Two Homologous Enzymes of the GalU Family in Rhodococcus opacus 1CP—RoGalU1 and RoGalU2



Abstract
:1. Introduction
2. Results
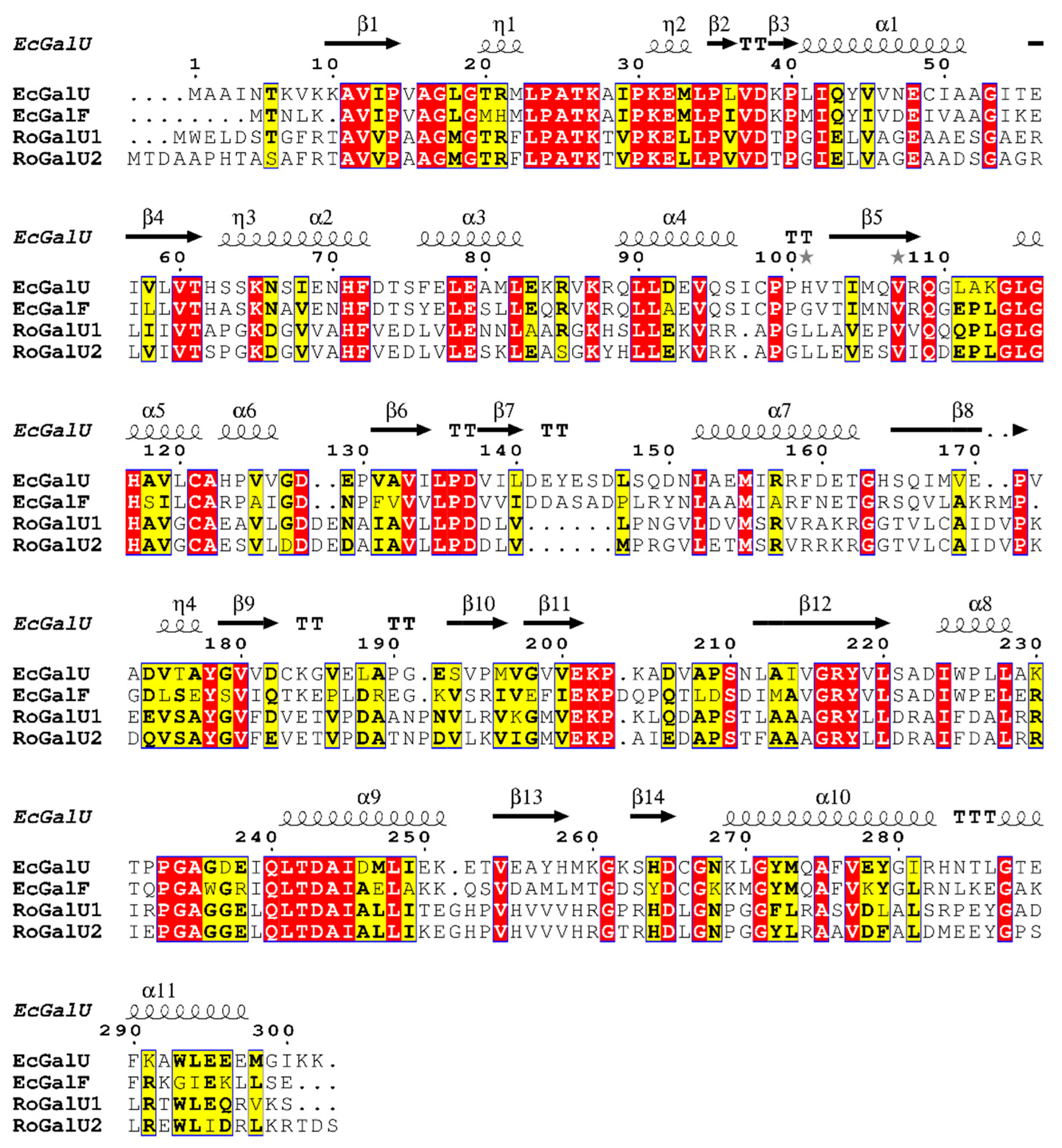
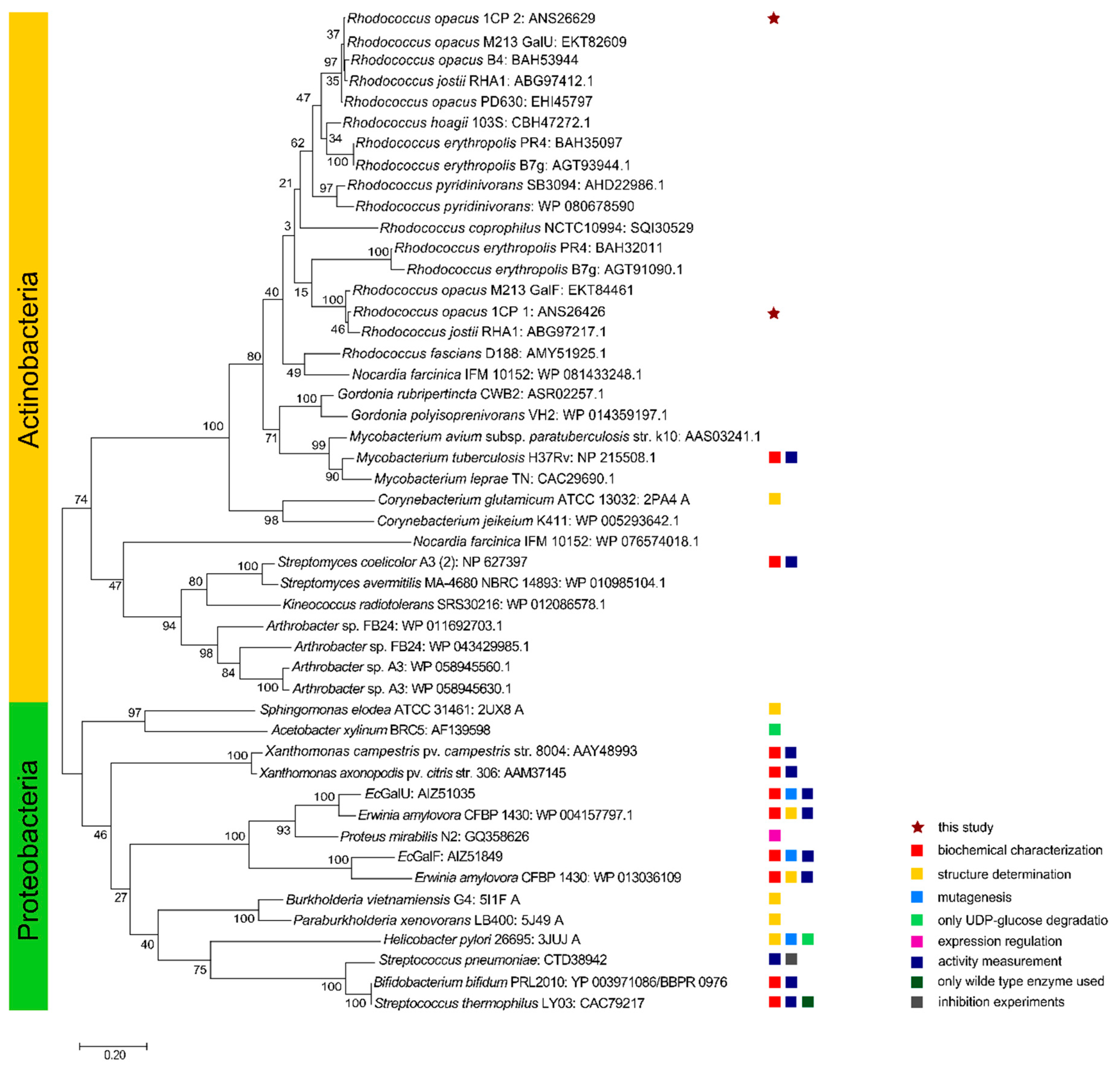
2.1. Genome Mining and Phylogenetic Analyses
2.2. Recombinant Expression of RogalU1 in E. coli, Purification, and Renaturation
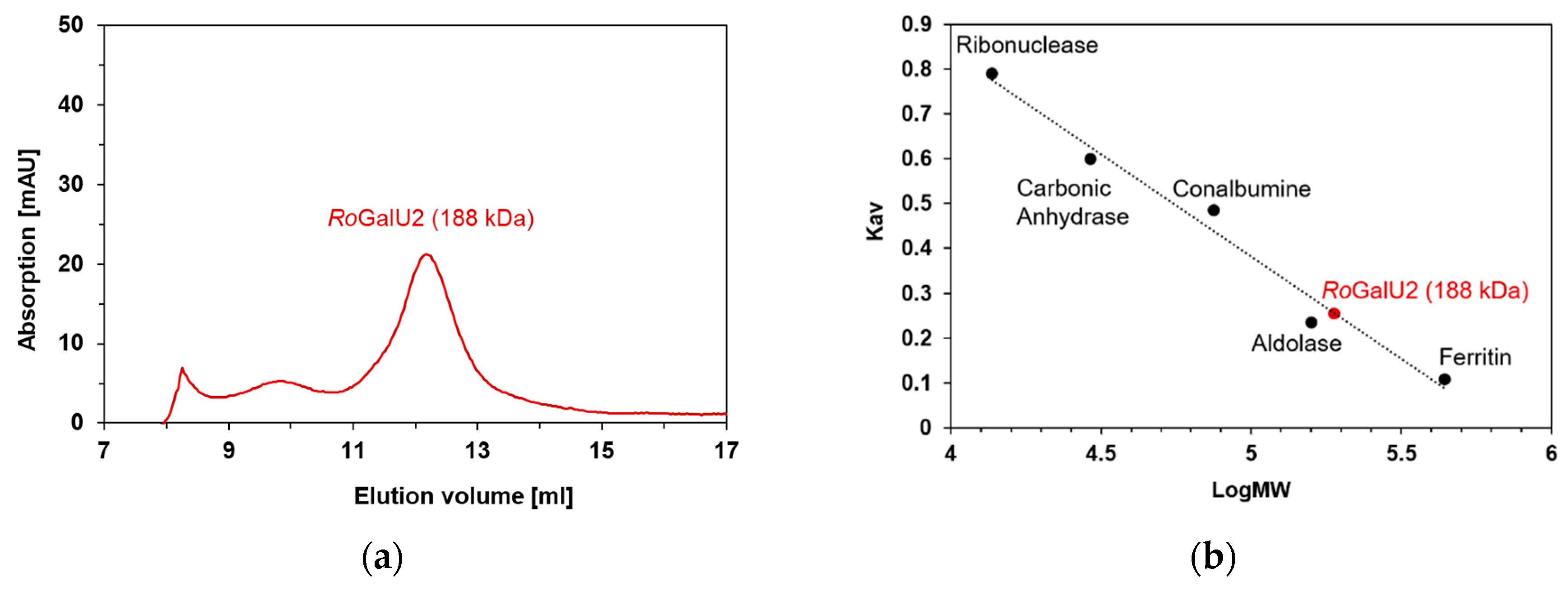
2.3. Recombinant Expression in E. coli, Purification and Production of Active RoGalU2
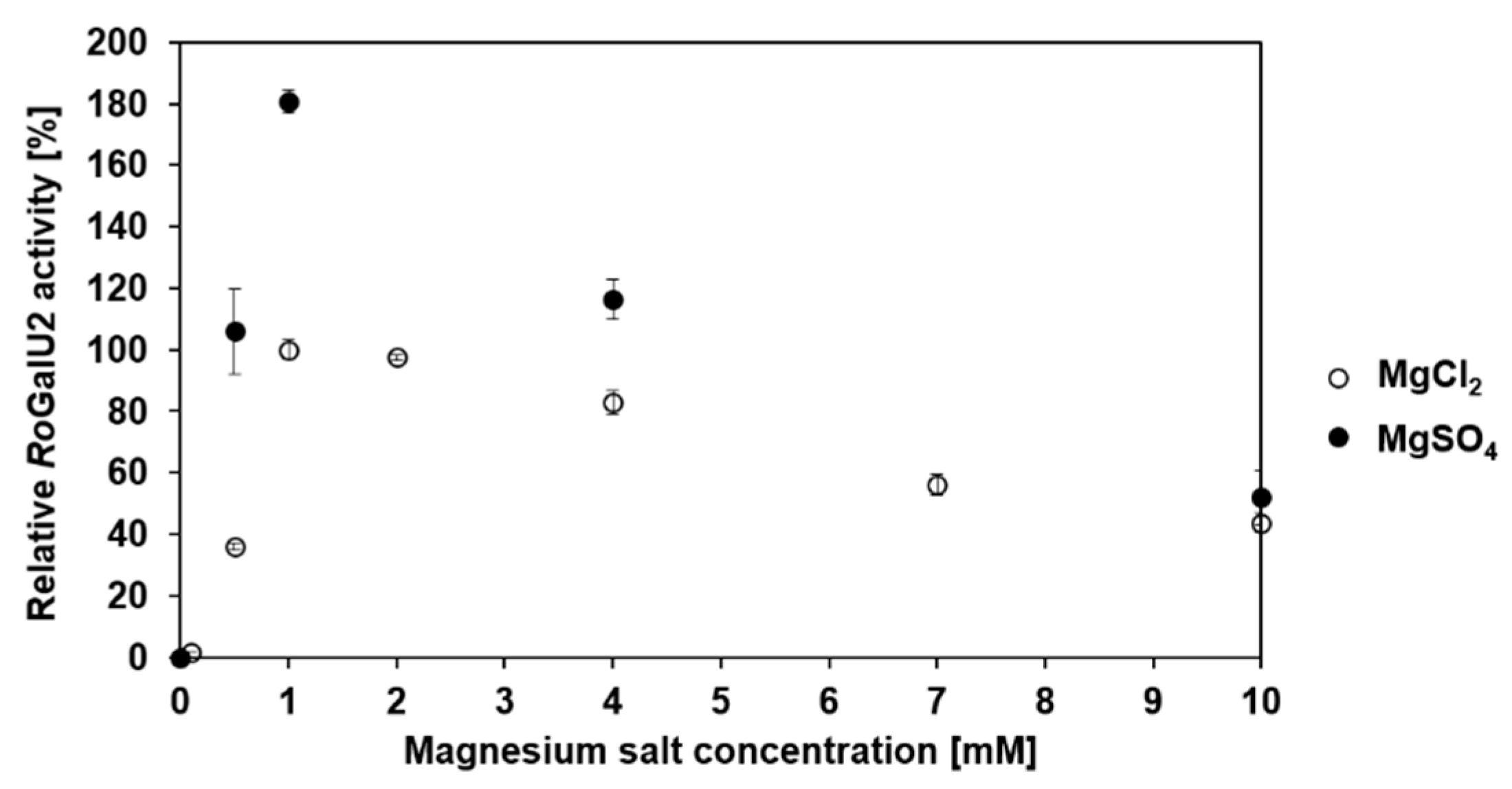
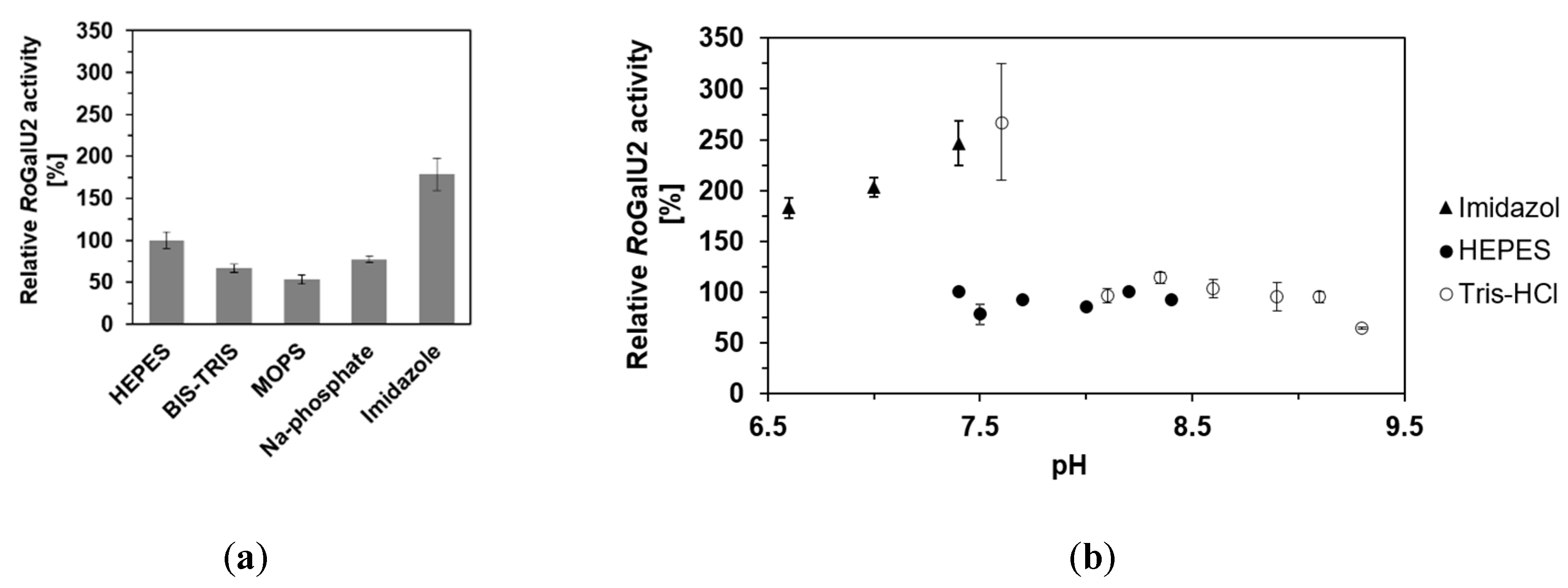
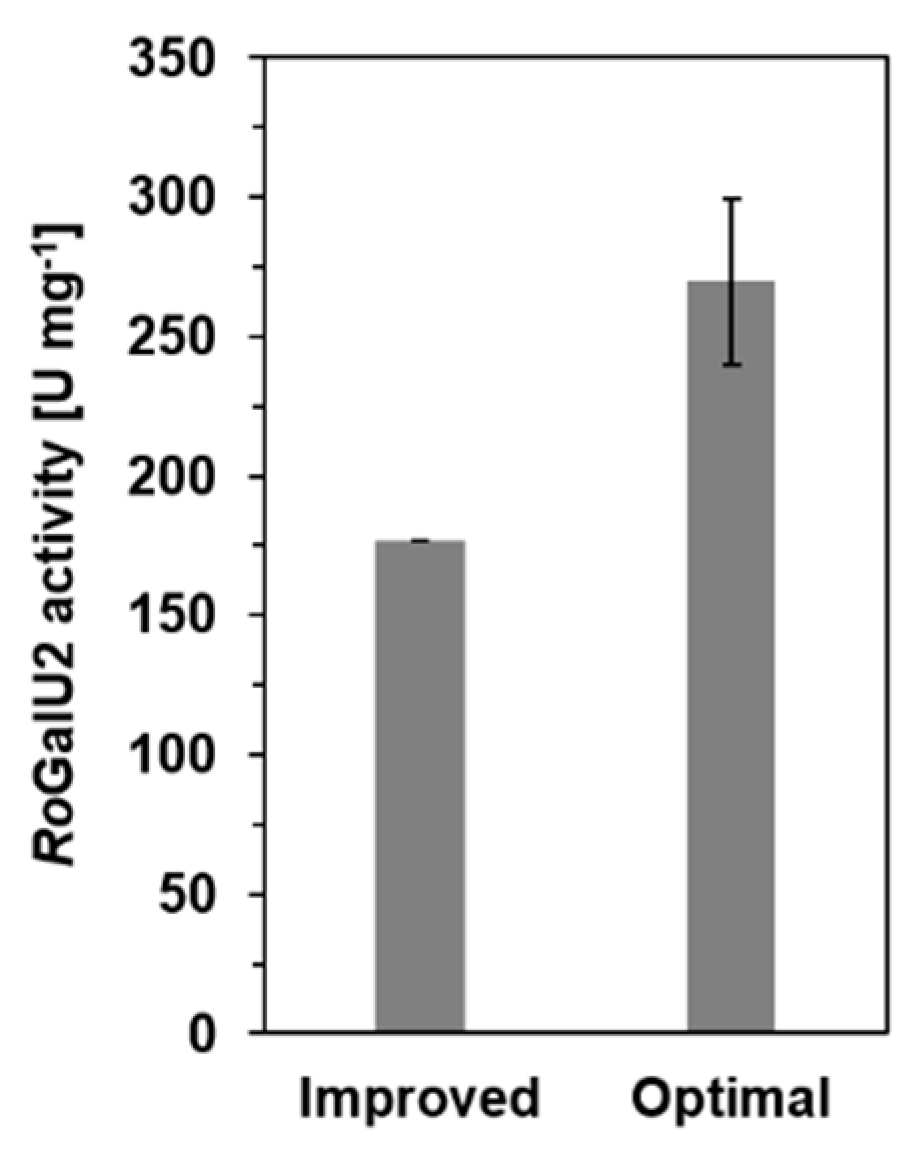
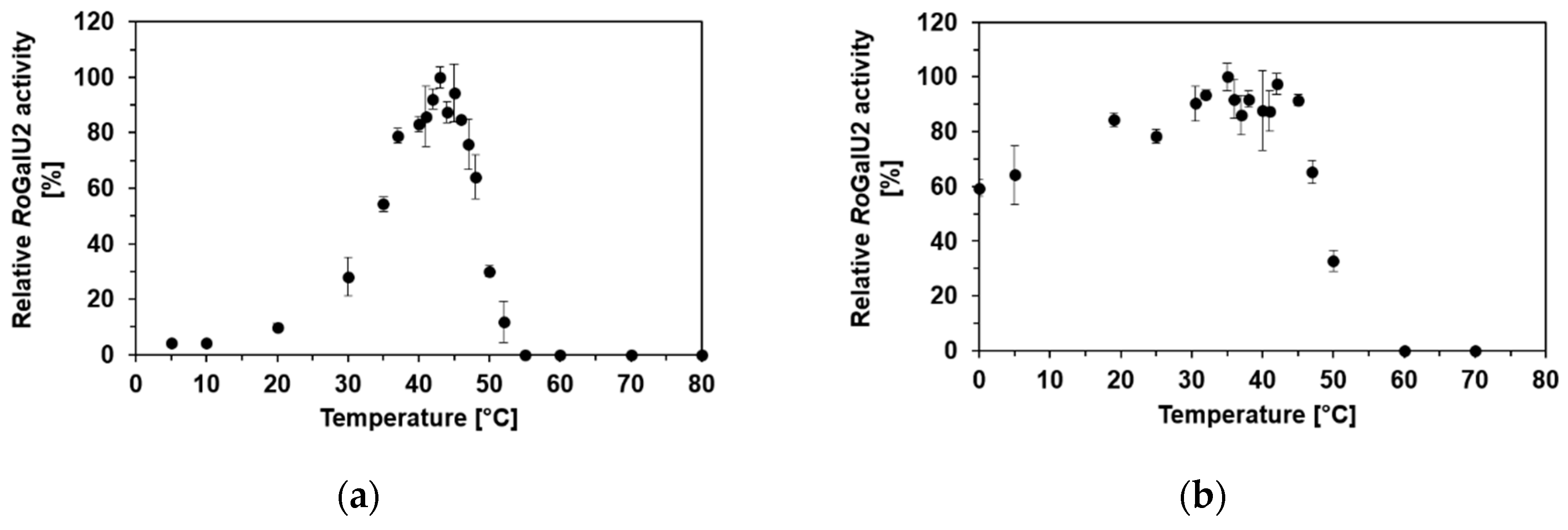
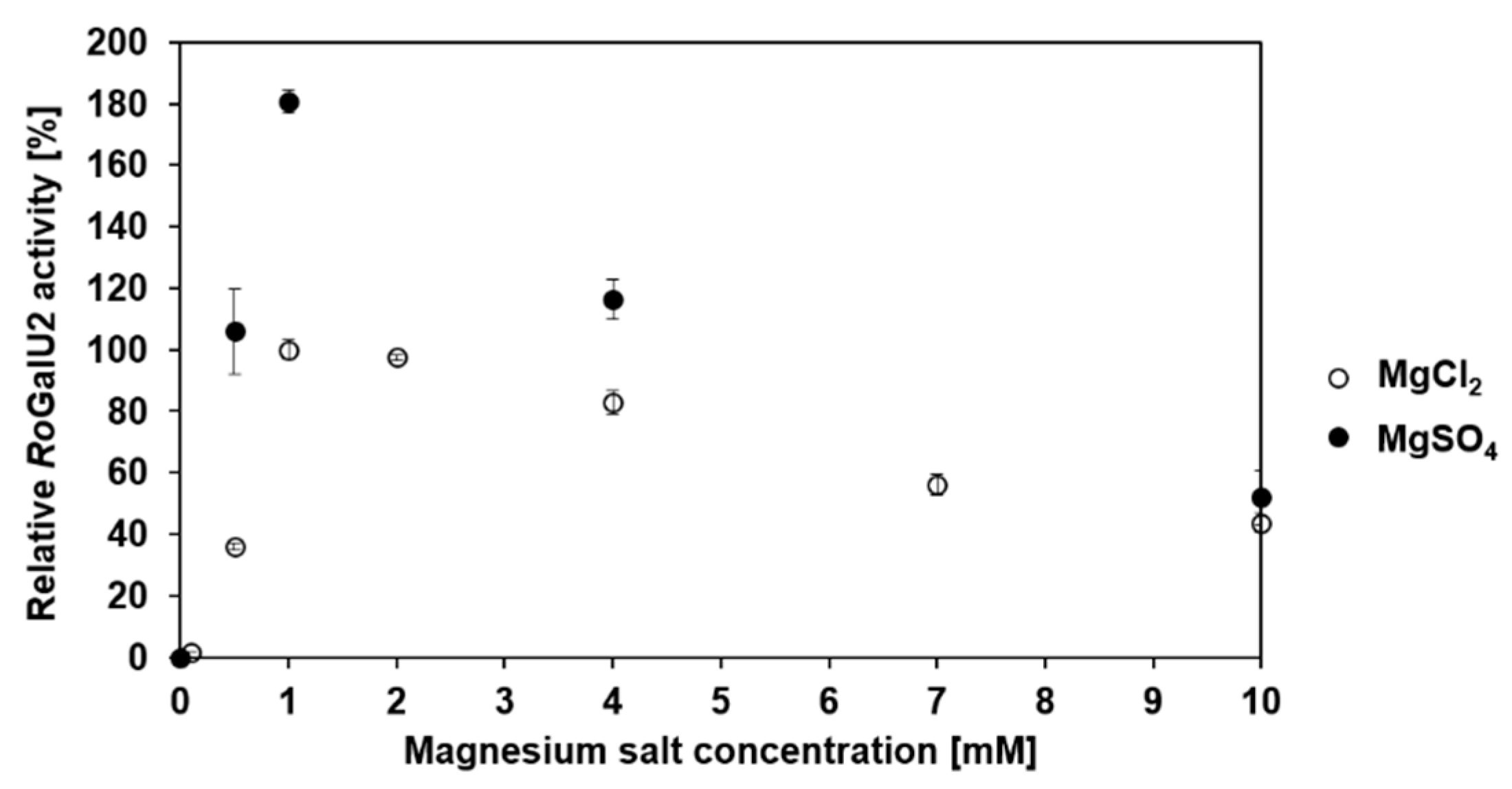
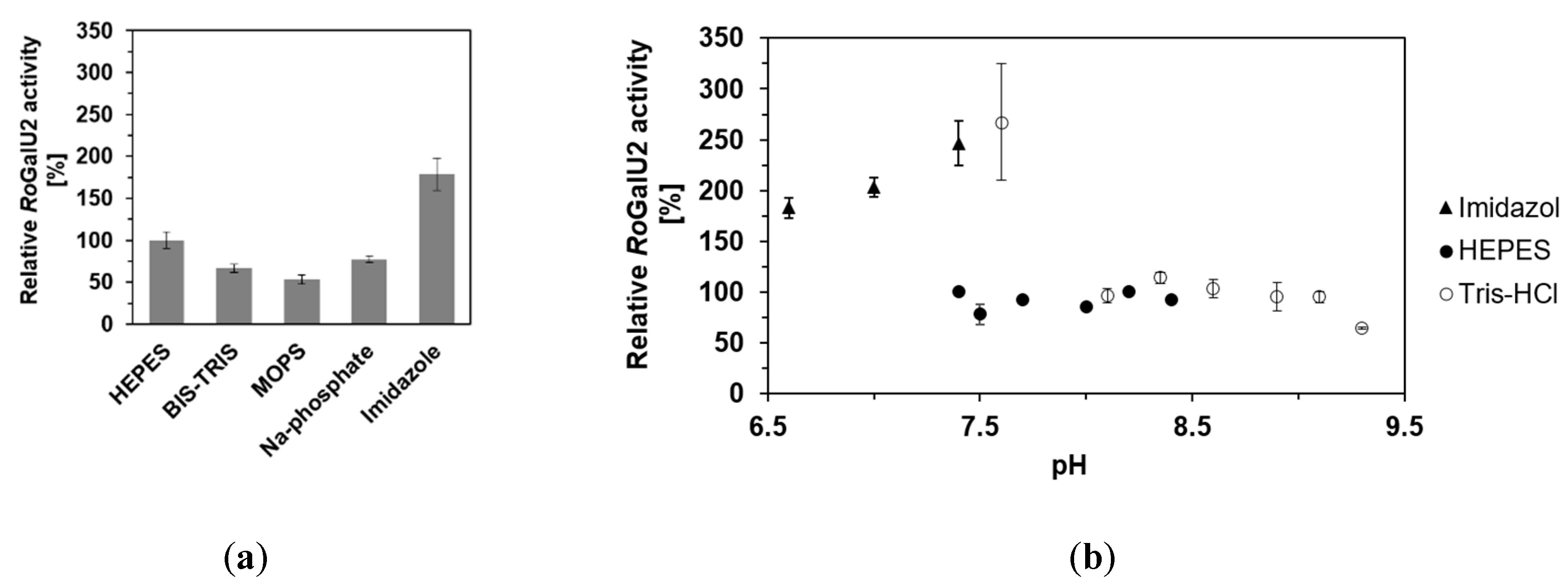
2.4. Determining Optimum Reaction Conditions for RoGalU2
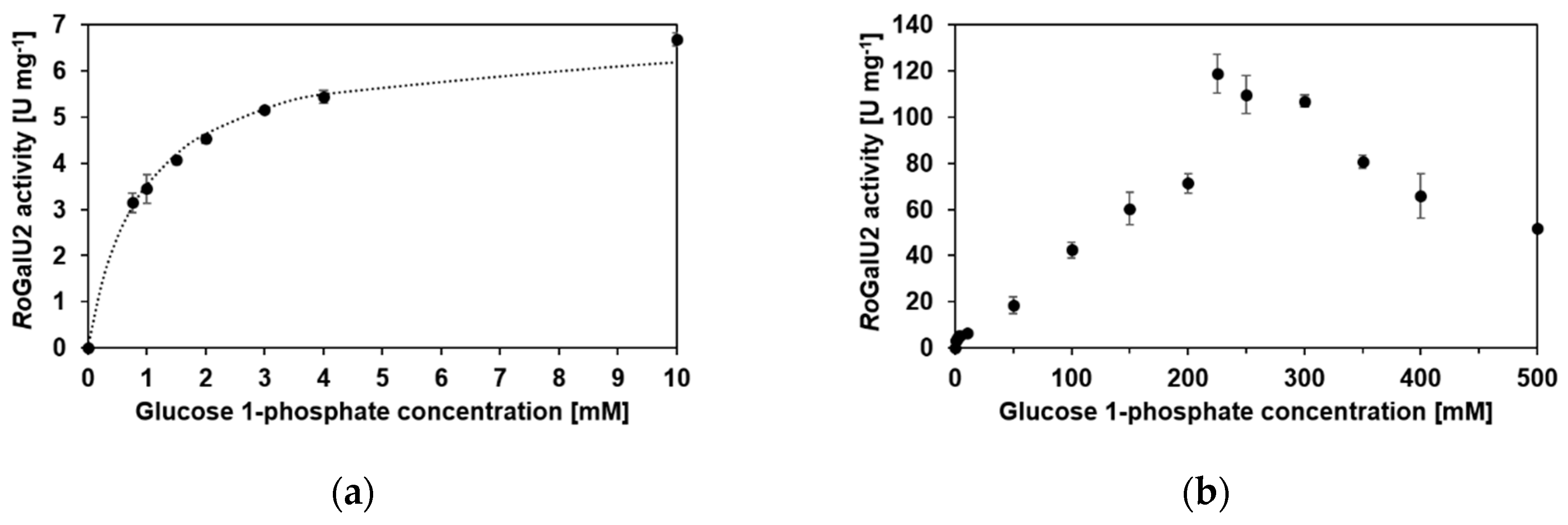
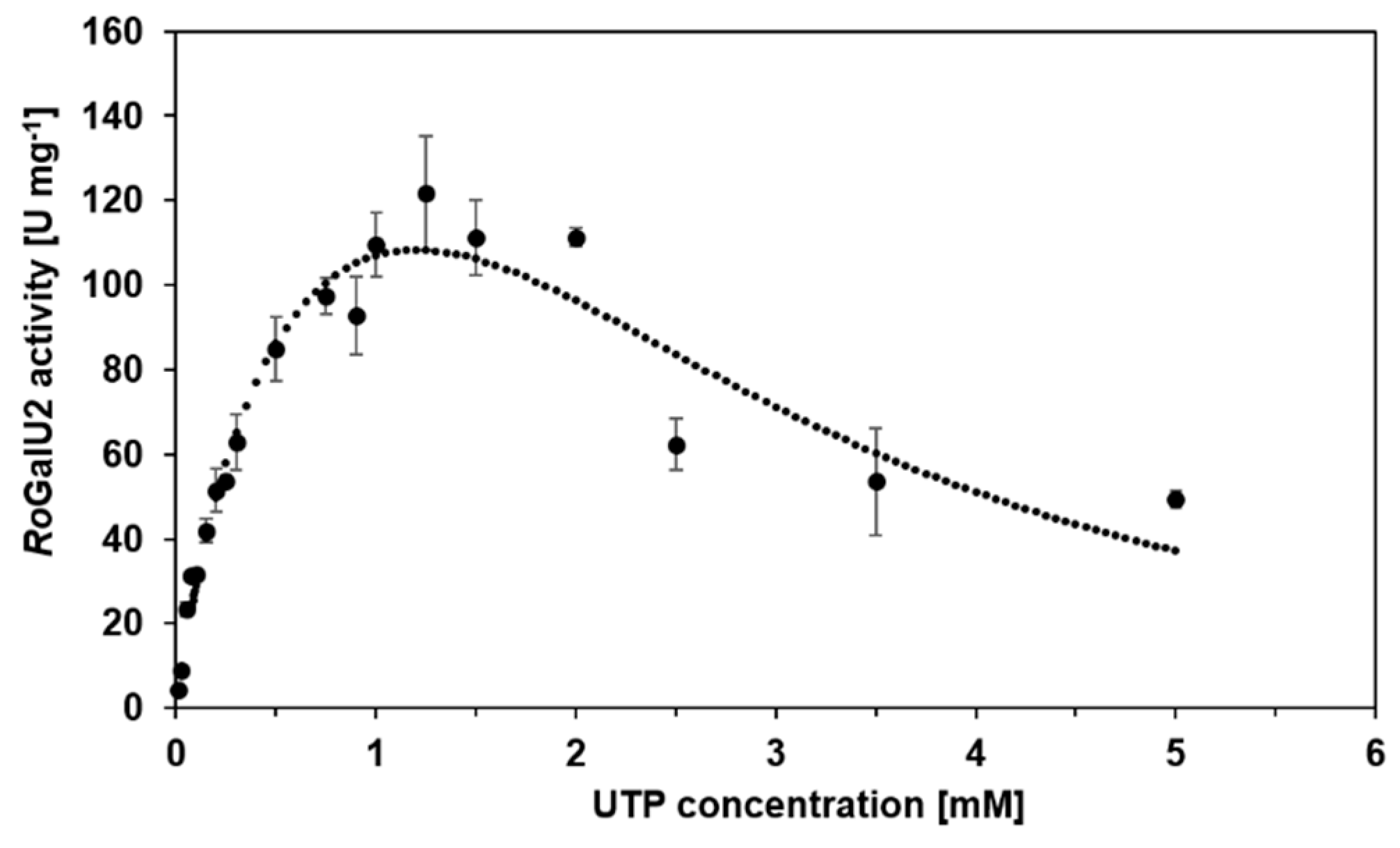
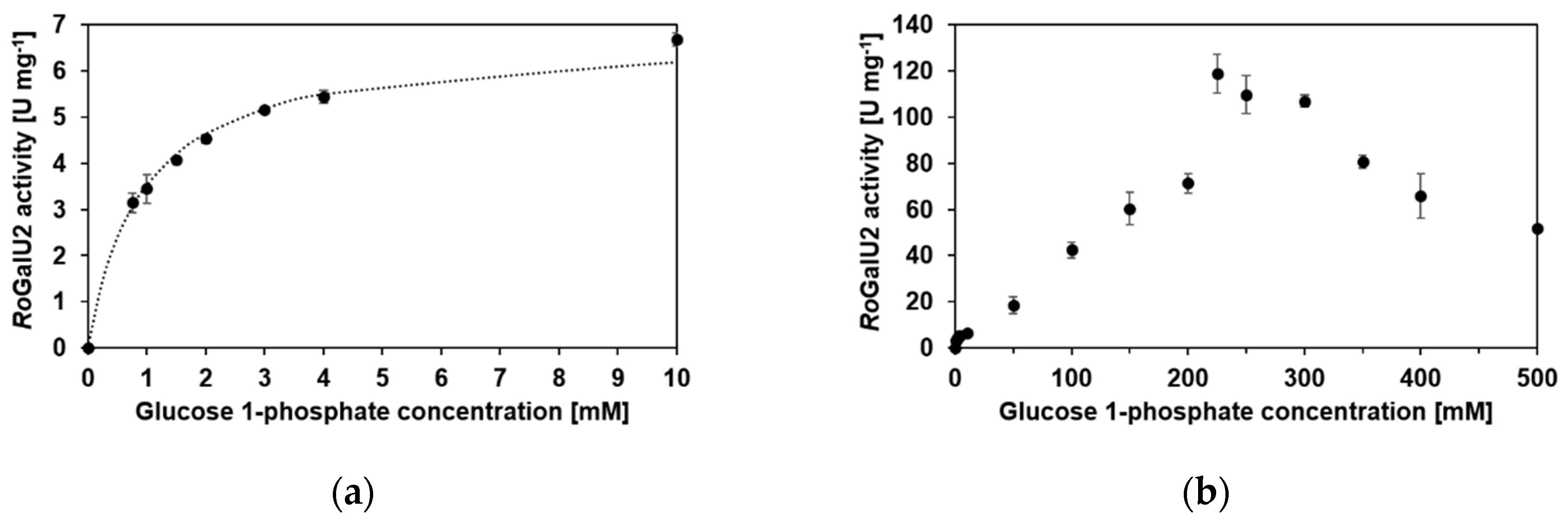
2.5. Enzyme Kinetics with RoGalU2
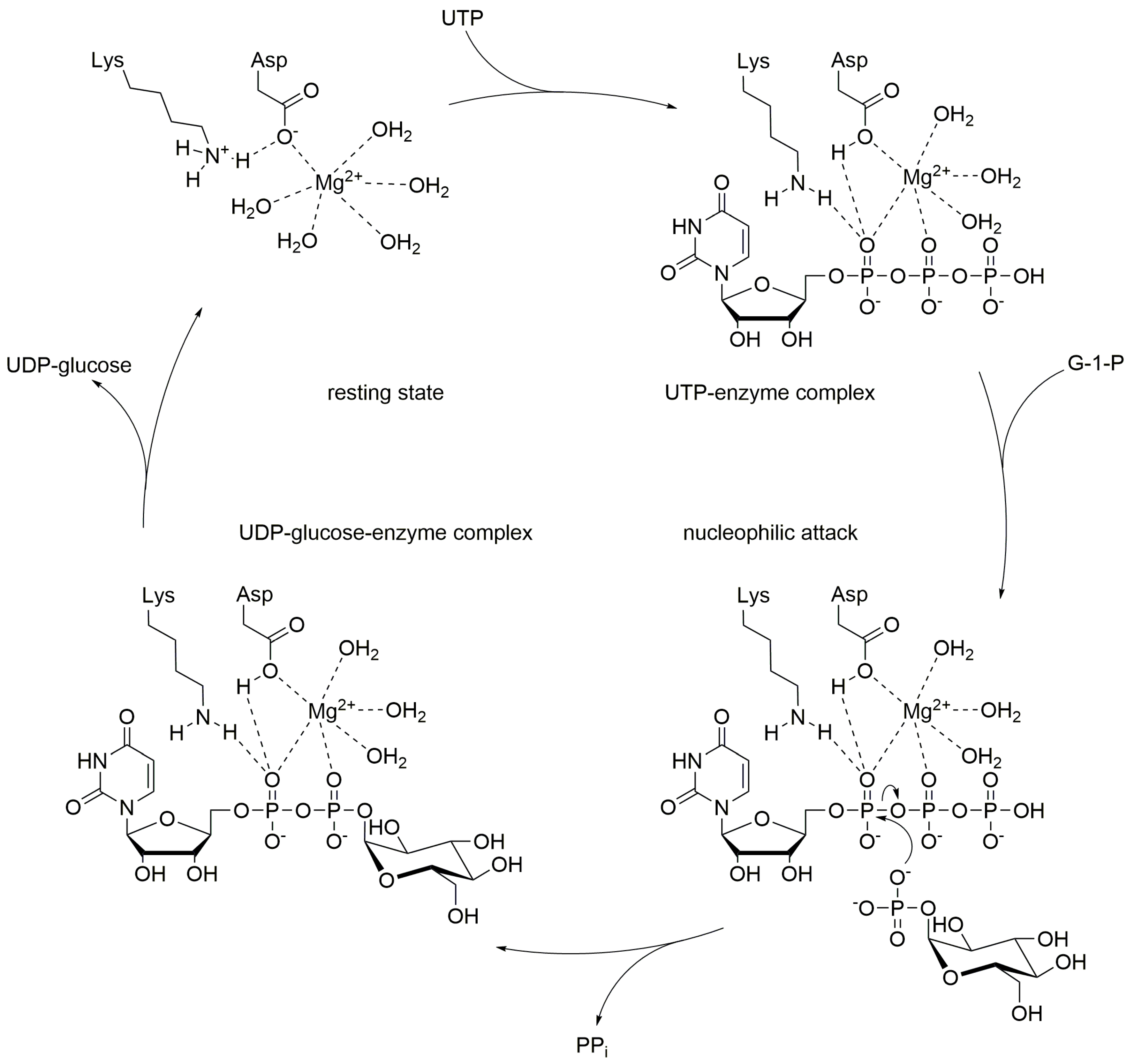
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Plasmids, and Gene Synthesis
4.2. Protein Production
4.3. Purification of Recombinant Proteins
4.4. Renaturation of RoGalU1
4.5. Protein Determination
4.6. Enzyme Activity Assay
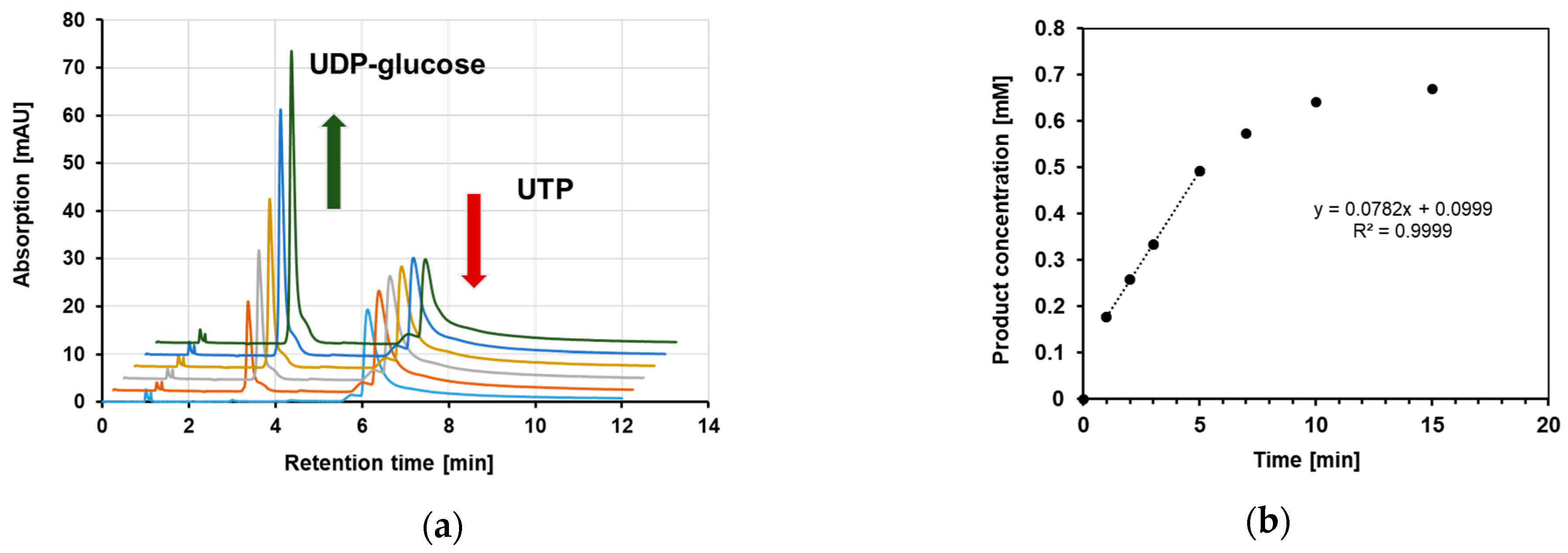
4.7. Product Determination by HPLC for Specific Enzyme Activity Evaluation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gutmann, A.; Nidetzky, B. Unlocking the potential of Leloir glycosyltransferases for applied biocatalysis: Efficient synthesis of Uridine 5’-diphosphate-glucose by sucrose synthase. Adv. Synth. Catal. 2016, 358, 3600–3609. [Google Scholar] [CrossRef]
- Mestrom, L.; Marsden, S.R.; Dieters, M.; Achterberg, P.; Stolk, L.; Bento, I.; Hanefeld, U.; Hagedoorn, P.-L. Artificial fusion of mCherry enhances trehalose transferase solubility and stability. Appl. Environ. Microbiol. 2019, 85. [Google Scholar] [CrossRef] [PubMed]
- Schmölzer, K.; Lemmerer, M.; Gutmann, A.; Nidetzky, B. Integrated process design for biocatalytic synthesis by a Leloir glycosyltransferase: UDP-glucose production with sucrose synthase. Biotechnol. Bioeng. 2017, 114, 924–928. [Google Scholar] [CrossRef] [PubMed]
- Bungaruang, L.; Gutmann, A.; Nidetzky, B. Leloir glycosyltransferases and natural product glycosylation: Biocatalytic synthesis of the C-glucoside Nothofagin, a major antioxidant of redbush herbal tea. Adv. Synth. Catal. 2013, 355, 2757–2763. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Li, J.; Yao, P.; Zhu, Y.; Men, Y.; Zeng, Y.; Yang, J.; Sun, Y. Exploiting the aglycon promiscuity of glycosyltransferase Bs-YjiC from Bacillus subtilis and its application in synthesis of glycosides. J. Biotechnol. 2017, 248, 69–76. [Google Scholar] [CrossRef]
- Diricks, M.; Gutmann, A.; Debacker, S.; Dewitte, G.; Nidetzky, B.; Desmet, T. Sequence determinants of nucleotide binding in sucrose synthase: Improving the affinity of a bacterial sucrose synthase for UDP by introducing plant residues. Protein Eng. Des. Sel. 2017, 30, 141–148. [Google Scholar] [CrossRef]
