Cerium Oxide Nanoparticles Protect against Oxidant Injury and Interfere with Oxidative Mediated Kinase Signaling in Human-Derived Hepatocytes

 , , , , , ,
, , , , , ,
Abstract
:
1. Introduction
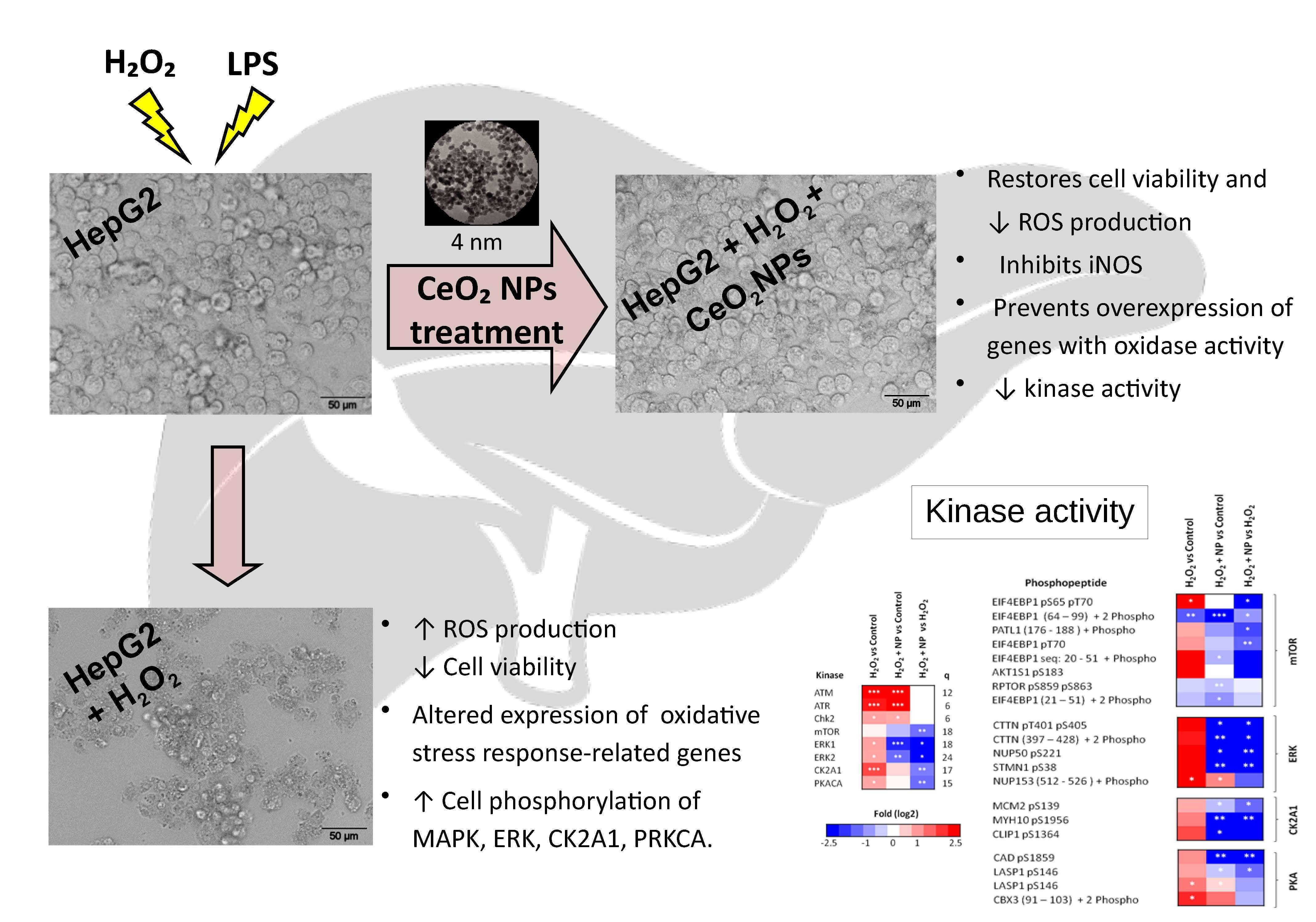
2. Results
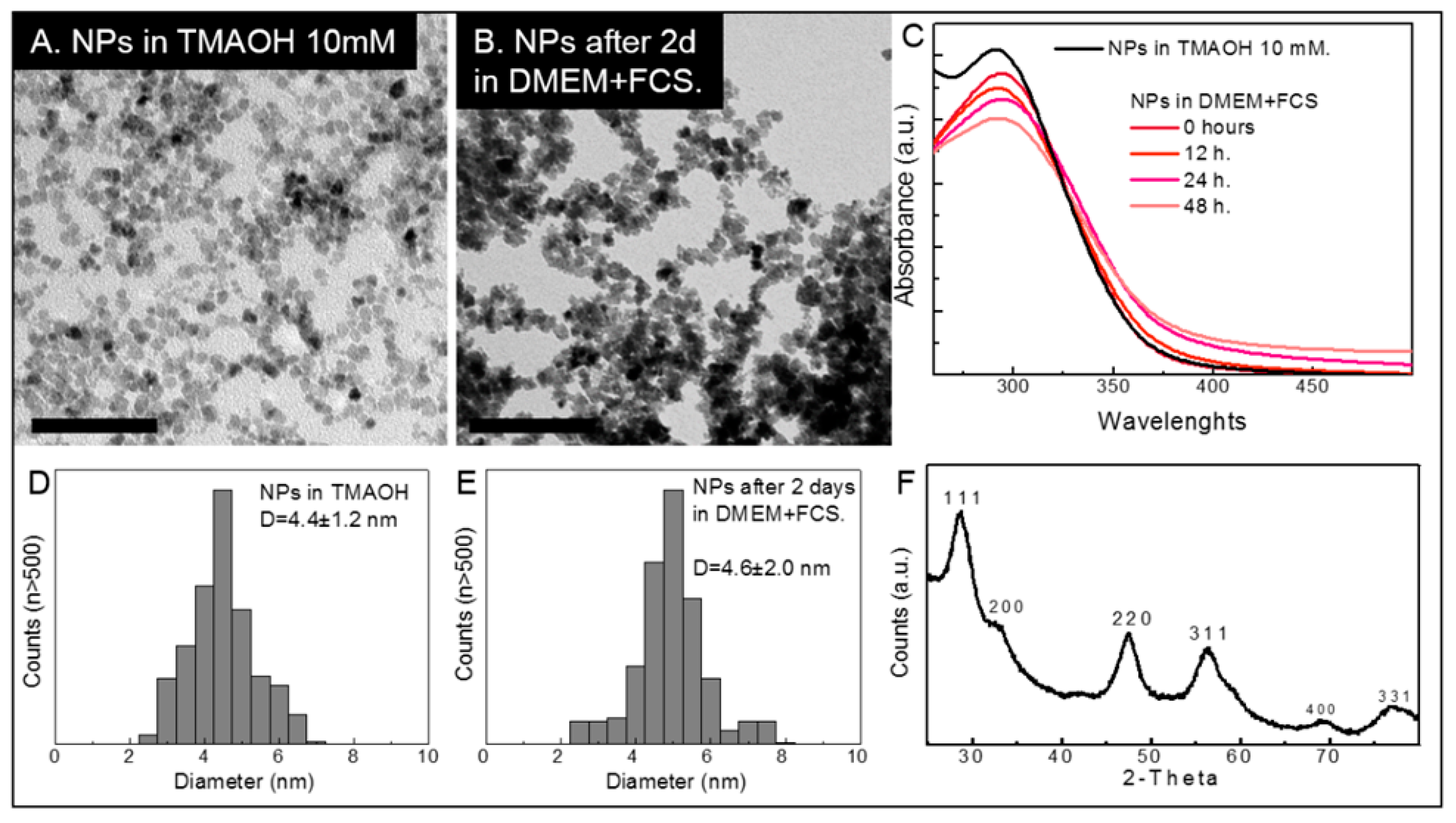
2.1. Cerium Oxide Nanoparticles Characterization
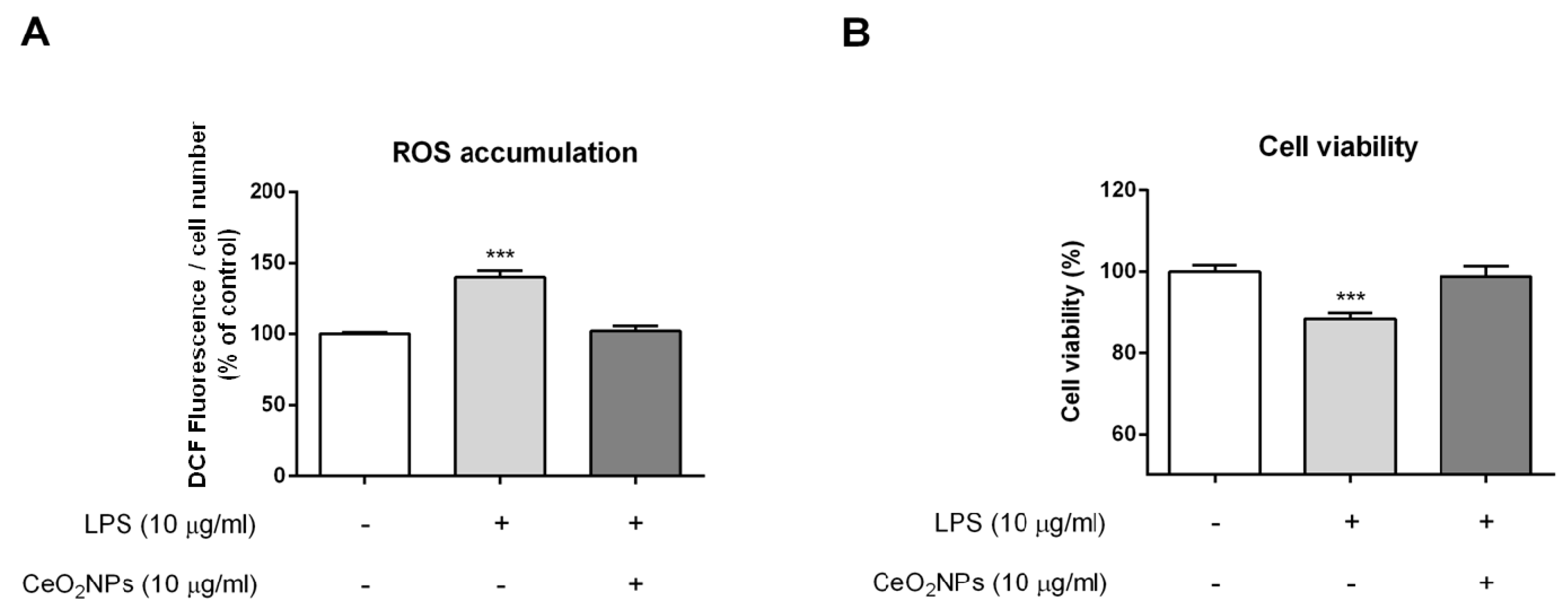
2.2. CeO2NPs Protect HepG2 Cells from H2O2- and LPS-induced Cytotoxicity
2.3. Expression Profile of Genes Related to Oxidative Stress in HepG2 Cells Exposed to H2O2
2.4. CeO2NPs Reduce H2O2-Induced Expression of iNOS in HepG2 cells
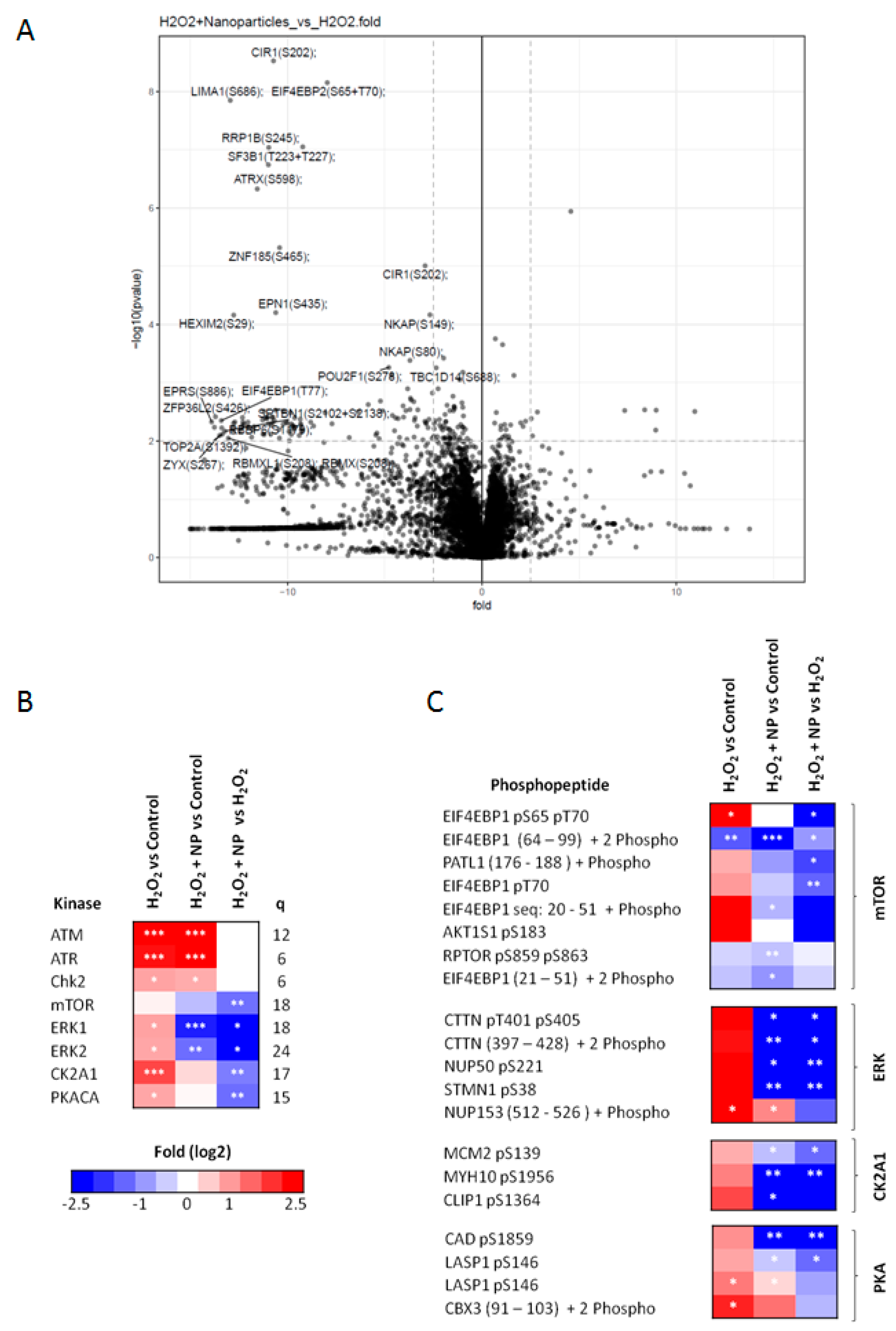
2.5. Identification of Signaling Networks Affected by Oxidative Stress in HepG2. Effect of CeO2NPs
3. Discussion
4. Material and Methods
4.1. Synthesis and Characterization of CeO2NPs
4.2. Cell Culture and Treatment
4.3. Oxidative Stress Gene Expression PCR Array in Cultured Cells
4.4. Statistical Analysis
4.5. Phosphoproteomic Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 2’,7’-DCF-DA | 2′,7′-dichlorofluorescin diacetate |
| CeO2NPs | Cerium Oxide Nanoparticles |
| DDT | Dithiothreitol |
| DMEM | Dulbecco’s Modified Eagle Medium |
| FCS | Fetal Calf Serum |
| HBSS | Hank’s Balanced Salt Solution |
| HCC | Hepatocellular Carcinoma |
| HCD | Higher Energy Collisional Dissociation |
| HR-TEM | High Resolution Transmission Electron Microscopy |
| KSEA | Kinase-Substrate Enrichment Analysis |
| LC-MS/MS | Liquid Chromatography Tandem Mass Spectrometry |
| LPS | Lipopolysaccharide |
| MS | Mass Spectrometry |
| MS/MS | Tandem mass spectrometry |
| NPs | Nanoparticles |
| PCR | Polymerase Chain Reaction |
| ROS | Radical Oxygen Species |
| RT-PCR | Real Time PCR |
| SOD | Superoxide Dismutase |
| UPLC | Ultrahigh Pressure Liquid Chromatography |
| UV-VIS | Ultraviolet-Visible |
| XRD | X-ray Diffraction |
References
- Laviña, B.; Gracia-Sancho, J.; Rodríguez-Vilarrupla, A.; Chu, Y.; Heistad, D.D.; Bosch, J.; García-Pagán, J.C. Superoxide dismutase gene transfer reduces portal pressure in CCl4 cirrhotic rats with portal hypertension. Gut 2009, 58, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhang, Y.; Li, Q.; Xu, M.; Bai, J.; Wu, S. Resveratrol improves alcoholic fatty liver disease by downregulating HIF-1α expression and mitochondrial ROS production. PLoS ONE 2017, 12, e0183426. [Google Scholar] [CrossRef] [PubMed]
- Rojkind, M.; Kershenobich, D. Effect of colchicine on collagen, albumin and transferrin synthesis by cirrhotic rat liver slices. Biochim. Biophys. Acta 1975, 378, 415–423. [Google Scholar] [CrossRef]
