Interplay between MicroRNAs and Oxidative Stress in Neurodegenerative Diseases

 ,
, 
Abstract
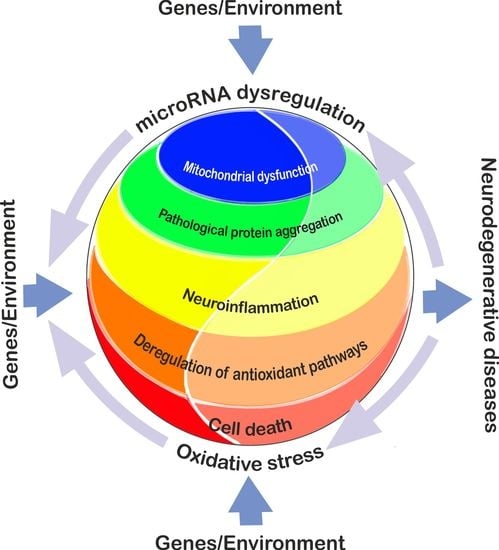
:
1. Introduction
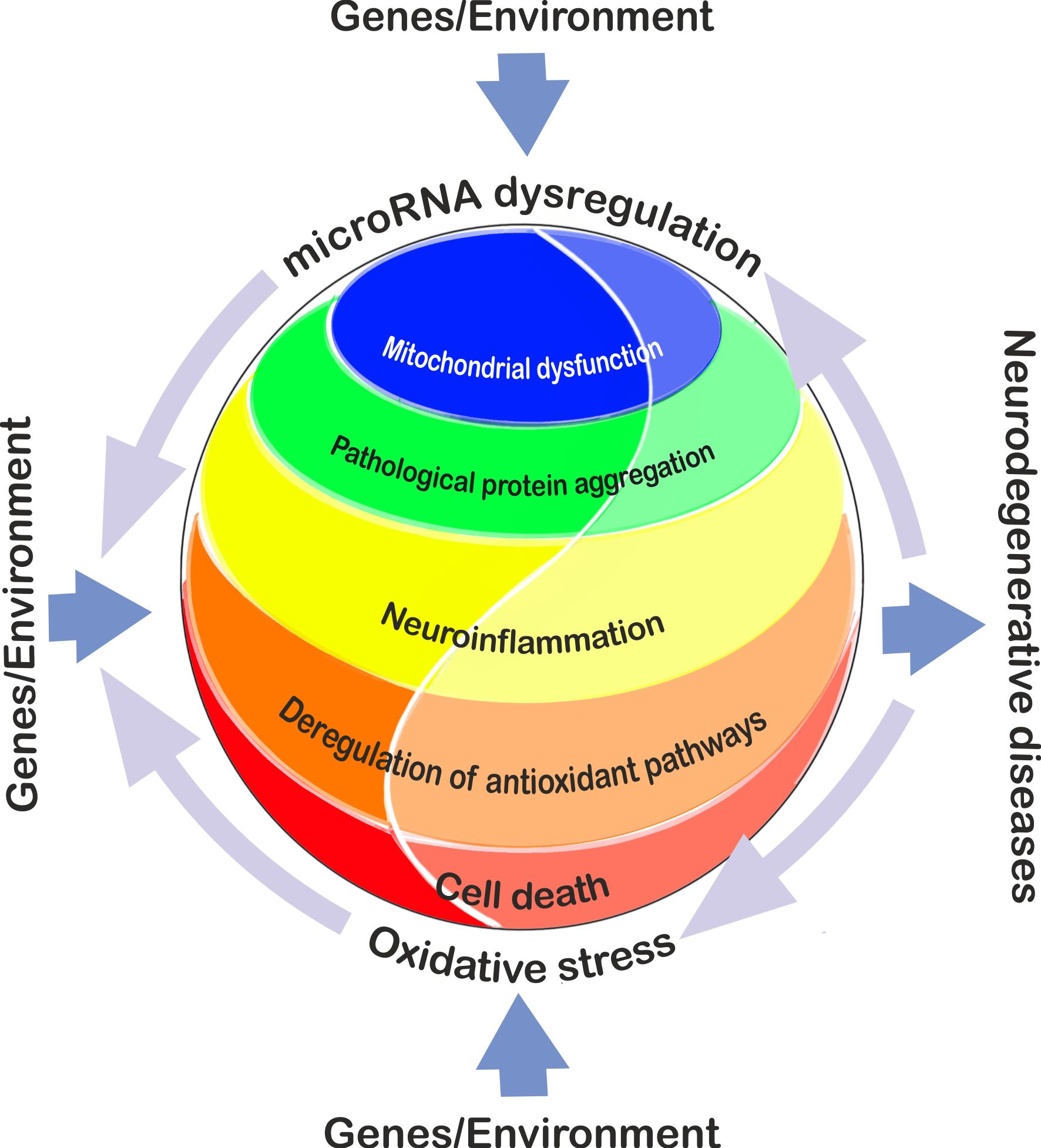
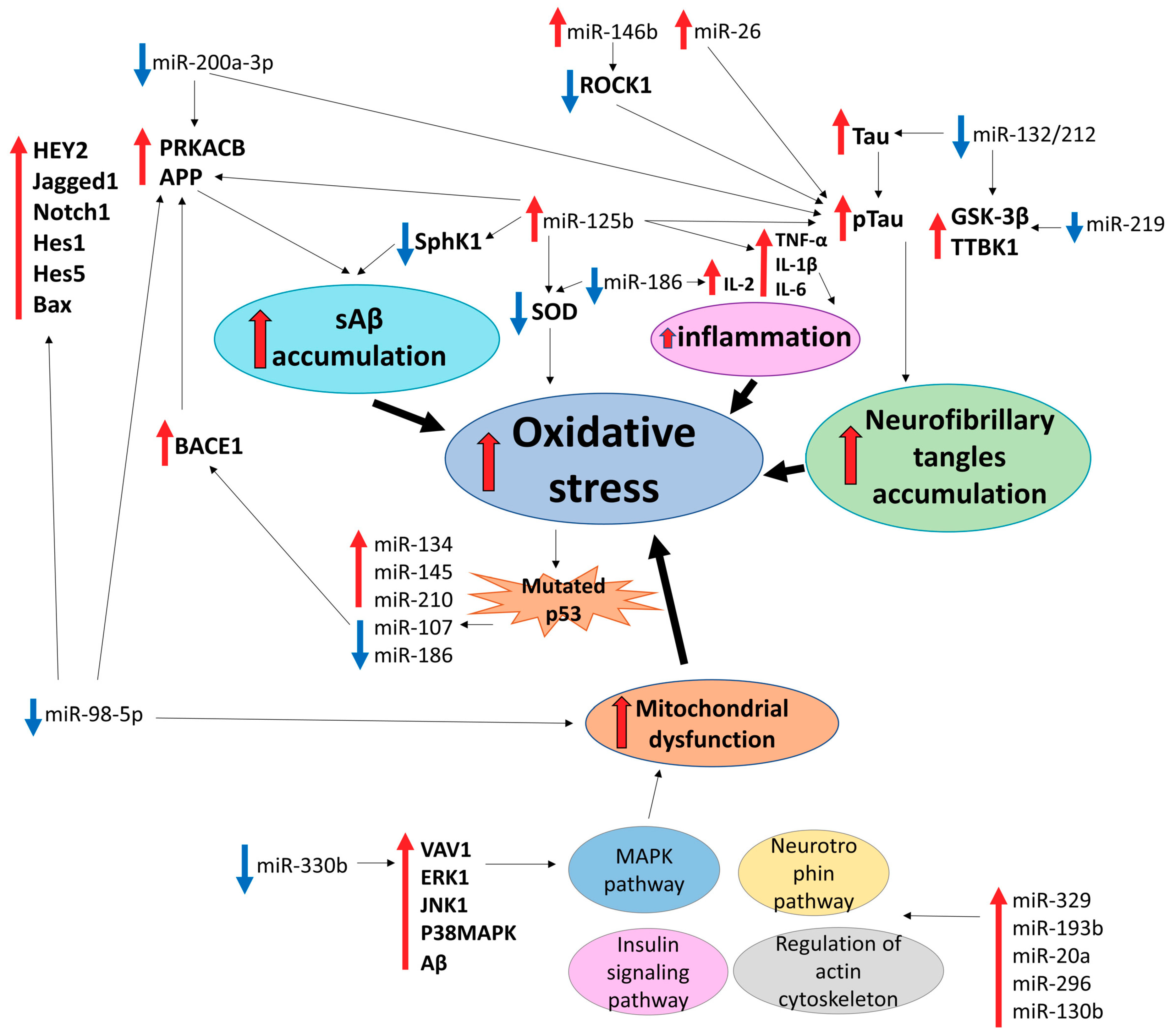
2. Alzheimer’s Disease
3. Parkinson’s Disease
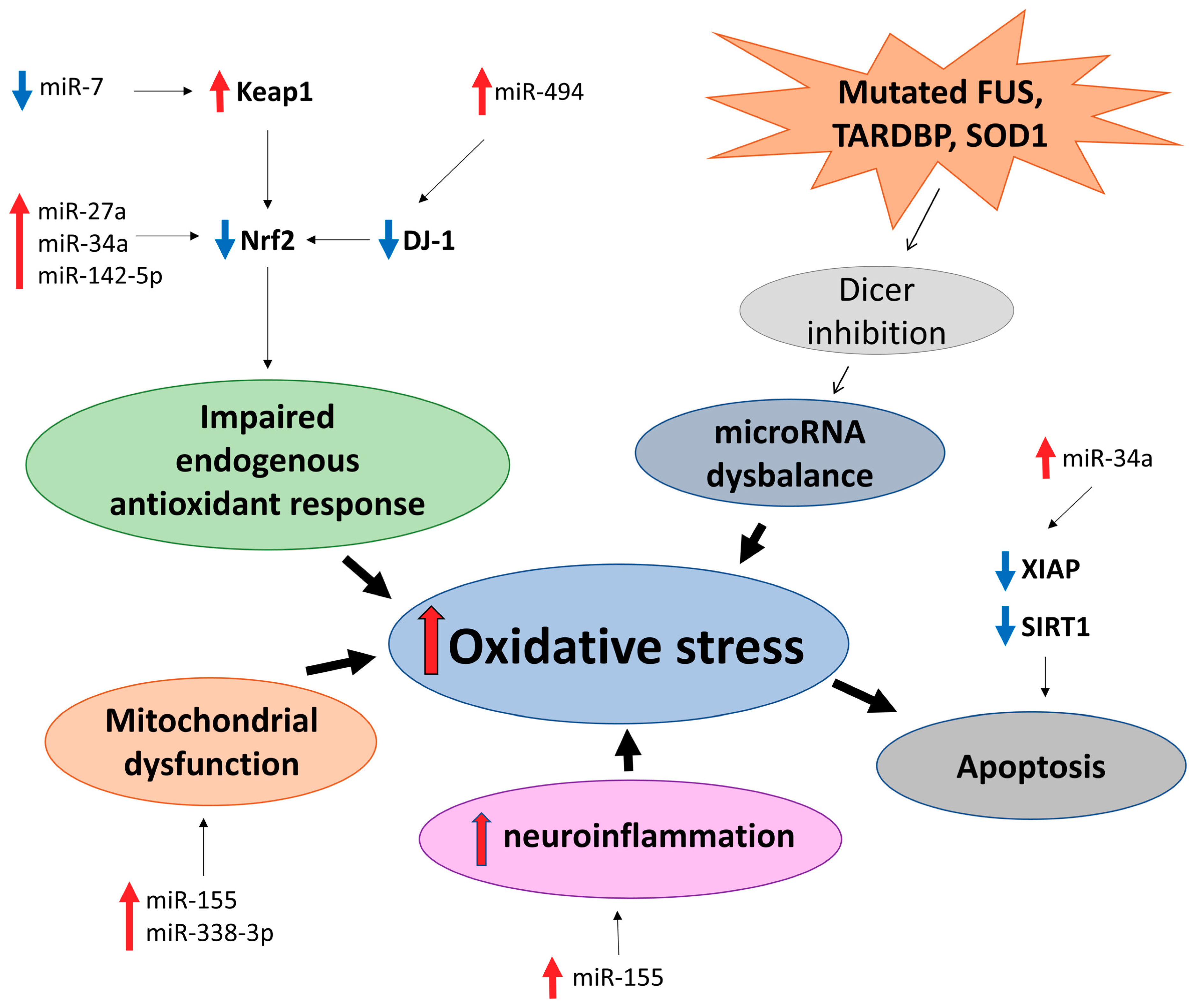
4. Amyotrophic Lateral Sclerosis
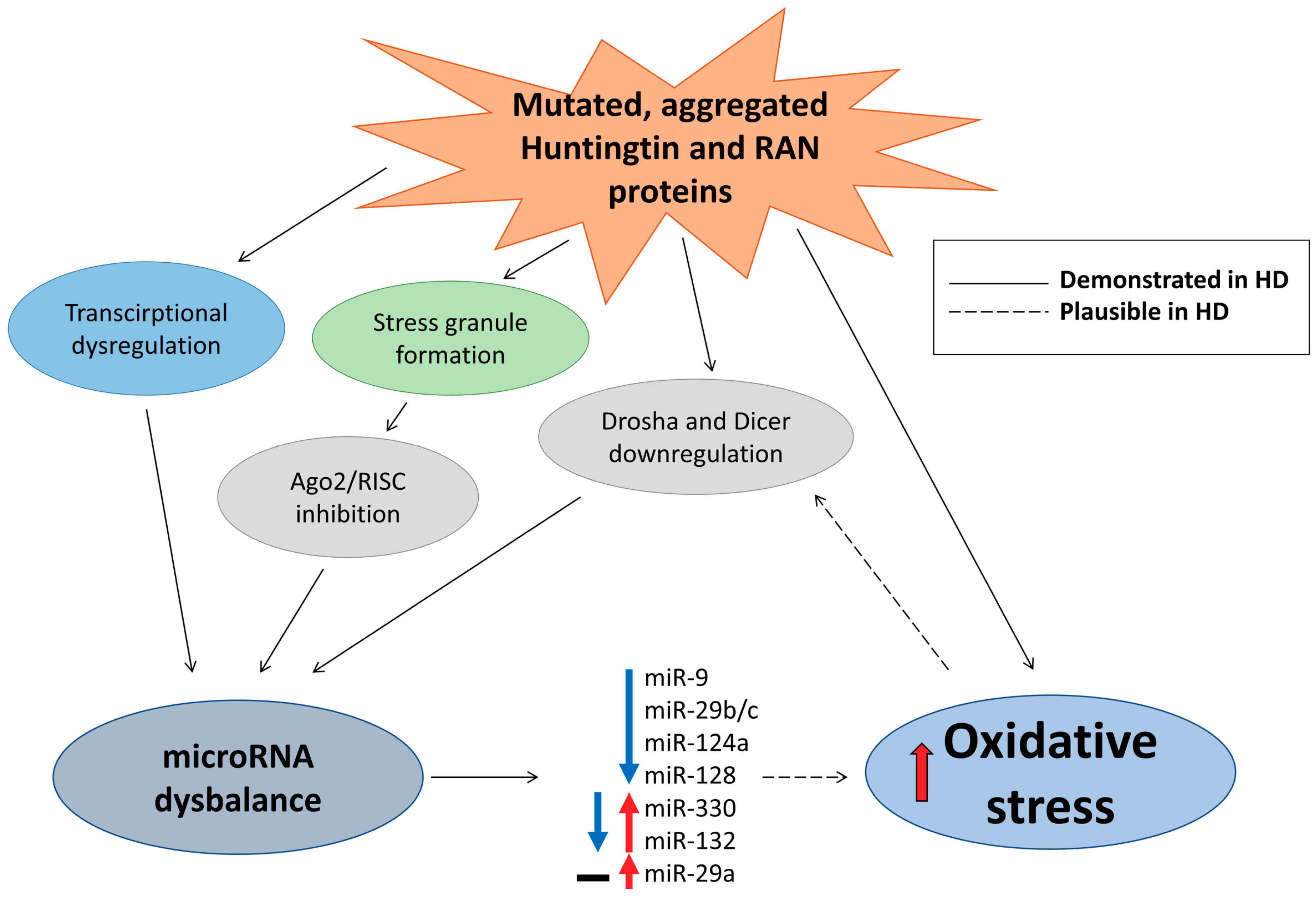
5. Huntington’s Disease
6. Common and Unique MicroRNAs Affecting Oxidative Stress in Neurodegenerative Diseases
7. Challenges and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Aβ | Amyloid-β |
| AD | Alzheimer’s disease |
| ALS | Amyotrophic Lateral Sclerosis |
| APP | Amyloid precursor protein |
| ARE | Antioxidant response element |
| ATP | Adenosine triphosphate |
| BACE1 | Beta-secretase 1 |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| DAT | Dopamine transporter |
| GSH | Glutathione |
| HD | Huntington’s disease |
| mHTT | mutant Huntingtin |
| PD | Parkinson’s disease |
| PSEN | Presenilin |
| RAN | Repeat associated non-ATG |
| REST | Repressor Element 1 Silencing Transcription Factor |
| RISC | RNA-induced silencing complex |
| ROS | Reactive oxygen species |
| sAβ | soluble amyloid-β |
| SNpc | Substantia nigra pars compacta |
| SOD | Superoxide dismutase |
| VMAT2 | Vesicular monoamine transporter 2 |
| UTR | Untranslated region |
References
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Collaborators, G.B.D.N. Global, regional, and national burden of neurological disorders, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Trigo-Damas, I.; Quiroga-Varela, A.; Jackson-Lewis, V.R. Oxidative stress and Parkinson’s disease. Front. Neuroanat. 2015, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Tonnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Kumar, A.; Ratan, R.R. Oxidative stress and Huntington’s disease: The good, the bad, and the ugly. J. Huntingtons Dis. 2016, 5, 217–237. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative stress in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, C.; Giguere, N.; Bourque, M.J.; Levesque, M.; Slack, R.S.; Trudeau, L.E. Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative stress in neurodegenerative diseases: From molecular mechanisms to clinical applications. Oxid. Med. Cell Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Kim, J.E.; Rhie, S.J.; Yoon, S. The role of oxidative stress in neurodegenerative diseases. Exp. Neurobiol. 2015, 24, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. Antioxidants as therapies: Can we improve on nature? Free Radic. Biol. Med. 2014, 66, 20–23. [Google Scholar] [CrossRef]
- Vasconcelos, A.R.; Dos Santos, N.B.; Scavone, C.; Munhoz, C.D. Nrf2/ARE pathway modulation by dietary energy regulation in neurological disorders. Front. Pharmacol. 2019, 10, 33. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef]
- Schmiedel, J.M.; Klemm, S.L.; Zheng, Y.; Sahay, A.; Bluthgen, N.; Marks, D.S.; van Oudenaarden, A. Gene expression. MicroRNA control of protein expression noise. Science 2015, 348, 128–132. [Google Scholar] [CrossRef]
- Rajman, M.; Schratt, G. MicroRNAs in neural development: From master regulators to fine-tuners. Development 2017, 144, 2310–2322. [Google Scholar] [CrossRef] [PubMed]
- Im, H.I.; Kenny, P.J. MicroRNAs in neuronal function and dysfunction. Trends Neurosci. 2012, 35, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, E.; Kim, S.Y.; Carmell, M.A.; Murchison, E.P.; Alcorn, H.; Li, M.Z.; Mills, A.A.; Elledge, S.J.; Anderson, K.V.; Hannon, G.J. Dicer is essential for mouse development. Nat. Genet. 2003, 35, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Horii, T.; Kimura, M.; Goto, Y.; Ochiya, T.; Hatada, I. One Argonaute family member, Eif2c2 (Ago2), is essential for development and appears not to be involved in DNA methylation. Genomics 2007, 89, 687–696. [Google Scholar] [CrossRef]
- Kawase-Koga, Y.; Otaegi, G.; Sun, T. Different timings of Dicer deletion affect neurogenesis and gliogenesis in the developing mouse central nervous system. Dev. Dyn. 2009, 238, 2800–2812. [Google Scholar] [CrossRef]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef]
- Huang, T.; Liu, Y.; Huang, M.; Zhao, X.; Cheng, L. Wnt1-cre-mediated conditional loss of Dicer results in malformation of the midbrain and cerebellum and failure of neural crest and dopaminergic differentiation in mice. J. Mol. Cell Biol. 2010, 2, 152–163. [Google Scholar] [CrossRef]
- Davis, T.H.; Cuellar, T.L.; Koch, S.M.; Barker, A.J.; Harfe, B.D.; McManus, M.T.; Ullian, E.M. Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J. Neurosci. 2008, 28, 4322–4330. [Google Scholar] [CrossRef]
- De Pietri Tonelli, D.; Pulvers, J.N.; Haffner, C.; Murchison, E.P.; Hannon, G.J.; Huttner, W.B. miRNAs are essential for survival and differentiation of newborn neurons but not for expansion of neural progenitors during early neurogenesis in the mouse embryonic neocortex. Development 2008, 135, 3911–3921. [Google Scholar] [CrossRef]
- Chmielarz, P.; Konovalova, J.; Najam, S.S.; Alter, H.; Piepponen, T.P.; Erfle, H.; Sonntag, K.C.; Schutz, G.; Vinnikov, I.A.; Domanskyi, A. Dicer and microRNAs protect adult dopamine neurons. Cell Death Dis. 2017, 8, e2813. [Google Scholar] [CrossRef]
