliosis, gathering a multifactorial genesis for white matter alterations [36,37,38]. Cerebral small vessel disease is what we define to as a “progressive disease” [26]. Lesions progress over time, and the long-term outcome and impact on brain damage vary. Recent studies indicated that the strongest predictor of white matter progression is the high density, at baseline, of the white matter hyperintensity, with a rapid confluence of the lesions [39,40,41,42]. Nonetheless, one of the most unsolved doubts is which is the determinant factor for an acceleration process that mediates the transition from small vessel disease toward the subcortical dementia process? The burden of lacunes and a profound amount of white matter alterations (WMLs) [43,44,45] are two suitable candidates.
The cognitive alterations are determined by the specific point-to-point impairment, but also by the interruptions of the frontal and prefrontal-thalamus and thalamus-frontal and basal forebrain networks [46,47], which can also lead to functional cortical deafferentation. The caudate nucleus is the most precociously affected region by chronic hypoperfusion, followed by the putamen, insula, precentral gyrus, inferior frontal gyrus, and middle frontal gyrus. All these regions require, at steady state, more than 20% of the metabolic request compared to other brain areas [48,49,50,51,52,53,54,55]. Reduced metabolic rate of oxygen (estimated of about 35% in white matter) [56,57] has been found in patients with small vessel disease-related dementia; moreover, an incongruity between the brain oxygen supply and its consumption has been described in sVaD, which determines an altered neurovascular coupling and altered vasomotor reactivity [26,58,59,60,61,62,63].
Neuropsychological pattern profiles of sVaD are related to the subcortical-cortical loop deafferentation and are distinguished by poor executive function, poor planning, working memory alterations, loss of inhibition, reduced mental flexibility, multitasking procedures invalidation, and decrease speed of executive process [64,65,66,67,68,69,70].
No specific treatment has been discovered, either as pathogenic or highly standard recommended for this condition.
3. State-Of-The-Art: What Do We Know about Nutrients and Brain Degenerative Diseases?
Decades have passed since the first descriptions of how vitamins can affect brain networks [71], but results remain compelling. Cummings [72] suggested that the nutraceutical approach might have an effect on synaptic integrity, and therefore, it could be useful in many different clinical conditions, such as AD, vascular disease, traumatic brain injury, and Parkinson’s disease. Choline is an essential nutrient for humans. It is a precursor of the synthesis of all the membrane phospholipids (e.g., phosphatidylcholine (PC)), the neurotransmitter acetylcholine, and via betaine, the methyl group donor S-adenosylmethionine. High choline intake during gestation and in the soon-after postnatal period, in animal models, improves cognitive function in adulthood. It has been associated with a prevention of age-related memory decline, and of the neuropathological changes associated with AD, epilepsy, fetal alcohol syndrome, and inherited conditions such as Down and Rett syndromes, but also with stroke and, recently, vascular-risk conditions [73,74]. Choline and its metabolites are differently associated with cardiometabolic risk factors, history of cardiovascular disease, and MRI-documented cerebrovascular disease in older adults. [75]. Choline might modify the DNA methylation in the brain and may induce alterations in the expression of genes that encode proteins essential for learning and memory processing, suggesting a possible epigenomic mechanism of action [75]. Prenatal choline supplementation has been demonstrated to show a three-day advancement in hippocampal development [76]. This effect on adult neurogenesis has been observed together with a local increment of other trophic factors: nerve growth factor, brain-derived neurotrophic factor, insulin-like growth factor 2, and vascular endothelial growth factor [77,78]. A concomitant enlarged dimension of the basal forebrain cholinergic neurons has also been observed [79], with a concomitant increase of acetylcholine production [80].
Choline influences, as a cofactor of methyl donors, the DNA methylation process (in addition to folate and vitamin B12; see elsewhere in this manuscript). Choline is a strong determining factor of transcriptional regulation and the methylation process of different regions of insulin growth factor 2 gene [81,82], and many others fundamental for synaptic plasticity, with the cascade of dynamic processes highly dependent on it, i.e., learning and memory [83,84,85,86,87,88].
Choline also is a precursor of phosphatidylcholine, the principal constituent of all biological membranes, including the polarized ones, such as neurons and astrocytes. Evidence that phospholipid metabolism is abnormal in AD originated with postmortem brain sample studies [75,89]. These studies showed reduced levels of phosphatidylcholine and phosphatidylethanolamine, and increased levels of their metabolites, glycerophosphocholine, and glycerophosphoethanolamine, in the cerebral cortex of AD patients compared to age-matched controls [89,90]. These variations have also been observed in brain regions free of plaques and tangles [90]. Therefore, this data suggests that the defect does not rely on an amyloid-related process but is widespread through the AD brain. More recently, reduced levels of phosphatidylcholine and phosphatidylethanolamine and increased levels of their metabolites, glycerophosphocholine and glycerophosphoethanolamine, have been reported in vascular-risk factors conditions, such as fatty liver disease [91], obesity, and insulin resistance [92,93].
Furthermore, a substantial reduction in molecular species of phosphatidylcholine containing docosahexaenoic acid (PC-DHA) levels has been observed in the temporal cortex gray matter of AD patients [94]. These markers have been observed in AD brains but also AD peripheral blood plasma [95,96,97]. All these findings support another theory for AD pathogenesis: Lipid altered or defective production [75,98,99]. Healthy old subjects with a lower level of erythrocyte phospholipid n-3 fatty acid (eicosapentaenoic (EPA, 20:5n-3)) are more prone to faster cognitive alteration, compared to healthy old subjects with normal levels of erythrocyte phospholipid n-3 fatty acid (eicosapentaenoic (EPA, 20:5n-3)) [100]. Docosahexaenoic acid and erythrocyte phospholipid n-3 fatty acid (eicosapentaenoic acid) are transported through the BBE as lysophosphatidylcholines by a specific transporter [101,102]. Therefore, it has been argued that low plasma levels of lysophosphatidylcholine might reduce fatty acids inside the brain in AD patients [97,103].
Hence, medical supplementation of food, including with phosphatide precursors (docosahexaenoic and eicosapentaenoic acid, phospholipids, choline, uridine monophosphate, vitamin E, vitamin C, and vitamins B12, B6, and B9) has been employed in different clinical trials [104,105]. The second study has two outcomes: The primary aim is to demonstrate an improvement of memory scores in specific tests in mild AD when compared to placebo. Secondary outcomes include different sub-scores of neuropsychological battery and EEG measures, including relative and absolute power in alpha, beta, theta, and delta frequency bands, associated with the phase lag index in each of the frequency bands. The study achieved its primary outcome, showing significantly better performance in memory in the treated mild AD group. It did not obtain any significant measure in the second neuropsychological outcome, but showed that peak frequency and phase lag index of the delta frequency showed a drug/placebo difference in favor of the treated group [105]. Three other studies continued from these finding [106,107,108]. Rijpma et al. [106] demonstrated that prolonged diet supplementation could increase circulating levels of fatty acid levels and uridine monophosphate. Uridine, together with docosahexaenoic and eicosapentaenoic acids, is a rate-limiting precursor via the Kennedy pathway for the synthesis of phospholipids in neuronal membranes.
A recent review of the studies on food supplementation [109] has drawn hopeful conclusions: early interventions work better, and mildly affected patients improve memory scores in specific tests. Moreover, their caregivers perceive an amelioration (even if not significantly) of AD behavior, and these supplementations might potentially delay or ameliorate the pathogenetic process in AD and small vessel disease [110,111,112,113].
4. Vitamin B1 (Thiamine)
Thiamine is probably one of the most studied vitamins related to Wernicke encephalopathy and Korsakoff syndrome, which have been thoroughly studied for decades [114,115]. Nevertheless, recent data emphasizes the role of thiamine, due to the massive increase of alcoholism in Western countries [116,117,118], in addition to the finding that thiamine defects are frequently found in association with dialyzed patients, hyperemesis gravidarum, malignancy with or without therapy, sleeve gastrectomy, magnesium depletion, AIDS, etc. [119,120,121,122,123].
Thiamine, also called vitamin B1, is a labile quaternary ammonia compound, not produced in humans. It is found as unphosphorylated thiamine (i.e., free TH) and as phosphorylated derivates, with magnesium as a cofactor: Thiamine monophosphate (THMP), thiamine diphosphate (THDP, aka TH pyrophosphate), and thiamine triphosphate (THTP) [124,125].
Thiamine acts as a cofactor in three key-enzymatic reactions: The conversion of pyruvate to acetyl-CoA, the conversion of alpha-ketoglutarate to succinate, in the Kreb’s cycle, and the catalysis by transketolase in the pentose monophosphate shunt [125]. Lack of alpha-ketoglutarate dehydrogenase interrupts the Kreb’s cycle, essential for ATP production in the brain; the loss of ATP induces a quick increment of the calcium inflow, with consequent induction of neuronal apoptosis and an abrupt growth of the glutamate currents [125,126]. The loss of alpha-ketoglutarate dehydrogenase causes a decrease of the aspartate and GABA brain concentration, a decrement of the reduced oxidative phosphorylation, and an increment of lactate [127,128,129,130]. Diminished transketolase activity leads to a loss of sphingolipid synthesis, an overproduction of branched-chain amino acids, and an altered pentose phosphate shunt [131,132,133,134]. Until the last decade, there were limited works describing the causative role of brain damage induced by thiamine loss per se; usually, thiamine defects have been tightly related to alcohol addiction, the former being recognized as the causative factor of the neural alterations, rather than the alcohol by itself.
Moreover, it has been observed that in alcohol-addiction there is an altered gene expression of two proteins, carriers for thiamine, THTR1 and THTR2 (not known if genetically or epigenetically transformed) [135,136,137], suggesting a possible superimposed damage factor. Thus, genetic predisposition of thiamine transporter reduced efficacy is currently more accepted as a potentiating effect of brain damage, even in well-nourished patients. Animal models have been used to prove that severe isolated thiamine deprivation brings a significant alteration of the inferior colliculus and the medial vestibular nucleus over a one-week period. More prolonged thiamine depletion caused severe basal ganglia impairment and, in the final period of observation, functional alteration of the mammillary bodies and the dorsal medial nucleus of the thalamus [138]. Some studies suggested a possible relationship between a loss of thiamine and a macroscopic decrease of the endorphinergic system [139], but this data remains isolated because it has been observed in many patients, with a contemporary loss of thiamine and heavy alcohol consumption. The principal cause of the observed data remains a mystery (alcohol induction of endorphin depletion has been positively documented) [130]. The loss of the activity of the three thiamine enzymes in the brain (the conversion of pyruvate to acetyl-CoA, the active participation in the conversion of alpha-ketoglutarate to succinate, in the Kreb’s cycle, and the catalysis by transketolase in the pentose monophosphate shunt) has been observed in many neurodegenerative conditions [140,141,142,143,144]. The administration of benfotiamine induced a mild inhibition of cholinesterase, leading to a significant amount of acetylcholine in the post-synaptic space and determining a reduction of amyloid plaques and tau tangles, probably reducing the cascade of neuroinflammation, which promotes the hyperphosphorylation process [145,146,147,148].
Thiamine also has a non-coenzymatic function [149]; its effects on the axonal conductance and the release of different neurotransmitters, such as acetylcholine, dopamine, and noradrenaline, has been described [149,150,151]. These data have been supported by the findings of a rapid change of thiamine status (the so-called mobile thiamine pool) [150,151] in different neural networks. In particular, oxythiamine stimulates potassium-evoked acetylcholine release in the presence of calcium [152,153]. By definition, thiamine exerts a refining system able to protect the hippocampal neurons cultured with an excess of glutamate [154]. On the contrary, thiamine deficiency also induces an excess of glutamate release, diminished by a glutamatergic blocking action with an N-methyl-D-aspartate (NMDA) antagonist (similar to memantine) [155,156]. These studies have not yet been applied in clinical practice [157].
5. Vitamin B2 (Riboflavin)
Riboflavin, or B2, is mainly dependent on dietary intake. It is active in different forms, as riboflavin, or as coenzymes for several reactions, namely, flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD). Riboflavin is involved in a large variety of processes and has a critical neuroprotective function [158]. It is a primary antioxidant; thus, B2 is involved in glutathione reduction, where FAD is a co-factor, forming reduced, active glutathione, which acts against oxidative stress and lipid peroxidation. Vitamin B2 increases the activity of antioxidant enzymes, such as the superoxide dismutase (SOD) and catalase [158,159,160,161], additionally. Riboflavin seems to also reduce the oxidative damage after reperfusion, through a direct action on free radicals, as shown by studies on rabbits’ hearts after infarction [162], mice ischemic liver cells [163], and rats’ brains [164]. In these cases, vitamin B2 reduces edema and neuronal death after traumatic brain injury [151,160,164,165]. Riboflavin, together with folate, lowers homocysteine levels, hence helping to avoid vascular and toxic damages: FAD is needed in the one-carbon metabolism cycle to reduce 5,10-methylenetetrahydrofolate (5,10-MTHF) to 5-methyl THF which, in turn, provides the methyl group for homocysteine re-methylation to methionine [158,161,166]. This vitamin might play a role in the reduction of glutamate excitotoxicity via direct inhibition of glutamate neuronal release, as evidenced in animal studies [167,168]; further, it is crucial for the tryptophan-kynurenine pathway, where neuroactive compounds that might influence glutamate receptors are produced [158]. In addition, B2 seems to have a direct anti-inflammatory ability, through inhibition of NF-kB and high-mobility group protein B1 (HMGB1) [158,169], and it is involved in mitochondrial functioning, FAD and FMN being co-factors for complex I and complex II of the electron transport chain [158,159]. Lastly, some of these functions are shared with the B6 vitamin; B6 requires the active form of B2, FMN, as a co-factor, in order to be transformed into its active form pyridoxal 5′ phosphate (PLP) [170].
Riboflavin deficiency derives from inadequate dietary intake, mainly derived from a rice-based diet, poor animal proteins [171], or in conditions of higher demand, e.g., pregnancy, childhood, aging [159], genetic disorders, or malabsorption conditions. Deficiency is also related to drug and substance interaction, e.g., furosemide or alcohol, contributes to riboflavin reduction [172]. Lack of riboflavin is recognized in starvation and lower income groups, but it has also been found in adolescents with low milk consumption [166], and a recent study showed that in the Western world, including Europe, North America, Australia, and New Zealand, 40% of people older than 65 years are below the estimated average requirement [173]. A suboptimal, subclinical level of B2 is likely more common than previously thought and might affect all ages [174]. Further studies, based on biomarkers more than simple dietary intake surveys, are required for more reliable data and a better knowledge of this crucial vitamin deficiency [170].
Ariboflavinosis may lead to several conditions, such as anemia, cheilosis, glossitis, angular stomatitis, cataract, and seborrheic dermatitis [159,174,175]. It is frequently associated with other vitamin deficiencies, and signs and symptoms are rarely isolated [170]. Impaired nerve function has also been correlated to riboflavin insufficiency, both as acquired [174,176,177] and genetic disorders, i.e., Brown–Vialetto–Van Laere neuropathy [178].
Subclinical levels of riboflavin might be related to different neurological diseases, such as Parkinson’s disease, migraines, and multiple sclerosis, due to the multiple roles discussed above [158,161]. Thus, when implemented, riboflavin seems to have neuroprotective functions, and clinical trials have reported how a high intake of this vitamin reduced motor impairment in patients with Parkinson’s disease (PD) and proved to be useful for migraine prophylaxis in adults [179,180,181].
On the contrary, the direct relationship between riboflavin and dementia, mainly the so-called small vessel disease/subcortical vascular dementia, has been rarely investigated, even if vitamin B2 is related to endothelial release of nitric oxide [166] and to the catabolism of homocysteine, both of which are related to cerebral small vessel disease [159,182,183,184,185].
Some studies have analyzed the relationship between B2 and cognitive decline [186,187,188,189] without investigating possible direct molecular mechanisms. In one study, the authors enrolled a cohort of 70-year-old subjects, who had previously been assessed for intelligent quotient at age 11 years. At 70 years, participants underwent a range of neuropsychological tests, including the Mini-Mental State Examination (MMSE), and the National Adult Reading Test (NART), and completed food questionnaires regarding the intake of several nutrients, including B vitamins and riboflavin [187]. No significant association was found between diet and cognitive performances [187]. In line with this, a Korean observational study [188] explored the association between intake of B vitamins, including riboflavin, and cognition in three groups of subjects over 60 years: Normal, mild cognitive impairment, and with Alzheimer’s disease. Riboflavin intake was positively correlated to cognitive test scores, e.g., MMSE for Koreans (MMSE-K) and the Boston Naming Test, in both the AD group and the MCI group, whereas no correlation was found in healthy subjects [188]. Of note, there was no significant difference in vitamin intake among the three groups when comparing AD patients with controls, as also noted by other authors [190]. Another Korean study found an association between poor cognitive performance, scored with MMSE-K, and riboflavin intake [189], although herein, the correlation was also positive in subjects with average scores. Two earlier studies [191,192] investigating subjects free of cognitive impairment reported a better memory function with higher riboflavin levels and an absence of correlation between cognitive performances and B2.
These results show how riboflavin might be related to cognition, although the reasons are yet to be proven. Two well-established risk factors for cerebrovascular damage and small vessel disease are hyperhomocysteinemia, acting via endothelial impairment, ischemia, and oxidative damage [182], and advanced glycation end product (AGE) formation, which can lead to micro and macrovascular damage, accelerating AD [193]. Riboflavin is an independent determinant of homocysteine levels [184,194], and its deficiency is related to higher homocysteine levels, increasing vascular complications. B2 is also essential for vitamin B6 activation. The latter is a powerful anti-glycation agent, preventing AGE formation, thus protecting blood vessels [195,196,197]. Furthermore, these findings suggest that B2 levels might differ even with a similar dietary intake [166,179]. However, additional studies, using more robust biomarkers, i.e., plasma levels instead of food intake questionnaires, and experimental approaches are required to better evaluate the role of B2 in cognitive decline.
6. Vitamin B3 (Niacin)
The vitamin B3 family consists of three different molecules: Niacin or nicotinic acid, nicotinamide, and nicotinamide riboside (NR) [198,199]. Together with tryptophan, they are essential for the synthesis of nicotinamide adenine dinucleotide (NAD+) and nicotinamide adenine dinucleotide phosphate (NADP+), and their reduced forms NADH and NADPH [198]. These co-factors play a fundamental role in several cellular processes and are essential for cells’ survival [198,199,200]. They are involved in vital redox reactions, such as glycolysis and gluconeogenesis, beta-fatty oxidation, and steroid anabolism, thus, protecting cells from oxidative damage. They are crucial for mitochondrial respiration and ATP formation, and are strongly demanded in the citric acid cycle. The vitamin B3 family interacts in the inflammatory cascade, promoting calcium signaling and acting as a direct neurotransmitter via the purine receptor.
Furthermore, the NAD+ pool influences genetic stability and epigenetic variability; it is the main co-factor of poly (ADP-ribose) polymerases (PARPs), ADP-ribose transferases (ATRDs), and sirtuins. These enzymes regulate the DNA repair process, transcription, chromatin expression, cellular death, and senescence regulating some vias, e.g., the telomere length [198,199,200,201].
Vitamin B3 compounds derive from different food sources, such as beans, meat, fish, milk, mushrooms, and enriched flour. However, malnutrition, alcohol abuse, and infectious/autoimmune processes might cause insufficient vitamin levels. The main consequence of acute B3 deficiency, leading to a lack of NAD, is pellagra. This disease is mostly present in emerging nations, where starvation or very poor corn-based diets are dominant. In countries with niacin-fortified food (i.e., flour and grain), pellagra can be only observed in alcohol addicted, immunosuppressed, and cachectic patients. Symptoms of pellagra include diarrhea, dermatitis, and neurological disorders such as anxiety, depression, and dementia [199,201,202].
Due to the above-explained different functions exerted by vitamin B3, suboptimal B3 levels might lead to cellular dysfunction, including early cell death, changes in cellular metabolism, and aging, increasing the chances of misfolding and telomeric alteration, even promoting neoplastic degeneration [199,203]. Some subgroups, such as the elderly, pregnant women, or patients undergoing DNA damaging treatments, are at a higher risk of suboptimal levels because of increased body requests, despite an adequate vitamin intake [199].
The role of B3 in the brain has been studied for more than 70 years [204,205,206], but only recently has the focus shifted from considering vitamin B3 as a determinant factor for cell survival, to the extension to be an essential neuronal and vessel protector [200,201,202,203,204,205,206,207,208,209]. Some of the most critical neuroprotective mechanisms have been primarily investigated in animal models but not yet reproduced in humans. It has been shown that B3, mainly as NAD+, might reduce the lesion size in global [210,211] or focal cerebral ischemia [199,200], as well as in transient cerebral ischemia [212,213], and might ameliorate the primary outcome, after traumatic brain injury [214,215,216,217,218]. Indeed, neuronal death, derived in acute conditions by necrosis and apoptosis, could be prevented via several mechanisms, such as oxidative stress protection [219,220,221], control over PARP-1 activity thus preserving energy levels [211,222], and direct inhibition of pro-inflammatory cytokines [223]. Further, B3, primarily as NR, protects neurons from axonal degeneration, derived from excitotoxicity, due to significant ischemic condition [224].
The same neuroprotective neuro-vascular pathways seem to also influence neurodegenerative pathologies [225]. Animal models show niacin’s role in Huntington’s disease (HD) [226], Parkinson’s disease [227], and Alzheimer’s disease [228]. It is likely that in PD and HD, nicotinamide supply might increase NAD+ and ATP levels, improving mitochondrial functioning, impaired in both pathologies [229]. Additionally, PD patients benefit from B3 supplementation, which reflects the improvement of some symptoms due to its interaction with the NIACR1 receptor, enhancing the availability of NAD+ by either the use of a diet supplemented with NAD+ precursors or the inhibition of NAD+-dependent enzymes, such as PARPs, which compete with mitochondria for NAD+. This could be a viable approach to preventing neurotoxicity associated with mitochondrial defects [201,230,231,232].
In dementias, the neuroprotective action of niacin is related to its effects on cerebral microvascular endothelial cells (ECs) and neurons. It protects both types of cells from oxidative damage, ischemic insults, and senescence, and influences inflammatory processes, primarily related to microglial functions. ECs are basal cells to brain homeostasis and regulate both blood flow and the blood–brain barrier [200,233]. They are injured in clinical stroke but are also altered in subclinical, chronic ischemic insults. Experimental studies on rats have shown how ischemic/hypoxic or direct damage from nitric oxide (NO) induces apoptosis in ECs [233,234]. Niacin protects ECs and neurons from apoptotic injury, preventing the exposure of membrane phosphatidylserine (PS) residues, which act as a phagocytosis signal, inducing the DNA fragmentation [200,207]. B3 interacts with different pathways that activate apoptosis, including caspase 1, 3, 8, PARP-1 activity and protein kinase B (Akt). The latter maintains mitochondrial polarization, avoiding cytochrome C release, and inhibiting the activation of the caspase [207,234].
More interestingly, exposure of PS leads to a loss of anticoagulant membrane components, increasing the risks of secondary thrombosis, clot aggregation, and inflammation [200,207,229]. Furthermore, it has been demonstrated that niacin also reduces atheroma formation, decreasing the total amount of lipoproteins and increasing the incidence of high-density lipoproteins (HDL) [235].
Dysfunctional ECs are determined more often by the normal processes of senescence. Indeed, dysfunction leads to an altered proliferation of ECs, associated with an increment of pro-inflammatory factors, impaired proliferation, higher rates of apoptosis, and increased permeability of the BBB due to the loss of the tight junctions [236,237,238]. Sirtuin1, a component of the sirtuins family, is an NAD+ enzyme involved in several functions, such as the senescence process, gene expression/regulation, and cell death, and it is primarily represented in neurons and ECs. A single study showed a decline in Sirt1 expression and activity in aged mice and aged humans’ ECs in vitro [236]. In this study, the authors demonstrated the underlying relationship between Sirt1 depletion and BBB increased permeability [236]. Since Sirt1 depends on NAD+ activity, one animal study showed that nicotinamide supplementation reversed age-related vascular EC dysfunction [239]. In humans, a phase II randomized clinical trial investigated the effects of Resveratrol, a Sirt1 agonist, in AD patients with promising results [240], but more studies are needed [241].