- Schmölzer, K.; Gutmann, A.; Diricks, M.; Desmet, T.; Nidetzky, B. Sucrose synthase: A unique glycosyltransferase for biocatalytic glycosylation process development. Biotechnol. Adv. 2016, 34, 88–111. [Google Scholar] [CrossRef]
- Thoden, J.B.; Holden, H.M. The molecular architecture of glucose-1-phosphate uridylyltransferase. Protein Sci. 2007, 16, 432–440. [Google Scholar] [CrossRef]
- Aragão, D.; Fialho, A.M.; Marques, A.R.; Mitchell, E.P.; Sa-Correia, I.; Frazao, C. The complex of Sphingomonas elodea ATCC 31461 glucose-1-phosphate uridylyltransferase with glucose-1-phosphate reveals a novel quaternary structure, unique among nucleoside diphosphate-sugar pyrophosphorylase members. J. Bacteriol. 2007, 189, 4520–4528. [Google Scholar] [CrossRef]
- Asención Diez, M.D.; Peirú, S.; Demonte, A.M.; Gramajo, H.; Iglesias, A.A. Characterization of recombinant UDP- and ADP-glucose pyrophosphorylases and glycogen synthase to elucidate glucose-1-phosphate partitioning into oligo- and polysaccharides in Streptomyces coelicolor. J. Bacteriol. 2012, 194, 1485–1493. [Google Scholar] [CrossRef]
- Ebrecht, A.C.; Asencion Diez, M.D.; Piattoni, C.V.; Guerrero, S.A.; Iglesias, A.A. The UDP-glucose pyrophosphorylase from Giardia lamblia is redox regulated and exhibits promiscuity to use galactose-1-phosphate. Biochim. Biophys. Acta 2015, 1850, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Koo, H.M.; Yim, S.-W.; Lee, C.-S.; Pyun, Y.R.; Kim, Y.S. Cloning, sequencing, and expression of UDP-glucose pyrophosphorylase gene from Acetobacter xylinum BRC5. Biosci. Biotechnol. Biochem. 2014, 64, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.; Wu, J.; Chen, S.; Zhang, X.; Wang, H. Expression, purification, and characterization of a functionally active Mycobacterium tuberculosis UDP-glucose pyrophosphorylase. Protein Expr. Purif. 2008, 61, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Wilczynska, M.; Kleczkowski, L.A. Molecular and kinetic characterization of two UDP-glucose pyrophosphorylases, products of distinct genes, from Arabidopsis. Biochim. Biophys. Acta 2008, 1784, 967–972. [Google Scholar] [CrossRef]
- Okazaki, Y.; Shimojima, M.; Sawada, Y.; Toyooka, K.; Narisawa, T.; Mochida, K.; Tanaka, H.; Matsuda, F.; Hirai, A.; Hirai, M.Y.; et al. A chloroplastic UDP-glucose pyrophosphorylase from Arabidopsis is the committed enzyme for the first step of sulfolipid biosynthesis. Plant Cell 2009, 21, 892–909. [Google Scholar] [CrossRef]
- Roeben, A.; Plitzko, J.M.; Korner, R.; Bottcher, U.M.; Siegers, K.; Hayer-Hartl, M.; Bracher, A. Structural basis for subunit assembly in UDP-glucose pyrophosphorylase from Saccharomyces cerevisiae. J. Mol. Biol. 2006, 364, 551–560. [Google Scholar] [CrossRef]