- Kaneko, H.; Dridi, S.; Tarallo, V.; Gelfand, B.D.; Fowler, B.J.; Cho, W.G.; Kleinman, M.E.; Ponicsan, S.L.; Hauswirth, W.W.; Chiodo, V.A.; et al. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature 2011, 471, 325–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, X.; Hogan, E.M.; Casserly, A.; Gao, G.; Gardner, P.D.; Tapper, A.R. Dicer expression is essential for adult midbrain dopaminergic neuron maintenance and survival. Mol. Cell Neurosci. 2014, 58, 22–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hebert, S.S.; Papadopoulou, A.S.; Smith, P.; Galas, M.C.; Planel, E.; Silahtaroglu, A.N.; Sergeant, N.; Buee, L.; De Strooper, B. Genetic ablation of Dicer in adult forebrain neurons results in abnormal tau hyperphosphorylation and neurodegeneration. Hum. Mol. Genet. 2010, 19, 3959–3969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, A.; O’Carroll, D.; Tan, C.L.; Hillman, D.; Sugimori, M.; Llinas, R.; Greengard, P. Cerebellar neurodegeneration in the absence of microRNAs. J. Exp. Med. 2007, 204, 1553–1558. [Google Scholar] [CrossRef] [Green Version]
- Konopka, W.; Kiryk, A.; Novak, M.; Herwerth, M.; Parkitna, J.R.; Wawrzyniak, M.; Kowarsch, A.; Michaluk, P.; Dzwonek, J.; Arnsperger, T.; et al. MicroRNA loss enhances learning and memory in mice. J. Neurosci. 2010, 30, 14835–14842. [Google Scholar] [CrossRef]
- Vinnikov, I.A.; Hajdukiewicz, K.; Reymann, J.; Beneke, J.; Czajkowski, R.; Roth, L.C.; Novak, M.; Roller, A.; Dörner, N.; Starkuviene, V.; et al. Hypothalamic miR-103 protects from hyperphagic obesity in mice. J. Neurosci. 2014, 34, 10659–10674. [Google Scholar] [CrossRef] [Green Version]
- Cuellar, T.L.; Davis, T.H.; Nelson, P.T.; Loeb, G.B.; Harfe, B.D.; Ullian, E.; McManus, M.T. Dicer loss in striatal neurons produces behavioral and neuroanatomical phenotypes in the absence of neurodegeneration. Proc. Natl. Acad. Sci. USA 2008, 105, 5614–5619. [Google Scholar] [CrossRef] [Green Version]
- Mang, G.M.; Pradervand, S.; Du, N.H.; Arpat, A.B.; Preitner, F.; Wigger, L.; Gatfield, D.; Franken, P. A neuron-specific deletion of the microRNA-processing enzyme DICER induces severe but transient obesity in mice. PLoS ONE 2015, 10, e0116760. [Google Scholar] [CrossRef]
- Schaefer, A.; Im, H.I.; Veno, M.T.; Fowler, C.D.; Min, A.; Intrator, A.; Kjems, J.; Kenny, P.J.; O’Carroll, D.; Greengard, P. Argonaute 2 in dopamine 2 receptor-expressing neurons regulates cocaine addiction. J. Exp. Med. 2010, 207, 1843–1851. [Google Scholar] [CrossRef] [Green Version]
- Schratt, G. microRNAs at the synapse. Nat. Rev. Neurosci 2009, 10, 842–849. [Google Scholar] [CrossRef]
- Antoniou, A.; Khudayberdiev, S.; Idziak, A.; Bicker, S.; Jacob, R.; Schratt, G. The dynamic recruitment of TRBP to neuronal membranes mediates dendritogenesis during development. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Thomas, K.T.; Gross, C.; Bassell, G.J. microRNAs Sculpt Neuronal Communication in a Tight Balance That Is Lost in Neurological Disease. Front. Mol. Neurosci. 2018, 11, 455. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Nicotera, P. MicroRNAs in age-related diseases. EMBO Mol. Med. 2013, 5, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Eacker, S.M.; Dawson, T.M.; Dawson, V.L. Understanding microRNAs in neurodegeneration. Nat. Rev. Neurosci. 2009, 10, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Eacker, S.M.; Dawson, T.M.; Dawson, V.L. The interplay of microRNA and neuronal activity in health and disease. Front. Cell Neurosci. 2013, 7, 136. [Google Scholar] [CrossRef] [Green Version]
- Heman-Ackah, S.M.; Hallegger, M.; Rao, M.S.; Wood, M.J. RISC in PD: The impact of microRNAs in Parkinson’s disease cellular and molecular pathogenesis. Front. Mol. Neurosci. 2013, 6, 40. [Google Scholar] [CrossRef] [Green Version]
- Mouradian, M.M. MicroRNAs in Parkinson’s disease. Neurobiol. Dis. 2012, 46, 279–284. [Google Scholar] [CrossRef]
- Sonntag, K.C. MicroRNAs and deregulated gene expression networks in neurodegeneration. Brain Res. 2010, 1338, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Kurzynska-Kokorniak, A.; Koralewska, N.; Pokornowska, M.; Urbanowicz, A.; Tworak, A.; Mickiewicz, A.; Figlerowicz, M. The many faces of Dicer: The complexity of the mechanisms regulating Dicer gene expression and enzyme activities. Nucleic Acids Res. 2015, 43, 4365–4380. [Google Scholar] [CrossRef]
- Leggio, L.; Vivarelli, S.; L’Episcopo, F.; Tirolo, C.; Caniglia, S.; Testa, N.; Marchetti, B.; Iraci, N. microRNAs in Parkinson’s disease: From pathogenesis to novel diagnostic and therapeutic approaches. Int. J. Mol. Sci. 2017, 18, 2698. [Google Scholar] [CrossRef] [Green Version]
- Emde, A.; Hornstein, E. miRNAs at the interface of cellular stress and disease. EMBO J. 2014, 33, 1428–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emde, A.; Eitan, C.; Liou, L.L.; Libby, R.T.; Rivkin, N.; Magen, I.; Reichenstein, I.; Oppenheim, H.; Eilam, R.; Silvestroni, A.; et al. Dysregulated miRNA biogenesis downstream of cellular stress and ALS-causing mutations: A new mechanism for ALS. EMBO J. 2015, 34, 2633–2651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engedal, N.; Zerovnik, E.; Rudov, A.; Galli, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Monsurro, V.; Betti, M.; Albertini, M.C. From oxidative stress damage to pathways, networks, and autophagy via MicroRNAs. Oxid. Med. Cell Longev. 2018, 2018, 4968321. [Google Scholar] [CrossRef] [PubMed]