To the best of our knowledge, we might hypothesize that low B3 levels are frequent in the elderly, and this may influence the development of small vessel disease and subcortical vascular dementia, switching ECs towards a procoagulant state, increasing plaque deposition, reducing Sirt1 expression, and promoting senescence mechanisms, such as apoptosis, neuroinflammation (through Sirt1 and PARP, 200, 242, 243), ECs, and neuronal death. Niacin modulates neuroinflammation in several ways. It regulates microglial activation, known to promote oxidative stress and inflammation, avoiding PS exposure, a trigger of microglial function and proliferation [200]. It modulates PARP-1 activity, involved in NF-kb transcription, which, in turn, regulates the secretion of cytokines and chemokines, and it may also inhibit some pro-inflammatory cytokines, such as interleukin (IL)-1β, IL 6, IL8, and TNF-α directly [223,242].
Nicotinamide restriction has been shown to increase NADPH oxidase, and reactive oxygen species (ROS) release in human keratinocytes [243]. High ROS levels are associated with the oxidative cascade of events, determining many neurodegenerative disorders, due to neuronal and EC loss [244,245]. Nicotinamide has proved to be a powerful antioxidant in animal models, maintaining brain mitochondrial homeostasis and reducing neuronal and EC oxidative stress [220,239,242].
Nicotinamide supplementation has been tested in animals and humans with AD yielding promising but still incomplete results. In AD model mice, NR prevented Aβ accumulation and hippocampal astrocyte loss [228,246,247], attenuating cognitive decline and improving selective cognitive impairment [228,246]. The same benefits have not been confirmed in clinical trials yet, despite past and ongoing attempts [242]. Further, dietary intake of B3 was associated with lower cognitive decline and seemed to have protective effects on the development of AD [248]. More clinical trials are currently investigating nicotinamide supplementation in AD patients and will soon add new knowledge regarding its efficacy in neurodegenerative diseases. However, since vitamin B3 is involved in vital pathways of both neurons and cerebrovascular endothelial cells, an association with dementia is likely, and its supplementation in subclinical conditions might improve not only AD but also vascular dementia outcomes and disease progression [249].
7. Vitamin B5 (Pantothenic Acid)
Vitamin B5, also called pantothenic acid/pantothenate, is the primary precursor of coenzyme A (CoA), crucial in several cellular processes. As wither the direct form (CoA) or the acetylated form (acetyl-CoA), this coenzyme is involved in energy production and respiration, via the citric acid cycle. It is essential for fatty acid synthesis and β-oxidation, and for cholesterol, lipid, and sphingolipid biosynthesis, as well as for the production of steroid hormones and neurotransmitters, i.e., acetylcholine [195,201,250,251]. Further, acetyl-CoA plays a role in global histone acetylation, modulating gene expression, cell growth, and proliferation [252,253]. Pantothenic acid, due to the increase of CoA, exerts an antioxidative property [254,255,256] and influences inflammatory factors, such as C-reactive protein (CRP) [257]. Indeed, it has been shown in vitro that pantothenic acid might enhance glutathione levels [254,255,256] and, in animal models, high concentrations protect neurons from radiation damages [255].
Moreover, pantothenic acid exerts an antioxidative pathway through cysteamine, a product of CoA degradation. This catabolic reaction is handled by a family of enzymes, called vanins, and vanin-1 is upregulated in oxidative stress. Hence, it seems plausible that the presence of high CoA turnover in some tissues demonstrates a protective role against oxidative injury [253].
Some pantothenic acid derivatives, such as panthenol and pantetheine, the dimeric form of endogenous pantetheine produced from pantothenic acid and cysteamine, modulate and interact with the cholesterol metabolism, lowering low-density lipoproteins (LDL) and enhancing HDL [258,259,260]. The exact mechanism underlying these processes is still unknown, and a correlation with homocysteine has been hypothesized, involving both CoA biosynthesis and atherosclerosis [259].
Vitamin B5 is present in almost every food, being an essential compound for all forms of life [253,261]. Significant sources of pantothenic acid are broccoli, meat, whole grains, cereals, and avocado. It is worth mentioning that commensal bacteria, such as Escherichia coli, can produce and secrete pantothenate directly in the gut, increasing the availability of this vitamin [195,253,255]. Therefore, despite low oral intake, B5 deficiencies have been rarely described in humans. The described symptoms of pantothenic acid deficiency include numbness and paresthesia, insomnia, and irritability, as well as nausea, vomiting, and non-specific cardiovascular alterations [195,201,261].
Pantothenic acid is well represented in the central nervous system (CNS) and is more concentrated in the brain than in the plasma [255,262]. Several old studies focused on its transportation and function in the CNS [263,264,265,266,267], but it has not been investigated as much as the other B vitamins, likely because very few studies described its deprivation.
Vitamin B5 is known to be related to a rare neurodegenerative autosomal recessive disorder, called pantothenate kinase-associated neurodegeneration (PKAN), due to genetic mutations in the pantothenate kinase-2 (PANK-2), an enzyme responsible for the phosphorylation of pantothenate, the first reaction of the CoA biosynthetic pathway [250,268,269,270,271].
Nevertheless, a recent study demonstrated that low levels of B5 were found in post-mortem brain tissue of Huntington’s disease (HD) patients as opposed to matched healthy controls [252]. The authors hypothesized that a B5 deficiency might be involved in the alteration of citric acid cycles, leading to inadequate energy levels, as frequently reported in HD patients.
Additionally, investigators have speculated on the role of acetyl-CoA. Acetyl-CoA is fundamental to several cellular pathways, including gene expression, which is strictly dependent on CoA levels, and its activity might be impaired due to a B5 insufficiency [252].
Only one study has tried to assess the role of pantothenic acid in cognition with unexpected results [272]. Indeed, the authors found that a higher dietary intake of B5 correlates to an increased burden of cerebral amyloid in patients with subjective cognitive impairment and MCI, both conditions that might precede AD development. The reason might be related to inappropriate usage of defense factors, such as pro-inflammatory cytokines, as a result of normally adequate levels of CoA [257,272]. Nonetheless, CoA has high turnover rates in several tissues [253], and its cytosolic and plasma levels might differ from pantothenic acid dietary intake, i.e., a high oral intake of vitamin B5 does not necessarily correspond to higher CoA cellular levels, also determined by the feedback inhibition regulating CoA production [273]. Additionally, it seems more plausible that insufficient CoA levels, essential for acetyl-CoA and, hence, acetylcholine synthesis, might influence cholinergic neurons and in such a way be associated with neurological diseases, including AD [252,274].
In conclusion, a lack of CoA seems to be related to neurodegenerative disorders, due to its antioxidative and anti-inflammatory properties, but its clinical effect has never been demonstrated.
8. Vitamin B6 (Pyridoxine)
Vitamin B6 has six different forms in mammals, including pyridoxamine, pyridoxal, and pyridoxine. These three compounds are converted into pyridoxal 5′-phosphate (PLP), which is the biologically active derivative of vitamin B6. PLP is a co-factor for more than 140 enzymes and accounts for almost 4% of humans’ enzymatic activities [275,276]. It is involved in amino acid, glucose, and lipid metabolism, as well as heme and hormone synthesis. In the brain, it is essential for sphingolipid formation for myelin and several neurotransmitters, such as serotonin, norepinephrine, dopamine, and GABA [201,275]. PLP is mostly known for its involvement in the one-carbon metabolism and its link to homocysteine regulation [170,195,276]. Further, B6 is also interacting with the immune systems, through NF-kB downregulation, CRP levels, and peripheric lymphocyte proliferation [195,277,278]. It has antioxidative activities, acting both as a scavenger of reactive oxygen species and as a coenzyme for cysteine, a precursor of glutathione, and it protects blood vessels from AGE accumulation [195,201,276].
Vitamin B6 is mostly available from natural sources, and a varied diet is a sufficient condition for its adequate intake. It is present in meat, fish, nuts, potatoes, bananas, and fortified cereals [275,279].
A severe B6 deficiency is a rare medical condition, leading to microcytic anemia due to an impaired hemoglobin synthesis [201,275]. General symptoms include irritability, depression, migraines, neuropathy, and sleep disorders, and are likely associated with impaired synthesis of neurotransmitters and hormones, especially GABA and melatonin [170,201]. In infants, an inborn error affecting PLP production pathways, might generate acute B6 insufficiency and provoke neonatal epileptic encephalopathies [275]. Marginal and subclinical B6 deficiency is rather common in the general population, and it has been associated with higher cardiovascular risk and chronic inflammatory diseases, including rheumatoid arthritis, chronic bowel disease, diabetes, psychiatric disorders, and cognitive decline [170,277,280]. Several subgroups are at risk of suboptimal B6 levels, independent of their dietary intake, due to increased requests, reduced absorption, or metabolic interactions. Indeed, an inadequate status is frequent in the elderly, as shown by extensive population studies in Europe, UK, and the US, even though body changes more than age itself seem to be determinant, and in alcoholics and women taking low-dosage of oral contraceptives [170,276,281,282].
Vitamin B6 and PLP are deeply associated with normal brain function. Beyond the synthesis of neurotransmitters, B6 compounds are involved in protecting neurons and ECS from oxidative damages and atherosclerosis, maintaining the BBB integrity, regulating inflammatory processes, and regulating homocysteine synthesis [282,283,284,285,286].
Several neurological conditions involve B6 compounds to various extents. Infants and children might develop the so-called pyridoxine-responsive epilepsies, originating mostly from rare recessive mutations in genes codifying for B6 metabolism’s enzymes [287,288]. In animal models with induced Streptococcus pneumoniae-related meningitis, B6 pre-treatment might reduce hippocampal neuronal loss [289,290]. Low levels of B6 seem to be associated with a higher risk of developing PD; it has been argued that PLP is a coenzyme of dopa-decarboxylase, which converts levodopa to dopamine [284,291,292].
The relationship between vitamin B6 and cardiovascular disease (CVD), including significant stroke and small vessel disease, both possibly inducing vascular dementia, depends on various processes in which B6 actively participates. These are inflammation, oxidative metabolism, glycation, and the B6 capacity to lower homocysteine. The latter is a well-known risk factor for endothelial damage and atherosclerosis, and seems to be involved in SVD and subcortical vascular dementia through endothelial damage [293,294]. Indeed, hyperhomocysteinemia in mice, resulting from a diet poor in B vitamins (namely, B6, folate, and B12) led to a vascular dementia model, characterized by microhemorrhages and neuroinflammation [280], and to severe neuronal degeneration, vascular dysfunction, BBB leakage, and memory loss [295]. In addition to lowering homocysteine [296,297,298], B6 might have an independent role in CVD through antioxidative and anti-inflammatory proprieties.
Vitamin B6 protects cerebral endothelial cells from oxidative damage and prevents atherosclerosis and BBB dysfunction. B6 avoids EC apoptosis induced by homocysteine in animal models and reduces endothelial progenitor cells’ apoptosis in stroke patients, likely through inhibition of caspase activity (which in the ischemic condition is enhanced by higher homocysteine levels) [299,300]. Pyridoxamine, pyridoxine, and pyridoxal phosphate reduce superoxide anion and lipid peroxidation, generated by hydrogen peroxide even if it does not prevent the direct cellular damage of these reactive compounds [301]. Pyridoxine interacts with LDL-induced endothelial dysfunction, restoring eNOS activity, NO levels, and cGMP [302,303]. One study reported that subjects receiving supplementation of several vitamins, including B6, had less oxidative stress in the brain measured by the established neural biomarkers [304]. Furthermore, all B6 compounds, particularly pyridoxamine, are active in the degradation of AGE products, reducing the risk of atherosclerosis and the production of free radicals, and perhaps, positively influencing Aβ deposition in AD [193,195,304,305,306].
In the context of inflammation, PLP levels are inversely related to inflammatory markers in several inflammation-related pathologies, such as rheumatoid arthritis and CVD [277]. In particular, low PLP has been related to high CRP in the general population, and high CRP, in turn, is a known risk factor of CVD [285,307,308]. Several studies reviewed by Lotto et al. [277] have found low B6 status in patients with coronary artery disease, myocardial infarction, and stroke, strengthening the hypothesis of an inflammatory link between atherosclerosis-based pathologies and B6. Furthermore, higher intakes of this vitamin have shown protective effects against inflammation. This is not surprising since PLP (B6 active form) influences the cytokine release and the lymphocyte proliferation [278,307,309].
A recent human study showed that in acute ischemic stroke, patients receiving supplementation of B vitamins, including B6, within 12 h after the beginning event, had decreased lipid peroxidation and inflammation, measured as CRP levels, independent from homocysteine levels [283].
Several authors have investigated the association between CVD and low B6 in humans. In a case-control study, low B6 levels were independently associated with the risk of stroke or transient ischemic attacks [310]. In a cohort of patients with silent brain impairments and cognitive decline, brain atrophy was significantly related to low PLP levels, while homocysteine had no significant relationship [311]. Further, in AD patients, white matter hyperintensities (WMH), markers of SVD, were associated with B6 deficiencies, independently of homocysteine concentrations [312,313]. Miller et al. [314] also found reduced levels of PLP in AD patients as opposed to healthy controls. Alternatively, in contrast with these results, Malaguarnera et al. [315] did not find any difference in B6 levels between VaD patients, AD, and healthy controls, but samples were small, and the mean levels of the three groups were not reported [315]. Another study by Nelson et al. [316] did not find any significant relationship between dietary B6 intake and AD risk, but plasma biomarkers were not investigated [285]. Hence, despite some negative findings, a relationship between B6 and dementia progression cannot be excluded.
Several clinical trials tried to assess the benefits of supplementation of B6, B12, and folate on the risk of major vascular diseases, including stroke, coronary artery disease, myocardial infarction, and neurodegenerative conditions, such as AD and VaD, due to SVD.
In the VISP study, Toole et al. [317] enrolled 3680 subjects from Canada, the US, and Scotland with previous non-disabling stroke. Participants received either a low or a high dose of vitamins B6, B12, and folic acid supplementation in a randomized, double-blind procedure. After two years of follow up, the trial was stopped for ineffectiveness: Vitamins, despite the lowering effect on homocysteine, did not reduce the rate of recurrent stroke, coronary artery disease, and death following vascular events [317]. However, several factors might have affected these results, including the deficient baseline homocysteine level, the short follow up time, and the small sample size for the conditions of study [318]. Another multi-centric randomized, double-blind placebo-controlled trial was carried out by the VITATOPS group [319]. Here, 8164 participants with ischemic stroke, TIA, or intracerebral hemorrhage, and from four different continents, were included in the final analysis and had randomly received either placebo or a vitamin supplementation of B6, B12, and folic acid. The primary outcomes were a non-fatal stroke, non-fatal myocardial infarction, and death of vascular origin. There was no significative difference in the two groups after a mean follow up of 3–4 years. However, the authors observed that in a subgroup analysis of patients with symptomatic SVD, the group who received the vitamins seemed to have better outcomes. According to this result, Cavalieri et al. [320] analyzed the effect of the same vitamin formulation versus placebo on MRI changes in patients with cerebral SVD. They did not find any difference in terms of lacunar infarct and WMH in the general group; yet, in a specific subgroup with severe SVD at baseline, the supplementation slowed the progression of WMH [320]. According to this, two other studies on healthy elderly showed that supplementation of B6 and B12 [321] or higher intake of vitamin B6 [322] were related to a higher gray matter volume, in specific brain regions, and also showed better cortical preservation. Another study on healthy and relatively young subjects (mean age around 45 years) investigated possible MRI differences in terms of WMH and brain atrophy, according to the assumption of B6 and folic acid or placebo, over two years. The authors noticed a mild but not significant improvement in MRI parameters in the group with the supplementation [323].
Other studies, involving different vitamin formulation (not including B6), have been conducted and meta-analyzed by the VITATOPS group [319]. Supplementary vitamins do not seem to reduce the risk of CVD in the general population with major vascular events. Nevertheless, particular subgroups, e.g., with severe cerebral SVD, might benefit from vitamin intake and, in the healthy elderly, an adequate vitamin level might help preserve brain integrity.
The association between cognitive function and B6 has been well investigated, with discordant results.
Low PLP levels have been associated with cognitive decline over two years in a Boston-Puerto Rican population, with a stronger link in the older participants [324]. Higher intakes of B6 showed a positive correlation with memory, psychomotor abilities, and verbal fluency [325,326,327]. Conversely, one study on healthy elderly subjects, despite an association between low B6 levels and poor baseline cognition, reported a loss of correlation after adjustment for covariates and other vitamins. Jannusch et al. [322] did not find any link between B6 and cognition in healthy subjects with normal to high levels. Hence, they suggested the existence of a “ceiling effect” [322]: When normal-high vitamin B6 levels have been reached, the effect on cognition after a supplementation might vanish.
Supplementation studies with cognitive function as primary outcome yielded similarly inconsistent results. In patients with mild-moderate AD, a high dosage of B6–B9 and B12, as opposed to a placebo, did not improve or slow down cognitive decline [328,329], despite a definite lowering of homocysteine levels. Similarly, patients with past ischemic vascular events who received B6 supplements did not show any change in cognitive function over 12 months [330]. A recent metanalysis, from 2017 [331], reported the lack of evidence on the therapeutic effects of vitamin supplementation on cognitive function in subjects with cognitive impairment due to AD or other dementias [331]. In healthy subjects, a significant effect on cognition was not reported either, despite a slight improvement in memory, and particularly of long-term memory [332,333,334].
More studies focusing on single vitamins, specific subgroup analysis, and longer follow-ups are still required to prove B6 involvement and potential benefits on cognitive decline. To the best of our knowledge, no study has yet reported breakthrough results, but, in light of current findings, there is a strong need for more specific and dedicated investigation [335].
9. Vitamin B7 (Biotin)
Vitamin B7, also called biotin or vitamin H (hair), is widely known for its role as co-factor for four carboxylases: acetyl-CoA, pyruvate carboxylase, beta-methylcrotonyl-CoA carboxylase, and propionyl-CoA carboxylase, essential for the synthesis of fatty acid, amino acids catabolism, the citric acid cycle, energy production, and gluconeogenesis [336,337,338].
Recently, the role of biotin has been extended to epigenetic modulation through histones biotinylation, gene expression, and cellular signaling [338,339,340]. In particular, it has been found that biotin regulates the synthesis of enzymes involved in the glucose homeostasis, such as glucokinase and phosphoenolpyruvate carboxykinase, and stimulates soluble guanylate cyclase (sGC), boosting cGMP concentrations [201,339,341].
Biotin is also involved in inflammatory processes and is a critical factor for NF-κB expression in lymphoid cells [336]. Further, it seems to have a role as an antioxidant, likely interacting with PARP1, reducing pro-inflammatory cytokines and apoptosis, as shown in hippocampal neurons receiving γ-irradiation [342,343].
Biotin levels depend on dietary intake and partially on gut flora production. Various foods are rich in this vitamin, such as liver, egg yolks, soybeans, fish, and leafy vegetables [201,337]. Severe biotin deficiency is uncommon and is usually related to malnutrition or a lack of biotinidase, involved in biotin recycling [338]. Clinical symptoms include alopecia, dermatitis, ketolactic acidosis, skin infection, ataxia, hypotonia, sensory loss, hallucination, and lethargy [337,338,344]. Indeed, neuronal cells and skin cells seem more sensitive to biotin deficiency than other cells, like fibroblasts [345]. Low biotin levels might be found in individuals with type II diabetes or poor glucose-regulation and pregnancy, perhaps due to constant increased metabolic requests [201,337]. Interestingly, unlike other B vitamins, B7 levels in the elderly seem to be in range or even higher as opposed to youngsters [346,347]; hence, more investigations are needed.
Biotin concentrations in the CNS are higher than in the plasma and are located in specific brain regions, mostly the diencephalic area and cerebellar motor nuclei [348]. A saturable transport system handles the transportation of B7 through the BBB, depending on a free carboxylic acid group, which, if necessary, can enhance biotin concentration in the cerebrospinal fluid to levels of 50%–250% more than the plasma [348,349,350]. Indeed, moderate B7 deficiency does not typically lead to neurological symptoms and is related to healthy brain carboxylase function [338].
The most common severe conditions related to biotin lack are the genetically determinant defects in biotin metabolism or transportation enzymes, e.g., biotin-responsive basal ganglia disease. These disorders are rare, mostly affecting children and adolescents, and could be potentially fatal if not adequately replaced with a high dosage of biotin [338,351].
In addition to genetic alterations, biotin might play a role in some chronic diseases and have neuroprotective functions. Indeed, in the 1980s, biotin was reported to improve diabetic neuropathy, as well as uremic neurological complications in a small number of subjects [352,353]. More recently, a study showed that biotin administration in multiple sclerosis patients slows the disability progression and ameliorates the global clinical impression compared with placebo [336]. No benefit was reported in a cohort of older patients with different patterns of progressive MS [354], and more investigations are certainly required.
Two mechanisms might be behind the biotin effect observed in MS. Through the potentiation of the carboxylases activity, biotin might enhance fatty acid synthesis and, thus, increase re-myelination and energy production in neurons, protecting against myelin loss and hypoxia-driven degeneration [354,355]. By a direct stimulation of the sGC, biotin might increase the brain production of cGMP, which is involved in survival, plasticity, and protective pathways [341].
To the best of our knowledge, biotin might play a role in chronic neurodegenerative disorders, such as AD, where patients have low cerebrospinal fluid cGMP levels, as well as in vascular dementia associated with SVD due to cGMP having an anti-inflammatory activity on the cerebral microvasculature, and in stroke due to cGMP anti-atherosclerotic proprieties [341,356,357]. Interestingly, one study on stroke-prone rats reported a decreased systolic blood pressure and a lower stroke incidence (0%) in the animals that received a saline solution with biotin, as opposed to the group that received the solution without biotin (20% of stroke incidence) [344]. Furthermore, B7 might play a role in stroke induction, through the regulation of NF-kB expression, and neuronal apoptosis [338,358].
Biotin might be involved and improve several acute and chronic neurological conditions, but the interest in its neuroprotective function is recent and more studies investigating possible pathways and mechanisms of action in vitro, as well as in vivo, are needed to clarify its role in the brain [359].
