Caspase-8 Regulates Endoplasmic Reticulum Stress-Induced Necroptosis Independent of the Apoptosis Pathway in Auditory Cells

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
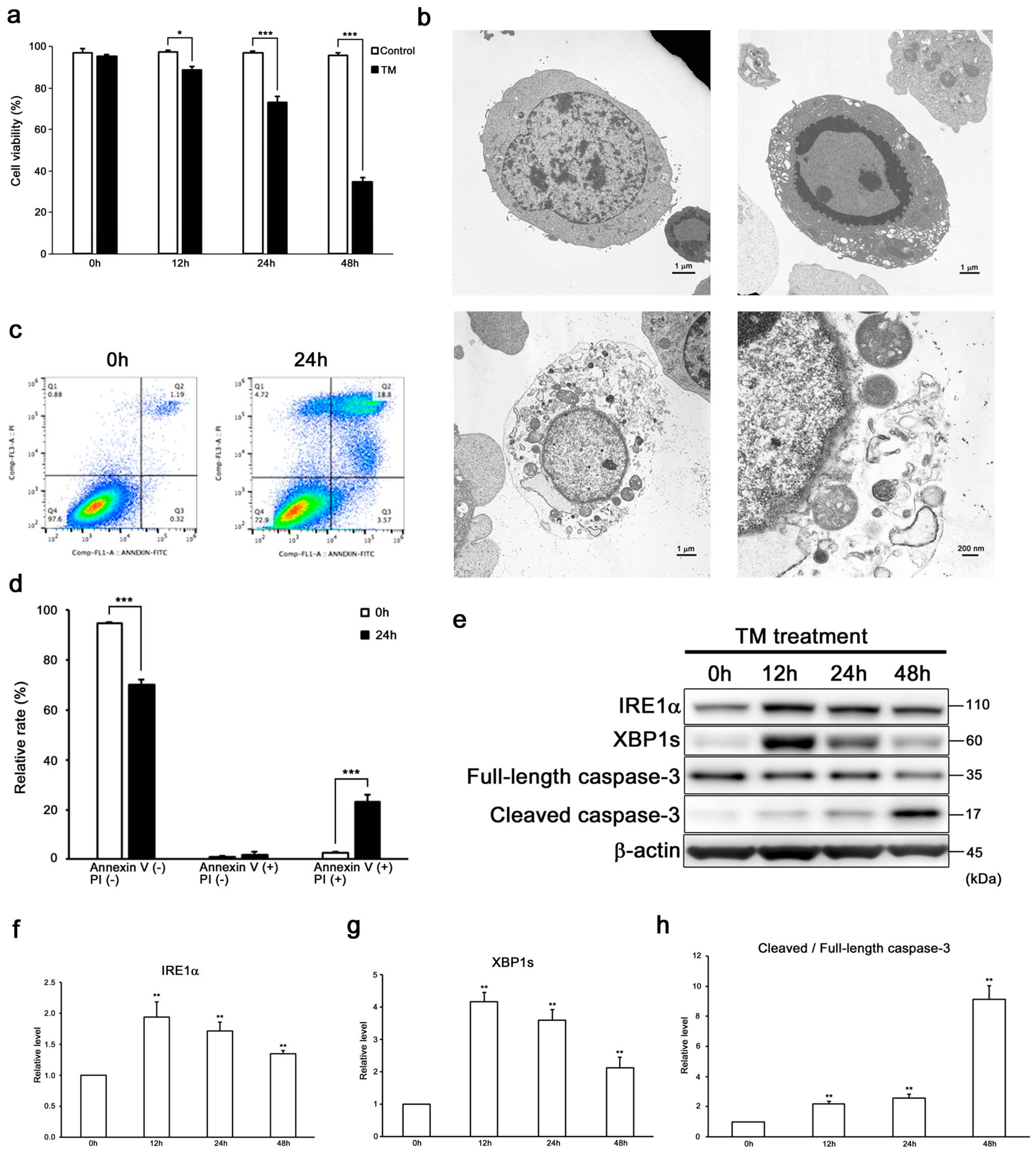
2.1. ER Stress Induces Not Only Apoptosis but also Necroptosis in HEI-OC1 Cells
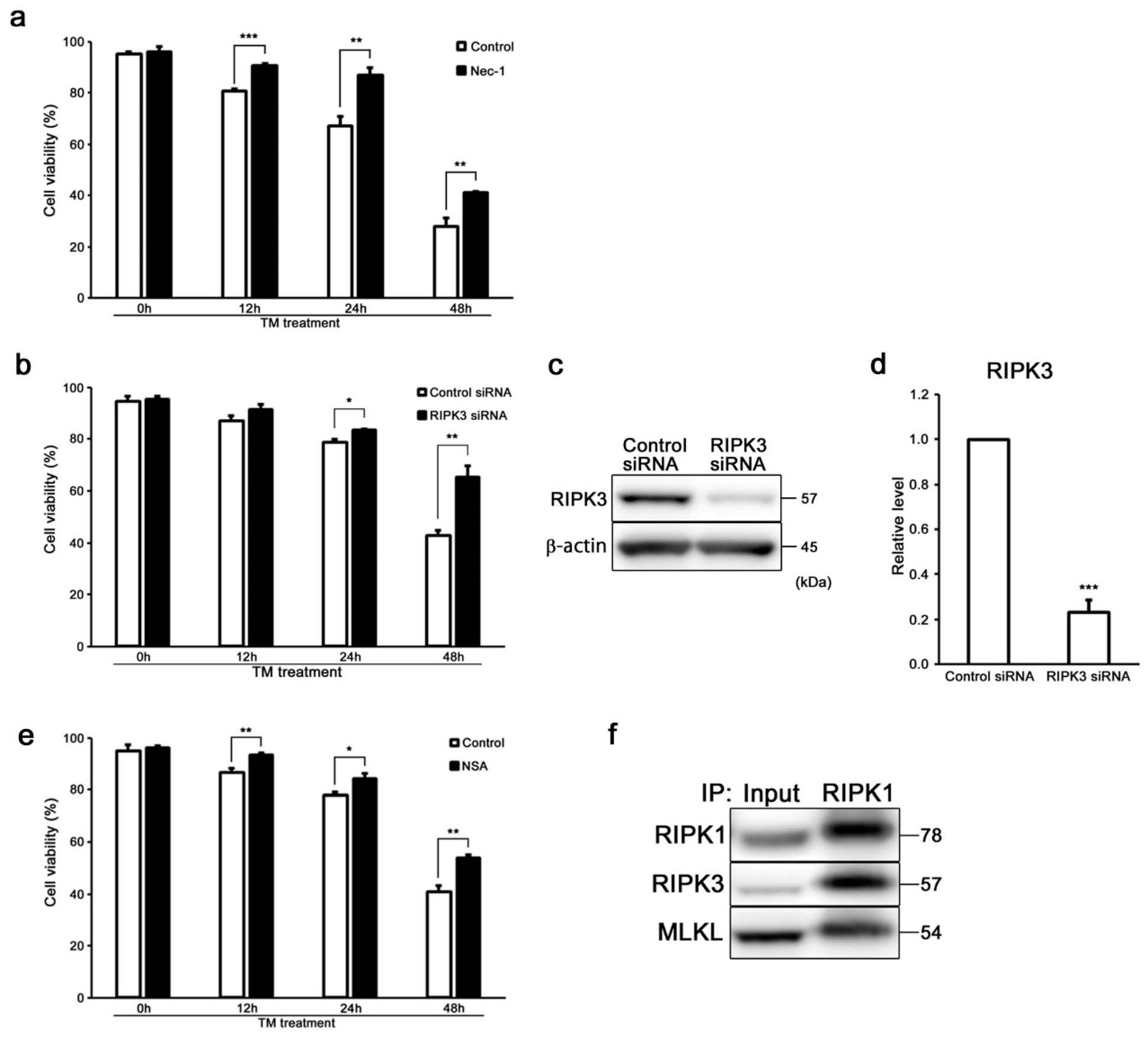
2.2. ER Stress Induces RIPK1-Dependent Necroptosis in HEI-OC1 Cells
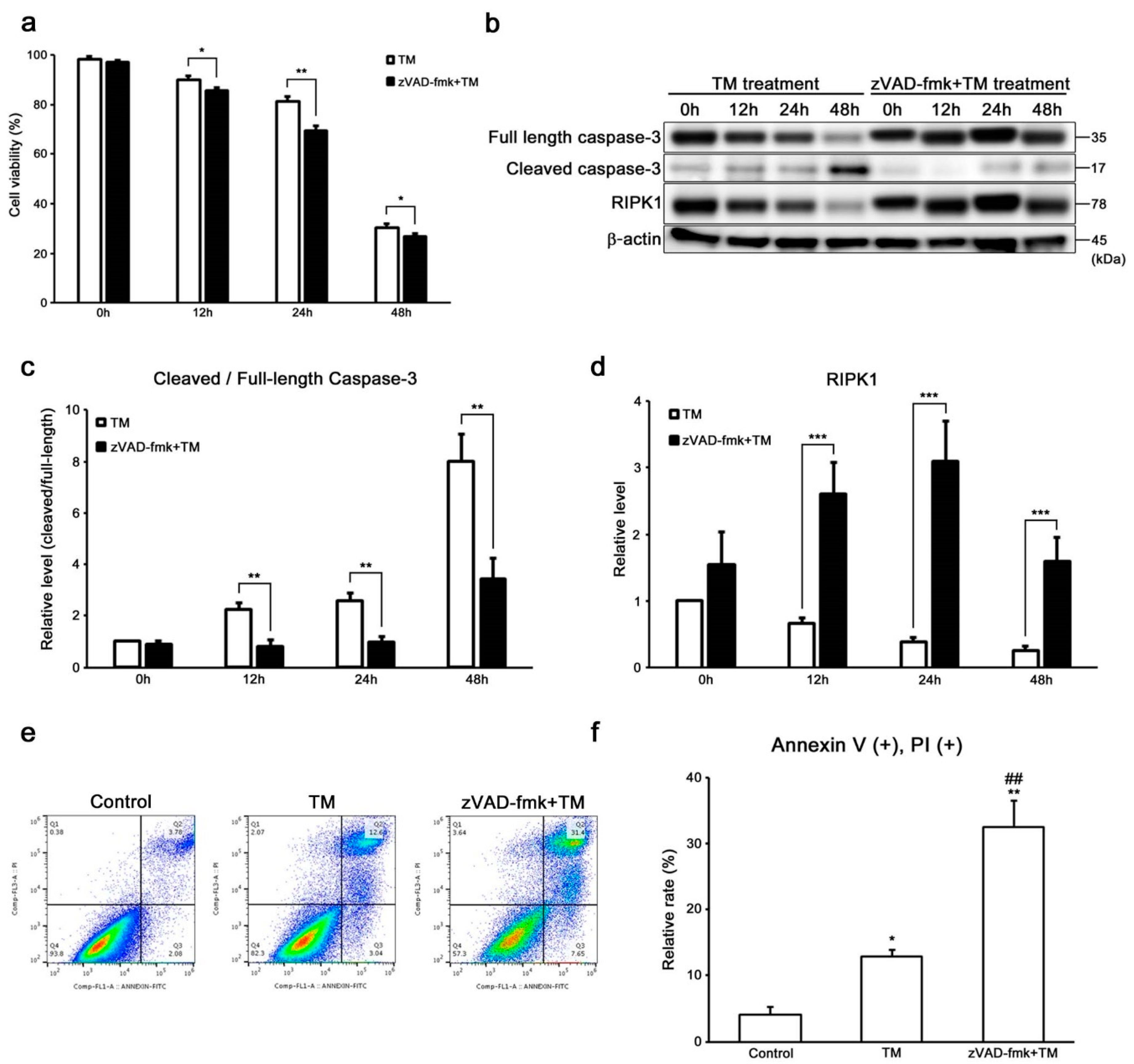
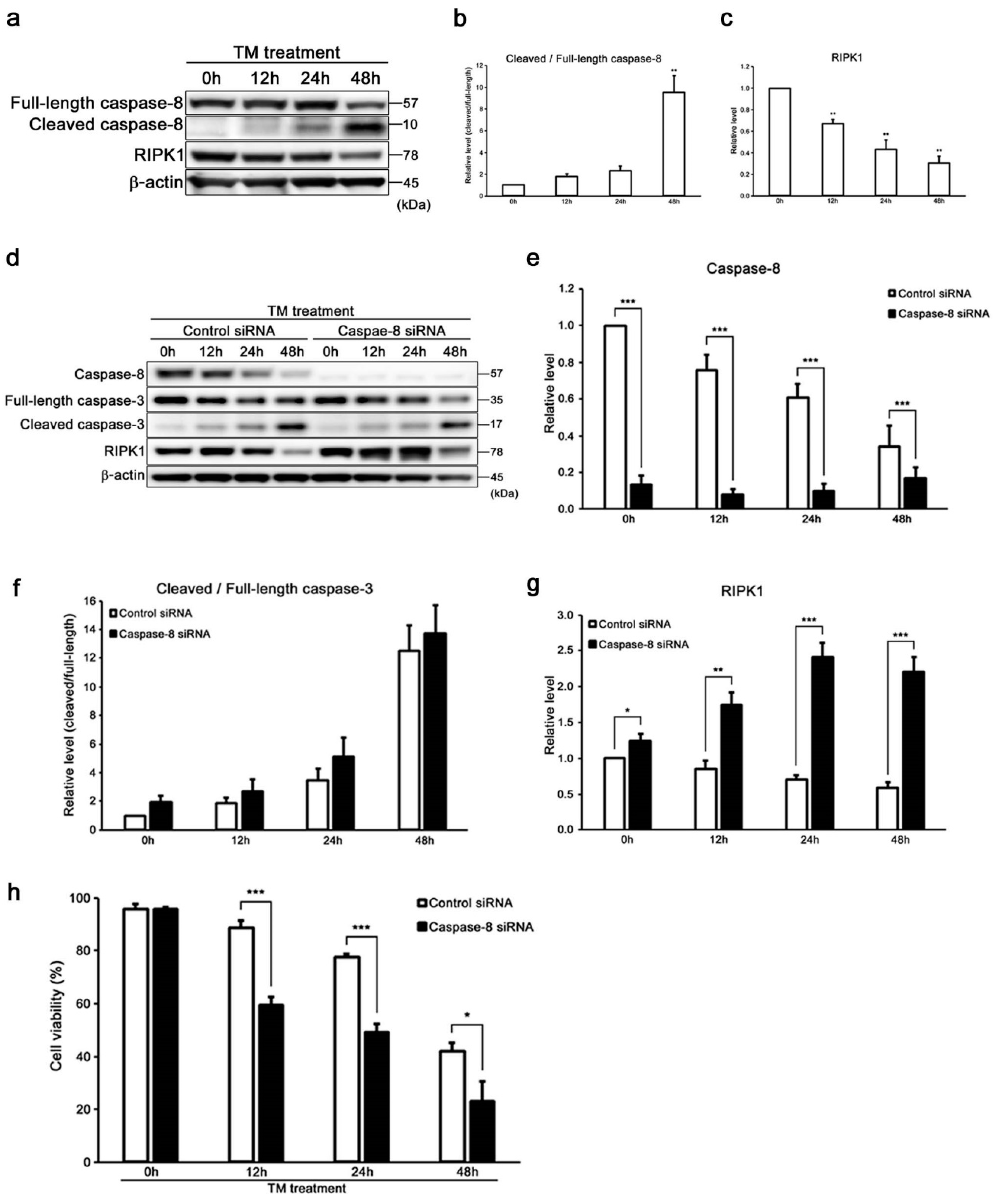
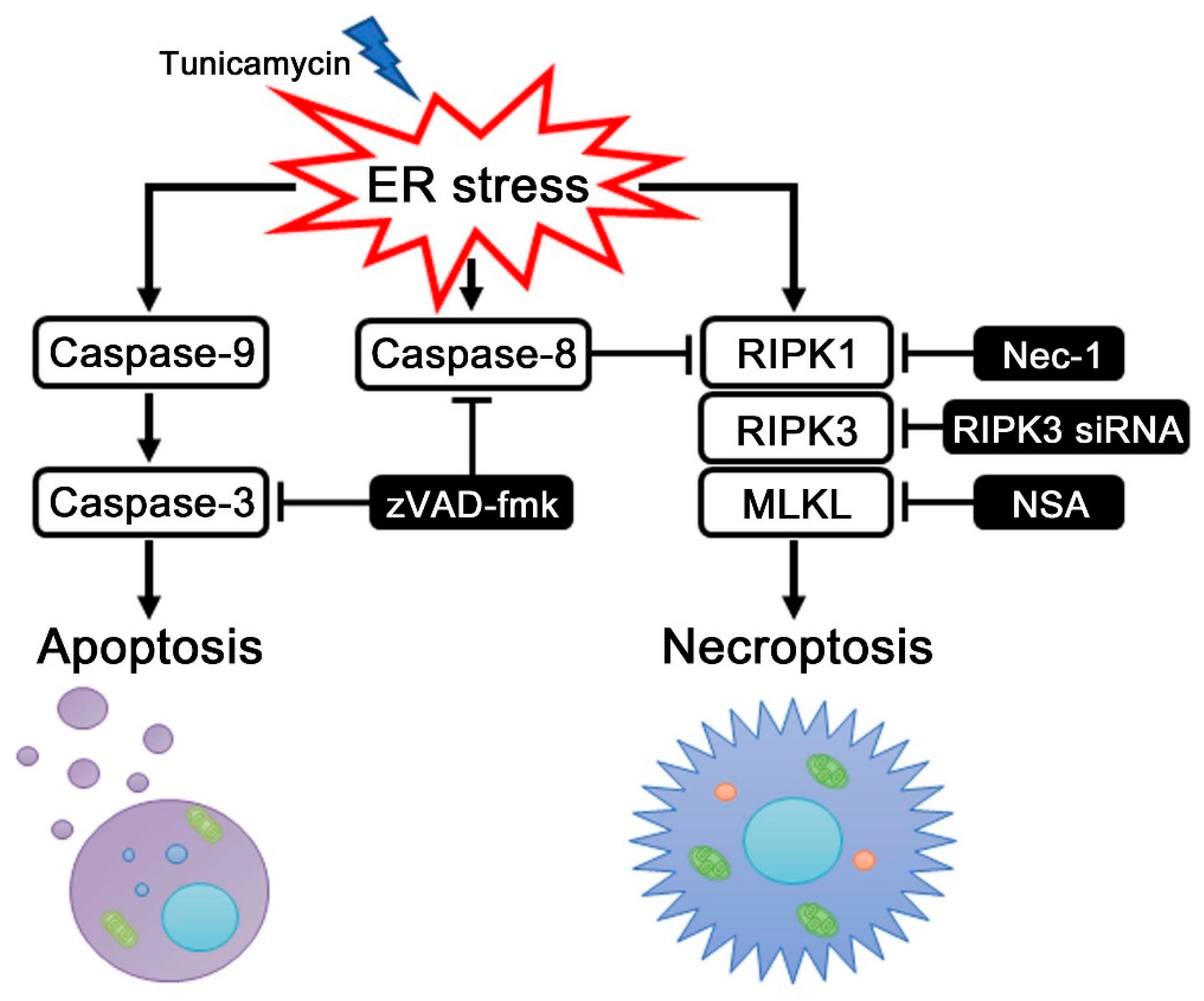
2.3. Caspase-8 Regulates ER Stress-Induced Necroptosis in HEI-OC1 Cells
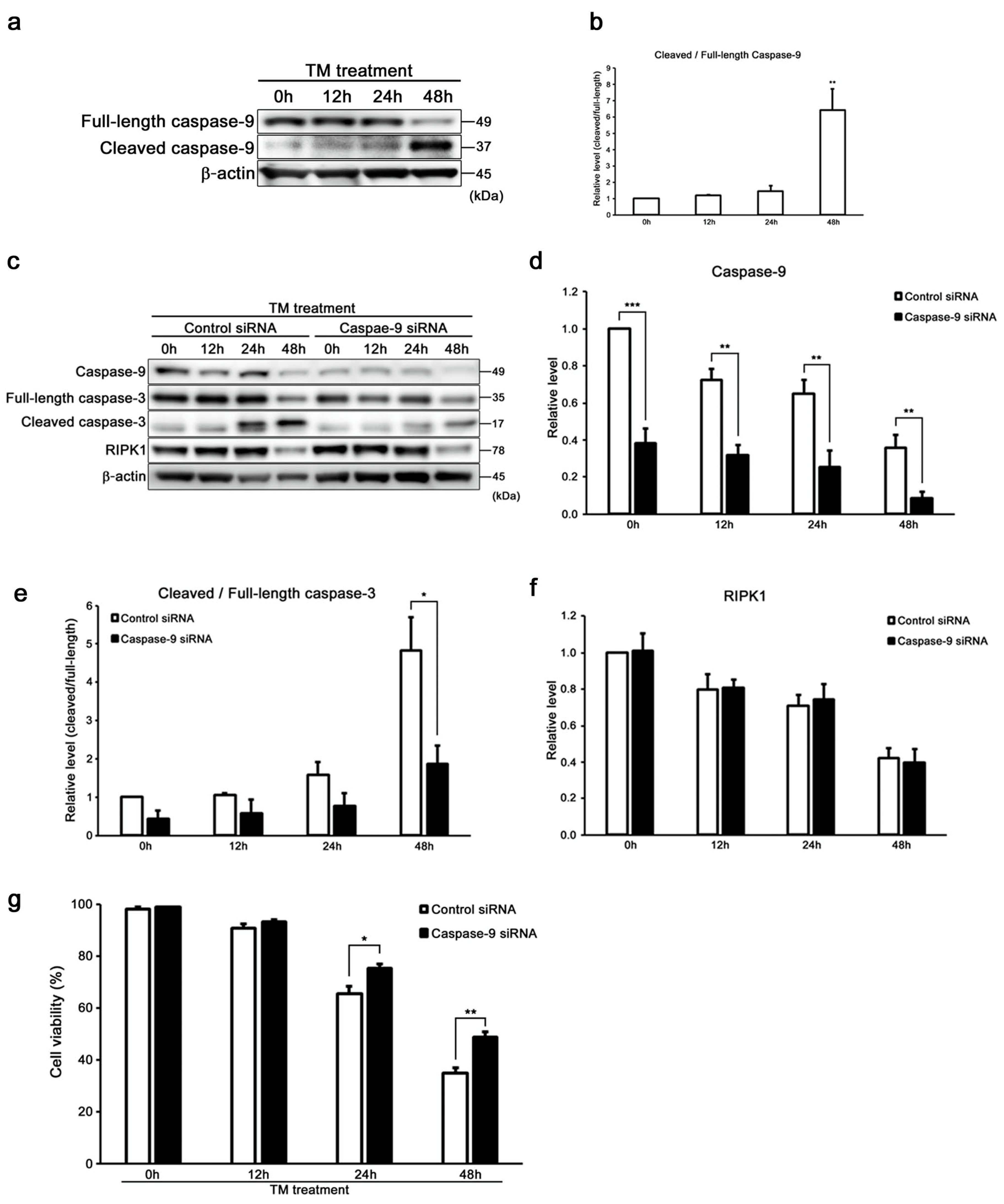
2.4. Caspase-9 Influences the Induction of Intrinsic Apoptosis with Caspase-3 in HEI-OC1 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture and Culture Condition
4.3. Cell Viability Assay
4.4. Western Blot Analysis
4.5. Co-Immunoprecipitation
4.6. Transmission Electron Microscopy
4.7. Flow Cytometry Analysis
4.8. Transient Small Interfering RNA (siRNA) Transfection
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schronder, M.; Kaufman, R.J. The mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Marniciniak, S.J.; Ron, D. Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef] [PubMed]
- Traves, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analysis reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Gorman, A.M.; Healy, S.J.; Jäger, R.; Samali, A. Stress management at the ER: Regulators of ER stress-induced apoptosis. Pharmacol. Ther. 2012, 134, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Scheper, W.; Nijholt, D.A.; Hoozemans, J.J. The unfolded protein response and proteostasis in Alzheimer disease: Preferential activation of autophagy by endoplasmic reticulum stress. Autophagy 2011, 7, 910–911. [Google Scholar] [CrossRef]