- Vinnikov, I.A.; Domanskyi, A. Can we treat neurodegenerative diseases by preventing an age-related decline in microRNA expression? Neural Regen. Res. 2017, 12, 1602–1604. [Google Scholar] [CrossRef]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Sumazin, P.; Yang, X.; Chiu, H.S.; Chung, W.J.; Iyer, A.; Llobet-Navas, D.; Rajbhandari, P.; Bansal, M.; Guarnieri, P.; Silva, J.; et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell 2011, 147, 370–381. [Google Scholar] [CrossRef] [Green Version]
- Prince, M.; Ali, G.-C.; Guerchet, M.; Prina, A.M.; Albanese, E.; Wu, Y.-T. Recent global trends in the prevalence and incidence of dementia, and survival with dementia. Alzheimer’s Res. Ther. 2016, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Tramutola, A.; Lanzillotta, C.; Perluigi, M.; Butterfield, D.A. Oxidative stress, protein modification and Alzheimer disease. Brain Res. Bull. 2017, 133, 88–96. [Google Scholar] [CrossRef]
- Van Cauwenberghe, C.; Van Broeckhoven, C.; Sleegers, K. The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genetics Med. 2015, 18, 421. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; De Ronchi, D.; Fratiglioni, L. The epidemiology of the dementias: An update. Curr. Opin. Psychiatry 2007, 20, 380–385. [Google Scholar] [CrossRef]
- Prasad, K.N. Oxidative stress and pro-inflammatory cytokines may act as one of the signals for regulating microRNAs expression in Alzheimer’s disease. Mech. Ageing Dev. 2017, 162, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Lanoiselée, H.-M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.-C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 mutations in early-onset Alzheimer disease: A genetic screening study of familial and sporadic cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, N.; Hoang, D.; Miller, N.; Ansaloni, S.; Huang, Q.; Rogers, J.T.; Lee, J.C.; Saunders, A.J. MicroRNAs can regulate human APP levels. Mol. Neurodegener. 2008, 3, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Bergmans, B.; Papadopoulou, A.S.; Delacourte, A.; De Strooper, B. MicroRNA regulation of Alzheimer’s Amyloid precursor protein expression. Neurobiol. Dis. 2009, 33, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.G.; Wang, J.L.; Li, L.; Xue, L.X.; Zhang, Y.Q.; Wang, P.C. MicroRNA-135a and -200b, potential Biomarkers for Alzheimer׳s disease, regulate β secretase and amyloid precursor protein. Brain Res. 2014, 1583, 55–64. [Google Scholar] [CrossRef]
- Kang, Q.; Xiang, Y.; Li, D.; Liang, J.; Zhang, X.; Zhou, F.; Qiao, M.; Nie, Y.; He, Y.; Cheng, J.; et al. MiR-124-3p attenuates hyperphosphorylation of Tau protein-induced apoptosis via caveolin-1-PI3K/Akt/GSK3β pathway in N2a/APP695swe cells. Oncotarget 2017, 8, 24314–24326. [Google Scholar] [CrossRef]
- Long, J.M.; Ray, B.; Lahiri, D.K. MicroRNA-153 physiologically inhibits expression of amyloid-β precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J. Biol. Chem. 2012, 287, 31298–31310. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Tan, L.; Lu, Y.; Peng, J.; Zhu, Y.; Zhang, Y.; Sun, Z. MicroRNA-138 promotes tau phosphorylation by targeting retinoic acid receptor alpha. FEBS Lett. 2015, 589, 726–729. [Google Scholar] [CrossRef] [Green Version]
- Prasad, K.N. Simultaneous activation of Nrf2 and elevation of antioxidant compounds for reducing oxidative stress and chronic inflammation in human Alzheimer’s disease. Mech. Ageing Dev. 2016, 153, 41–47. [Google Scholar] [CrossRef]
- Amakiri, N.; Kubosumi, A.; Tran, J.; Reddy, P.H. Amyloid beta and microRNAs in Alzheimer’s disease. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Varadarajan, S.; Yatin, S.; Aksenova, M.; Butterfield, D.A. Review: Alzheimer’s amyloid β-peptide-associated free radical oxidative stress and neurotoxicity. J. Struct. Biol. 2000, 130, 184–208. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Dolios, G.; Wang, R.; Liao, F.-F. Soluble beta-amyloid peptides, but not insoluble fibrils, have specific effect on neuronal icroRNA expression. PLoS ONE 2014, 9, e90770. [Google Scholar] [CrossRef]
- Wang, W.-X.; Rajeev, B.W.; Stromberg, A.J.; Ren, N.; Tang, G.; Huang, Q.; Rigoutsos, I.; Nelson, P.T. The expression of microRNA miR-107 decreases early in Alzheimer’s disease and may accelerate disease progression through regulation of β-site amyloid precursor protein-cleaving enzyme 1. J. Neurosci. 2008, 28, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Monte, S.; Wands, J. Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska, J.; Oczkowska, A.; Suwalska, M.; Rozycka, A.; Florczak-Wyspianska, J.; Dezor, M.; Lianeri, M.; Jagodzinski, P.P.; Kowalczyk, M.J.; Prendecki, M.; et al. Mutations in the exon 7 of Trp53 gene and the level of p53 protein in double transgenic mouse model of Alzheimer’s disease. Folia Neuropathol. 2014, 52, 30–40. [Google Scholar] [CrossRef]
- Dorszewska, J.; Oczkowska, A.; Suwalska, M.; Rozycka, A.; Florczak, J.; Dezor, M.; Lianeri, M.; Jagodzinski, P.; Kowalczyk, M.; Prendecki, M.; et al. Mutations of TP53 gene and oxidative stress in Alzheimer’s disease patients. Adv. Alzheimer’s Dis. 2014, 3, 24–32. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Zhang, R.; Li, P.; Liu, Y.; Qin, K.; Fa, Z.Q.; Liu, Y.J.; Ke, Y.Q.; Jiang, X.D. P53-induced microRNA-107 inhibits proliferation of glioma cells and down-regulates the expression of CDK6 and Notch-2. Neurosci. Lett. 2013, 534, 327–332. [Google Scholar] [CrossRef]