10. Vitamin B9 (Folic Acid) and Vitamin B12: Separate or Coexistent Realities?
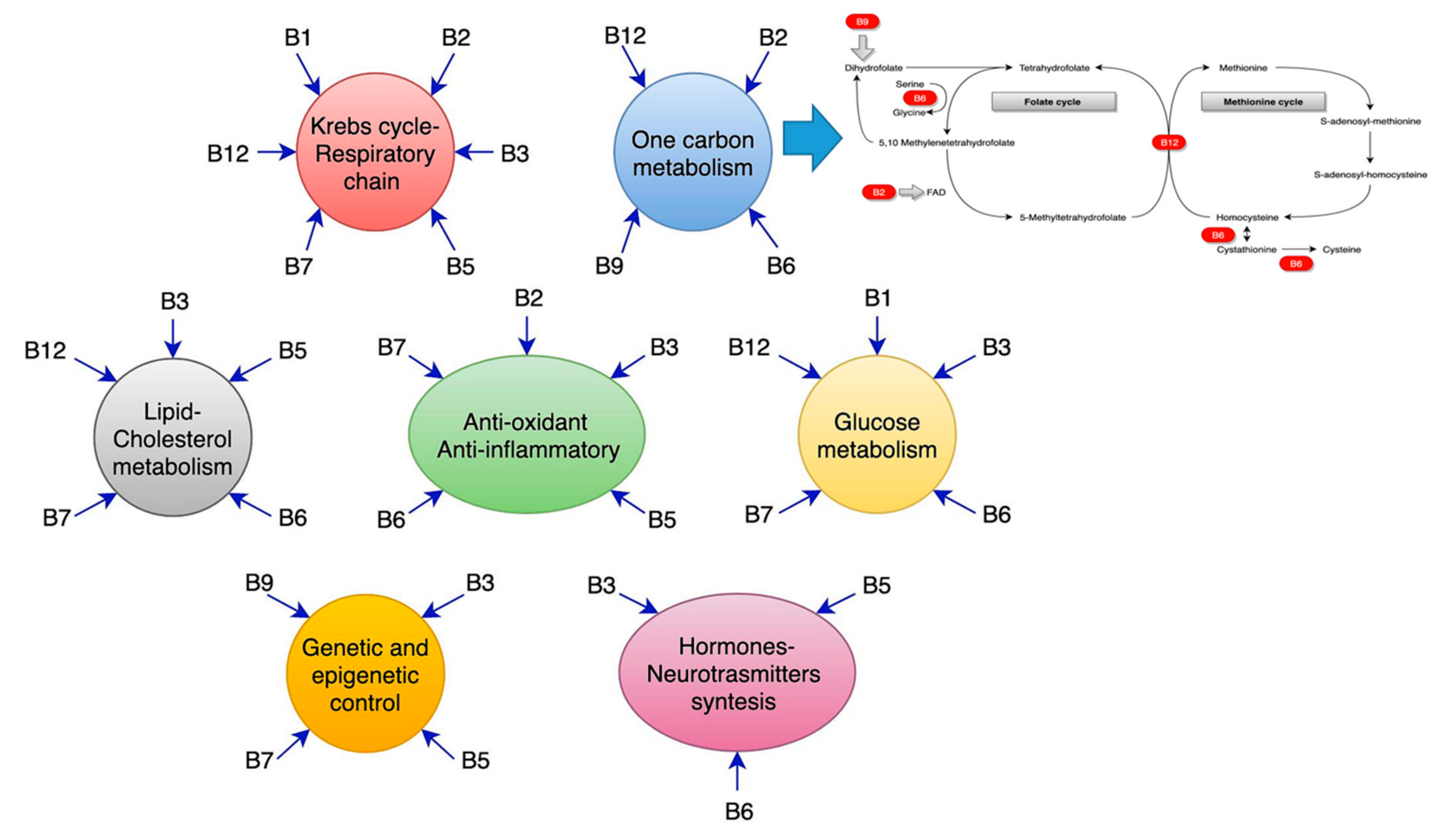
Folic acid derives its name from the Latin word, folium, meaning leaf, and the active form of vitamin B9 is folate known as levomefolic acid or 5-methyltetrahydrofolate (5-MTHF). Dietary folic acid predominantly exists as polyglutamate, which have to be hydrolyzed to mono-glutamates in order to be available [182,360]. Folic acid is hydrolyzed in the gut, and mono-glutamylated folates are absorbed in the duodenum and the first part of the jejunum by a high-affinity receptor, (PCFT1) [361,362]. Folic acid exerts its action mainly through its participation in the so-called histidine cycle (deamination of histidine, production of urocanic acid, and generation of fomiminoglutamate, fundamental for all the glutamic acid cycles), serine and glycine cycle, thymidylate cycle, and purine cycle. A quantity of 5–10 methylene tetrahydrofolate provides a hydroxymethyl group to glycine residues in order to produce serine, the principal donor of the one-carbon unit. The methylation process will be further explained in the text below. Folate is involved in the thymidylate synthase, which is fundamental in cell replication [363]. Finally, tetrahydrofolate derivatives are employed in two reaction steps of the de novo synthesis of purine.
There are different conditions which cause folate depletion; some are physiological, such as aging or pregnancy, while some are para-physiological, such as the recovery from wounds or systemic illness [364,365,366,367]. Dietary low intake, alcoholism, malabsorption, diffuse inflammatory disease of the small intestine, Crohn’s disease, coeliac disease, chronic liver disease, kidney failure, and medications (i.e., phenytoin, carbamazepine, metformin, methotrexate, and sulfasalazine) reduce the activity of pteroyl polyglutamate, a specific hydrolase required for folate absorption, and thereby leading to folate deficiency [368,369,370]. Some specific hematological conditions (occurring as an isolated deficit of homocysteine methyltransferase or a combined B12-homocysteine methyltransferase defect) lead to a clinical situation defined as methyl trap of tetrahydrofolate (THF). This active metabolic form of folic acid is converted to act as a donor of methyl-THF, and acid folic cannot be employed differently [368].
Curiously enough, even vitamin B12, also called cobalamin, is not produced by human beings. Approximately 20 human genes are known to be involved in the absorption, transport, and employment of vitamin B12 acquired from the diet [371]. This aspect gives the reason for the two fundamental cofactor roles of vitamin B12. It is required for the correct functions of the cytosolic methionine synthase and the mitochondrial methyl malonyl-CoA mutase. In particular, cobalamin helps the catalysis process, fundamental for the conversion of L-methyl malonyl-CoA to succinyl-CoA [371]. This reaction is fundamental in the catabolism of the side chain of cholesterol, of the catabolism of the branched-chain amino acids and odd-chain fatty acids. The cytosolic methionine synthase employs the methylated cobalamin for the remethylation of homocysteine to methionine, which we discuss later [372].
A congenital deficit of vitamin B12 is associated with destructive alteration of brain functions [373,374], although different causes precipitate vitamin B12 levels (the most normal aging process, per se) [360]. Some authors converge the aging process with a consequent reduction of intrinsic factor, primary or secondary to atrophic gastritis or hypochlorhydria [373,374]. Other clinical conditions of cobalamin defects are: Genetic deficiency of transcobalamin II, an inadequate intake (vegans, starvation, alcohol addiction, sleeve gastrectomy), malabsorption (inadequate production of intrinsic factors, terminal ileum inflammation, Crohn’s disease, small bowel syndrome, celiac disease), and drugs (pump proton inhibitors, metformin, antiepileptic drugs, chemotherapies, etc.) [369,375]. Vitamin B12 is used by the body in two forms, either as methylcobalamin or five deoxyadenosine cobalamin. The enzyme methionine synthase needs methylcobalamin as a cofactor. This enzyme is generally involved in the conversion of the amino acid homocysteine into methionine, while methionine, in turn, is required for DNA methylation [371]. The cofactor 5 deoxyadenosyl cobalamin is needed by the enzyme that converts l methyl malonyl CoA to succinyl CoA. This stage leads to an extraction of energy from proteins and fats. In addition, succinyl CoA is necessary for the production of hemoglobin, which is the vehicle for oxygen in red blood cells [376]. The loss of vitamin B12 leads to an impairment of Kreb’s cycle, with a reduction of efficacy of the conversion of succinate to fumarate, malate, and the end product of the cycle, rendered less efficacious in energy production [377,378]. Due to the lack of vitamin B12, there is an impairment in gluconeogenesis [379].
Folate and B12 are intimately bound in the methylation reactions, which we discuss in the following section. It is an old axiomatic teaching that treating a B12 deficient patient with folate or conversely a folate-deficient patient with B12 may exacerbate the neurologic consequences of either deficiency; therefore, cyanocobalamin deficiencies should be excluded before folate supplementation is commenced or, if necessary, it should be appropriate to supplement folate and vitamin B12 together [360,380,381,382,383,384].
This axiom is not fully understood; nevertheless, it has been recently rewritten by the NIH [385]. The recommendation implies that supplementation of large amounts of folic acid can mask the damaging effects of vitamin B12 deficiency and, therefore, NIH recommends that folic acid intake from fortified food and supplements should not exceed 1000 μg daily in healthy adults. Other recent data suggested that although an estimated 9–12% of older people in the UK suffering from folate deficiency, due to their everyday dietary intake, there is an excess of folic acid intake in young people, due to promotional and commercial purposes, even if there is “insufficient evidence that folic fortification could promote cancer” [170]. No precise rule has emerged, although as concluded by Porter et al., “those contemplating public health issues worldwide deed to consider a balanced approach and should endeavor to achieve the optimal status of all relevant B-vitamins, throughout all stages of life” [170]. The other emerging problem is new diet habits, i.e., vegetarians, vegans, and patients with an inferior diet (polished rice and nothing more) [386]. In the USA, good clinical practice, as stated by the Institute of Medicine and the Harvard TH Chan School of Public Health [387], recommends a general intake of 400 micrograms per day of folate, suggesting that people who regularly drink alcohol should intake at least 600 micrograms per day. Food sources of folate are fruits and vegetables, whole grains, beans, cereals, and fortified grain products. The recommended intake of vitamin B12 is 2.4 micrograms per day. It is found in fish, poultry, meat, eggs, dairy, fortified cereals, and enriched soy or rice milk [360].
Although many studies have been dedicated to the implementation of folic acid, vitamin B12, or both, in vascular condition, AD or advanced dementia patients, or the old population, de facto results have never been reproducible, and the debate thus remains open [388,389,390,391,392,393,394,395,396,397,398,399,400,401,402,403,404,405].
In conclusion, although the complex metabolic process in which vitamin B12 and folate are bound is well known, there is a lack of knowledge of implementation and time onset of supplementation. It appears as if more evidence would result from more advanced study, but strict lab measures, exclusion criteria, and frequent observation should be ethically recommended rules in clinical practice [360,406].
11. Homocysteine: The Sharing Process of Vitamin B12 and Folate.
Homocysteine (Hcy) is related to the production of 5,10-methylenetetrahydrofolate, a fundamental step for the synthesis of thymidylate, purines and methionine, employing vitamin B12 and folate as cofactors [407,408,409,410]. The S-adenosyl-methionine (SAM) to S-adenosyl-L-homocysteine (SAH) ratio defines the methylation potential of a cell [411]. If homocysteine is allowed to accumulate in normal conditions, it will be rapidly metabolized to SAH [412]. Whenever there is a methionine deficit, Hcy can be re-methylated to form methionine, by the employment of N5, N10-methylenetetrahydrofolate [412]. If there is an adequate amount of methionine, Hcy is employed for the production of cysteine, mediated by cystathionine–beta-synthase, with pyridoxine and folate as a cofactor [182]. Therefore, the accumulation of Hcy is dangerous when it occurs in the absence of folate and vitamin B12 as a cofactor. The causative factors of accumulation of Hcy in healthy adult life can be diverse, due to various genetic defects, or to the defects of vitamin B12 and folate [182]. A physiological increase of Hcy occurs in the brain (and CSF) and the plasma, within the aging process [296]. Evidence has showed that the adult life hyperhomocysteine condition is associated with cardiovascular and cerebrovascular diseases [413,414]. It has been reported that hyperhomocysteine (HHcy) promotes cerebro/cardiovascular atherosclerosis and instable/ruptured atherothrombotic plaques, upregulating the expression of matrix metalloproteinases-9 (MMP-9) expression [415,416].
Homocysteine is a common final pathway of the lack of folate and vitamin B12. Its role has been widely discussed elsewhere [296]; here, we report the significant and more innovative lines of study of homocysteine in brain vascular alteration, underlying small vessel diseases.
Undoubtedly, the methylation reactions are necessary for the brain, SAM being the sole donor in numerous methylation reactions involving proteins, phospholipids, and biogenic amines [375], and for packaging of many phospholipids [417]. Most of the polyunsaturated phosphatidylcholines (PC) in mammals are synthesized by phosphatidylethanolamine N-methyltransferase (PEMT) [418,419,420], which methylates phosphatidylethanolamine to generate phosphatidylcholine using SAM as the methyl donor. The leading site of the PEMT-catalyzed synthesis of PC-DHA is liver, whereas the PEMT activity in the brain is relatively low [421], and the polyunsaturated species of PC [422] that are synthesized there are adequate for local needs only. PEMT activity in the brain is higher during the perinatal period and declines in adulthood [421].
The PEMT reaction consumes three molecules of SAM for every PC molecule produced and generates three molecules of S-adenosyl-L-homocysteine (SAH), which act as an inhibitor of PEMT [423]. Some of the SAH-derived hepatic homocysteine enters the circulation, determining a link between Hcy and PEMT [424]. Some studies of AD patients related higher plasma concentration of Hcy to reduced levels of erythrocyte phosphatidylcholine-docosahexaenoic acid [425]. The role of phosphatidylcholine has been debated in vascular dementia, and citicoline has demonstrated neuroprotective effects in acute stroke and has been shown to improve cognition in patients with chronic cerebrovascular disease and some patients with Alzheimer’s disease. Citicoline has prevented cognitive decline after stroke, exhibiting a neuroprotective effect [426,427,428,429,430].
It has been proven that Hcy could be linked to neurodegeneration; Hcy, together with high levels of glycine in the brain, is an agonist of the endogenous NMDA receptors [431], influencing calcium influx [432], and exerting a direct activation of the group I metabotropic glutamate receptors [433]. Hyperhomocysteine (HHcy) upregulates Presenilin 1, which promotes APP synthesis [434]. The protein phosphatase methyltransferase 1, whose methylation is SAM-dependent, regulates the activity of the protein phosphatase methyltransferase 2A, which acts as a dephosphorylating system for tau protein [435]. Hence, the reduced methylation capacity increases the hyperphosphorylation of tau protein, determining microtubule disaggregation, and the deposition of the neurofibrillary tangles. Moreover, Hcy potentiates the toxicity of Abeta 42 deposition [436], in particular, increasing its deleterious effects on the smooth vascular cells in the brain [437].
Hcy acts as a pro-inflammatory and pro-oxidative factor. The SAM-to SAH ratio is the expression of the methylation potential of a cell; as a consequence, “HHcy tends to decrease the methylation potential” [412]. Therefore, Hcy can induce a global DNA hypomethylation and suppress the transcription of cyclin A in endothelial cells. Apoptosis of human endothelial cells after growth factor deprivation is associated with rapid and dramatic up-regulation of cyclin A–associated cyclin-dependent kinase 2 activity. Simultaneously, Hcy leads to up-regulation of other genes, causing an increase of p66shc expression in endothelial cells, inducing oxidant stress [412]. HHcy leads to an induction of mRNA of C-reactive protein (CRP), augmenting the NR1 subunit of NMDA receptor expression. Therefore, Hcy can promote a pro-inflammatory response in vascular smooth muscle cells of small brain arteries by stimulating CRP production, usually enhanced by a combined NMDA-ROS-erk1/2/p38-nfKBeta signal pathway [438].
Recently, a well-conducted study [439] demonstrated that cultured cell incubation with Hcy determined cell death at 80 microM Hcy exposure after five days; impressively, cell exposure to Hcy at lower concentrations for five days increased reactive oxygen species (ROS) production 4.4-fold. HHcy induced, in the beginning, endothelial cells to produce nitric oxide (NO) and to yield S-nitrose-Hcy, which acts as a protector of endothelium, although the chronic exposure to Hcy induces a final diminishment of NO [440]. HHcy acts in a multistep process against the endothelium. Its accumulation leads to a disruption of the cell’s integrity and, then, the HHcy-dependent reduced NO bioavailability induces an altered endothelium relaxation and an urgent inflammatory response of muscle cell arteries, testified by an increase of C-reactive proteins and cysteinyl-leukotrienes and of HMG-CoA reductase [441]. Finally, HHcy tends to reduce the efficiency of the cystathionine-beta-synthase, which induces altered redox homeostasis, with macroscopic alteration of the oxidative repairing process [442,443]. An accelerated lipid peroxidation sequela is the main result, with fatal outcomes for neuronal cells, astrocytes, and neurovascular coupling [444,445,446,447,448,449].
Recently, HHcy by itself (without folate and B12 deficiencies) has been associated with secondary septic status and conditioned poor outcome [450]. It has been argued that it leads to a direct increase of the macrophage response, with an induced release of ROS, and an altered oxidative repair [450]. More recently, an induction of B-lymphocyte has been demonstrated by a HHcy, with a macroscopic stimulation of the activity of the pyruvate kinase muscle isozyme 2, which seems to be involved in the acceleration of the atherosclerosis process [451,452] and probably the alteration of the transcriptional repression of fibroblast growth factor 2 [428]. As explained [296], the promoted HHcy activity on NMDA receptors has been demonstrated not only on neurons but also on neutrophils and macrophages. In this way, HHcy upregulates the nuclear factor-kappa B, one of “the master regulators of the expression of inflammatory genes” [453,454].
Even all these positive data, clinical trials, and studies failed to demonstrate conclusive results, either considering HHcy as a deterrence target, or preventing HHcy through supplementation in patients or a healthy population with vitamin B12, folate, or both. Many criticisms may be made of the trials that have been implemented [455,456,457,458].
12. Conclusions
The conclusions of the present review can be stated as follows: Vitamins of the B group are tightly related to gene control for endothelium protection and act as antioxidants. They play a co-enzymatic role in a number of ways: conversion of pyruvate to acetyl-CoA; conversion of alpha-ketoglutarate to succinate, in the Kreb’s cycle; catalysis by transketolase in the pentose monophosphate shunt, superoxide dismutase (SOD) and catalase; and, in the one-carbon metabolism cycle, to reduce 5,10-methylenetetrahydrofolate (5,10-MTHF) to 5-methyl THF, which, in turn, provides the methyl group for homocysteine re-methylation to methionine pantothenate kinase-2, acetyl-CoA, pyruvate carboxylase, beta-methylcrotonyl-CoA carboxylase, and propionyl-CoA carboxylase). Often, their role is the most critical in many different biochemical reactions inside the brain, interacting with many other constituents, such as participating in the synthesis of polyunsaturated phosphatidylcholine, through the SAM methyl donation (Figure 1). They have anti-inflammatory properties and play a protective role against neurodegenerative mechanisms, such as by modifying the glutamate currents and reducing calcium currents, as well as showing important antioxidant properties. It has even been argued that they play a direct neuropeptide synthesis promoter role.
However, there remain many coincidences and a significant number of unanswered questions [459,460,461]. Laboratory data are far from providing a clinical resolution of these mysteries, such as in the cases of AD or small vessel disease dementia. However, “one coincidence is just a coincidence, two coincidences are a clue, three coincidences are a proof”; at the moment, all the laboratory analyses are directed without any hesitation towards the data mentioned above. The questions are:
- Should we apply them to clinical practice?
- When should we employ them? Earlier is better, but when is “early” not intelligent or too wasteful?
- How can we manage clinical trials to be simultaneously efficacious and objective?
- Which are the markers of the evolution from healthy aging towards pathology? In this case, a question that remains open concerns identifying when the small vessel alteration of white matter becomes dementia.