- Yogalakshmi, B.; Viswanathan, P.; Anuradha, C.V. Investigation of antioxidant, anti-inflammatory and DNA-protective properties of eugenol in thioacetamide-induced liver injury in rats. Toxicology 2010, 268, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Houglum, K.; Venkataramani, A.; Lyche, K.; Chojkier, M. A pilot study of the effects of d-alphatocopherol on hepatic stellate cell activation in chronic hepatitis C. Gastroenterology 1997, 113, 1069–1073. [Google Scholar] [CrossRef]
- Singal, A.K.; Jampana, S.C.; Weinman, S.A. Antioxidants as therapeutic agents for liver disease. Liver Int. 2011, 31, 1432–1448. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2017, 67, 328–357. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL–EASD–EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Wason, M.S.; Zhao, J. Cerium oxide nanoparticles: Potential applications for cancer and other diseases. Am. J. Transl Res. 2013, 5, 126–131. [Google Scholar]
- Heckert, E.G.; Karakoti, A.S.; Seal, S.; Self, W.T. The role of cerium redox state in the SOD mimetic activity of nanoceria. Biomaterials 2008, 29, 2705–2709. [Google Scholar] [CrossRef]
- Barkam, S.; Ortiz, J.; Saraf, S.; Eliason, N.; Mccormack, R.; Das, S.; Gupta, A.; Neal, C.; Petrovici, A.O.; Hanson, C.; et al. Modulating the Catalytic Activity of Cerium Oxide Nanoparticles with the Anion of the Precursor Salt. J. Phys. Chem. C 2017, 121, 20039–20050. [Google Scholar] [CrossRef]
- Oró, D.; Yudina, T.; Fernández-Varo, G.; Casals, E.; Reichenbach, V.; Casals, G.; de la Presa, B.G.; Sandalinas, S.; Carvajal, S.; Puntes, V.; et al. Cerium oxide nanoparticles reduce steatosis, portal hypertension and display anti-inflammatory properties in rats with liver fibrosis. J. Hepatol. 2016, 64, 691–698. [Google Scholar] [CrossRef]
- Carvajal, S.; Perramón, M.; Oró, D.; Casals, E.; Fernández-Varo, G.; Casals, G.; Parra, M.; de la Presa, B.G.; Ribera, J.; Pastor, Ó.; et al. Cerium oxide nanoparticles display antilipogenic effect in rats with non-alcoholic fatty liver disease. Sci. Rep. 2019, 9, 12848. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Qu, X. Cerium oxide nanoparticle: A remarkably versatile rare earth nanomaterial for biological applications. NPG Asia Mater. 2014, 6, e90. [Google Scholar] [CrossRef]
- Raza, H.; John, A.; Benedict, S. Acetylsalicylic acid-induced oxidative stress, cell cycle arrest, apoptosis and mitochondrial dysfunction in human hepatoma HepG2 cells. Eur. J. Pharm. 2011, 668, 15–24. [Google Scholar] [CrossRef]