- Prendecki, M.; Florczak-Wyspianska, J.; Kowalska, M.; Ilkowski, J.; Grzelak, T.; Bialas, K.; Kozubski, W.; Dorszewska, J. APOE genetic variants and apoE, miR-107 and miR-650 levels in Alzheimer’s disease. Folia Neuropathol. 2019, 57, 106–116. [Google Scholar] [CrossRef]
- Kim, J.; Yoon, H.; Chung, D.E.; Brown, J.L.; Belmonte, K.C.; Kim, J. miR-186 is decreased in aged brain and suppresses BACE1 expression. J. Neurochem. 2016, 137, 436–445. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Wu, Y.; Gu, M.; Zhang, Y. MiR-342-5p decreases ankyrin G levels in Alzheimer’s disease transgenic mouse models. Cell Rep. 2014, 6, 264–270. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Deng, W.; Liu, Y.; Qin, C. MicroRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Res. 2012, 1455, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.D.; Sun, G.L.; Zhou, T.T.; Wang, Y.Y.; Xu, X.; Shi, X.F.; Zhu, Z.Y.; Rukachaisirikul, V.; Hu, L.H.; Shen, X. LX2343 alleviates cognitive impairments in AD model rats by inhibiting oxidative stress-induced neuronal apoptosis and tauopathy. Acta Pharmacol. Sin. 2017, 38, 1104–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between oxidative stress and SIRT1: Impact on the aging process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dias-Santagata, D.; Fulga, T.A.; Duttaroy, A.; Feany, M.B. Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J. Clin. Investig. 2007, 117, 236–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liu, J.; Wang, Q.; Jiang, H.; Zeng, L.; Li, Z.; Liu, R. MicroRNA-200a-3p mediates neuroprotection in Alzheimer-related deficits and attenuates amyloid-beta overproduction and tau hyperphosphorylation via coregulating BACE1 and PRKACB. Front. Pharmacol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chen, W.; Yi, Y.; Tong, Q. miR-219-5p inhibits tau phosphorylation by targeting TTBK1 and GSK-3β in Alzheimer’s disease. J. Cell. Biochem. 2019, 120, 9936–9946. [Google Scholar] [CrossRef]
- El Fatimy, R.; Li, S.; Chen, Z.; Mushannen, T.; Gongala, S.; Wei, Z.; Balu, D.T.; Rabinovsky, R.; Cantlon, A.; Elkhal, A.; et al. MicroRNA-132 provides neuroprotection for tauopathies via multiple signaling pathways. Acta Neuropathol. 2018, 136, 537–555. [Google Scholar] [CrossRef] [Green Version]
- Smith, P.Y.; Hernandez-Rapp, J.; Jolivette, F.; Lecours, C.; Bisht, K.; Goupil, C.; Dorval, V.; Parsi, S.; Morin, F.; Planel, E.; et al. MiR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum. Mol. Gen. 2015, 24, 6721–6735. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Huang, Y.; Wang, L.-L.; Zhang, Y.-F.; Xu, J.; Zhou, Y.; Lourenco, G.F.; Zhang, B.; Wang, Y.; Ren, R.-J.; et al. MicroRNA-146a suppresses ROCK1 allowing hyperphosphorylation of tau in Alzheimer’s disease. Sci. Rep. 2016, 6, 26697. [Google Scholar] [CrossRef]
- Absalon, S.; Kochanek, D.M.; Raghavan, V.; Krichevsky, A.M. MiR-26b, upregulated in Alzheimer’s disease, activates cell cycle entry, tau-phosphorylation, and apoptosis in postmitotic neurons. J. Neurosci. 2013, 33, 14645–14659. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, Q.; Niu, J.; Lu, K.; Xie, B.; Cui, D.; Xu, S. Screening of microRNAs associated with Alzheimer’s disease using oxidative stress cell model and different strains of senescence accelerated mice. J. Neurol. Sci. 2014, 338, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, Z.-F.; Li, W.; Hong, H.; Chen, J.; Tian, Y.; Liu, Z.-Y. Protective effects of microRNA-330 on amyloid β-protein production, oxidative stress, and mitochondrial dysfunction in Alzheimer’s disease by targeting VAV1 via the MAPK signaling pathway. J. Cell. Biochem. 2018, 119, 5437–5448. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Foruria, I.; Santulli, P.; Chouzenoux, S.; Carmona, F.; Chapron, C.; Batteux, F. Dysregulation of the ADAM17/Notch signalling pathways in endometriosis: From oxidative stress to fibrosis. Mol. Hum. Reprod. 2017, 23, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Jiao, W.E.; Wei, J.F.; Kong, Y.G.; Xu, Y.; Tao, Z.Z.; Chen, S.M. Notch signaling promotes development of allergic rhinitis by suppressing Foxp3 expression and treg cell differentiation. Int. Arch. Allergy Immunol. 2019, 178, 33–44. [Google Scholar] [CrossRef]
- Zhu, P.; Yang, M.; He, H.; Kuang, Z.; Liang, M.; Lin, A.; Liang, S.; Wen, Q.; Cheng, Z.; Sun, C. Curcumin attenuates hypoxia/reoxygenationinduced cardiomyocyte injury by downregulating Notch signaling. Mol. Med. Rep. 2019, 20, 1541–1550. [Google Scholar] [CrossRef]
- Chen, F.-Z.; Zhao, Y.; Chen, H.-Z. MicroRNA-98 reduces amyloid β-protein production and improves oxidative stress and mitochondrial dysfunction through the Notch signaling pathway via HEY2 in Alzheimer’s disease mice. Int. J. Mol. Med. 2019, 43, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Dong, H.; Si, Y.; Wu, N.; Cao, H.; Mei, B.; Meng, B. miR-125b promotes tau phosphorylation by targeting the neural cell adhesion molecule in neuropathological progression. Neurobiol. Aging 2019, 73, 41–49. [Google Scholar] [CrossRef]
- Jin, Y.; Tu, Q.; Liu, M. MicroRNA125b regulates Alzheimer’s disease through SphK1 regulation. Mol. Med. Rep. 2018, 18, 2373–2380. [Google Scholar] [CrossRef]
- Wu, D.-M.; Wen, X.; Wang, Y.-J.; Han, X.-R.; Wang, S.; Shen, M.; Fan, S.-H.; Zhuang, J.; Zhang, Z.-F.; Shan, Q.; et al. Effect of microRNA-186 on oxidative stress injury of neuron by targeting interleukin 2 through the janus kinase-signal transducer and activator of transcription pathway in a rat model of Alzheime’s disease. J. Cell. Physiol. 2018, 233, 9488–9502. [Google Scholar] [CrossRef]