- Kato, T.; Kitamura, K.; Maeda, M.; Kimura, Y.; Katayama, T.; Ashida, H.; Yamamoto, K. Free oligosaccharides in the cytosol of Caenorhabditis elegans are generated through endoplasmic reticulum-golgi trafficking. J. Biol. Chem. 2007, 282, 22080–22088. [Google Scholar] [CrossRef]
- Duan, Q.; Ni, L.; Wang, P.; Chen, C.; Yang, L.; Ma, B.; Gong, W.; Cai, Z.; Zou, M.H.; Wang, D.W. Deregulation of XBP1 expression contributes to myocardial vascular endothelial growth factor-A expression and angiogenesis during cardiac hypertrophy in vivo. Aging Cell 2016, 15, 625–633. [Google Scholar] [CrossRef]
- Fujinami, Y.; Mutai, H.; Kamiya, K.; Mizutari, K.; Fujii, M.; Matsunaga, T. Enhanced expression of C/EBP homologous protein (CHOP) precedes degeneration of fibrocytes in the lateral wall after acute cochlear mitochondrial dysfunction induced by 3-nitropropionic acid. Neurochem. Int. 2010, 56, 487–494. [Google Scholar] [CrossRef]
- Fujinami, Y.; Mutai, H.; Mizutari, K.; Nakagawa, S.; Matsunaga, T. A novel animal model of hearing loss caused by acute endoplasmic reticulum stress in the cochlea. J. Pharmacol. Sci. 2012, 118, 363–372. [Google Scholar] [CrossRef]
- Oishi, N.; Duscha, S.; Boukari, H.; Meyer, M.; Xie, J.; Wei, G.; Schrepfer, T.; Roschitzki, B.; Boettger, E.C.; Schacht, J. XBP1 mitigates aminoglycoside-induced endoplasmic reticulum stress and neuronal cell death. Cell Death Dis. 2015, 6, e1763. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.G. Developmental cell death: Morphological diversity and multiple mechanisms. Anat. Embryol. 1990, 181, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Edinger, A.L.; Thompson, C.B. Death by design: Apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 2004, 16, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Bredesen, D.E.; Rao, R.V.; Mehlen, P. Cell death in the nervous system. Nature 2006, 443, 796–802. [Google Scholar] [CrossRef]