- Melgar-Lesmes, P.; Pauta, M.; Reichenbach, V.; Casals, G.; Ros, J.; Bataller, R.; Morales-Ruiz, M.; Jiménez, W. Hypoxia and proinflammatory factors upregulate apelin receptor expression in human stellate cells and hepatocytes. Gut 2011, 60, 1404–1411. [Google Scholar] [CrossRef]
- Casals, E.; Pfaller, T.; Duschl, A.; Oostingh, G.J.; Puntes, V.F. Hardening of the nanoparticle-protein corona in metal (Au, Ag) and oxide (Fe3O4, CoO, and CeO2) nanoparticles. Small 2011, 7, 3479–3486. [Google Scholar] [CrossRef]
- Casado, P.; Rodriguez-Prados, J.C.; Cosulich, S.C.; Guichard, S.; Vanhaesebroeck, B.; Joel, S.; Cutillas, P.R. Kinase-Substrate Enrichment Analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal. 2012, 6, 268. [Google Scholar] [CrossRef]
- Angulo-Urarte, A.; Casado, P.; Castillo, S.D.; Kobialka, P.; Kotini, M.P.; Figueiredo, A.M.; Castel, P.; Rajeeve, V.; Milà-Guasch, M.; Millan, J.; et al. Endothelial cell rearrangements during vascular patterning require PI3-kinase-mediated inhibition of actomyosin contractility. Nat. Commun. 2018, 9, 4826–4842. [Google Scholar] [CrossRef]
- Niu, J.; Wang, K.; Kolattukudy, P.E. Cerium oxide nanoparticles inhibit oxidative stress and nuclear factor-κB activation in H9c2 cardiomyocytes exposed to cigarette smoke extract. J. Pharm. Exp. Ther. 2011, 338, 53–61. [Google Scholar] [CrossRef]
- Akhtar, M.J.; Ahamed, M.; Alhadlaq, H.A.; Khan, M.A.; Alrokayan, S.A. Glutathione replenishing potential of CeO2 nanoparticles in human breast and fibrosarcoma cells. J. Colloid Interface Sci. 2015, 453, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Hou, Y.; Cheng, G.; Zhang, C.; Wang, S.; Zhang, J. Cerium oxide nanoparticles protect endothelial cells from apoptosis induced by oxidative stress. Biol. Trace Elem. Res. 2013, 154, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Xia, T.; Kovochich, M.; Liong, M.; Mädler, L.; Gilbert, B.; Shi, H.; Yeh, J.I.; Zink, J.I.; Nel, A.E. Comparison of the mechanism of toxicity of zinc oxide and cerium oxide nanoparticles based on dissolution and oxidative stress properties. ACS Nano 2008, 2, 2121–2134. [Google Scholar] [CrossRef] [PubMed]
- Korsvik, C.; Patil, S.; Seal, S.; Self, W.T. Superoxide dismutase mimetic properties exhibited by vacancy engineered ceria nanoparticles. Chem. Commun. 2007, 1056–1058. [Google Scholar] [CrossRef] [PubMed]