- Jellinger, K.A. Neuropathobiology of non-motor symptoms in Parkinson disease. J. Neural Transm. 2015, 122, 1429–1440. [Google Scholar] [CrossRef]
- Tysnes, O.B.; Storstein, A. Epidemiology of Parkinson’s disease. J. Neural Transm. 2017, 124, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Li, X.; Li, X.; Liu, Q.; Cheng, Y. Oxidative stress in Parkinson’s disease: A systematic review and meta-analysis. Front. Mol. Neurosci. 2018, 11, 236. [Google Scholar] [CrossRef] [PubMed]
- Hwang, D.Y.; Hong, S.; Jeong, J.W.; Choi, S.; Kim, H.; Kim, J.; Kim, K.S. Vesicular monoamine transporter 2 and dopamine transporter are molecular targets of Pitx3 in the ventral midbrain dopamine neurons. J. Neurochem. 2009, 111, 1202–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, C.; Chen, Z.; He, J.; Tao, Z.; Yin, Z.Q. A microRNA, mir133b, suppresses melanopsin expression mediated by failure dopaminergic amacrine cells in RCS rats. Cell Signal. 2012, 24, 685–698. [Google Scholar] [CrossRef]
- Caudle, W.M.; Richardson, J.R.; Wang, M.Z.; Taylor, T.N.; Guillot, T.S.; McCormack, A.L.; Colebrooke, R.E.; Di Monte, D.A.; Emson, P.C.; Miller, G.W. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J. Neurosci. 2007, 27, 8138–8148. [Google Scholar] [CrossRef]
- Masoud, S.T.; Vecchio, L.M.; Bergeron, Y.; Hossain, M.M.; Nguyen, L.T.; Bermejo, M.K.; Kile, B.; Sotnikova, T.D.; Siesser, W.B.; Gainetdinov, R.R.; et al. Increased expression of the dopamine transporter leads to loss of dopamine neurons, oxidative stress and l-DOPA reversible motor deficits. Neurobiol. Dis. 2015, 74, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.; Wang, F.; Han, Y.; Geng, X.; Li, M.; Shi, Y.; Lu, L.; Chen, Y. MiR-137 and miR-491 negatively regulate dopamine transporter expression and function in neural cells. Neurosci. Bull. 2016, 32, 512–522. [Google Scholar] [CrossRef] [Green Version]
- Barodia, S.K.; Creed, R.B.; Goldberg, M.S. Parkin and PINK1 functions in oxidative stress and neurodegeneration. Brain Res. Bull. 2017, 133, 51–59. [Google Scholar] [CrossRef]
- Wang, H.L.; Chou, A.H.; Wu, A.S.; Chen, S.Y.; Weng, Y.H.; Kao, Y.C.; Yeh, T.H.; Chu, P.J.; Lu, C.S. PARK6 PINK1 mutants are defective in maintaining mitochondrial membrane potential and inhibiting ROS formation of substantia nigra dopaminergic neurons. Biochim. Biophys. Acta 2011, 1812, 674–684. [Google Scholar] [CrossRef] [Green Version]
- Wood-Kaczmar, A.; Gandhi, S.; Yao, Z.; Abramov, A.Y.; Miljan, E.A.; Keen, G.; Stanyer, L.; Hargreaves, I.; Klupsch, K.; Deas, E.; et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 2008, 3, e2455. [Google Scholar] [CrossRef]
- Kim, J.; Fiesel, F.C.; Belmonte, K.C.; Hudec, R.; Wang, W.X.; Kim, C.; Nelson, P.T.; Springer, W.; Kim, J. miR-27a and miR-27b regulate autophagic clearance of damaged mitochondria by targeting PTEN-induced putative kinase 1 (PINK1). Mol. Neurodegener. 2016, 11, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prajapati, P.; Sripada, L.; Singh, K.; Bhatelia, K.; Singh, R.; Singh, R. TNF-alpha regulates miRNA targeting mitochondrial complex-I and induces cell death in dopaminergic cells. Biochim. Biophys. Acta 2015, 1852, 451–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Ishimori, C.; Takahashi-Niki, K.; Taira, T.; Kim, Y.C.; Maita, H.; Maita, C.; Ariga, H.; Iguchi-Ariga, S.M. DJ-1 binds to mitochondrial complex I and maintains its activity. Biochem. Biophys. Res. Commun. 2009, 390, 667–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariga, H.; Takahashi-Niki, K.; Kato, I.; Maita, H.; Niki, T.; Iguchi-Ariga, S.M. Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid. Med. Cell Longev. 2013, 2013, 683920. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.; Sullards, M.C.; Olzmann, J.A.; Rees, H.D.; Weintraub, S.T.; Bostwick, D.E.; Gearing, M.; Levey, A.I.; Chin, L.S.; Li, L. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J. Biol. Chem. 2006, 281, 10816–10824. [Google Scholar] [CrossRef] [Green Version]
- Xiong, R.; Wang, Z.; Zhao, Z.; Li, H.; Chen, W.; Zhang, B.; Wang, L.; Wu, L.; Li, W.; Ding, J.; et al. MicroRNA-494 reduces DJ-1 expression and exacerbates neurodegeneration. Neurobiol. Aging 2014, 35, 705–714. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, C.; Sun, Q.; Pan, H.; Huang, P.; Ding, J.; Chen, S. MicroRNA-4639 is a regulator of DJ-1 expression and a potential early diagnostic marker for Parkinson’s disease. Front. Aging Neurosci. 2017, 9, 232. [Google Scholar] [CrossRef]
- Minones-Moyano, E.; Porta, S.; Escaramis, G.; Rabionet, R.; Iraola, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Marti, E. MicroRNA profiling of Parkinson’s disease brains identifies early downregulation of miR-34b/c which modulate mitochondrial function. Hum. Mol. Genet. 2011, 20, 3067–3078. [Google Scholar] [CrossRef]
- Ramsey, C.P.; Glass, C.A.; Montgomery, M.B.; Lindl, K.A.; Ritson, G.P.; Chia, L.A.; Hamilton, R.L.; Chu, C.T.; Jordan-Sciutto, K.L. Expression of Nrf2 in neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2007, 66, 75–85. [Google Scholar] [CrossRef]