Further studies should take into account all these questions, with well-designed and globally homogeneous trials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Karakis, I.; Pase, M.P.; Beiser, A.; Booth, S.L.; Jacques, P.F.; Rogers, G.; DeCarli, C.; Vasan, R.S.; Wang, T.J.; Himali, J.J.; et al. Association of serum vitamin D with the risk of incident dementia and subclinical indices of brain aging: The Framingham Heart Study. J. Alzheimer’s Dis. 2016, 51, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Román, G.C.; Tatemichi, T.K.; Erkinjuntti, T.; Cummings, J.L.; Masdeu, J.C.; Garcia, J.H.; Amaducci, L.; Orgogozo, J.M.; Brun, A.; Hofman, A. Vascular dementia: Diagnostic criteria for Research studies. Reports of the NINDS-AIREN International Workshop. Neurology 1993, 43, 250–260. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization for vascular dementia. The ICD-10 Classification of Mental and Behavioral Disorders; Diagnostic Criteria for Research; World Health Organization: Geneva, Switzerland, 1993; pp. 48–50. [Google Scholar]
- Chui, H. Vascular dementia, a new beginning: Shifting focus from clinical phenotype to ischemic brain injury. Neurol. Clin. 2000, 18, 951–978. [Google Scholar] [CrossRef]
- Román, G.C.; Goldstein, M. A population-based study of dementia in 85-year-olds. N. Engl. J. Med. 1993, 329, 63. [Google Scholar]
- Shi, Y.; Wardlaw, J.M. Update on cerebral small vessel disease. A dynamic whole-brain disease. Stroke Vasc. Neurol. 2016, 1, 83–92. [Google Scholar] [CrossRef]
- American Psychiatric Association. Major or Mild Vascular Neurocognitive disorders. In Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Publishing: Washington, DC, USA, 2013; pp. 612–615. [Google Scholar]
- Sinha, P.; Bharath, S.; Chandra, S.R. DSM-5 in vascular dementia. Comparison with other diagnostic criteria in a retrospective study. EC Neurol. 2015, 2, 135–143. [Google Scholar]
- Wardlaw, J.M.; Smith, E.E.; Biessels, G.J.; Cordonnier, C.; Fazekas, F.; Frayne, R.; Lindley, R.I.; O’Brien, J.T.; Barkhof, F.; Benavente, O.R.; et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013, 12, 822–832. [Google Scholar] [CrossRef]
- De Laat, K.F.; van Norden, A.G.; Gons, R.A.; van Oudheusden, L.J.; van Uden, I.W.; Bloem, B.R.; Zwiers, M.P.; de Leeuw, F.E. Gait in elderly with cerebral small vessel disease. Stroke 2010, 41, 1652–1658. [Google Scholar] [CrossRef]
- Jellinger, K.A. Pathology and pathogenesis of vascular cognitive impairment—Acritical update. Front. Aging Neurosci. 2013, 5, 17. [Google Scholar] [CrossRef]
- Patel, B.; Markus, H.S. Magnetic resonance imaging in cerebral small vessel disease and its use as a surrogate disease marker. Int. J. Stroke 2011, 6, 47–59. [Google Scholar] [CrossRef]
- Erkinjuntti, T.; Inzitari, D.; Pantoni, L.; Wallin, A.; Scheltens, P.; Rockwood, K.; Roman, G.C.; Chui, H.; Desmond, D.W. Resaerch criteria for subcortical vascular dementia in clinical trials. J. Neural Transm. Suppl. 2000, 59, 23–30. [Google Scholar] [PubMed]
- Roman, G.C.; Erkinjunnti, T.; Wallin, A.; Pantoni, L.; Chui, H.C. Subcortical ischemic vascular dementia. Lancet Neurol. 2002, 1, 426–436. [Google Scholar] [CrossRef]
- Jani, B.I.; Rajkumar, C. Ageing and vascular ageing. Postgrad. Med. J. 2006, 82, 357–362. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, J.C. Vascular basis of Alzheimer’s pathogenesis. Ann. N. Y. Acad. Sci. 2002, 977, 196–215. [Google Scholar] [CrossRef] [PubMed]
- Mathias, C.J.; Kimber, J.R. Postural hypotension: Causes, clinical features, investigation, and management. Annu. Rev. Med. 1999, 50, 317–336. [Google Scholar] [CrossRef] [PubMed]
- Roriz-Filho, J.S.; Bernardes-Silva-Filho, S.R.; Rosset, I.; Roriz-Cruz, M. Postural blood pressure dysregulation and dementia: Evidence for a vicious circle and implications for neurocardiovascular rehabilitation. In Cardiac Rehabilitation; Halliday, J.T., Ed.; Novascience Publisher Inc.: New York, NY, USA, 2009; pp. 1–37. ISBN 987-1-60741-918-1. [Google Scholar]
- Pantoni, L.; Garcia, J.H.; Gutierrez, J.A. Cerebral white matter is highly vulnerable to ischemia. Stroke 1996, 27, 1641–1647. [Google Scholar] [CrossRef]
- Schmidt, R.; Schmidt, H.; Haybaeck, J.; Loitfelder, M.; Weis, S.; Cavalieri, M.; Seiler, S.; Enzinger, C.; Ropele, S.; Erkinjuntti, T.; et al. Heterogeneity in age-related white matter changes. Acta Neuropathol. 2011, 122, 171–185. [Google Scholar] [CrossRef]
- Hommet, C.; Mondon, K.; Constans, T.; Beaufils, E.; Desmidt, T.; Camus, V.; Cottier, J.P. Review of cerebral microangiopathy and Alzheimer’s disease: Relation between white matter hyperintensities and microbleeds. Dement. Geriatr. Cogn. Disord. 2011, 32, 367–378. [Google Scholar] [CrossRef]
- Munoz, D.G.; Hastak, S.M.; Harper, B.; Lee, D.; Hachinski, V.C. Pathologic correlates of increased signals of the centrum ovale on magnetic resonance imaging. Arch. Neurol. 1993, 50, 492–497. [Google Scholar] [CrossRef]
- Salloway, S. Subcortical Vascular Dementia: Binswanger’s and CADASIL; American Academy of Neurology (AAN): Honolulu, HI, USA, 2003; pp. 1–29. [Google Scholar]
- Mirski, M.A. Pharmacology of Blood Pressure Management during Cerebral Ischemia; American Academy of Neurology (AAN): Miami, FL, USA, 2005; pp. 456–469. [Google Scholar]
- Wallin, A.; Blennow, K.; Gottfries, C.G. Neurochemical abnormalities in vascular dementia. Dementia 1989, 1, 120–130. [Google Scholar]
- Moretti, R.; Caruso, P. Small vessel disease to subcortical dementia: A dynamic model, which interfaces aging, cholinergic dysregulation and the neurovascular unit. Vasc. Health Risk Manag. 2019, in press. [Google Scholar]
- Bohnen, N.I.; Muller, M.L.T.M.; Kuwabara, H.; Ocnstantien, G.M.; Studentski, S.A. Age-associated leukoaraiosis and cortical cholinergic deafferentation. Neurology 2009, 72, 1411–1416. [Google Scholar] [CrossRef] [PubMed]
- Román, G.C. Brain hypoperfusion: A critical factor in vascular dementia. Neurol. Res. 2004, 26, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.S.; Beyreuther, K.; Schmitt, H.P. Synaptophysin immunoreactivity of the cortical neuropil in vascular dementia of Binswanger type compared with the dementia of Alzheimer type and non-demented controls. Dementia 1994, 5, 79–87. [Google Scholar] [CrossRef]
- Ahtiluoto, S.; Polvikoski, T.; Peltonen, M.; Solomon, A.; Tuomilehto, J.; Winblad, B.; Sulkava, R.; Kivipelto, M. Diabetes, Alzheimer disease, and vascular dementia: A population-based neuropathologic study. Neurology 2010, 75, 1195–1202. [Google Scholar] [CrossRef]
- Englund, E.A.; Person, B. Correlations between histopathologic white matter changes and proton MR relaxation times in dementia. Alzheimer Dis. Assoc. Disord. 1987, 1, 156–170. [Google Scholar] [CrossRef]
- Román, G.C. Senile dementia of the Binswanger type: A vascular form of dementia in the elderly. JAMA 1987, 258, 1782–1788. [Google Scholar] [CrossRef]
- Moody, D.M.; Brown, W.R.; Challa, V.R.; Anderson, R.L. Periventricular venous collagenosis: Association with leukoaraiosis. Radiology 1995, 194, 469–476. [Google Scholar] [CrossRef]
- Vinters, H.V.; Ellis, W.G.; Zarow, C.; Zaias, B.W.; Jagust, W.J.; Mack, W.J.; Chui, H.C. Neuropathological substrate of ischemic vascular dementia. J. Neuropathol. Exp. Neurol. 2000, 59, 931–945. [Google Scholar] [CrossRef]
- Garcia, J.H.; Lassen, N.A.; Weiller, C.; Sperling, B.; Nakagawara, J. Ischemic stroke and incomplete infarction. Stroke 1996, 27, 761–765. [Google Scholar] [CrossRef]
- Dalkara, T.; Alarcon-Martinez, L. Cerebral micro-vascular signaling in health and disease. Brain Res. 2015, 1623, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Giannakopoulos, P.; Gold, G.; Kövari, E.; von Gunten, A.; Imhof, A.; Bouras, C.; Hof, P.R. Assessing the cognitive impact of Alzheimer disease pathology and vascular burden in the aging brain: The Geneva experience. Acta Neuropathol. 2007, 113, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Launer, L.J.; Hughes, T.M.; White, L.R. Microinfarcts, brain atrophy, and cognitive function: The Honolulu Asia Aging Study Autopsy Study. Ann. Neurol. 2011, 70, 774–780. [Google Scholar] [CrossRef] [PubMed]
- Van der Veen, P.H.; Muller, M.; Vincken, K.L.; Hendrikse, J.; Mali, W.P.; van der Graaf, Y.; Geerlings, M.I. Longitudinal relationship between cerebral small vessel disease and cerebral blood flow. The second manifestations of arterial disease-magnetic resonance study. Stroke 2015, 46, 1233–1238. [Google Scholar] [CrossRef]
- Gouw, A.A.; van der Flier, W.M.; Fazekas, F.; van Straaten, E.C.; Pantoni, L.; Poggesi, A.; Inzitari, D.; Erkinjuntti, T.; Wahlund, L.O.; Waldemar, G.; et al. Progression of white matter hyperintensities and incidence of new lacunes over a 3-year period: The leukoaraiosis and disability study. Stroke 2008, 39, 1414–1420. [Google Scholar] [CrossRef]
- Schmidt, R.; Seiler, S.; Loitfelder, M. Longitudinal change of small vessel disease related brain abnormalities. J. Cereb. Blood Flow Metab. 2016, 36, 26–39. [Google Scholar] [CrossRef] [Green Version]
- Muñoz Maniega, S.; Chappell, F.M.; Valdés Hernández, M.C.; Armitage, P.A.; Makin, S.D.; Heye, A.K.; Thrippleton, M.J.; Sakka, E.; Shuler, K.; Dennis, M.S.; et al. Integrity of normal appearing white matter: Influence of age, visible lesion burden and hypertension in patients with small-vessel disease. J. Cereb. Blood Flow Metab. 2016, 37, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Smallwood, A.; Oulhaj, A.; Joachim, C.; Christie, S.; Sloan, C.; Smith, A.D.; Esiri, M. Cerebral subcortical small vessel disease and its relation to cognition in elderly subjects: A pathological study in the Oxford Project to Investigate Memory and Ageing (OPTIMA) cohort. Neuropathol. Appl. Neurobiol. 2012, 38, 337–343. [Google Scholar] [CrossRef]
- Kramer, J.H.; Reed, B.R.; Mungas, D.; Weiner, M.W.; Chui, H. Executive dysfunction in subcortical ischaemic vascular disease. J. Neurol. Neurosurg. Psychiatry 2002, 72, 217–220. [Google Scholar] [CrossRef] [Green Version]
- Burton, E.; Ballard, C.; Stephens, S.; Kenny, R.A.; Kalaria, R.; Barber, R.; O’Brien, J. Hyperintensities and fronto-subcortical atrophy on MRI are substrates of mild cognitive deficits after stroke. Dement. Geriatr. Cogn. Disord. 2003, 16, 113–118. [Google Scholar] [CrossRef]
- Tullberg, M.; Fletcher, E.; DeCarli, C.; Mungas, D.; Reed, B.R.; Harvey, D.J.; Weiner, M.W.; Chui, H.C.; Jagust, W.J. White matter lesions impair frontal lobe function regard-less of their location. Neurology 2004, 63, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Gold, G.; Kövari, E.; Herrmann, F.R.; Canuto, A.; Hof, P.R.; Michel, J.P.; Bouras, C.; Giannakopoulos, P. Cognitive consequences of thalamic, basal ganglia, and deep white matter lacunes in brain aging and dementia. Stroke 2005, 36, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Cheng, B.; Golsari, A.; Fiehler, J.; Rosenkranz, M.; Gerloff, C.; Thomalla, G. Dynamics of regional distribution of ischemic lesions in middle cerebral artery trunk occlusion relates to collateral circulation. J. Cereb. Blood Flow Metab. 2010, 31, 36–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijkhuizen, R.M.; Knollema, S.; van der Worp, H.B.; Ter Horst, G.J.; De Wildt, D.J.; Berkelbach van der Sprenkel, J.W.; Tulleken, K.A.; Nicolay, K. Dynamics of cerebral tissue injury and perfusion after temporary hypoxia-ischemia in the rat: Evidence for region-specific sensitivity and delayed damage. Stroke 1998, 29, 695–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, J.H.; Liu, K.F.; Ye, Z.R.; Gutierrez, J.A. Incomplete infarct and delayed neuronal death after transient middle cerebral artery occlusion in rats. Stroke 1997, 28, 2303–2309. [Google Scholar] [CrossRef]
- Konaka, K.; Miyashita, K.; Naritomi, H. Changes in diffusion-weighted magnetic resonance imaging findings in the acute and subacute phases of anoxic encephalopathy. J. Stroke Cerebrovasc. Dis. 2007, 16, 82–83. [Google Scholar] [CrossRef]
- Ravens, J.R. Vascular changes in the human senile brain. Adv. Neurol. 1978, 20, 487–501. [Google Scholar]
- Klassen, A.C.; Sung, J.H.; Stadlan, E.M. Histological changes in cerebral arteries with increasing age. J. Neuropathol. Exp. Neurol. 1968, 27, 607–623. [Google Scholar] [CrossRef]
- Cummings, J.L. Frontal-subcortical circuits and human behavior. Arch. Neurol. 1993, 50, 873–880. [Google Scholar] [CrossRef]
- Mega, M.S.; Cummings, J.L. Frontal-subcortical circuits and neuropsychiatric disorders. J. Neuropsychiatry Clin. Neurosci. 1994, 6, 358–370. [Google Scholar]
- Yao, H.; Sadoshima, S.; Kuwabara, Y.; Ichiya, Y.; Fujishima, M. Cerebral blood flow and oxygen metabolism in patients with vascular dementia of the Binswanger type. Stroke 1990, 21, 1694–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, A.; Ishii, N.; Nishihara, Y.; Horie, A. Medullary arteries in aging and dementia. Stroke 1991, 22, 442–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tak, S.; Yoon, S.J.; Jang, J.; Yoo, K.; Jeong, Y.; Ye, J.C. Quantitative analysis of hemodynamic and metabolic changes in subcortical vascular dementia using simulataneous near-infrared spectroscopy and FMRI measurements. Neuroimage 2011, 55, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Schroeter, M.; Cutini, S.; Wahl, M.; Scheid, R.; von Cramon, D.Y. Neurovascular coupling is impaired in cerebral microangiopathy an event related stroop study. Neuroimage 2007, 34, 26–34. [Google Scholar] [CrossRef]
- Bar, K.; Boettger, M.; Seidler, N.; Mentzelh, H.J.; Terborg, C.; Sauer, H. Influence of galantamine on vasomotor reactivity in AD and vascular dementia due to microangiopathy. Stroke 2007, 38, 3186–3192. [Google Scholar] [CrossRef] [Green Version]
- De Reuck, J.; Decoo, D.; Marchau, M.; Santens, P.; Lemahieu, I.; Strijckmans, K. Positron emission tomography in vascular dementia. J. Neurol. Sci. 1998, 154, 55–61. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Murase, K.; Oku, N.; Kitagawa, K.; Imaizumi, M.; Takasawa, M.; Nishikawa, T.; Matsumoto, M.; Hatazawa, J.; Hori, M. Statistical image analysis of cerebral blood flow in vascular dementia with small-vessel disease. J. Nucl. Med. 2003, 44, 505–511. [Google Scholar]
- Yang, D.; Kim, B.; Park, J.; Kim, S.; Kim, E.; Sohn, H. Analysis of cerebral blood flow of subcortical vascular dementia with single photon emission computed tomography: Adaptation of statistical parametric mapping. J. Neurol. Sci. 2002, 203, 199–205. [Google Scholar] [CrossRef]
- Ramirez-Gomez, C.; Zheng, C.; Reed, B.; Kramer, J.; Mungas, D.; Zarow, C.; Vinters, H.; Ringman, J.M.; Chui, H. Neuropsychological profiles differentiate Alzheimer Disease from Subcortical Ischemic vascular dementia in an autopsy-defined cohort. Dement. Geriatr. Cogn. Disord. 2017, 44, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.L. Vascular subcortical dementias: Clinical aspects. Dementia 1994, 5, 177–180. [Google Scholar] [CrossRef]
- Desmond, D.W.; Erkinjuntti, T.; Sano, M.; Cummings, J.L.; Bowler, J.V.; Pasquier, F.; Moroney, J.T.; Ferris, S.H.; Stern, Y.; Sachdev, P.S.; et al. The cognitive syndrome of vascular dementia: Implications for clinical trials. Alzheimer Dis. Assoc. Disord. 1999, 13, 21–29. [Google Scholar] [CrossRef]
- Sachdev, P.S.; Brodaty, H.; Valenzuela, M.J.; Lorentz, L.; Looi, J.C.; Wen, W.; Zagami, A.S. The neuropsychological profile of vascular cognitive impairment in stroke and TIA patients. Neurology 2004, 62. [Google Scholar] [CrossRef] [PubMed]
- Traykov, L.; Baudic, S.; Thibaudet, M.C.; Rigaud, A.S.; Samgghe, A.; Boller, F. Neuropsychological deficit in early subcortical vascular dementia: Comparison to AD. Dement. Geriatr. Cogn. Disord. 2002, 14, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.; Signori, R. Neural correlates for apathy: Frontal-prefrontal and parietal cortical-subcortical circuits. Front. Aging Neurosci. 2016, 9, 289. [Google Scholar] [CrossRef] [Green Version]
- Ishii, N.; Nashihara, Y.; Imamura, T. Why do frontal lobe symptoms predominate in vascular dementia with lacunes? Neurology 1986, 36, 340–345. [Google Scholar] [CrossRef]
- De Jager, C.A. Critical levels of brain atrophy associated with homocysteine and cognitive decline. Neurobiol. Ageing 2014, 35, S35–S39. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.L. Food for thought: Souvenaid in mild Alzheimer’s disease. J. Alzheimer’s Dis. 2012, 31, 237–238. [Google Scholar] [CrossRef]
- Peng, D. Geriatric Neurology Group; Chinese Society of Geriatrics. Clinical Practice Guideline for Cognitive Impairment of Cerebral Small Vessel Disease Writing Group. Clinical practice guideline for cognitive impairment of cerebral small vessel disease. Aging Med. 2019, 2, 64–73. [Google Scholar] [CrossRef]
- Roe, A.J.; Zhang, S.; Bhadelia, R.A.; Johnson, E.J.; Lichtenstein, A.H.; Rogers, G.T.; Rosenberg, I.H.; Smith, C.E.; Zeisel, S.H.; Scott, T.S. Choline and its metabolites are differently associated with cardiometabolic risk factors, history of cardiovascular disease, and MRI-documented cerebrovascular disease in older adults. Am. J. Clin. Nutr. 2017, 105, 1283–1290. [Google Scholar] [CrossRef]
- Blusztajn, J.K.; Slack, B.E.; Mellott, T.J. Neuroprotective Actions of Dietary Choline. Nutrients 2017, 9, 815. [Google Scholar] [CrossRef] [Green Version]
- Castro, C.A.; Rudy, J.W. Early-life malnutrition selectively retards the development of distal- but not proximal-cue navigation. Dev. Psychobiol. 1987, 20, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Mellott, T.J.; Huleatt, O.M.; Shade, B.N.; Pender, S.M.; Liu, Y.B.; Slack, B.E.; Blusztajn, J.K. Perinatal choline supplementation reduces amyloidosis and increases choline acetyltransferae expression in the hippocampus of the APPswePS1dE9 Alzheimer’s disease model mice. PLoS ONE 2017, 12, e0170450. [Google Scholar]
- Napoli, I.; Blusztajn, J.K.; Mellott, T.J. Prenatal choline supplementation in rats increases the expression of IGF2 and its receptor IGF2R and enhances IGF2-induced acetylcholine release in hippocampus and frontal cortex. Brain Res. 2008, 1237, 124–135. [Google Scholar] [CrossRef]
- Williams, C.L.; Meck, W.H.; Heyer, D.; Loy, R. Hypertrophy of basal forebrain neurons and enhanced visuospatial memory in perinatally choline-supplemented rats. Brain Res. 1998, 794, 225–238. [Google Scholar] [CrossRef]
- Cermak, J.M.; Holler, T.; Jackson, D.A.; Blusztajn, J.K. Prenatal availability of choline modifies development of the hippocampal cholinergic system. FASEB J. 1998, 12, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacheva, V.P.; Mellott, T.J.; Davison, J.M.; Wagner, N.; Lopez-Coviella, I.; Schnitzler, A.C.; Blusztajn, J.K. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J. Biol. Chem. 2007, 282, 31777–31788. [Google Scholar] [CrossRef] [Green Version]
- Lopes, S.; Lewis, A.; Hajkova, P.; Dean, W.; Oswald, J.; Forne, T.; Murrell, A.; Constancia, M.; Bartolomei, M.; Walter, J.; et al. Epigenetic modifications in an imprinting cluster are controlled by a hierarchy of DMRs suggesting long-range chromatin interactions. Hum. Mol. Genet. 2003, 12, 295–305. [Google Scholar] [CrossRef]
- Levenson, J.M.; Roth, T.L.; Lubin, F.D.; Miller, C.A.; Huang, I.C.; Desai, P.; Malone, L.M.; Sweatt, J.D. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 2006, 281, 15763–15773. [Google Scholar] [CrossRef] [Green Version]
- Yossifoff, M.; Kisliouk, T.; Meiri, N. Dynamic changes in DNA methylation during thermal control establishment affect CREB binding to the brain-derived neurotrophic factor promoter. Eur. J. Neurosci. 2008, 28, 2267–2277. [Google Scholar] [CrossRef]
- Lubin, F.D.; Roth, T.L.; Sweatt, J.D. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J. Neurosci. 2008, 28, 10576–10586. [Google Scholar] [CrossRef]
- Feng, J.; Zhou, Y.; Campbell, S.L.; Le, T.; Li, E.; Sweatt, J.D.; Silva, A.J.; Fan, G. Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 2010, 13, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Webb, W.M.; Sanchez, R.G.; Perez, G.; Butler, A.A.; Hauser, R.M.; Rich, M.C.; O’Bierne, A.L.; Jarome, T.J.; Lubin, F.D. Dynamic association of epigenetic H3K4me3 and DNA 5hmC marks in the dorsal hippocampus and anterior cingulate cortex following reactivation of a fear memory. Neurobiol. Learn. Mem. 2017, 142, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.J.; Rahn, E.J.; Paulukaitis, B.S.; Savell, K.E.; Kordasiewicz, H.B.; Wang, J.; Lewis, J.W.; Posey, J.; Strange, S.K.; Guzman-Karlsson, M.C.; et al. Tcf4 Regulates Synaptic Plasticity, DNA Methylation, and Memory Function. Cell Rep. 2016, 16, 2666–2685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barany, M.; Chang, Y.C.; Arus, C.; Rustan, T.; Frey, W.H. Increased glycerol-3-phosphorylcholine in post-mortem Alzheimer’s brain. Lancet 1985, 1, 517. [Google Scholar] [CrossRef]
- Blusztajn, J.K.; Gonzalez-Coviella, I.L.; Logue, M.; Growdon, J.H.; Wurtman, R.J. Levels of phospholipid catabolic intermediates, glycerophosphocholine and glycerophosphoethanolamine, are elevated in brains of Alzheimer’s disease but not of Down’s syndrome patients. Brain Res. 1990, 536, 240–244. [Google Scholar] [CrossRef]
- Corbin, K.D.; Zeisel, S.H. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr. Opin. Gastroeneterol. 2012, 28, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Karaca, U.; Schram, M.T.; Houben, A.J.; Muris, D.M.; Stehouwer, C.D. Microvascular dysfunction as a link between obesity, insulin resistance and hypertension. Diabetes Res. Clin. Pract. 2014, 103, 382–387. [Google Scholar] [CrossRef]
- Roberson, L.L.; Aneni, E.C.; Maziak, W.; Agatston, A.; Feldman, T.; Rouseff, M.; Tran, T.; Blaha, M.J.; Santos, R.D.; Sposito, A.; et al. Beyond BMI: The metabolically healthy obese phenotype and its association with clinical/subclinical cardiovascular disease all-cause mortality; a systematic review. BMC Public Health 2014, 14, 14. [Google Scholar] [CrossRef] [Green Version]
- Yuki, D.; Sugiura, Y.; Zaima, N.; Akatsu, H.; Takei, S.; Yao, I.; Maesako, M.; Kinoshita, A.; Yamamoto, T.; Kon, R.; et al. DHA-PC and PSD-95 decrease after loss of synaptophysin and before neuronal loss in patients with Alzheimer’s disease. Sci. Rep. 2014, 4, 7130. [Google Scholar] [CrossRef] [Green Version]
- Schaefer, E.J.; Bongard, V.; Beiser, A.S.; Lamon-Fava, S.; Robins, S.J.; Au, R.; Tucker, K.L.; Kyle, D.J.; Wilson, P.W.; Wolf, P.A. Plasma phosphatidylcholine docosahexaenoic acid content and risk of dementia and Alzheimer disease: The Framingham Heart Study. Arch. Neurol. 2006, 63, 1545–1550. [Google Scholar] [CrossRef]
- Trushina, E.; Dutta, T.; Persson, X.M.; Mielke, M.M.; Petersen, R.C. Identification of altered metabolic pathways in plasma and CSF in mild cognitive impairment and Alzheimer’s disease using metabolomics. PLoS ONE 2013, 8, e63644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Dominguez, R.; Garcia-Barrera, T.; Gomez-Ariza, J.L. Combination of metabolomic and phospholipid-profiling approaches for the study of Alzheimer’s disease. J. Proteom. 2014, 104, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Zhong, X.; Cheema, A.K.; Orquiza, M.H.; Chidambaram, S.; Tan, M.T.; Gresenz, C.R.; Fitzgerald, K.T.; Nalls, M.A.; Singleton, A.B.; et al. Plasma 24-metabolite Panel Predicts Preclinical Transition to Clinical Stages of Alzheimer’s Disease. Front. Neurol. 2015, 6, 237. [Google Scholar] [CrossRef] [PubMed]
- Proitsi, P.; Kim, M.; Whiley, L.; Simmons, A.; Sattlecker, M.; Velayudhan, L.; Lupton, M.K.; Soininen, H.; Kloszewska, I.; Mecocci, P.; et al. Association of blood lipids with Alzheimer’s disease: A comprehensive lipidomics analysis. Alzheimers Dement. 2016, 13, 140–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heude, B.; Ducimetiere, P.; Berr, C. Cognitive decline and fatty acid composition of erythrocyte membranes—The EVA Study. Am. J. Clin. Nutr. 2003, 77, 803–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, M.R.; Goh, E.L.; Silver, D.L. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 2014, 509, 503–506. [Google Scholar] [CrossRef]
- Guemez-Gamboa, A.; Nguyen, L.N.; Yang, H.; Zaki, M.S.; Kara, M.; Ben-Omran, T.; Akizu, N.; Rosti, R.O.; Rosti, B.; Scott, E.; et al. Inactivating mutations in MFSD2A, required for omega-3 fatty acid transport in brain, cause a lethal microcephaly syndrome. Nat. Genet. 2015, 47, 809–813. [Google Scholar] [CrossRef] [Green Version]
- Goedecke, L.; Fernadnez-Hernando, C. MicroRNAs: A connection between cholesterol metabolism and neurodegeneration. Neurobiol. Dis. 2014, 72, 48–53. [Google Scholar] [CrossRef] [Green Version]
- Scheltens, P.; Kamphuis, P.J.; Verhey, F.R.; Olde Rikkert, M.G.M.; Wurtman, R.J.; Wilkinson, D.; Twisk, J.W.R.; Kurz, A. Efficacy of a medical food in mild Alzheimer’s Disease: A randomized, controlled trial. Alzheimer’s Dement. 2010, 6, 1–10. [Google Scholar] [CrossRef]
- Scheltens, P.; Twisk, J.W.R.; Blesa, R.; Scarpini, E.; von Arnim, C.A.F.; Bongers, A.; Harrison, J.; Swinkels, S.H.N.; Stam, C.J.; de Waal, H.; et al. Efficacy of Souvenaid in mild Alzheimer’s disease: Results from a randomized, controlled trial. J. Alzheimer’s Dis. 2014, 31, 225–226. [Google Scholar] [CrossRef] [Green Version]
- Rijpma, A.; Meulenbroek, O.; van Hees, A.M.J.; Sijben, J.W.C.; Vellas, B.; Shah, R.C.; Bennett, D.A.; Scheltens, P.; Olde Rikkert, M.G.M. Effects of Souvenaid on plasma micronutiren levels and fatty acid profiles in mild and mild-to-mderate AD. Alzheimer’s Res. Ther. 2015, 7, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olde Rikkert, M.G.; Verhey, F.R.; Blesa, R.; von Arnim, C.A.; Bongers, A.; Harrison, J.; Sijben, J.; Scarpini, E.; Vandewoude, M.F.; Vellas, B.; et al. Tolerability and safety of Souvenaid in Patients with Mild Alzheimer’s Disease: Results of multi center, 24-week, open label extension study. J. Alzheimer’s Dis. 2015, 44, 471–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchetti, A.; Perotta, D.; Cravello, L.; Ranieri, P.; Trabucchi, M. Effectiveness of a specific nutritional supplement on cognitive, behavioral and functional sympotms in mild cognitive impairment and AD: caregivers’ judgements. Results of an observational survey. JGG 2018, 66, 68–74. [Google Scholar]
- Yanai, H. Effects of N-3 Polyunsaturated Fatty Acids on Dementia. J. Clin. Med. Res. 2017, 9, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.C.; Schneider, J.A.; Tangney, C.C. Thoughts on B-vitamins and dementia. J. Alzheimer’s Dis. 2006, 9, 429–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suwa, M.; Yamaguchi, S.; Komori, T.; Kajimoto, S.; Kino, M. The Association between Cerebral White Matter Lesions and Plasma Omega-3 to Omega-6 Polyunsaturated Fatty Acids Ratio to Cognitive Impairment Development. Biomed Res. Int. 2015, 2015, 153437. [Google Scholar] [CrossRef] [Green Version]
- Bowman, G.; Silbert, L.C.; Dodge, H.H.; Lahna, D.; Hagen, K.; Murchison, C.F.; Howieson, D.; Kaye, J.; Quinn, J.F.; Shinto, L. Randomized Trial of Marine n-3 Polyunsaturated Fatty Acids for the Prevention of Cerebral Small Vessel Disease and Inflammation in Aging (PUFA Trial): Rationale, Design and Baseline Results. Nutrients 2019, 11, 735. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Ren, H.; Yao, X.; Shi, Z.; Liang, F.; Kang, J.X.; Wan, J.B.; Pei, Z.; Su, K.P.; Su, H. Enriched Brain Omega-3 Polyunsaturated Fatty Acids Confer Neuroprotection against Microinfarction. EBioMedicine 2018, 32, 50–61. [Google Scholar] [CrossRef]
- Victor, M.; Adam, R.D.; Collins, G.H. The Wernicke-Korsakoff Syndrome and Related Neurologic Disorders Due to Alcoholism and Malnutrition, 2nd ed.; Davis Publications: Philadelphia, PA, USA, 1989. [Google Scholar]
- Victor, M.; Adams, R.D.; Collins, G.H. The Wernicke-Korsakoff syndrome. A clinical and pathological study of 245 patients, 82 with post-mortem examinations. Contemp. Neurol. Ser. 1971, 7, 201–206. [Google Scholar]
- Alzheimer’s Society. What is alcohol-related brain damage. Factsheet 438LP. 2015. Available online: https://www.alzheimers.org.uk (accessed on 15 November 2019).