- Pirmohamed, T.; Dowding, J.M.; Singh, S.; Wasserman, B.; Heckert, E.; Karakoti, A.S.; King, J.E.; Seal, S.; Self, W.T. Nanoceria exhibit redox state-dependent catalase mimetic activity. Chem. Commun. 2010, 46, 2736–2738. [Google Scholar] [CrossRef] [PubMed]
- Nelson, B.C.; Walker, M.L.; Riley, K.R.; Sims, C.M. Antioxidant Cerium Oxide Nanoparticles in Biology and Medicine. Antioxidants 2016, 5, 15. [Google Scholar] [CrossRef]
- Cichoz-Lach, H.; Michalak, A. Oxidative stress as a crucial factor in liver diseases. World J. Gastroenterol. 2014, 20, 8082–8091. [Google Scholar] [CrossRef]
- Zhao, G.; Yu, R.; Deng, J.; Zhao, Q.; Li, Y.; Joo, M.; van Breemen, R.B.; Christman, J.W.; Xiol, L. Pivotal role of reactive oxygen species in differential regulation of lipopolysaccharide-induced prostaglandins production in macrophages. Mol. Pharm. 2013, 83, 167–178. [Google Scholar] [CrossRef]
- Pflüger, P.; Viau, C.M.; Coelho, V.R.; Berwig, N.A.; Staub, R.B.; Pereira, P.; Saffi, J. Gamma-decanolactone inhibits iNOS and TNF-alpha production by lipopolysaccharide-activated microglia in N9 cells. Eur. J. Pharm. 2016, 780, 38–45. [Google Scholar] [CrossRef]
- Pulli, B.; Ali, M.; Iwamoto, Y.; Zeller, M.W.; Schob, S.; Linnoila, J.J.; Chen, J.W. Mieloperoxidase-hepatocyte-stellate cell cross talk promotes hepatocyte injury and fibrosis in experimental nonalcoholic steatohepatitis. Antioxid. Redox Signal. 2015, 23, 1255–1269. [Google Scholar] [CrossRef]
- Rensen, S.S.; Slaats, Y.; Nijhuis, J.; Jans, A.; Bieghs, V.; Driessen, A.; Malle, E.; Greve, J.W.; Buurman, W.A. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am. J. Pathol. 2009, 175, 1473–1482. [Google Scholar] [CrossRef] [PubMed]
- Langenbach, R.; Morham, S.G.; Tiano, H.F.; Loftin, C.D.; Ghanayem, B.I.; Chulada, P.C.; Mahler, J.F.; Lee, C.A.; Goulding, E.H.; Kluckman, K.D.; et al. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell 1995, 83, 483–492. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.S.; Bosetti, F. Cyclooxygenase-1 null mice show reduced neuroinflammation in response to β-amyloid. Aging 2009, 1, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Tyml, K.; Wilson, J.X. iNOS expression requires NADPH oxidase-dependent redox signaling in microvascular endothelial cells. J. Cell Physiol. 2008, 217, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawson, T.; Linding, R. Network medicine. FEBS Lett. 2008, 582, 1266–1270. [Google Scholar] [CrossRef] [Green Version]