- Kim, H.; Choi, J.; Kim, T.; Lokanath, N.K.; Ha, S.C.; Suh, S.W.; Hwang, H.-Y.; Kim, K.K. Structural basis for the reaction mechanism of UDP-glucose pyrophosphorylase. Mol. Cells 2010, 29, 397–405. [Google Scholar] [CrossRef]
- Thoden, J.B.; Holden, H.M. Active site geometry of glucose-1-phosphate uridylyltransferase. Protein Sci. 2007, 16, 1379–1388. [Google Scholar] [CrossRef]
- Ebrecht, A.C.; Orlof, A.M.; Sasoni, N.; Figueroa, C.M.; Iglesias, A.A.; Ballicora, M.A. On the Ancestral UDP-Glucose Pyrophosphorylase Activity of GalF from Escherichia coli. Front. Microbiol. 2015, 6, 1253. [Google Scholar] [CrossRef]
- Balan, D.; Tokas, J.; Singal, H.R. UDP-glucose pyrophosphorylase: Isolation, purification and characterization from developing thermotolerant wheat (Triticum aestivum) grains. Protein Expr. Purif. 2018, 148, 68–77. [Google Scholar] [CrossRef]
- Chen, R.; Zhao, X.; Shao, Z.; Zhu, L.; He, G. Multiple isoforms of UDP-glucose pyrophosphorylase in rice. Physiol Plant 2007, 129, 725–736. [Google Scholar] [CrossRef]
- Cotrim, C.A.; Soares, J.S.M.; Kobe, B.; Menossi, M. Crystal structure and insights into the oligomeric state of UDP-glucose pyrophosphorylase from sugarcane. PLoS ONE 2018, 13, e0193667. [Google Scholar] [CrossRef] [PubMed]
- McCoy, J.G.; Bitto, E.; Bingman, C.A.; Wesenberg, G.E.; Bannen, R.M.; Kondrashov, D.A.; Phillips, G.N., Jr. Structure and dynamics of UDP-glucose pyrophosphorylase from Arabidopsis thaliana with bound UDP-glucose and UTP. J. Mol. Biol. 2007, 366, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Meng, M.; Fitzek, E.; Gajowniczek, A.; Wilczynska, M.; Kleczkowski, L.A. Domain-specific determinants of catalysis/substrate binding and the oligomerization status of barley UDP-glucose pyrophosphorylase. Biochim. Biophys. Acta 2009, 1794, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.K. UDPglucose pyrophosphorylase from Ehrlich ascites carcinoma cell-purification and characterization. Indian J. Biochem. Biophys. 1985, 22, 203–207. [Google Scholar] [PubMed]
- Granzow, C.; Kopun, M.; Zimmermann, H.P. Role of nuclear glycogen synthase and cytoplasmic UDP glucose pyrophosphorylase in the biosynthesis of nuclear glycogen in HD33 Ehrlich-Lettré ascites tumor cells. J. Cell Biol. 1981, 89, 475–484. [Google Scholar] [CrossRef]
- Reynolds, T.H.; Pak, Y.; Harris, T.E.; Manchester, J.; Barrett, E.J.; Lawrence, J.C. Effects of insulin and transgenic overexpression of UDP-glucose pyrophosphorylase on UDP-glucose and glycogen accumulation in skeletal muscle fibers. J. Biol. Chem. 2005, 280, 5510–5515. [Google Scholar] [CrossRef]
- Führing, J.I.; Cramer, J.T.; Schneider, J.; Baruch, P.; Gerardy-Schahn, R.; Fedorov, R. A quaternary mechanism enables the complex biological functions of octameric human UDP-glucose pyrophosphorylase, a key enzyme in cell metabolism. Sci. Rep. 2015, 5, 9618. [Google Scholar] [CrossRef]