- UK Chief Medical Officers’ Alcohol Guidelines Review. Available online: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/489795/summary.pdf (accessed on 15 September 2016).
- Gupta, S.; Warner, J. Alcohol-related dementia: A 21st-century silent epidemic? Br. J. Psychiatry 2008, 193, 351–353. [Google Scholar] [CrossRef] [Green Version]
- Sechi, G.; Serra, A. Wernicke’s encephalopathy: New clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007, 6, 442–455. [Google Scholar] [CrossRef]
- Pech, N.; Meyer, F.; Lippert, H.; Manger, T.; Stroh, C. Complications, reoperations, and nutrient deficiencies two years after sleeve gastrectomy. J. Obes. 2012, 2012, 828737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, C.G.; Sheedy, D.L.; Lara, A.I.; Garrick, T.M.; Hilton, J.M.; Raisanen, J. Prevalence of Wernicke-Korsakoff syndrome in Australia: Has thiamine fortification made a difference? Med. J. Aust. 1998, 168, 542–545. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.D. Mechanisms of vitamin deficiency in chronic alcohol misusers and the development of the Wernicke-Korsakoff syndrome. Alcohol Alcohol. Oxf. Suppl. 2000, 35, 2–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majchrzak, D.; Singer, I.; Männer, M.; Rust, P.; Genser, D.; Wagner, K.H.; Elmadfa, I. B-vitamin status and concentrations of homocysteine in Austrian omnivores, vegetarians and vegans. Ann. Nutr. Metab. 2006, 50, 485–491. [Google Scholar] [CrossRef]
- Moretti, R.; Caruso, P.; Dal Ben, M.; Gazzin, S.; Tiribelli, C. Thiamine and alcohol for brain pathology: Super-imposing or different causative factors for brain damage? Curr. Drug Abuse Rev. 2017, 10, 44–51. [Google Scholar] [CrossRef]
- Butterworth, R.F.; Giguère, J.F.; Besnard, A.M. Activities of thiamine-dependent enzymes in two experimental models of thiamine-deficiency encephalopathy 2 alpha-Ketoglutarate dehydrogenase. Neurochem. Res. 1986, 11, 567–577. [Google Scholar] [CrossRef]
- Dreyfus, P.M. Clinical application of blood transketolase determinations. N. Engl. J. Med. 1962, 267, 596–598. [Google Scholar] [CrossRef]
- Hazell, A.S.; Todd, K.G.; Butterworth, R.F. Mechanisms of neuronal cell death in Wernicke’s encephalopathy. Metab. Brain Dis. 1998, 13, 97–122. [Google Scholar] [CrossRef]
- Butterworth, R.F.; Kril, J.J.; Harper, C.G. Thiamine-dependent enzyme changes in the brains of alcoholics: Relationship to the Wernicke-Korsakoff syndrome. Alcohol. Clin. Exp. Res. 1993, 17, 1084–1088. [Google Scholar] [CrossRef]
- Peterson, C.; Héroux, M.; Lavoie, J.; Butterworth, R.F. Loss of (3H) kainate and of NMDA-displaceable (3H)glutamate binding sites in brain in thiamine deficiency: Results of a quantitative autoradiographic study. Neurochem. Res. 1995, 20, 1155–1160. [Google Scholar] [CrossRef] [PubMed]
- Mousseau, D.D.; Rao, V.L.; Butterworth, R.F. Alterations in serotonin parameters in brain of thiamine-deficient rats are evident prior to the appearance of neurological symptoms. J. Neurochem. 1996, 67, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Todd, K.G.; Butterworth, R.F. Evaluation of the role of NMDA-mediated excitotoxicity in the selective neuronal loss in experimental Wernicke encephalopathy. Exp. Neurol. 1998, 149, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Jhala, S.S.; Wang, D.; Hazell, A.S. Thiamine deficiency results in release of soluble factors that disrupt mitochondrial membrane potential and downregulate the glutamate transporter splice-variant GLT-1b in cultured astrocytes. Biochem. Biophys. Res. Commun. 2014, 6, 335–341. [Google Scholar] [CrossRef]
- Sheng, M.; McFadden, G.; Greenberg, M.E. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron 1990, 4, 571–582. [Google Scholar] [CrossRef]
- Estus, S.; Zaks, W.J.; Freeman, R.S.; Gruda, M.; Bravo, R.; Johnson, E.M. Altered gene expression in neurons during programmed cell death: Identification of c-jun as necessary for neuronal apoptosis. J. Cell. Biol. 1994, 127, 1717–1727. [Google Scholar] [CrossRef] [Green Version]
- Guerrini, I.; Thomson, A.D.; Cook, C.C.; McQuillin, A.; Sharma, V.; Kopelman, M.; Reynolds, G.; Jauhar, P.; Harper, C.; Gurling, H.M. Direct genomic PCR sequencing of the high affinity thiamine transporter (SLC19A2) gene identifies three genetic variants in Wernicke Korsakoff syndrome (WKS). Am. J. Med. Genet B Neuropsychiatr. Genet. 2005, 137, 17. [Google Scholar] [CrossRef]
- Guerrini, I.; Cook, C.C.; Kest, W.; Devitgh, A.; McQuillin, A.; Curtis, D.; Gurling, H.M. Genetic linkage analysis supports the presence of two susceptibility loci for alcoholism and heavy drinking on chromosome 1p22.1-11.2 and 1q21.3-24.2. BMC Genet. 2005, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Guerrini, I.; Thomson, A.D.; Gurling, H.M. Molecular genetics of alcohol related brain damage. Alcohol Alcohol. 2009, 44, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Witt, E.D.; Goldman-Rakic, P.S. Intermittent thiamine deficiency in the rhesus monkey. I. Progression of neurological signs and neuroanatomical lesions. Ann. Neurol. 1983, 13, 376–395. [Google Scholar] [CrossRef]
- Summers, J.A.; Pullan, P.T.; Kril, J.J.; Harper, C.G. Increased central immunoreactive beta-endorphin content in patients with Wernicke-Korsakoff syndrome and in alcoholics. J. Clin. Pathol. 1991, 44, 126–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G.E.; Hirsch, J.H.; Fonzetti, P.; Jordon, B.D.; Cirio, R.T.; Elder, J. Vitamin B1 and dementia. Ann. N. Y. Acad. Sci. 2016, 1367, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.; Hauser, R.; Chen, M. Plasma thiamine deficiency associated with Alzheimer’s disease but not Parkinson’s Disease. Met. Brain Dis. 1998, 13, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Fei, G.; Lu, J.; Jin, L.; Pan, S.; Chen, Z.; Wang, C.; Sang, S.; Liu, H.; Hu, W.; et al. Measurement of blood thiamine metabolites for Alzheimer’s Disease Diagnosis. Ebiomedicine 2016, 3, 155–162. [Google Scholar] [CrossRef] [Green Version]
- Chari, D.; Ali, R.; Gupta, R. Reversible dementia in elderly: Really uncommon? J. Geriatr. Ment. Health 2015, 2, 30–37. [Google Scholar]
- Morris, M.S. The Role of B Vitamins in Preventing and Treating Cognitive Impairment and Decline. Adv. Nutr. 2012, 3, 801–812. [Google Scholar] [CrossRef] [Green Version]
- Bunik, V.I. Thiamin-dependent enzymes: New perspectives from the interface between chemistry and biology. FEBS J. 2013, 280, 6373. [Google Scholar] [CrossRef] [Green Version]
- Szutowicz, A.; Bielarczyk, H.; Jankowska-Kulawy, A.; Pawelczyk, T.; Ronowska, A. Acetyl CoA, the key factor for survival or death of cholinergic neurons in course of neurodegenerative diseases. Neurochem. Res. 2013, 38, 1523–1542. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.; Gong, N.; Zhao, J.; Yu, Z.; Gu, F.; Chen, J.; Sun, X.; Lei, Z.; Yu, M.; Xu, Z.; et al. Powerful beneficial effects of benfotiamine on cognitive impairment and b-amyloid deposition in amyloid precursor protein/presenilin-1transgenic mice. Brain 2010, 133, 1342–1352. [Google Scholar] [CrossRef]
- Calingasan, N.Y.; Gandy, S.E.; Baker, H.; Sheu, K.F.; Kim, K.S.; Wisniewski, H.M.; Gibson, G.E. Accumulation of amyloid precursor protein-like immunoreactivity in rat brain in response to thiamine deficiency. Brain Res. 1995, 677, 50–60. [Google Scholar] [CrossRef]
- Bettendorff, L.; Wins, P. Biological functions of thiamine derivatives: Focus on non-coenzyme roles. Biochemistry 2013, 1, 10. [Google Scholar]
- Parkhomenko, Y.M.; Pavlova, A.S.; Mezhenskaya, O.A. Mechanisms responsible for the high sensitivity of neural cells to vitamin B1 deficiency. Neurophysiology 2016, 48, 429–448. [Google Scholar] [CrossRef]
- Mkrtchyan, G.; Aleshin, V.; Parkhomenko, Y.M.; Kaehne, T.; Disalvo, M.L.; Parroni, A.; Contestabile, R.; Vovk, A.; Bettendorf, L.; Bunik, V. Molecular mechanisms of the non-coenzyme action of thiamin in brain: Biochemical, structural and pathway analysis. Sci. Rep. 2015, 27, 12583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frederich, M.; Delvaux, D.; Gigliobianc, T.; Gangolf, M.; Dive, G.; Mazzucchelli, G. Thiaminylated adenine nucleotides. Chemical synthesis, structural characterization and natural occurrence. FEBS J. 2009, 276, 3256–3268. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, J.A.; Parrott, J. New considerations on the neuromodulatory role of thiamine. Pharmcology 2012, 89, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Geng, M.Y.; Saito, H.; Katsuki, H. The effects of thiamine and oxythiamine on the survival of cultured brain neurons. Jpn. J. Pharmacol. 1995, 68, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Langlais, P.J.; Zhang, S.X. Extracellular Glutamate Is Increased in Thalamus During Thiamine Deficiency-Induced Lesions and Is Blocked by MK-801. J. Neurochem. 1993, 61, 2175–2182. [Google Scholar] [CrossRef]
- Hazell, A.S.; Butterworth, R.F.; Hakim, A.M. Cerebral vulnerability is associated with selective increase in extracellular glutamate concentration in experimental thiamine deficiency. J. Neurochem. 1993, 61, 1155–1158. [Google Scholar] [CrossRef]
- Chou, W.P.; Chang, Y.H.; Lin, H.C.; Chang, Y.H.; Chen, Y.Y.; Ko, C.H. Thiamine for preventing dementia development among patients with alcohol use disorder: A nationwide population-based cohort study. Clin. Nutr. 2019, 38, 1269–1273. [Google Scholar] [CrossRef]
- Marashly, E.T.; Bohlega, S.A. Riboflavin has neuroprotective potential: Focus on Parkinson’s disease and migraine. Front. Neurol. 2017, 8, 333. [Google Scholar] [CrossRef]
- Thakur, K.; Tomar, S.K.; Singh, A.K.; Mandal, S.; Arora, S. Riboflavin and health: A review of recent human Research. Crit. Rev. Food Sci. Nutr. 2017, 57, 3650–3660. [Google Scholar] [CrossRef] [PubMed]
- Ashoori, M.; Saedisomeolia, A. Riboflavin (Vitamin B2) and Oxidative Stress: A Review. Br. J. Nutr. 2014, 111, 1985–1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saedisomeolia, A.; Ashoori, M. Riboflavin in Human Health: A Review of Current Evidences. Adv. Food Nutr. Res. 2018, 83, 57–81. [Google Scholar] [CrossRef] [PubMed]
- Nadh, T.; To, H.; Christensen, H.N. Riboflavin Can Protect Tissue from Oxidative Injury. Nutr. Rev. 1993, 51, 149–150. [Google Scholar] [CrossRef]
- Sanches, S.C.; Ramalho, L.N.Z.; Mendes-Braz, M.; Terra, V.A.V.A.; Cecchini, R.; Augusto, M.J.; Ramalho, F.S. Riboflavin (Vitamin B-2) Reduces Hepatocellular Injury Following Liver Ischaemia and Reperfusion in Mice. Food Chem. Toxicol. 2014, 67, 65–71. [Google Scholar] [CrossRef]
- Betz, A.L.; Ren, X.D.; Ennis, S.R.; Hultquist, D.E. Riboflavin Reduces Edema in Focal Cerebral Ischemia. Brain Edema IX 1994, 60, 314–317. [Google Scholar]
- Peraza, A.V.; Guzmán, D.C.; Brizuela, N.O.; Herrera, M.O.; Olguín, H.J.; Silva, M.L.; Tapia, B.J.; Mejía, G.B. Riboflavin and pyridoxine restore dopamine levels and reduce oxidative stress in brain of rats. BMC Neurosci. 2018, 19, 71. [Google Scholar] [CrossRef]
- Moat, S.J.; Ashfield-watt, P.A.L.; Powers, H.J.; Newcombe, R.G.; Mcdowell, I.F.W. Effect of Riboflavin Status on the Homocysteine-Lowering Effect of Folate in Relation to the MTHFR (C677T) Genotype. Clin. Chem. 2003, 302, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.J.; Wu, W.M.; Yang, F.L.; Hsu, G.S.W.; Huang, C.Y. Vitamin B2 Inhibits Glutamate Release from Rat Cerebrocortical Nerve Terminals. Neuroreport 2008, 19, 1335–1338. [Google Scholar] [CrossRef]
- Lin, Y.; Desbois, A.; Jiang, S.; Hou, S.T. Group B Vitamins Protect Murine Cerebellar Granule Cells from Glutamate/NMDA Toxicity. Neuroreport 2004, 15, 2241–2244. [Google Scholar] [CrossRef]
- Mazur-Bialy, A.I.; Pocheć, E. HMGB1 Inhibition During Zymosan-Induced Inflammation: The Potential Therapeutic Action of Riboflavin. Arch. Immunol. Ther. Exp. 2016, 64, 171–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porter, K.; Hoey, L.; Hughes, C.F.; Ward, M.; McNulty, H. Causes, Consequences and Public Health Implications of Low B-Vitamin Status in Ageing. Nutrients 2016, 8, 725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boisvert, W.A.; Castaneda, C.; Mendoza, I.; Langeloh, G.; Solomons, N.W.; Gershoff, S.N.; Russell, R.M.; Soloinons, W. Prevalence of Riboflavin Deficiency among Guatemalan Elderly People and Its Relationship to Milk Intake. Am. J. Clin. Nutr. 1993, 58, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Essama-Tjani, J.C.; Guilland, J.C.; Fuchs, F.; Lombard, M.; Richard, D. Changes in Thiamin, Riboflavin, Niacin, Beta-Carotene, Vitamins, C, A, D and E Status of French Elderly Subjects during the First Year of Institutionalization. Int. J. Vitam. Nutr. Res. 2000, 70, 54–64. [Google Scholar] [CrossRef]
- Ter Borg, S.; Verlaan, S.; Hemsworth, J.; Mijnarends, D.M.; Schols, J.M.; Luiking, Y.C.; De Groot, L.C. Micronutrient Intakes and Potential Inadequacies of Community-Dwelling Older Adults: A Systematic Review. Br. J. Nutr. 2015, 113, 1195–1206. [Google Scholar] [CrossRef]
- Powers, H.J. Riboflavin (Vitamin B-2) and Health. Am. J. Clin. Nutr. 2003, 77, 1352–1360. [Google Scholar] [CrossRef]
- Skalka, H.W.; Prchal, J.T. Cataracts and Riboflavin Deficiency. Am. J. Clin. Nutr. 1981, 34, 861–863. [Google Scholar] [CrossRef]
- Leshner, R.T. Riboflavin deficiency—A reversible neurodegenerative disease. Ann. Neurol. 1981, 10, 294–295. [Google Scholar]
- Norton, W.N.; Daskal, I.; Savage, H.E.; Seibert, R.A.; Lane, M. Effects of Riboflavin Deficiency on the Ultrastructure of Rat Sciatic Nerve Fibers. Am. J. Pathol. 1976, 85, 651–660. [Google Scholar] [CrossRef] [Green Version]
- Manole, A.; Jaunmuktane, Z.; Hargreaves, I.; Ludtmann, M.H.R.; Salpietro, V.; Bello, O.D.; Pope, S.; Pandraud, A.; Horga, A.; Scalco, R.S.; et al. Clinical, Pathological and Functional Characterization of Riboflavin-Responsive Neuropathy. Brain 2017, 140, 2820–2837. [Google Scholar] [CrossRef] [Green Version]
- Coimbra, C.G.; Junqueira, V.B.C. High Doses of Riboflavin and the Elimination of Dietary Red Meat Promote the Recovery of Some Motor Functions in Parkinson’s Disease Patients. Braz. J. Med. Biol. Res. 2003, 36, 1409–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, D.F.; Saluja, H.S. Prophylaxis of Migraine Headaches with Riboflavin: A Systematic Review. J. Clin. Pharm. Ther. 2017, 42, 394–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boehnke, C.; Reuter, U.; Flach, U.; Schuh-Hofer, S.; Einhaupl, K.M.; Arnold, G. High-Dose Riboflavin Treatment Is Efficacious in Migraine Prophylaxis: An Open Study in a Tertiary Care Centre. Eur. J. Neurol. 2004, 11, 475–477. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Refsum, H. Homocysteine, B Vitamins, and Cognitive Impairment. Annu. Rev. Nutr. 2016, 36, 211–239. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, A.H.; Yeo, N.E.; Weekman, E.M.; Wilcock, D.M. Homocysteine, Hyperhomocysteinemia and Vascular Contributions to Cognitive Impairment and Dementia (VCID). Biochim. Biophys. Acta 2015, 1862, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Hustad, S.; Ueland, P.M.; Vollset, S.E.; Zhang, Y.; Bjorke-Monsen, A.L.; Schneede, J. Riboflavin as a Determinant of Plasma Total Homocysteine: Effect Modification by the Methylenetetrahydrofolate Reductase C677T Polymorphism. Clin. Chem. 2000, 46, 1065–1071. [Google Scholar]
- Udhayabanu, T.; Manole, A.; Rajeshwari, M.; Varalakshmi, P.; Houlden, H.; Ashokkumar, B. Riboflavin Responsive Mitochondrial Dysfunction in Neurodegenerative Diseases. J. Clin. Med. 2017, 6, 52. [Google Scholar] [CrossRef] [Green Version]
- Xiu, L.; Lee, M.; Wahlqvist, M.L.; Chen, R.C.; Huang, Y.; Chen, K.; Li, D. Low and High Homocysteine Are Associated with Mortality Independent of B Group Vitamins but Interactive with Cognitive Status in a Free-Living Elderly Cohort. Nutr. Res. 2012, 32, 928–939. [Google Scholar] [CrossRef]
- McNeill, G.; Jia, X.; Whalley, L.J.; Fox, H.C.; Corley, J.; Gow, A.J.; Brett, C.E.; Starr, J.M.; Deary, I.J. Antioxidant and B Vitamin Intake in Relation to Cognitive Function in Later Life in the Lothian Birth Cohort 1936. Eur. J. Clin. Nutr. 2011, 65, 619–626. [Google Scholar] [CrossRef]
- Kim, H.; Kim, G.; Jang, W.; Kim, S.Y.; Chang, N. Association between Intake of B Vitamins and Cognitive Function in Elderly Koreans with Cognitive Impairment. Nutr. J. 2014, 13, 118. [Google Scholar] [CrossRef] [Green Version]
- Lee, L.; Kang, S.A.; Lee, H.O.; Lee, B.H.; Park, J.S.; Kim, J.H.; Jung, I.K.; Park, Y.J.; Lee, J.E. Relationships between Dietary Intake and Cognitive Function Level in Korean Elderly People. Public Health 2001, 115, 133–138. [Google Scholar] [CrossRef]
- Cunha, N.M.D.; Georgousopoulou, E.N.; Boyd, L.; Sturm, J.; Brien, B.O.; Lucock, M.; Mckune, A.J.; Mellor, D.; Roach, P.D.; Naumovski, N.; et al. Relationship Between B-Vitamin Biomarkers and Dietary Intake with Apolipoprotein E Є4 in Alzheimer’s Disease Dietary Intake with Apolipoprotein E ¾ 4 in. J. Nutr. Gerontol. Geriatr. 2019, 38, 173–195. [Google Scholar] [CrossRef] [PubMed]
- Tucker, D.M.; Penland, J.G.; Sandstead, H.H.; Milne, D.B.; Heck, D.G.; Klevay, L.M.; Tucker, D.M.; Pen, J.G.; Heck, G.; Klevay, L.M. Nutrition Status and Brain Function in Aging. Am. J. Clin. Nutr. 1990, 52, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Ortega, R.M.; Requejo, A.M.; Andrés, P.; Sobaler, A.M.L.; Quintas, M.E.; Redondo, M.R.; Navia, B.; Rivas, T. Dietary Intake and Cognitive Function in A Group of Elderly People. Am. J. Clin. Nutr. 1997, 66, 803–809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubitz, I.; Ricny, J.; Atrakchi-Baranes, D.; Shemesh, C.; Kravitz, E.; Liraz-zaltsman, S.; Maksin-matveev, A.; Cooper, I.; Leibowitz, A.; Uribarri, J.; et al. High Dietary Advanced Glycation End Products Are Associated with Poorer Spatial Learning and Accelerated Aβ Deposition in an Alzheimer Mouse Model. Aging Cell 2016, 15, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Jacques, P.F.; Kalmbach, R.; Bagley, P.J.; Russo, G.T.; Rogers, G.; Wilson, P.W.F.; Rosenberg, I.H.; Selhub, J. The Relationship between Riboflavin and Plasma Total Homocysteine in the Framingham Offspring Cohort Is Influenced by Folate Status and the C677T Transition in the Methylenetetrahydrofolate Reductase Gene. J. Nutr. 2002, 132, 283–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikkelsen, K.; Apostolopoulos, V. B Vitamins and Ageing. Subcell. Biochem. 2018, 90, 451–470. [Google Scholar] [CrossRef]
- Pantoni, L. Cerebral Small Vessel Disease: From Pathogenesis and Clinical Characteristics to Therapeutic Challenges. Lancet Neurol. 2010, 9, 689–701. [Google Scholar] [CrossRef]
- Yin, F.; Sancheti, H.; Patil, I.; Cadenas, E. Energy Metabolism and Inflammation in Brain Aging and Alzheimer’s Disease. Free Radic. Biol. Med. 2016, 100, 108–122. [Google Scholar] [CrossRef] [Green Version]
- Makarov, M.V.; Trammell, S.A.J.; Migaud, M.E. The Chemistry of the Vitamin B3 Metabolome. Biochem. Soc. Trans. 2019, 47, 131–147. [Google Scholar] [CrossRef]
- Kirkland, J.B.; Meyer-Ficca, M.L. Niacin. Adv. Food Nutr. Res. 2018, 83, 83–149. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chong, Z.Z.; Maiese, K. Navigating novel mechanisms of cellular plasticity with the nad+ precursor and nutrient nicotinamide. Front. Biosci. 2004, 9, 2500–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, D.O. B Vitamins and the Brain: Mechanisms, Dose and Efficacy—A Review. Nutrients 2016, 8, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, B.; Sannegowda, R.B.; Jain, R.; Dubey, P.; Prakash, S. A rare case of alcoholic pellagra encephalopathy with startle myoclonus and marked response to niacin therapy: Time for a new dictum? BMJ Case Rep. 2013. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.W.; Lee, J.H.; Moon, J.H.; Nazim, U.M.; Lee, Y.J.; Seol, J.W.; Hur, J.; Eo, S.K.; Lee, J.H.; Park, S.Y. Niacin alleviates TRAIL-mediated colon cancer cell death via autophagy flux activation. Oncotarget 2016, 7, 4356–4368. [Google Scholar] [CrossRef] [Green Version]