- Lockshin, R.A.; Zakeri, Z. Apoptosis, autophagy, and more. Int. J. Biochem. Cell Biol. 2004, 36, 2405–2419. [Google Scholar] [CrossRef]
- Xu, C.; Bailly-Maitre, B.; Reed, J.C. Endoplasmic reticulum stress: Cell life and death decisions. J. Clin. Investig. 2005, 115, 2656–2664. [Google Scholar] [CrossRef]
- Elbein, A.D. Inhibitors of the biosynthesis and processing of N-linked oligosaccharides. CRC Crit. Rev. Biochem. 1984, 16, 21–49. [Google Scholar] [CrossRef]
- Takatsuki, A.; Arima, K.; Tamura, G. Tunicamycin, a new antibiotic. I. Isolation and characterization of tunicamycin. J. Antibiot. 1971, 24, 215–223. [Google Scholar] [CrossRef]
- Tait, S.W.; Ichim, G.; Green, D.R. Die another way-non-apoptotic mechanisms of cell death. J. Cell Sci. 2014, 127, 2135–2144. [Google Scholar] [CrossRef]
- Yang, X.S.; Yi, T.L.; Zhang, S.; Xu, Z.W.; Yu, Z.Q.; Sun, H.T.; Yang, C.; Tu, Y.; Cheng, S.X. Hypoxia-inducible factor-1 alpha is involved in RIP-induced necroptosis caused by in vitro and in vivo ischemic brain injury. Sci. Rep. 2017, 7, 5818. [Google Scholar] [CrossRef]
- Challa, S.; Chan, F.K. Going up in flames: Necrotic cell injury and inflammatory diseases. Cell Mol. Life Sci. 2010, 67, 3241–3253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Tang, M.B.; Luo, H.Y.; Shi, C.H.; Xu, Y.M. Necroptosis in neurodegenerative diseases: A potential therapeutic target. Cell Death Dis. 2017, 8, e2905. [Google Scholar] [CrossRef]