- Linding, R.; Jensen, L.J.; Ostheimer, G.J.; Van Vugt, M.A.; Jørgensen, C.; Miron, M.I.; Diella, F.; Colwill, K.; Taylor, L.; Elder, K.; et al. Systematic discovery of in vivo phosphorylation networks. Cell 2007, 129, 1415–1426. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, D.; Gygi, S.P. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat. Biotechnol. 2005, 23, 1391–1398. [Google Scholar] [CrossRef]
- Kim, H.; Yoo, J.E.; Cho, J.Y.; Oh, B.K.; Yoon, Y.S.; Han, H.S.; Lee, H.S.; Jang, J.J.; Jeong, S.H.; Kim, J.W.; et al. Telomere length; TERT and shelterin complex proteins in hepatocellular carcinomas expressing “stemness”-related markers. J. Hepatol. 2013, 59, 746–752. [Google Scholar] [CrossRef]
- Oh, B.K.; Kim, Y.J.; Park, C.; Park, Y.N. Up-regulation of telomere-binding proteins; TRF1; TRF2; and TIN2 is related to telomere shortening during human multistep hepatocarcinogenesis. Am. J. Pathol. 2005, 166, 73–80. [Google Scholar] [CrossRef] [Green Version]
- Gu, D.L.; Chen, Y.H.; Shih, J.H.; Lin, C.H.; Jou, Y.S.; Chen, C.F. Target genes discovery through copy number alteration analysis in human hepatocellular carcinoma. World J. Gastroenterol. 2013, 19, 8873–8879. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wang, S.C.; Wei, Y.; Luo, X.; Jia, Y.; Li, L.; Gopal, P.; Zhu, M.; Nassour, I.; Chuang, J.C.; et al. Arid1a has context-dependent oncogenic and tumor suppressor functions in liver cancer. Cancer Cell 2017, 32, 574–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaiser, B.K.; Fernandes, T.F.; Jepson, M.A.; Lead, J.R.; Tyler, C.R.; Baalousha, M.; Biswas, A.; Britton, G.J.; Cole, P.A.; Johnston, B.D.; et al. Interspecies comparisons on the uptake and toxicity of silver and cerium dioxide nanoparticles. Environ. Toxicol. Chem. 2012, 31, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Singh, S. Redox-dependent catalase mimetic cerium oxide-based nanozyme protect human hepatic cells from 3-AT induced acatalasemia. Colloids Surf. B Biointerfaces 2019, 175, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Ni, D.; Wei, H.; Chen, W.; Bao, Q.; Rosenkrans, Z.T.; Barnhart, T.E.; Ferreira, C.A.; Wang, Y.; Yao, H.; Sun, T.; et al. Ceria Nanoparticles Meet Hepatic Ischemia-Reperfusion Injury: The Perfect Imperfection. Adv. Mater. 2019, 31, e1902956. [Google Scholar] [CrossRef]