- Xie, Y.; Chen, Y. MicroRNAs: Emerging targets regulating oxidative stress in the models of Parkinson’s disease. Front. Neurosci. 2016, 10, 298. [Google Scholar] [CrossRef] [Green Version]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Jain, M.R.; Li, H.; Junn, E. MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radic. Biol. Med. 2015, 89, 548–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMillan, K.J.; Murray, T.K.; Bengoa-Vergniory, N.; Cordero-Llana, O.; Cooper, J.; Buckley, A.; Wade-Martins, R.; Uney, J.B.; O’Neill, M.J.; Wong, L.F.; et al. Loss of MicroRNA-7 regulation leads to alpha-synuclein accumulation and dopaminergic neuronal loss in vivo. Mol. Ther. 2017, 25, 2404–2414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narasimhan, M.; Patel, D.; Vedpathak, D.; Rathinam, M.; Henderson, G.; Mahimainathan, L. Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PLoS ONE 2012, 7, e51111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slota, J.A.; Booth, S.A. MicroRNAs in neuroinflammation: Implications in disease pathogenesis, biomarker discovery and therapeutic applications. Noncoding RNA 2019, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Farrer, M.J. Genetics of Parkinson disease: Paradigm shifts and future prospects. Nat. Rev. Genet. 2006, 7, 306–318. [Google Scholar] [CrossRef]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar] [CrossRef]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Perfeito, R.; Ribeiro, M.; Rego, A.C. Alpha-synuclein-induced oxidative stress correlates with altered superoxide dismutase and glutathione synthesis in human neuroblastoma SH-SY5Y cells. Arch. Toxicol. 2017, 91, 1245–1259. [Google Scholar] [CrossRef]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [Green Version]
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra378. [Google Scholar] [CrossRef] [Green Version]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.H.; Zhang, J.L.; Duan, Y.L.; Zhang, Q.S.; Li, G.F.; Zheng, D.L. MicroRNA-214 participates in the neuroprotective effect of Resveratrol via inhibiting alpha-synuclein expression in MPTP-induced Parkinson’s disease mouse. Biomed. Pharmacother. 2015, 74, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Doxakis, E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Mouradian, M.M.; Junn, E. Inhibition of miR-34b and miR-34c enhances alpha-synuclein expression in Parkinson’s disease. FEBS Lett. 2015, 589, 319–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Rodriguez-Oroz, M.C.; Obeso, J.A.; Cooper, J.M. Influence of microRNA deregulation on chaperone-mediated autophagy and alpha-synuclein pathology in Parkinson’s disease. Cell Death Dis. 2013, 4, e545. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yang, H.; Zhu, D.; Huang, H.; Liu, G.; Lun, P. Targeted suppression of chaperone-mediated autophagy by miR-320a promotes alpha-synuclein aggregation. Int. J. Mol. Sci. 2014, 15, 15845–15857. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Cheng, Y. miR-16-1 promotes the aberrant alpha-synuclein accumulation in parkinson disease via targeting heat shock protein 70. Sci. World J. 2014, 2014, 938348. [Google Scholar] [CrossRef] [Green Version]
- Recasens, A.; Perier, C.; Sue, C.M. Role of microRNAs in the Regulation of alpha-Synuclein Expression: A Systematic Review. Front. Mol. Neurosci. 2016, 9, 128. [Google Scholar] [CrossRef] [Green Version]
- Dias, V.; Junn, E.; Mouradian, M.M. The role of oxidative stress in Parkinson’s disease. J. Parkinsons Dis. 2013, 3, 461–491. [Google Scholar] [CrossRef] [Green Version]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Thome, A.D.; Harms, A.S.; Volpicelli-Daley, L.A.; Standaert, D.G. microRNA-155 regulates alpha-synuclein-induced inflammatory responses in models of Parkinson disease. J. Neurosci. 2016, 36, 2383–2390. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lu, M.; Du, R.H.; Qiao, C.; Jiang, C.Y.; Zhang, K.Z.; Ding, J.H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener. 2016, 16, 11–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caggiu, E.; Paulus, K.; Mameli, G.; Arru, G.; Sechi, G.P.; Sechi, L.A. Differential expression of miRNA 155 and miRNA 146a in Parkinson’s disease patients. eNeurologicalSci 2018, 13, 1–4. [Google Scholar] [CrossRef]
- Yao, L.; Ye, Y.; Mao, H.; Lu, F.; He, X.; Lu, G.; Zhang, S. MicroRNA-124 regulates the expression of MEKK3 in the inflammatory pathogenesis of Parkinson’s disease. J. Neuroinflamm. 2018, 15, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilhelmus, M.M.; Nijland, P.G.; Drukarch, B.; de Vries, H.E.; van Horssen, J. Involvement and interplay of Parkin, PINK1, and DJ1 in neurodegenerative and neuroinflammatory disorders. Free Radic. Biol. Med. 2012, 53, 983–992. [Google Scholar] [CrossRef]
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Volk, A.E.; Weishaupt, J.H.; Andersen, P.M.; Ludolph, A.C.; Kubisch, C. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med. Genet. 2018, 30, 252–258. [Google Scholar] [CrossRef] [Green Version]
- Guerrero, E.N.; Wang, H.; Mitra, J.; Hegde, P.M.; Stowell, S.E.; Liachko, N.F.; Kraemer, B.C.; Garruto, R.M.; Rao, K.S.; Hegde, M.L. TDP-43/FUS in motor neuron disease: Complexity and challenges. Prog. Neurobiol. 2016, 145–146, 78–97. [Google Scholar] [CrossRef] [Green Version]
- Belzil, V.V.; Katzman, R.B.; Petrucelli, L. ALS and FTD: An epigenetic perspective. Acta Neuropathol. 2016, 132, 487–502. [Google Scholar] [CrossRef]