- Cai, Z.; Jitkaew, S.; Zhao, J.; Chiang, H.C.; Choksi, S.; Liu, J.; Ward, Y.; Wu, L.G.; Liu, Z.G. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol. 2014, 16, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, W.; Ren, J.; Huang, D.; He, W.T.; Song, Y.; Yang, C.; Li, W.; Zheng, X.; Chen, P.; et al. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 2014, 24, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed Lineage Kinase Domain-like Protein MLKL Causes Necrotic Membrane Disruption upon Phosphorylation by RIP3. Mol. Cell. 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL Compromises Plasma Membrane Integrity by Binding to Phosphatidylinositol Phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef]
- Ofengeim, D.; Yuan, J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 2013, 14, 727–736. [Google Scholar] [CrossRef]
- Duprez, L.; Takahashi, N.; Van Hauwermeiren, F.; Vandendriessche, B.; Goossens, V.; Vanden Berghe, T.; Declercq, W.; Libert, C.; Cauwels, A.; Vandenabeele, P. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity 2011, 35, 908–918. [Google Scholar] [CrossRef]
- Tamai, K.; Toyoshima, M.; Tanaka, N.; Yamamoto, N.; Owada, Y.; Kiyonari, H.; Murata, K.; Ueno, Y.; Ono, M.; Shimosegawa, T.; et al. Loss of hrs in the central nervous system causes accumulation of ubiquitinated proteins and neurodegeneration. Am. J. Pathol. 2008, 173, 1806–1817. [Google Scholar] [CrossRef]
- Yamanaka, T.; Tosaki, A.; Kurosawa, M.; Akimoto, K.; Hirose, T.; Ohno, S.; Hattori, N.; Nukina, N. Loss of aPKCλ in differentiated neurons disrupts the polarity complex but does not induce obvious neuronal loss or disorientation in mouse brains. PLoS ONE 2013, 8, e84036. [Google Scholar] [CrossRef]
- Tamai, K.; Tanaka, N.; Nara, A.; Yamamoto, A.; Nakagawa, I.; Yoshimori, T.; Ueno, Y.; Shimosegawa, T.; Sugamura, K. Role of Hrs in maturation of autophagosomes in mammalian cells. Biochem. Biophys. Res. Commun. 2007, 360, 721–727. [Google Scholar] [CrossRef] [PubMed]