- Dickmanns, A.; Damerow, S.; Neumann, P.; Schulz, E.-C.; Lamerz, A.-C.; Routier, F.H.; Ficner, R. Structural basis for the broad substrate range of the UDP-sugar pyrophosphorylase from Leishmania major. J. Mol. Biol. 2011, 405, 461–478. [Google Scholar] [CrossRef]
- Benini, S.; Toccafondi, M.; Rejzek, M.; Musiani, F.; Wagstaff, B.A.; Wuerges, J.; Cianci, M.; Field, R.A. Glucose-1-phosphate uridylyltransferase from Erwinia amylovora: Activity, structure and substrate specificity. Biochim. Biophys. Acta Proteins Proteom. 2017, 1865, 1348–1357. [Google Scholar] [CrossRef]
- Bosco, M.B.; Machtey, M.; Iglesias, A.A.; Aleanzi, M. UDPglucose pyrophosphorylase from Xanthomonas spp. Characterization of the enzyme kinetics, structure and inactivation related to oligomeric dissociation. Biochimie 2009, 91, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Degeest, B.; de Vuyst, L. Correlation of activities of the enzymes alpha-phosphoglucomutase, UDP-galactose 4-epimerase, and UDP-glucose pyrophosphorylase with exopolysaccharide biosynthesis by Streptococcus thermophilus LY03. Appl. Environ. Microbiol. 2000, 66, 3519–3527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padilla, L.; Morbach, S.; Krämer, R.; Agosin, E. Impact of heterologous expression of Escherichia coli UDP-glucose pyrophosphorylase on trehalose and glycogen synthesis in Corynebacterium glutamicum. Appl. Environ. Microbiol. 2004, 70, 3845–3854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toccafondi, M.; Cianci, M.; Benini, S. Expression, purification, crystallization and preliminary X-ray analysis of glucose-1-phosphate uridylyltransferase (GalU) from Erwinia amylovora. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 1249–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawano, Y.; Sekine, M.; Ihara, M. Identification and characterization of UDP-glucose pyrophosphorylase in cyanobacteria Anabaena sp. PCC 7120. J. Biosci. Bioeng. 2014, 117, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.S.; Koh, S.; Shin, H.J.; Lee, D.S.; Lee, S.Y. Biochemical characterization of a UDP-sugar pyrophosphorylase from Thermus caldophilus GK24. Biotechnol. Appl. Biochem. 1999, 29 (Pt 1), 11–17. [Google Scholar]
- Holden, H.M.; Rayment, I.; Thoden, J.B. Structure and function of enzymes of the Leloir pathway for galactose metabolism. J. Biol. Chem. 2003, 278, 43885–43888. [Google Scholar] [CrossRef] [Green Version]
- Marques, A.R.; Ferreira, P.B.; Sá-Correia, I.; Fialho, A.M. Characterization of the ugpG gene encoding a UDP-glucose pyrophosphorylase from the gellan gum producer Sphingomonas paucimobilis ATCC 31461. Mol. Genet. Genomics 2003, 268, 816–824. [Google Scholar] [CrossRef]
- Berbis, M.; Sanchez-Puelles, J.; Canada, F.; Jimenez-Barbero, J. Structure and function of prokaryotic UDP-glucose pyrophosphorylase, a drug target candidate. CMC 2015, 22, 1687–1697. [Google Scholar] [CrossRef] [Green Version]