- Wang, Z.; Bai, Z.; Qin, X.; Cheng, Y. Aberrations in oxidative stress markers in amyotrophic lateral sclerosis: A systematic review and meta-analysis. Oxid. Med. Cell Longev. 2019, 2019, 1712323. [Google Scholar] [CrossRef]
- Joilin, G.; Leigh, P.N.; Newbury, S.F.; Hafezparast, M. An overview of MicroRNAs as biomarkers of ALS. Front. Neurol. 2019, 10, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovanda, A.; Leonardis, L.; Zidar, J.; Koritnik, B.; Dolenc-Groselj, L.; Ristic Kovacic, S.; Curk, T.; Rogelj, B. Differential expression of microRNAs and other small RNAs in muscle tissue of patients with ALS and healthy age-matched controls. Sci. Rep. 2018, 8, 5609. [Google Scholar] [CrossRef] [PubMed]
- Ricci, C.; Marzocchi, C.; Battistini, S. MicroRNAs as biomarkers in amyotrophic lateral sclerosis. Cells 2018, 7, 219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzuti, M.; Filosa, G.; Melzi, V.; Calandriello, L.; Dioni, L.; Bollati, V.; Bresolin, N.; Comi, G.P.; Barabino, S.; Nizzardo, M.; et al. MicroRNA expression analysis identifies a subset of downregulated miRNAs in ALS motor neuron progenitors. Sci. Rep. 2018, 8, 10105. [Google Scholar] [CrossRef]
- Waller, R.; Wyles, M.; Heath, P.R.; Kazoka, M.; Wollff, H.; Shaw, P.J.; Kirby, J. Small RNA sequencing of sporadic amyotrophic lateral sclerosis cerebrospinal fluid reveals differentially expressed miRNAs related to neural and glial activity. Front. Neurosci. 2017, 11, 731. [Google Scholar] [CrossRef]
- Borel, F.; Gernoux, G.; Sun, H.; Stock, R.; Blackwood, M.; Brown, R.H., Jr.; Mueller, C. Safe and effective superoxide dismutase 1 silencing using artificial microRNA in macaques. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Koval, E.D.; Shaner, C.; Zhang, P.; du Maine, X.; Fischer, K.; Tay, J.; Chau, B.N.; Wu, G.F.; Miller, T.M. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum. Mol. Genet. 2013, 22, 4127–4135. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wei, Q.; Gu, X.; Chen, Y.; Chen, X.; Cao, B.; Ou, R.; Shang, H. Decreased glycogenolysis by miR-338-3p promotes regional glycogen accumulation within the spinal cord of amyotrophic lateral sclerosis mice. Front. Mol. Neurosci. 2019, 12, 114. [Google Scholar] [CrossRef] [Green Version]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Hanson, P.S.; Morris, C.M. SIRT1 ameliorates oxidative stress induced neural cell death and is down-regulated in Parkinson’s disease. BMC Neurosci. 2017, 18, 46. [Google Scholar] [CrossRef]
- Paladino, S.; Conte, A.; Caggiano, R.; Pierantoni, G.M.; Faraonio, R. Nrf2 pathway in age-related neurological disorders: Insights into MicroRNAs. Cell Physiol. Biochem. 2018, 47, 1951–1976. [Google Scholar] [CrossRef] [PubMed]
- Ba, Q.; Cui, C.; Wen, L.; Feng, S.; Zhou, J.; Yang, K. Schisandrin B shows neuroprotective effect in 6-OHDA-induced Parkinson’s disease via inhibiting the negative modulation of miR-34a on Nrf2 pathway. Biomed. Pharmacother. 2015, 75, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhang, L.; Lu, Y.; Zhang, M.; Zhang, Z.; Wang, K.; Lv, J. Down-regulation of microRNA-142-5p attenuates oxygen-glucose deprivation and reoxygenation-induced neuron injury through up-regulating Nrf2/ARE signaling pathway. Biomed. Pharmacother. 2017, 89, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Aschrafi, A.; Kar, A.N.; Natera-Naranjo, O.; MacGibeny, M.A.; Gioio, A.E.; Kaplan, B.B. MicroRNA-338 regulates the axonal expression of multiple nuclear-encoded mitochondrial mRNAs encoding subunits of the oxidative phosphorylation machinery. Cell Mol. Life Sci. 2012, 69, 4017–4027. [Google Scholar] [CrossRef]
- Shioya, M.; Obayashi, S.; Tabunoki, H.; Arima, K.; Saito, Y.; Ishida, T.; Satoh, J. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol. Appl. Neurobiol. 2010, 36, 320–330. [Google Scholar] [CrossRef]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Banez-Coronel, M.; Ayhan, F.; Tarabochia, A.D.; Zu, T.; Perez, B.A.; Tusi, S.K.; Pletnikova, O.; Borchelt, D.R.; Ross, C.A.; Margolis, R.L.; et al. RAN translation in huntington disease. Neuron 2015, 88, 667–677. [Google Scholar] [CrossRef] [Green Version]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Moumne, L.; Betuing, S.; Caboche, J. Multiple aspects of gene dysregulation in Huntington’s disease. Front. Neurol. 2013, 4, 127. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Winderickx, J.; Franssens, V.; Liu, B. A Mitochondria-associated oxidative stress perspective on Huntington’s disease. Front. Mol. Neurosci. 2018, 11, 329. [Google Scholar] [CrossRef]
- Johnson, R.; Zuccato, C.; Belyaev, N.D.; Guest, D.J.; Cattaneo, E.; Buckley, N.J. A microRNA-based gene dysregulation pathway in Huntington’s disease. Neurobiol. Dis. 2008, 29, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Packer, A.N.; Xing, Y.; Harper, S.Q.; Jones, L.; Davidson, B.L. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington’s disease. J. Neurosci. 2008, 28, 14341–14346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savas, J.N.; Makusky, A.; Ottosen, S.; Baillat, D.; Then, F.; Krainc, D.; Shiekhattar, R.; Markey, S.P.; Tanese, N. Huntington’s disease protein contributes to RNA-mediated gene silencing through association with Argonaute and P bodies. Proc. Natl. Acad. Sci. USA 2008, 105, 10820–10825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pircs, K.; Petri, R.; Madsen, S.; Brattas, P.L.; Vuono, R.; Ottosson, D.R.; St-Amour, I.; Hersbach, B.A.; Matusiak-Bruckner, M.; Lundh, S.H.; et al. Huntingtin aggregation impairs autophagy, leading to argonaute-2 accumulation and global MicroRNA dysregulation. Cell Rep. 2018, 24, 1397–1406. [Google Scholar] [CrossRef] [Green Version]