- Reichenbach, V.; Ros, J.; Fernández-Varo, G.; Casals, G.; Melgar-Lesmes, P.; Campos, T.; Makriyannis, A.; Morales-Ruiz, M.; Jiménez, W. Prevention of fibrosis progression in CCl4-treated rats: Role of the hepatic endocannabinoid and apelin systems. J. Pharm. Exp. Ther. 2012, 340, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Melgar-Lesmes, P.; Casals, G.; Pauta, M.; Ros, J.; Reichenbach, V.; Bataller, R.; Morales-Ruiz, M.; Jimenez, W. Apelin mediates the induction of profibrogenic genes in human hepatic stellate cells. Endocrinology 2010, 151, 5306–5314. [Google Scholar] [CrossRef] [Green Version]
- Perkins, D.N.; Pappin, D.J.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence database using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Casado, P.; Cutillas, P.R. A self-validating quantitative mass spectrometry method for assessing the accuracy of high-content phosphoproteomic experiments. Mol. Cell. Proteom. 2011, 10, M110.003079. [Google Scholar] [CrossRef] [Green Version]
- Cutillas, P.R.; Venhaesebroeck, B. Quantitative profile of five murine core proteomes using label-free functional proteomics. Mol. Cell. Proteom. 2007, 6, 1560–1573. [Google Scholar] [CrossRef] [Green Version]
- Casado, P.; Bilanges, B.; Rajeeve, V.; Vanhaesebroeck, B.; Cutillas, P.R. Environmental stress affects the activity of metabolic and growth factor signaling networks and induces autophagy markers in MCF7 breast cancer cells. Mol. Cell. Proteom. 2014, 13, 836–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CeO2NPs (TEM = 4–5 nm) | ||
|---|---|---|
| Table Header | DLS (Z-average, nm) | Z-Pot (mV) |
| NPs after purification (in TMAOH 10 mM) | 33.0 | −47.0 |
| 0 d in cCCM | 70 | −35.4 |
| 1 d in cCCM | 70 | −36.4 |
| 2 d in cCCM | 72.9 | −35.0 |
| 15 d in cCCM | 79 | −16.9 |
| 30 d in cCCM | 100.2 | −9.6 |
| Genes | H2O2 (n = 5) | H2O2 + CeO2NPs (n = 6) |
|---|---|---|
| Antioxidants | ||
| CAT | −2.06 ** | −2.67 *** |
| GPX7 | −2.01 * | −2.25 *** |
| LPO | 3.54 | 1.72 |
| MPO | 5.32 * | 1.54 *† |
| TTN | 3.13 | 1.78 |
| PTGS1 | 3.69 * | 1.07 † |
| SOD3 | −3.28 ** | −6.30 *** |
| SRNX1 | 3.99 ** | 2.91 ** |
| TXNRD1 | 3.07 * | 2.16 |
| Genes involved in ROS metabolism | ||
| NCF1 | 2.59 * | 1.38 * |
| NCF2 | 1.11 | −1.89 † |
| UCP2 | −2.30 * | −4.04 *** |
| EPHX2 | −3.51 *** | −4.36 *** |
| Oxidative stress responsive genes | ||
| DCHR24 | −2.66 ** | −3.36 *** |
| DUSP1 | 8.30 * | 9.39 * |
| FOXM1 | −3.20 ** | −3.44 *** |
| GCLC | 3.29 *** | 3.42 *** |
| GCLM | 3.86 ** | 2.30 * |
| HMOX1 | 2.68 | 3.15 |
| HSPA1A | 5.28 * | 2.42 |
| MBL2 | −3.32 * | −2.6 ** |
| OXR1 | −2.08 *** | −2.64 *** |
| SCARA3 | −5.11 ** | −7.08 ** |
| SEPP1 | −4.19 *** | −4.41 *** |
| Oxygen transporters | ||
| MB | −5.88 * | −5.84 ** |