- Silva, E.; Marques, A.R.; Fialho, A.M.; Granja, A.T.; Sá-Correia, I. Proteins encoded by Sphingomonas elodea ATCC 31461 rmlA and ugpG genes, involved in gellan gum biosynthesis, exhibit both dTDP- and UDP-glucose pyrophosphorylase activities. Appl. Environ. Microbiol. 2005, 71, 4703–4712. [Google Scholar] [CrossRef] [Green Version]
- Gröning, J.A.D.; Eulberg, D.; Tischler, D.; Kaschabek, S.R.; Schlömann, M. Gene redundancy of two-component (chloro)phenol hydroxylases in Rhodococcus opacus 1CP. FEMS Microbiol. Lett. 2014, 361, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letek, M.; González, P.; Macarthur, I.; Rodríguez, H.; Freeman, T.C.; Valero-Rello, A.; Blanco, M.; Buckley, T.; Cherevach, I.; Fahey, R.; et al. The genome of a pathogenic Rhodococcus: Cooptive virulence underpinned by key gene acquisitions. PLoS Genet. 2010, 6, e1001145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizanur, R.M.; Pohl, N.L. A thermostable promiscuous glucose-1-phosphate uridyltransferase from Helicobacter pylori for the synthesis of nucleotide sugars. J. Mol. Catal. B Enzymatic 2008, 50, 13–19. [Google Scholar] [CrossRef]
- Tischler, D.; Gröning, J.A.D.; Kaschabek, S.R.; Schlömann, M. One-component styrene monooxygenases: An evolutionary view on a rare class of flavoproteins. Appl. Biochem. Biotechnol. 2012, 167, 931–944. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320-4. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Yano, T.; Koga, S. Dynamic behavior of the chemostat subject to substrate inhibition. Biotechnol. Bioeng. 1969, 11, 139–153. [Google Scholar] [CrossRef]
- Asención Diez, M.D.; Demonte, A.M.; Syson, K.; Arias, D.G.; Gorelik, A.; Guerrero, S.A.; Bornemann, S.; Iglesias, A.A. Allosteric regulation of the partitioning of glucose-1-phosphate between glycogen and trehalose biosynthesis in Mycobacterium tuberculosis. Biochim. Biophys. Acta 2015, 1850, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, H.I.; Pratap, A.P. Production and quantitative analysis of trehalose lipid biosurfactants using high-performance liquid chromatography. J. Surfactants Deterg. 2018, 21, 553–564. [Google Scholar] [CrossRef]
- Tischler, D.; Niescher, S.; Kaschabek, S.R.; Schlömann, M. Trehalose phosphate synthases OtsA1 and OtsA2 of Rhodococcus opacus 1CP. FEMS Microbiol. Lett. 2013, 342, 113–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, B.; Crombet, L.; Loppnau, P.; Cossar, D. A screening strategy for heterologous protein expression in Escherichia coli with the highest return of investment. Protein Expr. Purif. 2012, 81, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Strandberg, L.; Enfors, S.O. Factors influencing inclusion body formation in the production of a fused protein in Escherichia coli. Appl. Environ. Microbiol. 1991, 57, 1669–1674. [Google Scholar] [PubMed]