- Kelley, P. Niacin and niacinamide accumulation by rabbit brain slices and choroid plexus. J. Neurochem. 1979, 33, 291–298. [Google Scholar]
- Murthy, M.R.; Rappoport, D.A. Biochemistry of the developing rat brain. Iv. Effect of nicotinamide on brain and liver mitochondria. Biochim. Biophys. Acta 1963, 78, 71–76. [Google Scholar] [CrossRef]
- Deguchi, T.; Ichiyama, A.; Nishizuka, Y.; Hayaishi, O. Studies on the biosynthesis of nicotinamide-adenine dinucleotide in the brain. Biochim. Biophys. Acta 1968, 158, 382–393. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z. Nicotinamide: Necessary Nutrient Emerges as a Novel Cytoprotectant for the Brain. Trends Pharmacol. Sci. 2003, 24, 228–232. [Google Scholar] [CrossRef]
- Park, J.H.; Long, A.; Owens, K.; Kristian, T. Nicotinamide Mononucleotide Inhibits Post-Ischemic NAD+ Degradation and Dramatically Ameliorates Brain Damage Following Global Cerebral Ischemia. Neurobiol. Dis. 2016, 95, 102–110. [Google Scholar] [CrossRef] [Green Version]
- Klaidman, L.; Morales, M.; Kem, S.; Yang, J.; Adams, J.D. Nicotinamide Offers Multiple Protective Mechanisms in Stroke as a Precursor for NAD+, as a PARP Inhibitor and by Partial Restoration of Mitochondrial Function. Pharmacology 2003, 69, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Sadanaga-Akiyoshi, F.; Yao, H.; Tanuma, S.; Nakahara, T.; Hong, J.S.; Ibayashi, S.; Uchimura, H.; Fujishima, M. Nicotinamide Attenuates Focal Ischemic Brain Injury in Rats: With Special Reference to Changes in Nicotinamide and NAD+ Levels in Ischemic Core and Penumbra. Neurochem. Res. 2003, 28, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Wu, T.; Chang, G.; Li, C.; Chen, T.; Chen, H.; Maynard, K.I. Delayed Treatment with Nicotinamide Inhibits Brain Energy Depletion, Improves Cerebral Microperfusion, Reduces Brain Infarct Volume, but Does Not Alter Neurobehavioral Outcome Following Permanent Focal Cerebral Ischemia in Sprague Dawley Rats. Curr. Nurovasc. Res. 2006, 3, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Klaidman, L.K.; Ling, M.; Kem, S.; Sugawara, T.; Chan, P.; Adams, J.D. Nicotinamide Therapy Protects against Both Necrosis and Apoptosis in a Stroke Model. Pharmacol. Biochem. Behav. 2002, 73, 901–910. [Google Scholar] [CrossRef]
- Yang, J.; Klaidman, L.K.; Nalbandian, A.; Oliver, J.; Chang, M.L.; Chan, P.H.; Adams, J.D. The effects of nicotinamide on energy metabolism following transient focal cerebral ischemia in Wistar rats. Neurosci. Lett. 2002, 333, 91–94. [Google Scholar] [CrossRef]
- Hoane, M.R.; Akstulewicz, S.L.; Toppen, J. Treatment with Vitamin B 3 Improves Functional Recovery and Reduces GFAP Expression Following Traumatic Brain Injury in Rats. J. Neurotrauma 2003, 20, 1189–1199. [Google Scholar] [CrossRef]
- Shear, D.A.; Dixon, C.E.; Bramlett, H.M.; Mondello, S.; Dietrich, W.D.; Deng-Bryant, Y.; Schmid, K.E.; Wang, K.K.W.; Hayes, R.L.; Povlishock, J.T.; et al. Nicotinamide Treatment in Traumatic Brain Injury: Operation Brain Trauma Therapy. J. Neurotrauma 2015, 33, 523–537. [Google Scholar] [CrossRef]
- Hoane, M.R.; Gilbert, D.R.; Holland, M.A.; Pierce, J.L. Nicotinamide Reduces Acute Cortical Neuronal Death and Edema in the Traumatically Injured Brain. Neurosci. Lett. 2006, 408, 35–39. [Google Scholar] [CrossRef]
- Goffus, A.M.; Anderson, G.D.; Hoane, M.R. Sustained Delivery of Nicotinamide Limits Cortical Injury and Improves Functional Recovery Following Traumatic Brain Injury. Oxidative Med. Cell. Longev. 2010, 3, 145–152. [Google Scholar] [CrossRef] [Green Version]
- Haar, C.V.; Anderson, G.D.; Hoane, M.R. Continuous Nicotinamide Administration Improves Behavioral Recovery and Reduces Lesion Size Following Bilateral Frontal Controlled Cortical Impact Injury. Behav. Brain Res. 2011, 224, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Klaidman, L.K.; Mukherjee, S.K.; Adams, J.D., Jr. Oxidative Changes in Brain Pyridine Nucleotides and Neuroprotection Using Nicotinamide. Biochim. Biophys. Acta 2001, 1525, 136–148. [Google Scholar] [CrossRef]
- Kamat, J.P.; Devasagayam, T.P.A. Nicotinamide (Vitamin B 3) as an Effective Antioxidant against Oxidative Damage in Rat Brain Mitochondria. Redox Rep. 1999, 4, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shetty, P.K.; Galef, F.; Turner, D.A. Neurobiology of Disease Nicotinamide Pre-Treatment Ameliorates NAD (H) Hyperoxidation and Improves Neuronal Function after Severe Hypoxia. Neurobiol. Dis. 2014, 62, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, E.; Okuda, H.; Nishida, K.; Fujimoto, S.; Nagasawa, K. Protective Effect of Nicotinamide against Poly (ADP-Ribose) Polymerase-1-Mediated Astrocyte Death Depends on Its Transporter-Mediated Uptake. Life Sci. 2010, 86, 676–682. [Google Scholar] [CrossRef]
- Ungerstedt, J.S.; Blombäck, M.; Södeström, T. Nicotinamide Is a Potent Inhibitor of Proinflammatory Cytokines. Clin. Exp. Immunol. 2003, 6, 48–52. [Google Scholar] [CrossRef]
- Vaur, P.; Brugg, B.; Mericskay, M.; Li, Z.; Schmidt, M.S.; Vivien, D.; Orset, C.; Jacotot, E.; Brenner, C.; Duplus, E. Nicotinamide Riboside, a Form of Vitamin B3, Protects against Excitotoxicity-Induced Axonal Degeneration. FASEB J. 2017, 31, 5440–5452. [Google Scholar] [CrossRef] [Green Version]
- Gasperi, V.; Sibilano, M.; Savini, I.; Catani, M.V. Niacin in the Central Nervous System: An Update of Biological Aspects and Clinical Applications. Int. J. Mol. Sci. 2019, 20, 974. [Google Scholar] [CrossRef] [Green Version]
- Hathorn, T.; Synder-Keller, A.; Messer, A. Nicotinamide Improves Motor Deficits and Upregulates PGC-1α and BDNF Gene Expression in a Mouse Model of Huntington’s Disease. Neurobiol. Disord. 2011, 41, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Fukushima, T.; Kaetsu, A.; Lim, H.; Moriyama, M. Possible Role of 1-Methylnicotinamide in the Pathogenesis of Parkinson’s Disease. Exp. Toxic Pathol. 2001, 43, 469–473. [Google Scholar] [CrossRef]
- Gong, B.; Pan, Y.; Vempati, P.; Zhao, W.; Knable, L.; Ho, L.; Wang, J.; Sastre, M.; Ono, K.; Sauve, A.A.; et al. Nicotinamide Riboside Restores Cognition through an Upregulation of Proliferator-Activated Receptor-g Coactivator 1 a Regulated b -Secretase 1 Degradation and Mitochondrial Gene Expression in Alzheimer’s Mouse Models. Neurobiol. Aging 2013, 34, 1581–1588. [Google Scholar] [CrossRef] [Green Version]
- Fricker, R.A.; Green, E.L.; Jenkins, S.I.; Griffin, S.M. The Influence of Nicotinamide on Health and Disease in the Central Nervous System. Int. J. Tryptophan. Res. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakade, C.; Chong, R.; Bradley, E.; Morgan, J.C. Low-Dose Niacin Supplementation Modulates GPR109A, Niacin Index and Ameliorates Parkinson’s Disease Symptoms without Side Effects. Clin. Case Rep. 2015, 3, 635–637. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; Loh, S.H.; Martins, L.M. Enhancing NAD+ salvage metabolism is neuroprotective in a PINK1 model of Parkinson’s disease. Biol. Open 2017, 6, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aaseth, J.; Dusek, P.; Roos, P.M. Prevention of progression in Parkinson’s disease. BioMetals 2018, 31, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Tan, Z.; Tran, N.D. Chemical Hypoxia-Ischemia Induces Apoptosis in Cerebromicrovascular Endothelial Cells. Brain Res. 2000, 877, 134–140. [Google Scholar] [CrossRef]
- Chong, Z.Z.; Lin, S.H.; Maiese, K. Nicotinamide modulates membrane potential and cysteine protease activity during cerebral vascular endothelial cell injury. J. Vasc. Res. 2002, 39, 131–147. [Google Scholar] [CrossRef]
- Niu, N.; Yu, Y.H.; Wang, Y.; Wang, L.J.; Li, Q.; Guo, L.M. Combined Effects of Niacin and Chromium Treatment on Vascular Endothelial Dysfunction in Hyperlipidemic Rats. Mol. Biol. Rep. 2009, 36, 1275–1281. [Google Scholar] [CrossRef]
- Stamatovic, S.M.; Martinez-revollar, G.; Hu, A.; Choi, J. Decline in Sirtuin-1 Expression and Activity Plays a Critical Role in Blood-Brain Barrier Permeability in Aging Neurobiology of Disease. Neurobiol. Dis. 2018, 126, 105–116. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, A.; Wang, Y. SIRT1 in Endothelial Cells as a Novel Target for the Prevention of Early Vascular Aging. J. Cardiovasc. Pharmacol. 2016, 67, 465–473. [Google Scholar] [CrossRef]
- Xu, J.; Jackson, C.W.; Khoury, N.; Escobar, I.; Perez-pinzon, M.A. Brain SIRT1 Mediates Metabolic Homeostasis and Neuroprotection. Front. Endocrinol. 2018, 9, 702. [Google Scholar] [CrossRef]
- De Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; Imai, S.; Seals, D.R. Nicotinamide Mononucleotide Supplementation Reverses Vascular Dysfunction and Oxidative Stress with Aging in Mice. Aging Cell 2016, 15, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Sawda, C.; Moussa, C.; Turner, R.S. Resveratrol for Alzheimer’s Disease. Ann. N. Y. Acad. Sci. 2017, 1403, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Chung, J.W. Associations of Dietary Riboflavin, Niacin, and Retinol with Age-related Hearing Loss: An Analysis of Korean National Health and Nutrition Examination Survey Data. Nutrients 2019, 11, 896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayrakdar, E.T.; Uyanikgil, Y.; Kanit, L.; Koylu, E.; Yalcin, A. Nicotinamide Treatment Reduces the Levels of Oxidative Stress, Apoptosis, and PARP-1 Activity in Aβ (1–42)-Induced Rat Model of Alzheimer’s Disease. Free Rad. Res. 2014, 48, 146–158. [Google Scholar] [CrossRef]
- Benavente, C.A.; Jacobson, E.L. Niacin Restriction Upregulates NADPH Oxidase and ROS in Human Keratinocytes. Free Radic. Biol. Med. 2009, 44, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef] [Green Version]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Gao, Y.; Zeng, M.; Wang, Y.; Wei, T.F.; Lu, Y.B.; Zhang, W.P. Nicotinamide Ribose Ameliorates Cognitive Impairment of Aged and Alzheimer’s Disease Model Mice. Metab. Brain Dis. 2019, 34, 353–366. [Google Scholar] [CrossRef]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD+ Supplementation Normalizes Key Alzheimer’s Features and DNA Damage Responses in a New AD Mouse Model with Introduced DNA Repair Deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Scherr, P.A.; Tangney, C.C.; Hebert, L.E.; Bennett, D.A.; Wilson, R.S.; Aggarwal, N. Dietary Niacin and the Risk of Incident Alzheimer’s Disease and of Cognitive Decline. J. Neurol. Neurosur. Psychiatry 2004, 75, 1093–1100. [Google Scholar] [CrossRef] [Green Version]
- Csiszar, A.; Tarantini, S.S.; Yabluchanskiy, A.; Balasubramanian, P.; Kiss, T.; Farkas, E.; Baur, J.A.; Ungvari, Z. Role of endothelial NAD+ deficiency in age-related vascular dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2019, 316, H1253–H1266. [Google Scholar] [CrossRef] [PubMed]
- Venco, P.; Dusi, S.; Valletta, L.; Tiranti, V. Alteration of the Coenzyme a Biosynthetic Pathway in Neurodegeneration with Brain Iron Accumulation Syndromes. Biochem. Soc. Trans. 2014, 42, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Kunugi, H.; Ali, A.M. Royal Jelly and Its Components Promote Healthy Aging and Longevity: From Animal Models to Humans. Int. J. Mol. Sci. 2019, 20, 4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patassini, S.; Begley, P.; Xu, J.; Church, S.J.; Kureishy, N.; Reid, S.J.; Waldvogel, H.J.; Faull, R.L.M.; Snell, R.G.; Unwin, R.D.; et al. Cerebral Vitamin B5 (D-Pantothenic Acid) Deficiency as a Potential Cause of Metabolic Perturbation and Neurodegeneration in Huntington’s Disease. Metabolites 2019, 9, 113. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, R.; Zhang, Y.; Rock, C.O.; Jackowski, S. Progress in Lipid Research Coenzyme A: Back in Action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef]
- Slyshenkov, V.S.; Dymkowska, D.; Wojtczak, L. Pantothenic Acid and Pantothenol Increase Biosynthesis of Glutathione by Boosting Cell Energetics. FEBS Lett. 2004, 569, 169–172. [Google Scholar] [CrossRef] [Green Version]
- Shedid, S.M.; Saada, H.N.; Eltahawy, N.A.; Hammad, A.S. Curative Role of Pantothenic Acid in Brain Damage of Gamma Irradiated Rats. Indian J. Clin. Biochem. 2018, 33, 314–321. [Google Scholar] [CrossRef]
- Wojtczak, L.; Slyshenkov, V.S. Protection by Pantothenic Acid against Apoptosis and Cell Damage by Oxygen Free Radicals—The Role of Glutathione. Biofactors 2003, 17, 61–73. [Google Scholar] [CrossRef]
- Jung, S.; Kim, M.K.; Choi, B.Y. The Long-Term Relationship between Dietary Pantothenic Acid (Vitamin B 5) Intake and C-Reactive Protein Concentration in Adults Aged 40 Years and Older. Nutr. Metab. Cardiovasc. Dis. 2017, 27, 806–816. [Google Scholar] [CrossRef]
- Naruta, E.; Buko, V. Hypolipidemic Effect of Pantothenic Acid Derivatives in Mice with Hypothalamic Obesity Induced by Aurothioglucose. Exp. Toxic Pathol. 2001, 53, 393–398. [Google Scholar] [CrossRef] [Green Version]
- Evans, M.; Rumberger, J.A.; Azumano, I.; Napolitano, J.J.; Citrolo, D.; Kamiya, T. Pantethine, a Derivative of Vitamin B5, Favorably Alters Total, LDL and Non-HDL Cholesterol in Low to Moderate Cardiovascular Risk Subjects Eligible for Statin Therapy: A Triple-Blinded Placebo and Diet-Controlled Investigation. Vasc. Health Risk Manag. 2014, 10, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mcrae, M.P. Treatment of Hyperlipoproteinemia with Pantethine: A Review and Analysis of Efficacy and Tolerability. Nutr. Res. 2005, 25, 319–333. [Google Scholar] [CrossRef]
- Lanska, D.J. The Discovery of Niacin, Biotin, and Pantothenic Acid. Ann. Nutr. Metab. 2012, 61, 246–253. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Ito, K.; Ohtsuki, S.; Kubo, Y.; Suzuki, T.; Terasaki, T. Major Involvement of Na+ -Dependent Multivitamin Transporter (SLC5A6/SMVT) in Uptake of Biotin and Pantothenic Acid by Human Brain Capillary Endothelial Cells. J. Neurochem. 2015, 134, 97–112. [Google Scholar] [CrossRef]
- Spector, R. Development and Characterization of Pantothenic Acid Transport in Brain. J. Neurochem. 1986, 47, 563–568. [Google Scholar] [CrossRef]
- Spector, R.; Boose, B. Accumulation of Pantothenic Acid by the Isolated Choroid Plexus and Brain Slices In Vitro. J. Neurochem. 1984, 43, 472–478. [Google Scholar] [CrossRef]
- Spector, R. Pantothenic Acid Transport and Metabolism in the Central Nervous System. Am. J. Physiol. 1986, 250, 292–297. [Google Scholar] [CrossRef]
- Spector, R.; Sivesind, C.; Kinzenbaw, D. Pantothenic Acid Transport Through the Blood-Barrier. J. Neurochem. 1986, 47, 966–971. [Google Scholar] [CrossRef]
- Rajalakshmi, R.; Nakhasi, H.L. Effects of neonatal pantothenic acid deficiency on brain lipid composition in rats. J. Neurochem. 1975, 24, 979–981. [Google Scholar] [CrossRef]
- Elbaum, D.; Beconi, M.G.; Monteagudo, E.; Di Marco, A.; Quinton, S.; Lyons, K.A.; Vaino, A.; Harper, S. Fosmetpantotenate (RE-024), a Phosphopantothenate Replacement Therapy for Pantothenate Kinase-Associated Neurodegeneration: Mechanism of Action and Efficacy in Nonclinical Models. PLoS ONE 2018, 13, e0192028. [Google Scholar] [CrossRef] [Green Version]
- Hayflick, S.J. Defective Pantothenate Metabolism and Neurodegenration. Biochem. Soc. Trans. 2018, 42, 1063–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Meo, I.; Carecchio, M.; Tiranti, V. Inborn Errors of Coenzyme a Metabolism and Neurodegeneration. J. Inherit. Metab. Dis. 2019, 42, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zizioli, D.; Tiso, N.; Guglielmi, A.; Saraceno, C.; Busolin, G.; Giuliani, R.; Khatri, D.; Monti, E.; Borsani, G.; Argenton, F.; et al. Neurobiology of Disease Knock-down of Pantothenate Kinase 2 Severely Affects the Development of the Nervous and Vascular System in Zebra Fish, Providing New Insights into PKAN Disease. Neurobiol. Dis. 2016, 85, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ahn, S.; Lee, H.A.; Won, K.S.; Chang, H.W.; Oh, J.S.; Kim, H.W. Dietary Intake of Pantothenic Acid Is Associated with Cerebral Amyloid Burden in Patients with Cognitive Impairment. Food Nutr. Res. 2018, 62. [Google Scholar] [CrossRef]
- Leonardi, R.; Jackowski, S. Biosynthesis of Pantothenic Acid and Coenzyme A. EcoSal Plus 2007, 2. [Google Scholar] [CrossRef] [Green Version]
- Nakahiro, M.; Mochizuki, D.; Uchida, S.; Yoshida, H. Effect of the ‘antidementia drug’ pantoyl-GABA on high affinity transport of choline and on the contents of choline and acetylcholine in rat brain. Br. J. Pharmacol. 1988, 95, 1303–1307. [Google Scholar] [CrossRef] [Green Version]
- Stover, P.J.; Fields, M.S. Vitamin B-6. Adv. Nutr. 2015, 6, 132–133. [Google Scholar] [CrossRef]
- Ueland, P.M.; Mccann, A.; Midttun, Ø.; Ulvik, A. Inflammation, Vitamin B6 and Related Pathway. Mol. Asp. Med. 2016, 53, 20–27. [Google Scholar] [CrossRef]
- Lotto, V.; Choi, S.; Friso, S. Vitamin B6: A Challenging Link between Nutrition and Inflammation in CVD. Br. J. Nutr. 2011, 106, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Meydani, N.S.; Ribaya-mercado, J.; Russel, M.R.; Sahyoun, N.; Morrow, D.F.; Gershoff, N.S. Vitamin B-6 Deficiency Impairs Interleukin 2 Production and Lymphocyte Proliferation in Elderly Adults. Am. J. Clin. Nutr. 1991, 53, 1275–1280. [Google Scholar] [CrossRef]
- Brown, M.; Beier, K. Vitamin B6 Deficiency (Pyridoxine); StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Sudduth, T.L.; Powell, D.K.; Smith, C.D.; Greenstein, A.; Wilcock, D.M. Induction of Hyperhomocysteinemia Models Vascular Dementia by Induction of Cerebral Microhemorrhages and Neuroinflammation. J. Cereb. Blood Flow Metab. 2013, 33, 708–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungert, A.; Neuhauser-Berthold, M. Longitudinal Analysis on Determinants of Vitamin B6 Status in Community-Dwelling Older Adults over a Period of 18 Years. J. Gerontol. A Biol. Sci. Med. Sci. 2019. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.S.; Picciano, M.F.; Jacques, P.F.; Selhub, J. Plasma Pyridoxal 5′-Phosphate in the US Population: The National Health and Nutrition Examination Survey, 2003–2004. Am. J. Clin. Nutr. 2008, 87, 1446–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullegaddi, R.; Powers, H.J.; Gariballa, S.E. B-Group Vitamin Supplementation Mitigates Oxidative Damage after Acute Ischaemic Stroke. Clin. Sci. 2004, 484, 477–484. [Google Scholar] [CrossRef] [Green Version]
- Shen, L. Associations between B Vitamins and Parkinson’s Disease. Nutrients 2015, 7, 7197–7208. [Google Scholar] [CrossRef]
- Friso, S.; Jacques, P.F.; Wilson, P.W.F.; Rosenberg, I.H.; Selhub, J. Clinical Investigation and Reports Low Circulating Vitamin B 6 Is Associated with Elevation of the Inflammation Marker C-Reactive Protein Independently of Plasma Homocysteine Levels. Circulation 2015, 103, 2788–2791. [Google Scholar] [CrossRef] [Green Version]
- Höhn, A.; Weber, D.; Jung, T.; Ott, C.; Hugo, M.; Kochlik, B.; Kehm, R.; König, J.; Grune, T.; Castro, J.P. Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 2017, 11, 482–501. [Google Scholar] [CrossRef]
- Potter, M.C.; Wozniak, K.M.; Callizot, N.; Slusher, B.S. Glutamate carboxypeptidase II inhibition behaviorally and physiologically improves pyridoxine-induced neuropathy in rats. PLoS One. 2014, 9, e102936. [Google Scholar] [CrossRef]