- Roudier, N.; Lefebvre, C.; Legouis, R. CeVPS-27 is an endosomal protein required for the molting and the endocytic trafficking of the low-density lipoprotein receptor-related protein 1 in Caenorhabditis elegans. Traffic 2005, 6, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Saveljeva, S.; Mc Laughlin, S.L.; Vandenabeele, P.; Samali, A.; Bertrand, M.J. Endoplasmic reticulum stress induces ligand-independent TNFR1-mediated necroptosis in L929 cells. Cell Death Dis. 2015, 6, e1587. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Wang, Z.; Ma, F.; Tran, B.; Zhong, R.; Xiong, Y.; Dai, T.; Wu, J.; Xin, X.; Guo, W.; et al. ZYZ-803 Mitigates Endoplasmic Reticulum Stress-Related Necroptosis after Acute Myocardial Infarction through Downregulating the RIP3-CaMKII Signaling Pathway. Oxid. Med. Cell. Longev. 2019, 2019, 6173685. [Google Scholar] [CrossRef] [PubMed]
- Salvesen, G.S.; Dixit, V.M. Caspases: Intracellular signaling by proteolysis. Cell 1997, 91, 443–446. [Google Scholar] [CrossRef] [Green Version]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [Green Version]
- Kishino, A.; Hayashi, K.; Hidai, C.; Masuda, T.; Nomura, Y.; Oshima, T. XBP1-FoxO1 interaction regulates ER stress-induced autophagy in auditory cells. Sci. Rep. 2017, 6, 24652. [Google Scholar] [CrossRef] [Green Version]
- Maeda, A.; Fadeel, B. Mitochondria released by cells undergoing TNF-α-induced necroptosis act as danger signals. Cell Death Dis. 2014, 5, e1312. [Google Scholar] [CrossRef] [Green Version]
- Tian, F.; Yao, J.; Yan, M.; Sun, X.; Wang, W.; Gao, W.; Tian, Z.; Guo, S.; Dong, Z.; Li, B.; et al. 5-Aminolevulinic Acid-Mediated Sonodynamic Therapy Inhibits RIPK1/RIPK3-Dependent Necroptosis in THP-1-Derived Foam Cells. Sci. Rep. 2016, 6, 21992. [Google Scholar] [CrossRef] [Green Version]