- Aragão, D.; Marques, A.R.; Frazão, C.; Enguita, F.J.; Carrondo, M.A.; Fialho, A.M.; Sá-Correia, I.; Mitchell, E.P. Cloning, expression, purification, crystallization and preliminary structure determination of glucose-1-phosphate uridylyltransferase (UgpG) from Sphingomonas elodea ATCC 31461 bound to glucose-1-phosphate. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2006, 62, 930–934. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.K.; Sowokinos, J.R.; Hahn, I.-S. Regulation of UDP-glucose pyrophosphorylase isozyme UGP5 associated with cold-sweetening resistance in potatoes. J. Plant Physiol. 2008, 165, 679–690. [Google Scholar] [CrossRef]
- Steiner, T.; Lamerz, A.C.; Hess, P.; Breithaupt, C.; Krapp, S.; Bourenkov, G.; Huber, R.; Gerardy-Schahn, R.; Jacob, U. Open and closed structures of the UDP-glucose pyrophosphorylase from Leishmania major. J. Biol. Chem. 2007, 282, 13003–13010. [Google Scholar] [CrossRef] [Green Version]
- De Bruyn, F.; Beauprez, J.; Maertens, J.; Soetaert, W.; de Mey, M. Unraveling the Leloir pathway of Bifidobacterium bifidum: Significance of the uridylyltransferases. Appl. Environ. Microbiol. 2013, 79, 7028–7035. [Google Scholar] [CrossRef] [Green Version]
- Ma, Z.; Fan, H.-j.; Lu, C.-p. Molecular cloning and analysis of the UDP-glucose pyrophosphorylase in Streptococcus equi subsp. zooepidemicus. Mol. Biol. Rep. 2011, 38, 2751–2760. [Google Scholar] [CrossRef]
- Weissborn, A.C.; Liu, Q.; Rumley, M.K.; Kennedy, E.P. UTP: Alpha-D-glucose-1-phosphate uridylyltransferase of Escherichia coli: isolation and DNA sequence of the galU gene and purification of the enzyme. J. Bacteriol. 1994, 176, 2611–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, Y.; Zang, Q.; Shimizu, Y.; Dadashipour, M.; Zhang, Z.; Kawarabayasi, Y. Increasing the thermostable sugar-1-phosphate nucleotidylyltransferase activities of the archaeal ST0452 protein through site saturation mutagenesis of the 97th amino acid position. Appl. Environ. Microbiol. 2017, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Tsujimura, M.; Akutsu, J.-i.; Sasaki, M.; Tajima, H.; Kawarabayasi, Y. Identification of an extremely thermostable enzyme with dual sugar-1-phosphate nucleotidylyltransferase activities from an acidothermophilic archaeon, Sulfolobus tokodaii strain 7. J. Biol. Chem. 2005, 280, 9698–9705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischler, D.; Eulberg, D.; Lakner, S.; Kaschabek, S.R.; van Berkel, W.J.H.; Schlömann, M. Identification of a novel self-sufficient styrene monooxygenase from Rhodococcus opacus 1CP. J. Bacteriol. 2009, 191, 4996–5009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oelschlägel, M.; Gröning, J.A.D.; Tischler, D.; Kaschabek, S.R.; Schlömann, M. Styrene oxide isomerase of Rhodococcus opacus 1CP, a highly stable and considerably active enzyme. Appl. Environ. Microbiol. 2012, 78, 4330–4337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleczkowski, L.A. Glucose activation and metabolism through UDP-glucose pyrophosphorylase in plants. Phytochemistry 1994, 37, 1507–1515. [Google Scholar] [CrossRef]