| Protein | Phosphopeptide | H2O2 vs Control. Fold | H2O2 + NPs vs H2O2 Fold |
|---|---|---|---|
| AT-rich interactive domain-containing protein 1A | ARID1A seq: 1182–1202 + Phospho (ST) | 3.0099 * | −2.727 * |
| Charged multivesicular body protein 2b | CHMP2B pS199 | 9.963 * | −9.964 * |
| C-Jun-amino-terminal kinase-interacting protein 4 | SPAG9 seq: 223–241 + Phospho (ST) | 10.117 * | −10.11 * |
| Deoxynucleotidyltransferase terminal-interacting protein 2 | DNTTIP2 pS141 | 10.135 * | −10.13 * |
| DNA replication licensing factor MCM3 | MCM3 seq: 696–724 + Gln- > pyro-Glu (N-term Q); Phospho (ST) | 8.390 * | −8.391 * |
| DNA-dependent protein kinase catalytic subunit | PRKDC seq: 3197–3232 + Phospho (ST) | 8.6913 * | −8.692 * |
| Double-stranded RNA-binding protein Staufen homolog 1 | STAU1 pS390 | 8.9250 * | −8.926 * |
| E3 ubiquitin-protein ligase UBR5 | UBR5 seq: 636–654 + Phospho (ST) | 10.128 * | −10.12 * |
| Eukaryotic translation initiation factor 4E-binding protein 1 | EIF4EBP1 pS65 pT70 | 10.841 * | −10.84 * |
| Kanadaptin | SLC4A1AP pS466 | 9.2633 * | −9.264 * |
| Kanadaptin | SLC4A1AP seq: 324–362 + Oxidation (M); Phospho (ST) | 9.164 * | −9.165 * |
| MKL/myocardin-like protein 2 | MKL2 seq: 535–562 + Phospho (ST) | 9.971 * | −9.973 * |
| Pericentriolar material 1 protein | PCM1 seq: 1923–1972 + Phospho (ST); Phospho (Y) | 10.677 * | −10.67 * |
| Prolyl 3-hydroxylase OGFOD1 | OGFOD1 seq: 381–427 + Phospho (ST) | 9.012 * | −9.013 * |
| Protein PRRC2A | PRRC2A seq: 1103–1128 + Gln- > pyro-Glu (N-term Q); Phospho (ST) | 9.2358 * | −9.236 * |
| R3H domain-containing protein 1 | R3HDM1 seq: 295–314 + Phospho (ST) | 8.592 * | −8.593 * |
| Ras-responsive element-binding protein 1 | RREB1 seq: 1636–1665 + Phospho (ST) | 9.296 * | −9.297 * |
| Stress-70 protein. mitochondrial | HSPA9 pM370 pM389 | 11.353 * | −11.35 * |
| Telomeric repeat-binding factor 2 | TERF2 seq: 404–447 + Phospho (ST) | 8.8833 * | −8.884 * |
| Transcriptional coactivator YAP1 | YAP1 seq: 162–181 + Gln- > pyro-Glu (N-term Q); Phospho (ST) | 3.3580 * | −5.372 * |
| Apoptotic chromatin condensation inducer in the nucleus | ACIN1 seq: 463–506 + 3 Phospho (ST) | 5.9139 * | −11.89 ** |
| Paxillin | PXN seq: 298–317 + Phospho (ST) | 1.8510 * | −2.535 ** |
| Poly(rC)-binding protein 1 | PCBP1 pM186 pS190 | 1.9716 * | −2.646 ** |
| Nascent polypeptide-associated complex subunit alpha | NACA pS2029 | 1.284 * | −3.119 ** |
| RNA-binding protein 25 | RBM25 pS703 | 11.804 * | −4.246 ** |