- Brentnall, M.; Rodriguez-Menocal, L.; De Guevara, R.L.; Cepero, E.; Boise, L.H. Caspase-9, caspase-3 and caspase-7 have distinct roles during intrinsic apoptosis. BMC Cell Biol. 2013, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Sun, Y.; Chen, S.; Zhou, X.; Wu, X.; Kong, W.; Kong, W. Impaired unfolded protein response in the degeneration of cochlea cells in a mouse model of age-related hearing loss. Exp. Gerontol. 2015, 60, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Zong, S.; Liu, T.; Wan, F.; Chen, P.; Luo, P.; Xiao, H. Endoplasmic Reticulum Stress Is Involved in Cochlear Cell Apoptosis in a Cisplatin-Induced Ototoxicity Rat Model. Audiol. Neurootol. 2017, 22, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, S.G.; Ishigaki, S.; Oslowski, C.M.; Lu, S.; Lipson, K.L.; Ghosh, R.; Hayashi, E.; Ishihara, H.; Oka, Y.; Permutt, M.A.; et al. Wolfram syndrome 1 gene negatively regulates ER stress signaling in rodent and human cells. J. Clin. Investig. 2010, 120, 744–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco-Sánchez, B.; Clément, A.; Fierro, J., Jr.; Washbourne, P.; Westerfield, M. Complexes of Usher proteins preassemble at the endoplasmic reticulum and are required for trafficking and ER homeostasis. Dis. Model. Mech. 2014, 7, 547–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshima, R.; Hasegawa, T.; Tamai, K.; Sugeno, N.; Yoshida, S.; Kobayashi, J.; Kikuchi, A.; Baba, T.; Futatsugi, A.; Sato, I.; et al. ESCRT-0 dysfunction cpmpromises autophagic degradation of protein aggregates and facilitates ET stress-mediated neurodegeneration via apoptotic and necroptotic pathways. Sci. Rep. 2016, 6, 24997. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Vercammen, D.; Beyaert, R.; Denecker, G.; Goossens, V.; Van Loo, G.; Declercq, W.; Grooten, J.; Fiers, W.; Vandenabeele, P. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 1998, 187, 1477–1485. [Google Scholar] [CrossRef] [Green Version]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The Release of Damage-Associated Molecular Patterns and Its Physiological Relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Ros, U.; Peña-Blanco, A.; Hänggi, K.; Kunzendorf, U.; Krautwald, S.; Wong, W.W.; García-Sáez, A.J. Necroptosis Execution Is Mediated by Plasma Membrane Nanopores Independent of Calcium. Cell Rep. 2017, 19, 175–187. [Google Scholar] [CrossRef] [Green Version]
- Fernandes-Alnemri, T.; Litwack, G.; Alnemri, E.S. CPP32, a novel human apoptotic protein with homology to Caenorhabditis elegans cell death protein Ced-3 and mammalian interleukin-1 beta-converting enzyme. J. Biol. Chem. 1994, 269, 30761–30764. [Google Scholar]
- Glab, J.A.; Doerflinger, M.; Nedeva, C.; Jose, I.; Mbogo, G.W.; Paton, J.C.; Paton, A.W.; Kueh, A.J.; Herold, M.J.; Huang, D.C.; et al. DR5 and caspase-8 are dispensable in ER stress-induced apoptosis. Cell Death Differ. 2017, 24, 944–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, M.J.; Kang, H.; Lee, Y.Y.; Choo, O.S.; Jang, J.H.; Park, S.H.; Moon, J.S.; Choi, S.J.; Choung, Y.H. Cisplatin-Induced Ototoxicity in Rats Is Driven by RIP3-Dependent Necroptosis. Cells 2019, 8, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhl, D.; Du, T.T.; Wagner, E.L.; Choi, J.H.; Li, S.; Reed, R.; Kim, K.; Freeman, M.; Hashisaki, G.; Lukens, J.R.; et al. Necroptosis and Apoptosis Contribute to Cisplatin and Aminoglycoside Ototoxicity. J. Neurosci. 2019, 39, 2951–2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Li, B.; Apisa, L.; Yu, H.; Entenman, S.; Xu, M.; Stepanyan, R.; Guan, B.J.; Müller, U.; Hatzoglou, M.; et al. ER stress inhibitor attenuates hearing loss and hair cell death in Cdh23erl/erl mutant mice. Cell Death Dis. 2016, 7, e2485. [Google Scholar] [CrossRef]