- Li, Q.; Huang, Y.-Y.; Conway, L.P.; He, M.; Wei, S.; Huang, K.; Duan, X.-C.; Flitsch, S.L.; Voglmeir, J. Discovery and biochemical characterization of a thermostable glucose-1-phosphate nucleotidyltransferase from Thermodesulfatator indicus. Protein Pept. Lett. 2017, 24, 729–734. [Google Scholar] [CrossRef]
- Bernstein, R.L.; Robbind, P.W. Control aspects of Uridine 5’-diphosphate glucose and Thymidine 5’-diphosphate glucose synthesis by microbial enzymes. J. Biol. Chem. 1965, 240, 391–397. [Google Scholar]
- Lindquist, L.; Kaiser, R.; Reeves, P.R.; Lindberg, A.A. Purification, characterization and HPLC assay of Salmonella glucose-1-phosphate thymidylyltransferase from the cloned rfbA gene. Eur. J. Biochem. 1993, 211, 763–770. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W293–W303. [Google Scholar] [CrossRef] [Green Version]
- Kumpf, A.; Tischler, D. Structural investigations of GalU enzymes from Actinobacteria, Crystals. Unpublished work.
- Mestrom, L.; Przypis, M.; Kowalczykiewicz, D.; Pollender, A.; Kumpf, A.; Marsden, S.R.; Bento, I.; Jarzębski, A.B.; Szymańska, K.; Chruściel, A.; et al. Leloir glycosyltransferases in applied biocatalysis: A multidisciplinary approach. Int. J. Mol. Sci. 2019, 20, 5263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moiseeva, O.V.; Solyanikova, I.P.; Kaschabek, S.R.; Gröning, J.; Thiel, M.; Golovleva, L.A.; Schlömann, M. A new modified ortho cleavage pathway of 3-chlorocatechol degradation by Rhodococcus opacus 1CP: Genetic and biochemical evidence. J. Bacteriol. 2002, 184, 5282–5292. [Google Scholar] [CrossRef] [Green Version]
- Studier, F.W. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 2005, 41, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Varied Substrate | UTP 1 | Glucose 1-Phosphate 1 | |
|---|---|---|---|
| conc. [mM] | 0.01–5 | 0.05–10 | 0.05–500 |
| apparent Km [mM] | 0.51 | 0.9 | 150 |
| Vmax [U mg−1] 2 | 177 | 6.75 | - |
| Vmax,ob [U mg−1] 3 | 122 | 6.68 | 119 |
| apparent kcat [s−1] | 96.2 | 3.7 | 65 |
| KI [mM] | 2.61 | - | 225 |
| kcat/Km [µM−1 s−1] | 0.19 | 0.004 | 0.0004 |
| Organism and Protein | Vmax [U mg−1] | Km [mM] | kcat [s−1] | c [mM] | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|
| UTP | G1P | UTP | G1P | UTP | G1P | UTP | G1P | ||
| R. opacus 1CP GalU2 | 177 | 6.751 1192 | 0.51 | 0.91 1502 | 96.2 | 3.71 652 | 0.01–5 | 0.05–500 | This study |
| E. coli K-12 GalU | 340 | 0.17 | 0.035 | 187 | 2 | 2 | [19] | ||
| E. coli K-12 GalF | 0.015 | 0.36 | 0.52 | 0.008 | 2 | 2 | [19] | ||
| M. tuberculosis H37Rv GalU | 2.5–2.73 9.8 U4 | 5.8 U4 | 0.13 0.0124 | 0.133 0.0454 | 93.444 | 55.034 | 0.001–0.014 | 0.008–0.054 | [51,13] |
| E. amylovora CFBP 1430 GalU | 14.4 | 32.5 | 0.027 | 0.007 | 7.9 | 17.2 | 0.006–0.1 | 0.003–0.1 | [30] |
| S. coelicolor A3 (2) GalU | 270 | >10 | 0.06 | 149 | ≤20 | n.s. | [10] | ||