- Wilson, M.P.; Plecko, B.; Mills, P.B.; Clayton, P.T.; Medicine, G. Disorders Affecting Vitamin B 6 Metabolism. J. Inherit. Metab. Dis. 2019, 42, 629–646. [Google Scholar] [CrossRef] [Green Version]
- Barichello, T.; Generoso, J.S.; Simões, L.R.; Ceretta, R.A.; Dominguini, D.; Ferrari, P.; Gubert, C.; Jornada, L.K.; Budni, J.; Kapczinski, F.; et al. Vitamin B6 Prevents Cognitive Impairment in Experimental Pneumococcal Meningitis. Exp. Biol. Med. 2014, 239, 1360–1365. [Google Scholar] [CrossRef]
- Zysset-Burri, D.C.; Bellac, C.L.; Leib, S.L.; Wittwer, M. Vitamin B6 Reduces Hippocampal Apoptosis in Experimental Pneumococcal Meningitis. BMC Infect. Dis. 2013, 13, 393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, K.; Miyake, Y.; Sasaki, S.; Tanaka, K.; Fukushima, W.; Kiyohara, C.; Tsuboi, Y.; Yamada, T.; Oeda, T.; Miki, T.; et al. Dietary Intake of Folate, Vitamin B6, Vitamin B12 and Riboflavin and Risk of Parkinson’s Disease: A Case-Control Study in Japan. Br. J. Nutr. 2010, 104, 757–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lau, L.M.L.; Koudstaal, P.J.; Witteman, J.C.M.; Hofman, A. Dietary Folate, Vitamin B 12, and Vitamin B 6 and the Risk of Parkinson Disease. Neurology 2006, 67, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Hunt, B.J.; O’Sullivan, M.; Bell, R.; D’Souza, R.; Jeffery, S.; Bamford, J.M.; Markus, H.S. Homocysteine Is a Risk Factor for Cerebral Small Vessel Disease, Acting via Endothelial Dysfunction. Brain 2004, 127, 212–219. [Google Scholar] [CrossRef] [Green Version]
- Bertsch, T.; Mielke, O.; Höly, S.; Casarin, W.; Aufenanger, J.; Walter, S.; Muehlhauser, F.; Kuehl, S.; Ragoschke, A.; Fassbender, K. Homocysteine in Cerebrovascular Disease: An Independent Risk Factor for Subcortical Vascular Encephalopathy. Clin. Chem. 2001, 39, 721–724. [Google Scholar] [CrossRef] [Green Version]
- Nuru, M.; Muradashvili, N.; Kalani, A.; Lominadze, D.; Tyagi, N. High Methionine, Low Folate and Low Vitamin B6/B12 (HM-LF-LV) Diet Causes Neurodegeneration and Subsequent Short-Term Memory Loss. Metab. Brain Dis. 2018, 33, 1923–1924. [Google Scholar] [CrossRef]
- Moretti, R.; Caruso, P. The Controversial Role of Homocysteine in Neurology: From Labs to Clinical Practice. Int. J. Mol. Sci. 2019, 20, 231. [Google Scholar] [CrossRef] [Green Version]
- Ansari, R.; Mahta, A.; Mallack, E.; Luo, J. Hyperhomocysteinemia and Neurologic Disorders: A Review. J. Clin. Neurol. 2014, 10, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Toda, N. Hyperhomocysteinemia Impairs Regional Blood Flow: Involvements of Endothelial and Neuronal Nitric Oxide. Pflügers Arch. 2016, 468, 1517–1525. [Google Scholar] [CrossRef]
- Endo, N.; Nishiyama, K.; Okabe, M.; Matsumoto, M.; Kanouchi, H.; Oka, T. Vitamin B 6 Suppresses Apoptosis of NM-1 Bovine Endothelial Cells Induced by Homocysteine and Copper. Biochim. Biophys. Acta 2007, 1770, 571–577. [Google Scholar] [CrossRef]
- Alam, M.M.; Mohammad, A.A.; Shuaib, U.; Wang, C.; Ghani, U.; Schwindt, B.; Todd, K.G.; Shuaib, A. Homocysteine Reduces Endothelial Progenitor Cells in Stroke Patients through Apoptosis. J. Cereb. Blood Flow Metab. 2009, 29, 157–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahfouz, M.M.; Zhou, S.Q.; Kummerow, F.A. Vitamin B 6 Compounds Are Capable of Reducing the Superoxide Radical and Lipid Peroxide Levels Induced by H2O2 in Vascular Endothelial Cells in Culture. Int. J. Vitam. Nutr. Res. 2009, 79, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Liu, Z.; Lu, H.; Zhang, W.; Mi, Q. Pyridoxine Inhibits Endothelial NOS Uncoupling Induced by Oxidized Low-Density Lipoprotein via the PKC Signaling Pathway in Human Umbilical Vein Endothelial Cells. Br. J. Pharmacol. 2012, 165, 754–764. [Google Scholar] [CrossRef] [Green Version]
- Ji, Y.; Diao, J.; Han, Y.; Huang, Y.; Bai, H.; Chen, Q.; Fan, L.; Ferro, A. Pyridoxine Prevents Dysfunction of Endothelial Cell Nitric Oxide Production in Response to Low-Density Lipoprotein. Atherosclerosis 2006, 188, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Ford, T.C.; Downey, L.A.; Simpson, T.; Mcphee, G.; Oliver, C.; Stough, C. The Effect of a High-Dose Vitamin B Multivitamin Supplement on the Relationship between Brain Metabolism and Blood Biomarkers of Oxidative Stress: A Randomized Control Trial. Nutrients 2018, 10, 1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onorato, J.M.; Jenkins, A.J.; Thorpe, S.R.; Baynes, J.W. Pyridoxamine, an Inhibitor of Advanced Glycation Reactions, Also Inhibits Advanced Lipoxidation Reactions. J. Biol. Chem. 2000, 275, 21177–21184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelso, B.G.; Brower, J.B.; Targovnik, J.H.; Caplan, M.R. Pyridoxine Restores Endothelial Cell Function in High Glucose. Metab. Syndr. Relat. Disord. 2011, 9, 63–68. [Google Scholar] [CrossRef]
- Morris, M.S.; Sakakeeny, L.; Jacques, P.F.; Picciano, M.F.; Selhub, J. Vitamin B-6 Intake Is Inversely Related to, and the Requirement Is Affected by Inflammation Status. J. Nutr. 2009, 140, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Ifrea, R.A.T.; Ozlea, L.C.; Arașca, E.C. The Impact of C Reactive Protein on Global Cardiovascular Risk on Patients with Coronary Artery Disease. Curr. Health Sci. J. 2013, 39, 225–231. [Google Scholar]
- Rall, L.C.; Meydani, S.N. Vitamin B, and Immune Competence. Nutr. Rev. 1993, 51, 217–225. [Google Scholar] [CrossRef]
- Kelly, P.J.; Shih, V.E.; Kistler, J.P.; Barron, M.; Lee, H.; Mandell, R.; Furie, K.L. Low Vitamin B6 but Not Homocyst(e)ine Is Associated with Increased Risk of Stroke and Transient Ischemic Attack in the Era of Folic Acid Grain Fortification. Stroke 2003, 34, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Polyak, Z.; Berner, Y.N.; Sela, B.; Gomori, J.M.; Doolman, R. Hyperhomocysteinemia and Vitamin Score: Correlations with Silent Brain Ischemic Lesions and Brain Atrophy. Dement. Geriatr. Cogn. Disord. 2003, 16, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Mulder, C.; Scheltens, P.; Barkhof, F.; Gundy, C.; Verstraeten, R.A.; de Leeuw, F.E. Low B Vitamin B6 Levels Are Associated with White Matter Lesions in Alzheimer’s Disease. J. Am. Geriatr. Soc. 2005, 53, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Mulder, C.; van der Flier, W.M.; Veerhuis, R.; Bouwman, F.; Jakobs, C.; Verhoeven, N.M.; Barkhof, F.; Scheltens, P.; Blankenstein, M.A. Association Between Vitamin B6 and White Matter Hyperintensities in Patients with Alzheimer’s Disease Not Mediated by Homocysteine Metabolism. J. Am. Geriatr. Soc. 2007, 55, 956–958. [Google Scholar] [CrossRef]
- Miller, J.W.; Green, R.; Mungas, D.M.; Reed, B.R.; Jagust, W.J. Homocysteine, Vitamin B 6, and Vascular Disease in AD Patients. Neurology 2002, 58, 1471–1476. [Google Scholar] [CrossRef]
- Malaguarnera, M.; Ferri, R.; Alagona, G.; Carnemolla, A.; Pennisi, G. Homocysteine, Vitamin B 12 and Folate in Vascular Dementia and in Alzheimer Disease. Clin. Chem. Lab. Med. 2004, 42, 1032–1035. [Google Scholar] [CrossRef]
- Nelson, C.; Wengreen, H.J.; Munger, R.G.; Corcoran, C.D. Dietary folate, vitamin B-12, vitamin B-6 and incident Alzheimer’s disease: The cache county memory, health, and aging study. J. Nutr. Health Ageing 2009, 13, 899–905. [Google Scholar] [CrossRef]
- Toole, J.F.; Malinow, M.R.; Chambless, L.E.; Spence, J.D.; Pettigrew, L.C.; Howard, V.J.; Sides, E.G.; Wang, C.H.; Stampfer, M. Lowering Homocysteine in Patients with Ischemic Stroke to Prevent Recurrent Stroke, Myocardial Infarction, and Death. The Vitamin Intervention for Stroke Prevention (VISP) Randomized Controlled Trial. JAMA 2004, 291, 565–575. [Google Scholar] [CrossRef] [Green Version]
- Schwammenthal, Y.; Tanne, D. Homocysteine, B-Vitamin Supplementation, and Stroke Prevention: From Observational to Interventional Trials. Lancet Neurol. 2004, 3, 493–495. [Google Scholar] [CrossRef]
- The VITATOPS Trial Study Group. B Vitamins in Patients with Recent Transient Ischemic Attack or Stroke in the VITAmins TO Prevent Stroke (VITATOPS) Trial: A Randomized, Double-Blind, Parallel, Placebo-Controlled Trial. Lancet Neurol. 2010, 9, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Cavalieri, M.; Schmidt, R.; Chen, C.; Mok, V.; De Freitas, G.R.; Song, S.; Yi, Q.; Ropele, S.; Grazer, A.; Homayoon, N.; et al. B Vitamins and Magnetic Resonance Imaging—Detected Ischemic Brain Lesions in Patients with Recent Transient Ischemic Attack or Stroke. Stroke 2012, 43, 3266–3270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erickson, I.K.; Suever, L.B.; Prakash, S.R.; Colcombe, S.J.; McAuley, E.; Kramer, A.F. Greater Intake of Vitamins B6 and B12 Spares Gray Matter in Healthy Elderly: A Voxel-Based Morphometry Study. Brain Res. 2009, 1553, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jannusch, K.; Jockwitz, C.; Bidmon, H.; Moebus, S.; Amunts, K.; Caspers, S. A Complex Interplay of Vitamin B1 and B6 Metabolism with Cognition, Brain Structure, and Functional Connectivity in Older Adults. Front. Neurosci. 2017, 11, 596. [Google Scholar] [CrossRef] [PubMed]
- Twisk, J.W.R.; Prevoo, W.; Rauwerda, J.A. Effect of Homocysteine-Lowering Treatment with Folic Acid plus Vitamin B 6 on Cerebrovascular Atherosclerosis and White Matter Abnormalities as Determined by MRA and MRI: A Placebo-Controlled, Randomized Trial. Eur. J. Clin. Investig. 2004, 34, 256–261. [Google Scholar]
- Palacios, N.; Scott, T.; Sahasrabudhe, N.; Gao, X.; Tucker, K.L. Lower Plasma Vitamin B-6 Is Associated with 2-Year Cognitive Decline in the Boston Puerto Rican Health Study. J. Nutr. 2019, 149, 635–641. [Google Scholar] [CrossRef]
- Riggs, M. Relations of Vitamin B-12, Homocysteine to Cognitive Aging Study1 Performance in the Normative. Am. J. Clin. Nutr. 1996, 63, 306–314. [Google Scholar] [CrossRef]
- Qin, B.; Xun, P.; Jacobs, D.R., Jr.; Zhu, N.; Daviglus, M.L.; Reis, J.P.; Steffen, L.M.; van Horn, L.; Sidney, S.; He, K. Intake of Niacin, Folate, Vitamin B-6, and Vitamin B-12 through Young Adulthood and Cognitive Function in Midlife: The Coronary Artery Risk Development in Young Adults (CARDIA) Study. Am. J. Clin. Nutr. 2017, 106, 1032–1040. [Google Scholar] [CrossRef] [Green Version]
- Tucker, K.L.; Qiao, N.; Scott, T.; Rosenberg, I.; Iii, A.S. High Homocysteine and Low B Vitamins Predict Cognitive Decline in Aging Men: The Veterans Affairs Normative Aging Study. Am. J. Clin. Nutr. 2005, 82, 627–635. [Google Scholar] [CrossRef]
- Aisen, P.S.; Schneider, L.S.; Sano, M.; Diaz-Arrastia, R.; van Dyck, C.H.; Weiner, M.F.; Bottiglieri, T.; Jin, S.; Stokes, K.T.; Thomas, R.G.; et al. High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: A randomized controlled trial. JAMA 2012, 300, 1774–1783. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, C.J.; Chien, K.L.; Chen, S.T.; Chen, R.C. Efficacy of Multivitamin Supplementation Containing Vitamins B6 and B12 and Folic Acid as Adjunctive Treatment with a Cholinesterase Inhibitor in Alzheimer’s Disease: A 26-Week, Randomized, Double-Blind, Placebo-Controlled Study in Taiwanese Patients. Clin. Ther. 2007, 29. [Google Scholar] [CrossRef]
- Stott, D.J.; MacIntosh, G.; Lowe, G.D.; Rumley, A.; McMahon, A.D.; Langhorne, P.; Tait, R.C.; O’Reilly, D.S.J.; Spilg, E.G.; MacDonald, J.B.; et al. Randomized Controlled Trial of Homocysteine-Lowering Vitamin Treatment in Elderly Patients with Vascular Disease. Am. J. Clin. Nutr. 2005, 82, 1320–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Ye, J.; Mu, J. Efficacy of Vitamin B Supplementation on Cognition in Elderly Patients with Cognitive-Related Diseases: A Systematic Review and Meta-Analysis. J. Geriatr. Psychiatry 2017, 30, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Bryan, J.; Calvaresi, E.; Hughes, D. Short-Term Folate, Vitamin B-12 or Vitamin B-6 Supplementation Slightly Affects Memory Performance but Not Mood in Women of Various Ages. J. Nutr. 2002, 132, 1345–1356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deijen, J.B.; van der Beck, E.J.; Orlebeke, J.F.; van der Berg, H. Vitamin B-6 Supplementation in Elderly Men: Effects on Mood, Memory, Performance and Mental Effort. Psychopharmacology 1992, 109, 489–496. [Google Scholar] [CrossRef]
- Malouf, R.; Evans, J.G. The Effect of Vitamin B6 on Cognition. Cochrane Database Syst. Rev. 2003, 4, CD004393. [Google Scholar] [CrossRef]
- Hassel, B.; Rogne, A.G.; Hope, S. Intellectual Disability Associated with Pyridoxine-Responsive Epilepsies: The Need to Protect Cognitive Development. Front. Psychiatry 2019, 10, 116. [Google Scholar] [CrossRef]
- Tourbah, A.; Lebrun-Frenay, C.; Edan, G.; Clanet, M.; Papeix, C.; Vukusic, S.; De Sèze, J.; Debouverie, M.; Gout, O.; Clavelou, P.; et al. MD1003 (High-Dose Biotin) for the Treatment of Progressive Multiple Sclerosis: A Randomised, Double-Blind, Placebo-Controlled Study. Mult. Scler. J. 2016, 22, 1719–1731. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, K.; Stojanovska, L.; Apostolopoulos, V. The Effects of Vitamin B in Depression. Curr. Med. Chem. 2016, 23, 4317–4337. [Google Scholar] [CrossRef] [Green Version]
- Zempleni, J.; Wijeratne, S.S.K.; Hassan, Y.I. Biotin. Biofactors 2016, 35, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Dakshinamurti, K. Biotin—A Regulator of Gene Expression B. J. Nutr. Biochem. 2005, 16, 419–423. [Google Scholar] [CrossRef]
- Zempleni, J. Uptake, Localization and Noncarboxylase Roles of Biotin. Annu. Rev. Nutr. 2005, 25, 175–196. [Google Scholar] [CrossRef] [PubMed]
- Mccarty, M.F.; Dinicolantonio, J.J. Neuroprotective Potential of High-Dose Biotin. Med. Hypotheses 2017, 109, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Magied, N.; Shedid, S.M.; Ahmed, A.G. Mitigating Effect of Biotin against Irradiation-Induced Cerebral Cortical and Hippocampal Damage in the Rat Brain Tissue. Environ. Sci. Pollut. Res. 2019, 26, 13441–13452. [Google Scholar] [CrossRef] [PubMed]
- Madsen, C.T.; Sylvestersen, K.B.; Young, C.; Larsen, S.C.; Poulsen, J.W.; Andersen, M.A.; Palmqvist, E.A.; Hey-Mogensen, M.; Jensen, P.B.; Treebak, J.T.; et al. Biotin starvation causes mitochondrial protein hyperacetylation and partial rescue by the SIRT3-like deacetylase Hst4p. Nat. Commun. 2015, 6, 7726. [Google Scholar] [CrossRef] [Green Version]
- Watanabe-Kamiyama, M.; Kamiyama, S.; Horiuchi, K.; Ohinata, K.; Shirakawa, H.; Furukawa, Y.; Komai, M. Antihypertensive Effect of Biotin in Stroke-Prone Spontaneously Hypertensive Rats. Br. J. Nutr. 2008, 99, 756–763. [Google Scholar] [CrossRef] [Green Version]
- Suormala, T.; Wiesmann, U.N.; Cruz, F.; Wolf, A.; Daschner, M.; Limat, A.; Fowler, B.; Baumgartner, E.R. Biotin-Dependent Carboxylase Activities in Different CNS and Skin-Derived Cells, and Their Sensitivity to Biotin-Depletion. Int. J. Vitam. Nutr. Res. 2002, 72, 278–286. [Google Scholar] [CrossRef]
- Fukuwatari, T.; Wada, H.; Shibata, K.; Ukuwatari, T.F.; Ada, H.W.; Hibata, K.S. Age-Related Alterations of B-Group Vitamin Contents in Urine, Blood and Liver from Rats. J. Nutr. Sci. Vitaminol. 2008, 54, 357–362. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Yasumura, S.; Shibata, H. Biotin Status and Its Correlation with Other Biochemical Parameters in the Elderly People of Japan. J. Am. Coll. Nutr. 1998, 17, 37–41. [Google Scholar] [CrossRef]
- McKay, B.E.; Molineux, M.L.; Turner, R.W. Biotin Is Endogenously Expressed in Select Regions of the Rat Central Nervous System. J. Comp. Neurol. 2004, 473, 86–96. [Google Scholar] [CrossRef]
- Spector, R.; Mock, D. Biotin Transport Through the Blood-Brain Barrier. J. Neurochem. 1987, 48, 400–404. [Google Scholar] [CrossRef]
- Lo, W.; Kaldleck, T.; Packman, S. Biotin Transport in the Rat Central Nervous System. J. Nutr. Sci. Vitaminol. 1991, 37, 567–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kassem, H.; Wafaie, A.; Alsuhibani, S.; Farid, T. Biotin-Responsive Basal Ganglia Disease: Neuroimaging Features before and after Treatment. AJNR Am. J. Neuroradiol. 2014, 35, 1990–1995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yatzidis, H.; Kutsicos, D.; Agroyannis, B.; Papastephanidis, C.; Francos-Plemenos, M.; Delatola, Z. Biotin in the Managment of Uremic Neurologic Disorders. Nephron 1984, 36, 183–186. [Google Scholar] [CrossRef] [PubMed]
- Koutsikos, D.; Agroyannis, B.; Tzanatos-Exarchou, H. Biotin for Diabetic Peripheral Neuropathy. Biomed. Pharmacother. 1990, 44, 511–514. [Google Scholar] [CrossRef]
- Birnbaum, G.; Stulc, J. High Dose Biotin as Treatment for Progressive Multiple Sclerosis. Mult. Scler. Relat. Disord. 2017, 18, 141–143. [Google Scholar] [CrossRef]
- Sedel, F.; Bernard, D.; Mock, D.M.; Tourbah, A. Targeting Demyelination and Virtual Hypoxia with High-Dose Biotin as a Treatment for Progressive Multiple Sclerosis. Neuropharmacology 2015, 110, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Melichar, V.O.; Behr-Roussel, D.; Zabel, U.; Uttenthal, L.O.; Rodrigo, J.; Rupin, A.; Verbeuren, T.J.; Kumar, H.S.A.; Schmidt, H.H.H.W. Reduced cGMP Signaling Associated with Neointimal Proliferation and Vascular Dysfunction in Late-Stage Atherosclerosis. Proc. Natl. Acad. Sci. USA 2004, 101, 16671–16676. [Google Scholar] [CrossRef]
- Ahluwalia, A.; Foster, P.; Scotland, R.S.; Mclean, P.G.; Mathur, A.; Perretti, M.; Moncada, S.; Hobbs, A.J. Antiinflammatory Activity of Soluble Guanylate Cyclase: cGMP-Dependent Down-Regulation of P-Selectin Expression and Leukocyte Recruitment. Proc. Natl. Acad. Sci. USA 2004, 101, 1386–1391. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Liu, Y.; Huang, Q.; Su, Y.; Zhang, Y.; Wang, G.; Li, F. NF-kB Activation and Cell Death after Intracerebral Hemorrhage in Patients. Neurol. Sci. 2014, 35, 1097–1102. [Google Scholar] [CrossRef]
- Gonos, E.S.; Kapetanou, M.; Sereikaite, J.; Bartosz, G.; Naparło, K.; Grzesik, M.; Sadowska-Bartosz, I. Origin and pathophysiology of protein carbonylation, nitration and chlorination in age-related brain diseases and aging. Aging 2018, 10, 868–901. [Google Scholar] [CrossRef]
- Moretti, R.; Dal Ben, M.; Gazzin, S.; Tiribelli, C. Homocysteine in Neurology: From Endothelium to Neurodegeneration. Curr. Nutr. Food Sci. 2017, 13, 163–175. [Google Scholar] [CrossRef]
- Barber, R.C.; Lammer, E.J.; Shaw, G.M.; Greer, K.A.; Finnell, R.H. The role of folate transport and metabolism in neural tube defect risk. Mol. Genet. Metab. 1999, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kamen, B.A.; Smith, A.K. A review of folate receptor alpha cycling and 5-methyltetrahydrofolate accumulation with an emphasis on cell models in vitro. Adv. Drug Deliv. Rev. 2004, 56, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, L. Folic acid and vitamin B12 deficiency. J. Health Res. Rev. Dev. Ctries. 2014, 1, 5–9. [Google Scholar] [CrossRef]
- Wang, X.; Qin, X.; Demirtas, H.; Li, J.; Mao, G.; Huo, Y.; Sun, N.; Liu, L.; Xu, X. Efficacy of folic acid supplementation in stroke prevention: A meta-analysis. Lancet 2007, 369, 1876–1882. [Google Scholar] [CrossRef]
- McNulty, H. Folate requirements for health in different population groups. Br. J. Biomed. Sci. 1995, 52, 110–119. [Google Scholar]
- Stolzenberg, R. Possible folate deficiency with postsurgical infection. Nutr. Clin. Pract. Off. Publ. Am. Soc. Parenter. Nutr. 1994, 9, 247–250. [Google Scholar] [CrossRef]
- Pietrzik, K.F.; Thorand, B. Folate economy in pregnancy. Nutrition 1997, 13, 975–977. [Google Scholar] [CrossRef]
- Hoffbrand, A.V.; Weir, D.G. The history of folic acid. Br. J. Haematol. 2001, 113, 579–589. [Google Scholar] [CrossRef]
- Folate Evidence—Mayo Clinic, N.D. Available online. Available online: http://www.mayoclinic.org/drugs-supplements/folate/evidence/hrb-20059475 (accessed on 15 August 2019).