- Jia, Z.; He, Q.; Shan, C.; Li, F. Tauroursodeoxycholic acid attenuates gentamicin-induced cochlear hair cell death in vitro. Toxicol. Lett. 2018, 294, 20–26. [Google Scholar] [CrossRef]
- Fonseca, S.G.; Fukuma, M.; Lipson, K.L.; Nguyen, L.X.; Allen, J.R.; Oka, Y.; Urano, F. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J. Biol. Chem. 2005, 280, 39609–39615. [Google Scholar] [CrossRef] [Green Version]
- Kalinec, G.M.; Webster, P.; Lim, D.J.; Kalinec, F. A cochlear cell line as an in vitro system for drug ototoxicity screening. Audiol. Neurootol. 2003, 8, 177–189. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kishino, A.; Hayashi, K.; Maeda, M.; Jike, T.; Hidai, C.; Nomura, Y.; Oshima, T. Caspase-8 Regulates Endoplasmic Reticulum Stress-Induced Necroptosis Independent of the Apoptosis Pathway in Auditory Cells. Int. J. Mol. Sci. 2019, 20, 5896. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235896
Kishino A, Hayashi K, Maeda M, Jike T, Hidai C, Nomura Y, Oshima T. Caspase-8 Regulates Endoplasmic Reticulum Stress-Induced Necroptosis Independent of the Apoptosis Pathway in Auditory Cells. International Journal of Molecular Sciences. 2019; 20(23):5896. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235896
Chicago/Turabian StyleKishino, Akihiro, Ken Hayashi, Miyoko Maeda, Toyoharu Jike, Chiaki Hidai, Yasuyuki Nomura, and Takeshi Oshima. 2019. "Caspase-8 Regulates Endoplasmic Reticulum Stress-Induced Necroptosis Independent of the Apoptosis Pathway in Auditory Cells" International Journal of Molecular Sciences 20, no. 23: 5896. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235896