| B. bifidum PRL2010 GalU | 13 | 0.042 | 0.098 | 12 | 0–2 | 0–2 | [59] | ||
| X. capestris pv. campestris str. 8004 GalU | 60–90 | 0.21 | 0.06 | 40 | n.s. | 0-8 | [31] | ||
| X. axonopodis pv. citris str. 306 GalU | 60–90 | 0.11 | n.s. | 29 | n.s. | n.s. | [31] | ||
| S.thermophilus LY03 GalU | 0.2 | n.s. | n.s. | 0.11 | 1.25 | 1 | [32] | ||
| Sample | Relevant Characteristics | Source, Reference |
|---|---|---|
| Strains | ||
| R. opacus 1 CP | Benzoate+, 4-hydroxybenzoate+, 3-chlorobenzoate+, phenol+, 4-chlorophenol+, 2,4-dichlorophenol+, 2-chlorophenol+, 3-methylphenol+, 4-methylphenol+, phthalate+, isophthalate+, n-alkanes+ (C10-C16), styrene+ | [73,64] |
| E. coli DH5α | fhuA2 Δ(argF-lacZ) U169 phoA glnV44 Φ80 Δ(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | NEB 1 |
| E. coli BL21(DE3) pLysS | fhuA2 [lon] ompT gal (λ DE3) [dcm] ∆hsdS λ DE3 = λ sBamHIo ∆EcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 ∆nin5 | NEB 1 |
| Plasmids | ||
| pEX-A2 | multiple cloning site, Lac-Promoter, pUC origin, Ampr | Eurofins 2 |
| pET16bP | pET16b with additional multiple cloning site; allows production of recombinant proteins with N-terminal Histidine10-Tag and gene expression induction with IPTG | Wehmeyer 3 |
| pEX-A2-RogalU1 | pEX-A2 vector with recombinant UDP-glucose-pyrophosphorylase gene 1 of R. opacus 1 CP (RogalU1) 914 bp, NCBI protein accession: ANS26426 | This study |
| pEX-A2-RogalU2 | pEX-A2 vector with recombinant UDP-glucose-pyrophosphorylase gene 2 of R. opacus 1 CP (RogalU2) 932 bp, NCBI protein accession: ANS26629 | This study |
| pET16bP-RogalU1 | pET16bP vector with recombinant UDP-glucose-pyrophosphorylase gene 1 of R. opacus 1 CP (RogalU1) | This study |
| pET16bP-RogalU2 | pET16bP vector with recombinant UDP-glucose-pyrophosphorylase gene 2 of R. opacus 1 CP (RogalU2) | This study |
| Primer | ||
| pET16bP-fw | 5’ - CATCACAGCAGCGGCCATATCGAAG - 3’ | This study |
| pET16bP-rev | 5’ - CAGCTTCTTTTCGGGCTTTGTTAG - 3’ | This study |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumpf, A.; Partzsch, A.; Pollender, A.; Bento, I.; Tischler, D. Two Homologous Enzymes of the GalU Family in Rhodococcus opacus 1CP—RoGalU1 and RoGalU2. Int. J. Mol. Sci. 2019, 20, 5809. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225809
Kumpf A, Partzsch A, Pollender A, Bento I, Tischler D. Two Homologous Enzymes of the GalU Family in Rhodococcus opacus 1CP—RoGalU1 and RoGalU2. International Journal of Molecular Sciences. 2019; 20(22):5809. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225809
Chicago/Turabian StyleKumpf, Antje, Anett Partzsch, André Pollender, Isabel Bento, and Dirk Tischler. 2019. "Two Homologous Enzymes of the GalU Family in Rhodococcus opacus 1CP—RoGalU1 and RoGalU2" International Journal of Molecular Sciences 20, no. 22: 5809. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225809