- Bailey, L.B.; Stover, P.J.; McNulty, H.; Fenech, M.F.; Gregory, J.F.; Mills, J.L.; Pfeiffer, C.M.; Fazili, Z.; Zhang, M.; Ueland, P.M.; et al. Biomarkers of Nutrition for Development-Folate Review. J. Nutr. 2015, 145, 1636S–1680S. [Google Scholar] [CrossRef] [Green Version]
- Froese, D.S.; Fowler, B.; Baumgartner, M.R. Vitamin B12, folate and the methionine remethylation cycle-biochemistry, pathways and regulation. J. Inher. Metab. Disord. 2019, 42, 673–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Overbeek, E.C.; Staals, J.; van Oostenbrugge, R.J. Vitamin B12 and Progression of White Matter Lesions. A 2-Year Follow-Up Study in First-Ever Lacunar Stroke Patients. PLoS ONE 2013, 8, e78100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obeid, R.; Herrmann, W. Mechanisms of homocysteine neurotoxicity in neurodegenerative diseases with special reference to dementia. FEBS Lett. 2006, 580, 2994–3005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nilsson-Ehle, H. Age-related changes in cobalamin (vitamin B12) handling. Implications for therapy. Drugs Aging 1998, 12, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.M.; Allen, E.; Mills, K.; Clarke, R.; Uauy, R.; Dangour, A.D. Vitamin B-12 status and neurologic function in older people: A cross-sectional analysis of baseline trial data from the Older People and Enhanced Neurological Function (OPEN) study. Am. J. Clin. Nutr. 2016, 104, 790–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamparo, C.D.; Marcia, L.A. Diseases of the Human Body, 5th ed.; F. A. Davis Company: Philadelphia, PA, USA, 2011. [Google Scholar]
- Dowd, P.; Shapiro, M.; Kang, K. Letter: The mechanisms of action of vitamin B12. J. Am. Chem. Soc. 1975, 97, 4754–4757. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, E.H. Benefits and risks of folic acid to the nervous system. J. Neurol. Neurosurg. Psychiatry 2002, 72, 567–571. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Ford, E.S.; Li, C.; Greenlund, K.J.; Croft, J.B.; Balluz, L.S. Use of folic Acid and vitamin supplementation among adults with depression and anxiety: A cross-sectional, population-based survey. Nutr. J. 2011, 10, 102. [Google Scholar] [CrossRef] [Green Version]
- Moretti, R.; Torre, P.; Antonello, R.M.; Cazzato, G. Is isolated vitamin B12 deficiency a sufficient causative factor of dementia? Eur. J. Neurol. 2001, 8, 87–88. [Google Scholar] [CrossRef]
- Ubbink, J.B. Should all elderly people receive folate supplements? Drugs Aging 1998, 13, 415–420. [Google Scholar] [CrossRef]
- Mollin, D.L.; Ross, G.I. Serum vitamin B12 concentrations of patients with megaloblastic anemia after treatment with vitamin B12, folic acid, or folinic acid. Br. Med. J. 1953, 2, 640–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucock, M. Is folic acid the ultimate functional food component for disease prevention? BMJ 2004, 328, 211–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malnick, S.; Goland, S. Folic acid as ultimate in disease prevention: Beware of vitamin B12 deficiency. BMJ 2004, 328, 769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietary Supplement Fact Sheet: Vitamin B12—Health Professional Fact Sheet, N.D. Available online: https://ods.od.nih.gov/factsheets/VitaminB12-HealthProfessional/ (accessed on 15 August 2019).
- Elmadfa, I.; Singer, I. Vitamin B12 and homocysteine status among vegetarians: A global perspective. Am. J. Clin. Nut. 2009, 89, S1693–S1698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Institute of Medicine. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Panthotenic Acid, Biotin, and Choline; National Academy Press: Washington, DC, USA, 1999. [Google Scholar]
- Meadows, M.E.; Kaplan, R.F.; Bromfield, E.B. Cognitive recovery with vitamin B12 therapy: A longitudinal neuropsychological assessment. Neurology 1994, 44, 1764–1765. [Google Scholar] [CrossRef] [PubMed]
- Eastley, R.; Wilcock, G.K.; Bucks, R.S. Vitamin B12 deficiency in dementia and cognitive impairment: The effects of treatment on neuropsychological function. Int. J. Geriatr. Psychiatry 2000, 15, 226–233. [Google Scholar] [CrossRef]
- Teunisse, S.; Bollen, A.E.; van Gool, W.A.; Walstra, G.J. Dementia and subnormal levels of vitamin B12: Effects of replacement therapy on dementia. J. Neurol. 1996, 243, 522–529. [Google Scholar] [CrossRef]
- Wahlin, T.B.R.; Wahlin, A.; Winblad, B.; Bäckman, L. The influence of serum vitamin B12 and folate status on cognitive functioning in very old age. Biol. Psychol. 2001, 56, 247–265. [Google Scholar] [CrossRef]
- Fioravanti, M.; Ferrario, E.; Massaia, M.; Cappa, G.; Rivolta, G.; Grossi, E.; Buckley, A.E. Low folate levels in the cognitive decline of elderly patients and the efficacy of folate as a treatment for improving memory deficits. Arch. Gerontol. Geriatr. 1998, 26, 1–13. [Google Scholar] [CrossRef]
- Hassing, L.; Wahlin, A.; Winblad, B.; Bäckman, L. Further evidence on the effects of vitamin B12 and folate levels on episodic memory functioning: A population-based study of healthy very old adults. Biol. Psychiatry 1999, 45, 1472–1480. [Google Scholar] [CrossRef]
- Eussen, S.J.P.M.; Ferry, M.; Hininger, I.; Haller, J.; Matthys, C.; Dirren, H. Five year changes in mental health and associations with vitamin B12/folate status of elderly Europeans. J. Nutr. Health Aging 2002, 6, 43–50. [Google Scholar] [PubMed]
- Nilsson, K.; Gustafson, L.; Hultberg, B. Improvement of cognitive functions after cobalamin/folate supplementation in elderly patients with dementia and elevated plasma homocysteine. Int. J. Geriatr. Psychiatry 2001, 16, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, E.H. Folic acid, ageing, depression, and dementia. BMJ 2002, 324, 1512–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blom, H.J. Folic acid, methylation and neural tube closure in humans. Birth Defects Res. A Clin. Mol. Teratol. 2009, 85, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Pitkin, R.M. Folate and neural tube defects. Am. J. Clin. Nutr. 2007, 85, 285S–288S. [Google Scholar] [CrossRef] [Green Version]
- Frosst, P.; Blom, H.J.; Milos, R.; Goyette, P.; Sheppard, C.A.; Matthews, R.G.; Boers, G.J.; den Heijer, M.; Kluijtmans, L.A.; van den Heuvel, L.P.; et al. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 1995, 10, 111–113. [Google Scholar] [CrossRef]
- Bottiglieri, T.; Laundy, M.; Crellin, R.; Toone, B.K. Homocysteine, folate, methylation, and monoamine metabolism in depression. J. Neurol. Neurosurg. Psychiatry 2000, 69, 228–232. [Google Scholar] [CrossRef]
- Bottiglieri, T.; Crellin, R.; Reynolds, E.H. Folate and neuropsychiatry. In Folate Health Dis.; Marcel Dekker: New York, NY, USA, 1995; pp. 435–462. [Google Scholar]
- Botez, M.I.; Reynolds, E.H. Folic Acid in Neurology, Psychiatry and Internal Medicine; Raven Press: New York, NY, USA, 1979. [Google Scholar]
- Maxwell, C.J.; Hogan, D.B.; Ebly, E.M. Serum folate levels and subsequent adverse cerebrovascular outcomes in elderly persons. Dement. Geriatr. Cogn. Disord. 2002, 13, 225–234. [Google Scholar] [CrossRef]
- Snowdon, D.A.; Tully, C.L.; Smith, C.D.; Riley, K.P.; Markesbery, W.R. Serum folate and the severity of atrophy of the neocortex in Alzheimer disease: Findings from the Nun study. Am. J. Clin. Nutr. 2000, 71, 993–998. [Google Scholar] [CrossRef] [Green Version]
- Stanhewicz, A.E.; Kenney, W.L. Role of folic acid in nitric oxide bioavailability and vascular endothelial function. Nutr. Rev. 2017, 75, 61–70. [Google Scholar] [CrossRef]
- Ma, F.; Wu, T.; Zhao, J.; Song, A.; Liu, H.; Xu, W.; Huang, G. Folic acid supplementation improves cognitive function by reducing the levels of peripheral inflammatory cytokines in elderly Chinese subjects with MCI. Sci Rep. 2016, 6, 37486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudd, S.H.; Cantoni, G.L. Activation of methionine for transmethylation. III. The methionine-activating enzyme of Bakers’ yeast. J. Biol. Chem. 1958, 231, 481–492. [Google Scholar] [PubMed]
- Mato, J.M.; Alvarez, L.; Ortiz, P.; Pajares, M.A. S-adenosylmethionine synthesis: Molecular mechanisms and clinical implications. Pharmacol. Ther. 1997, 73, 265–280. [Google Scholar] [CrossRef] [Green Version]
- Blom, H.J.; Smulders, Y. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab. Dis. 2011, 34, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loscalzo, J.; Handy, D.E. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. 2013 Grover Conference Series. Pulm. Circ. 2014, 482, 169–174. [Google Scholar] [CrossRef] [Green Version]
- Weir, D.G.; Keating, S.; Molloy, A.; McPartlin, J.; Kennedy, S.; Blachflower, J.; Kennedy, D.G.; Rice, D.; Scott, J.M. Methylation deficiency causes vitamin B12-associated neuropathy in the pig. J. Neurochem. 1988, 51, 1949–1952. [Google Scholar] [CrossRef]
- Harvey, R.A.; Ferrier, D.R. Biochemistry. In Lippincott’s Illustrated Reviews, 5th ed.; Rhyner, S., Ed.; Wolters Kluwer Health: Philadelphia, PA, USA, 2011; pp. 264–265. [Google Scholar]
- Khan, U.; Crossley, C.; Kalra, L.; Rudd, A.; Wolfe, C.D.; Collinson, P.; Markus, H.S. Homocysteine and its relationship to stroke subtypes in a UK black population: The South London Ethnicity and Stroke Study. Stroke 2008, 39, 2943–2949. [Google Scholar] [CrossRef] [Green Version]
- Selhub, J.; Bagley, L.C.; Miller, J.; Rosenberg, I.H. B vitamins, homocysteine, and neurocognitive function in the elderly. Am. J. Clin. Nutr. 2000, 71, 614S–620S. [Google Scholar] [CrossRef]
- Li, Z.; Sun, L.; Zhang, H.; Liao, Y.; Wang, D.; Zhao, B.; Zhu, Z.; Zhao, J.; Ma, A.; Han, Y.; et al. Elevated plasma homocysteine was associated with hemorrhagic and ischemic stroke, but methylenetetrahydrofolate reductase gene c677t polymorphism was a risk factor for thrombotic stroke a multicenter case-control study in China. Stroke 2003, 34, 2085–2090. [Google Scholar] [CrossRef] [Green Version]
- Ali, Z.; Troncoso, J.C.; Fowler, D.R. Recurrent cerebral venous thrombosis associated with heterozygote methylenetetrahydrofolate reductase C677T mutation and sickle cell trait without homocysteinemia: An autopsy case report and review of literature. Forensic Sci. Int. 2014, 242, e52–e55. [Google Scholar] [CrossRef]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Määttä, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, K.A.; Sanders, L.M.; Fischer, L.M.; Zeisel, S.H. Docosahexaenoic acid in plasma phosphatidylcholine may be a potential marker for in vivo phosphatidylethanolamine N-methyltransferase activity in humans. Am. J. Clin. Nutr. 2011, 93, 968–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeLong, C.J.; Shen, Y.J.; Thomas, M.J.; Cui, Z. Molecular distinction of phosphatidylcholine synthesis between the CDP-choline pathway and phosphatidylethanolamine methylation pathway. J. Biol. Chem. 1999, 274, 29683–29688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pynn, J.; Henderson, N.G.; Clark, H.; Koster, G.; Bernhard, W.; Postle, A.D. The specificity and rate of human and mouse liver and plasma phosphatidylcholine synthesis analysed in vivo. J. Lipid Res. 2011, 52, 399–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blusztajn, J.K.; Zeisel, S.H.; Wurtman, R.J. Synthesis of lecithin (phosphatidylcholine) from phosphatidylethanolamine in bovine brain. Brain Res. 1979, 179, 319–327. [Google Scholar] [CrossRef]
- Blusztajn, J.K.; Holbrook, P.G.; Lakher, M.; Liscovitch, M.; Maire, J.-C.; Mauron, C.; Richardson, U.I.; Tacconi, M.T.; Wurtman, R.J. Relationships between acetylcholine release and membrane phosphatidylcholine turnover in brain and in cultured cholinergic neurons. In Phospholipids in the Nervous System: Biochemical and Molecular Pharmacology; Springer: Berlin, Germany, 1986; pp. 283–290. [Google Scholar]
- Vance, D.E. Phospholipid methylation in mammals: From biochemistry to physiological function. Biochim. Biophys. Acta 2014, 1838, 1477–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudd, S.H.; Brosnan, J.T.; Brosnan, M.E.; Jacobs, R.L.; Stabler, S.P.; Allen, R.H.; Vance, D.E.; Wagner, C. Methyl balance and transmethylation fluxes in humans. Am. J. Clin. Nutr. 2007, 85, 19–25. [Google Scholar] [CrossRef]
- Selley, M.L. A metabolic link between S-adenosylhomocysteine and polyunsaturated fatty acid metabolism in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1834–1839. [Google Scholar] [CrossRef]
- Clark, W.M.; Williams, B.J.; Selzer, K.A.; Zweifler, R.M.; Sabounjian, L.A.; Gammans, R.E. A randomized efficacy trial of citicoline in patients with acute ischemic stroke. Stroke 1999, 30, 2592–2597. [Google Scholar] [CrossRef] [Green Version]
- Clark, W.M.; Wechsler, L.R.; Sabounjian, L.A.; Schwiderski, U.E. Citicoline Stroke Study Group. A phase III randomized efficacy trial of 2000 mg citicolie in acute ischemic stroke patients. Neurology 2001, 57, 1595–1602. [Google Scholar] [CrossRef]
- Warach, S.; Pettigrew, L.C.; Dashe, J.F.; Pullicino, P.; Lefkowitz, D.M.; Sabounjian, L.; Harnett, K.; Schwiderski, U.; Gammans, R. Effect of citicoline on ischemic lesions as measured by diffusion-weighted magnetic resonance imaging. Citicoline 010 Investigators. Ann. Neurol. 2000, 48, 713–722. [Google Scholar] [CrossRef]
- Ivarez-Sabin, J.A.; Roman, G.C. Citicoline in Vascular Cognitive Impairment and Vascular Dementia After Stroke. Stroke 2011, 42, S40–S43. [Google Scholar] [CrossRef] [Green Version]
- Ylilauri, M.P.T.; Voutilainen, S.; Lönnroos, E.; Virtanen, H.E.K.; Tuomainen, T.P.; Salonen, J.T.; Virtanen, J.K. Associations of dietary choline intake with risk of incident dementia and with cognitive performance: The Kuopio Ischaemic Heart Disease Risk Factor Study. Am. J. Clin. Nutr. 2019. [Google Scholar] [CrossRef] [PubMed]
- Klancnik, J.M.; Cuénod, M.; Gähwiler, B.H.; Jiang, Z.P.; Do, K.Q. Release of endogenous amino acids, including homocysteic acid and cysteine sulphinic acid, from rat hippocampal slices evoked by electrical stimulation of Schaffer collateral-commissural fibres. Neuroscience 1992, 49, 557–570. [Google Scholar] [CrossRef]
- Lipton, S.A.; Kim, W.K.; Choi, Y.B.; Kumar, S.; D’Emilia, D.M.; Rayudu, P.V.; Arnelle, D.R.; Stamler, J.S. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar] [CrossRef] [Green Version]
- Zieminska, E.; Stafiej, A.; Lazarewicz, J.W. Role of group I metabotropic glutamate receptors and NMDA receptors in homocysteine-evoked acute neurodegeneration of cultured cerebellar granule neurons. Neurochem. Int. 2003, 43, 481–492. [Google Scholar] [CrossRef]
- Scarpa, S.; Fuso, A.; D’Anselmi, F.; Cavallaro, R.A. Presenilin 1 gene silencing by S-adenosylmethionine: A treatment for Alzheimer disease? FEBS Lett. 2003, 541, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.; Lu, Q.; Orecchio, L.; Kosik, K.S. Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar A beta. Mol. Cell. Neurosci. 1997, 9, 220–234. [Google Scholar] [CrossRef]
- Hasegawa, T.; Ukai, W.; Jo, D.G.; Xu, X.; Mattson, M.P.; Nakagawa, M.; Araki, W.; Saito, T.; Yamada, T. Homocysteic acid induces intraneuronal accumulation of neurotoxic Abeta42: Implications for the pathogenesis of Alzheimer’s disease. J. Neurosci. Res. 2005, 80, 869–876. [Google Scholar] [CrossRef]
- Sai, X.; Kawamura, Y.; Kokame, K.; Yamaguchi, H.; Hirohisa Shiraishi, H.; Suzuki, R.; Suzuki, T.; Kawaichi, M.; Miyata, T.; Kitamura, T.; et al. Endoplasmic reticulum stress-inducible protein, Herp, enhances presenilin-mediated generation of amyloid beta-protein. J. Biol. Chem. 2002, 277, 12915–12920. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Liu, J.; Zhao, J.; Mao, J.; Zhang, X.; Feng, L.; Han, C.; Li, M.; Wang, S.; Wu, D. Homocysteine induces the expression of C-reactive protein via NMDAr-ROS-MAPK-NF-KB signal pathway in rat vascular smooth muscle cells. Atherosclerosis 2014, 236, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Curro, M.; Gugliandolo, A.; Gangemi, C.; Risitano, R.; Ientile, R.; Caccamo, D. Toxic effects of mildy elevated homocysteine conncetrations in neuronal-like cells. Neurochem. Res. 2014, 39, 1485–1495. [Google Scholar] [CrossRef] [PubMed]
- Pushpakumar, S.; Kundu, S.; Sen, U. Endothelial Dysfunction: The Link Between Homocysteine and Hydrogen Sulfide. Curr. Med. Chem. 2014, 21, 3662–3672. [Google Scholar] [CrossRef]
- Vallance, P.; Chan, N. Endothelial function and nitric oxide: Clinical relevance. Heart 2001, 85, 342–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [CrossRef] [Green Version]
- Perna, A.F.; Ingrosso, D.; De Santo, N.G. Homocysteine and oxidative stress. Amino Acids 2003, 25, 409–417. [Google Scholar] [CrossRef]
- Hoffman, M. Hypothesis: Hyperhomocysteinemia is an indicator of oxidant stress. Med Hypotheses 2011, 77, 1088–1093. [Google Scholar] [CrossRef]
- Sawle, P.; Foresti, R.; Green, C.J.; Motterlini, R. Homocysteine attenuates endothelial heme oxygenase-1 induction by nitric oxide (NO) and hypoxia. FEBS Lett. 2001, 508, 403–406. [Google Scholar] [CrossRef] [Green Version]
- Stuhlinger, M.C.; Tsao, P.S.; Her, J.H.; Kimoto, M.; Balint, R.F.; Cooke, J.P. Homocysteine impairs the nitric oxide synthase pathway: Role of asymmetric dimethylarginine. Circulation 2001, 104, 2569–2575. [Google Scholar] [CrossRef]
- Wakita, H.; Tomimoto, H.; Akiguchi, I.; Kimura, J. Glial activation and white matter changes in the rat brain induced by chronic cerebral hypoperfusion: An immunoistochemical study. Acta Neuropathol. 1994, 87, 484–492. [Google Scholar] [CrossRef]
- Farkas, E.; Donka, G.; de Vous, R.A.I.; Mihaly, A.; Bari, F.; Luiten, P.G.M. Experimental cerebral hypoperfusion induces white matter injury and microglial activation in the rat brain. Acta Neuropathol. 2004, 108, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron 2017, 98, 17–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ploder, M.; Kurz, K.; Splitter, A.; Neurauter, G.; Roth, E.; Fuch, D. Early increase of plasma Hcy in sepsis patients with poor outcome. Mol. Med. 2010, 16, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Li, Q.; Du, H.P.; Wang, Y.L.; You, S.J.; Wang, F.; Xu, X.S.; Cheng, J.; Cao, Y.J.; Liu, C.F.; et al. Homocysteine Triggers inflammatory responses in macrophages through inhibiting CSE-H2S signaling via DNA hypermethylation of CSE promoter. Int. J. Mol. Sci. 2015, 16, 12560–12577. [Google Scholar] [CrossRef]
- Jiang, Y.-D.; Sun, T.; Zhang, H.-P.; Xiong, J.-T.; Cao, J.; Li, G.-Z.; Wang, S.-R. Folate and ApoE DNA methylation induced by homocysteine in human monocytes. DNA Cell Biol. 2007, 26, 737–744. [Google Scholar]
- Boldyrev, A.; Bryshkova, E.; Mashkina, A.; Vladychenskaya, E. Why is homocysteine toxic for the nervous and immune systems? Curr. Aging Sci. 2013, 6, 29–36. [Google Scholar] [CrossRef]
- Ying, G.; Wang, Y.; Cen, X.M.; Yang, M.; Liang, Y.; Xie, Q.B. Lipid peroxidation-mediated inflammation promotes cell apoptosis through activation of NFK-B pathway in rheumatoid arthritis synovial cells. Med. Infalmm. 2015, 2015, 460310. [Google Scholar]
- Chang, P.Y.; Lu, S.C.; Lee, C.M.; Chen, Y.J.; Dugan, T.A.; Huang, W.H.; Chang, S.F.; Liao, W.S.; Chen, C.H.; Lee, Y.T. Homocysteine inhibits arterial endothelial cell growth through transcriptional downregulation of fibroblast growth factor-2 involving G protein and DNA methylation. Circ. Res. 2008, 102, 933–941. [Google Scholar] [CrossRef]
- Nichols, J. Testing for homocysteine in clinical practice. Nutr. Health 2017, 23, 13–15. [Google Scholar] [CrossRef]
- Vidoni, M.L.; Gabriel, K.P.; Luo, S.T.; Simonsick, E.M.; Day, R.S. Vitamin B12 and Homocysteine Associations with Gait Speed in Older Adults: The Baltimore Longitudinal Study of Aging. J. Nutr. Health Aging 2017, 21, 1321–1328. [Google Scholar] [CrossRef] [Green Version]
- Kramer, C.S.; Szmidt, M.K.; Sicinska, E.; Brzozowska, A.; Santoro, A.; Franceschi, C.; de Groot, L.C.P.G.M.; Berendsen, A.A.M. The Elderly-Nutrient Rich Food Score Is Associated with Biochemical Markers of Nutritional Status in European Older Adults. Front. Nutr. 2019, 6, 150. [Google Scholar] [CrossRef] [PubMed]
- Hankey, G.J.; Ford, A.H.; Yi, Q.; Eikelboom, J.W.; Lees, K.R.; Chen, C.; Xavier, D.; Navarro, J.C.; Ranawaka, U.K.; Uddin, W.; et al. Effect of B-vitamins and lowering homocysteine on cognitive impairment in patients with previous stroke or transient ischemic attack: A prespecified secondary analysis of a randomized, placebo-controlled trial and meta-analysis. Stroke 2013, 44, 2232–2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ting, S.K.S.; Earnest, A.; Li, H.; Hameed, S.; Chang, H.M.; Chen, C.L.H.; Tan, E.K. B vitamins and cognition in subjects with small vessel disease: A substudy of VITATOPS, a randomized, placebo-controlled trial. J. Neurol. Sci. 2017, 379, 124–126. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Liu, K.; Chen, S.; Yu, H.; An, Y.; Wang, Y.; Zhang, X.; Wang, Y.; Qin, Z.; Xiao, R. Dietary Intake of Riboflavin and Unsaturated Fatty Acid Can Improve the Multi-Domain Cognitive Function in Middle-Aged and Elderly Populations: A 2-Year Prospective Cohort Study. Front. Aging Neurosci. 2019, 11, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
A synopsis of the different biochemical pathways supported by vitamins B.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Moretti, R.; Peinkhofer, C. B Vitamins and Fatty Acids: What Do They Share with Small Vessel Disease-Related Dementia? Int. J. Mol. Sci. 2019, 20, 5797. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225797
AMA Style
Moretti R, Peinkhofer C. B Vitamins and Fatty Acids: What Do They Share with Small Vessel Disease-Related Dementia? International Journal of Molecular Sciences. 2019; 20(22):5797. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225797
Chicago/Turabian StyleMoretti, Rita, and Costanza Peinkhofer. 2019. "B Vitamins and Fatty Acids: What Do They Share with Small Vessel Disease-Related Dementia?" International Journal of Molecular Sciences 20, no. 22: 5797. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225797
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.