| Phosphoribosyl pyrophosphate synthase-associated protein 1 | PRPSAP1 seq: 193–220 + Oxidation (M); Phospho (ST) | 12.440 ** | −4.758 * |
| Serine/arginine repetitive matrix protein 2 | SRRM2 seq: 2275–2301 + Phospho (ST) | 9.8054 ** | −9.80 * |
| Cell division cycle protein 23 homolog | CDC23 pT562 | 8.7452 ** | −8.746 ** |
| Centrosomal protein of 131 kDa | CEP131 seq: 45–56 + Phospho (ST) | 9.3197 ** | −9.320 ** |
| Deoxynucleotidyltransferase terminal-interacting protein 2 | DNTTIP2 pS381 | 9.412 ** | −9.41 ** |
| Nuclear receptor coactivator 2 | NCOA2 pS771 | 2.5932 ** | −2.630 ** |
| Oxysterol-binding protein 1 | OSBP seq: 377–395 + 2 Phospho (ST) | 2.8700 ** | −2.558 ** |
| RNA-binding motif protein. X chromosome | RBMX pS208 | 9.8237 ** | −9.824 ** |
| Transcription factor Sp5 | SP5 seq: 43–68 + Phospho (ST) | 4.5004 ** | −10.69 ** |
| Uncharacterized protein C6orf106 | C6orf106 seq: 264–287 + Phospho (ST) | 8.2914 ** | −2.875 ** |
| Epsin-1 | EPN1 seq: 412–445 + Phospho (ST) | 13.834 ** | −4.391 *** |
| Pinin | PNN pS66 | 8.894 *** | −4.875 * |
| Protein LYRIC | MTDH pS298 | 8.390 *** | −8.391 ** |
| POU domain. class 2. transcription factor 1 | POU2F1 seq: 273–293 + Phospho (ST) | 8.691 *** | −8.69 *** |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carvajal, S.; Perramón, M.; Casals, G.; Oró, D.; Ribera, J.; Morales-Ruiz, M.; Casals, E.; Casado, P.; Melgar-Lesmes, P.; Fernández-Varo, G.; et al. Cerium Oxide Nanoparticles Protect against Oxidant Injury and Interfere with Oxidative Mediated Kinase Signaling in Human-Derived Hepatocytes. Int. J. Mol. Sci. 2019, 20, 5959. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235959
Carvajal S, Perramón M, Casals G, Oró D, Ribera J, Morales-Ruiz M, Casals E, Casado P, Melgar-Lesmes P, Fernández-Varo G, et al. Cerium Oxide Nanoparticles Protect against Oxidant Injury and Interfere with Oxidative Mediated Kinase Signaling in Human-Derived Hepatocytes. International Journal of Molecular Sciences. 2019; 20(23):5959. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235959
Chicago/Turabian StyleCarvajal, Silvia, Meritxell Perramón, Gregori Casals, Denise Oró, Jordi Ribera, Manuel Morales-Ruiz, Eudald Casals, Pedro Casado, Pedro Melgar-Lesmes, Guillermo Fernández-Varo, and et al. 2019. "Cerium Oxide Nanoparticles Protect against Oxidant Injury and Interfere with Oxidative Mediated Kinase Signaling in Human-Derived Hepatocytes" International Journal of Molecular Sciences 20, no. 23: 5959. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20235959