- Banez-Coronel, M.; Porta, S.; Kagerbauer, B.; Mateu-Huertas, E.; Pantano, L.; Ferrer, I.; Guzman, M.; Estivill, X.; Marti, E. A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet. 2012, 8, e1002481. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.T.; Chu, K.; Im, W.S.; Yoon, H.J.; Im, J.Y.; Park, J.E.; Park, K.H.; Jung, K.H.; Lee, S.K.; Kim, M.; et al. Altered microRNA regulation in Huntington’s disease models. Exp. Neurol. 2011, 227, 172–179. [Google Scholar] [CrossRef]
- Marti, E.; Pantano, L.; Banez-Coronel, M.; Llorens, F.; Minones-Moyano, E.; Porta, S.; Sumoy, L.; Ferrer, I.; Estivill, X. A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 2010, 38, 7219–7235. [Google Scholar] [CrossRef]
- Wexler, N.S.; Lorimer, J.; Porter, J.; Gomez, F.; Moskowitz, C.; Shackell, E.; Marder, K.; Penchaszadeh, G.; Roberts, S.A.; Gayan, J.; et al. Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc. Natl. Acad. Sci. USA 2004, 101, 3498–3503. [Google Scholar] [CrossRef] [Green Version]
- Rosenblatt, A.; Liang, K.Y.; Zhou, H.; Abbott, M.H.; Gourley, L.M.; Margolis, R.L.; Brandt, J.; Ross, C.A. The association of CAG repeat length with clinical progression in Huntington disease. Neurology 2006, 66, 1016–1020. [Google Scholar] [CrossRef]
- Johri, A.; Beal, M.F. Antioxidants in Huntington’s disease. Biochim. Biophys. Acta 2012, 1822, 664–674. [Google Scholar] [CrossRef] [Green Version]
- Duran, R.; Barrero, F.J.; Morales, B.; Luna, J.D.; Ramirez, M.; Vives, F. Oxidative stress and plasma aminopeptidase activity in Huntington’s disease. J. Neural. Transm. 2010, 117, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Olejniczak, M.; Kotowska-Zimmer, A.; Krzyzosiak, W. Stress-induced changes in miRNA biogenesis and functioning. Cell Mol. Life Sci. 2018, 75, 177–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asada, S.; Takahashi, T.; Isodono, K.; Adachi, A.; Imoto, H.; Ogata, T.; Ueyama, T.; Matsubara, H.; Oh, H. Downregulation of Dicer expression by serum withdrawal sensitizes human endothelial cells to apoptosis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2512–H2521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inukai, S.; de Lencastre, A.; Turner, M.; Slack, F. Novel microRNAs differentially expressed during aging in the mouse brain. PLoS ONE 2012, 7, e40028. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stahler, C.; Meese, E.; et al. Distribution of miRNA expression across human tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4. [Google Scholar] [CrossRef]
- Witkos, T.M.; Koscianska, E.; Krzyzosiak, W.J. Practical aspects of microRNA target prediction. Curr. Mol. Med. 2011, 11, 93–109. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.Y.; Gonzalez-Martin, A.; Miletic, A.V.; Lai, M.; Knight, S.; Sabouri-Ghomi, M.; Head, S.R.; Macauley, M.S.; Rickert, R.C.; Xiao, C. Transfection of microRNA mimics should be used with caution. Front. Genet. 2015, 6, 340. [Google Scholar] [CrossRef] [Green Version]
- Jovicic, A.; Roshan, R.; Moisoi, N.; Pradervand, S.; Moser, R.; Pillai, B.; Luthi-Carter, R. Comprehensive expression analyses of neural cell-type-specific miRNAs identify new determinants of the specification and maintenance of neuronal phenotypes. J. Neurosci. 2013, 33, 5127–5137. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.W.; Rissland, O.S.; Koppstein, D.; Abreu-Goodger, C.; Jan, C.H.; Agarwal, V.; Yildirim, M.A.; Rodriguez, A.; Bartel, D.P. Global analyses of the effect of different cellular contexts on microRNA targeting. Mol. Cell 2014, 53, 1031–1043. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Liu, Y.; Wang, X.; Zhang, M.Q.; Hannon, G.J.; Huang, Z.J. Cell-type-based analysis of microRNA profiles in the mouse brain. Neuron 2012, 73, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svoboda, P. A toolbox for miRNA analysis. FEBS Lett. 2015, 589, 1694–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knauss, J.L.; Bian, S.; Sun, T. Plasmid-based target protectors allow specific blockade of miRNA silencing activity in mammalian developmental systems. Front. Cell Neurosci. 2013, 7, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef] [Green Version]
- Amin, N.D.; Bai, G.; Klug, J.R.; Bonanomi, D.; Pankratz, M.T.; Gifford, W.D.; Hinckley, C.A.; Sternfeld, M.J.; Driscoll, S.P.; Dominguez, B.; et al. Loss of motoneuron-specific microRNA-218 causes systemic neuromuscular failure. Science 2015, 350, 1525–1529. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Yi, B.; Ma, R.; Zhang, X.; Zhao, H.; Xi, Y. CRISPR/cas9, a novel genomic tool to knock down microRNA in vitro and in vivo. Sci. Rep. 2016, 6, 22312. [Google Scholar] [CrossRef] [Green Version]
- Kurata, J.S.; Lin, R.J. MicroRNA-focused CRISPR-Cas9 library screen reveals fitness-associated miRNAs. RNA 2018, 24, 966–981. [Google Scholar] [CrossRef] [Green Version]
- Back, S.; Necarsulmer, J.; Whitaker, L.R.; Coke, L.M.; Koivula, P.; Heathward, E.J.; Fortuno, L.V.; Zhang, Y.; Yeh, C.G.; Baldwin, H.A.; et al. Neuron-specific genome modification in the adult rat brain using CRISPR-Cas9 transgenic rats. Neuron 2019, 102, 105–119. [Google Scholar] [CrossRef] [Green Version]
- Hess, G.T.; Tycko, J.; Yao, D.; Bassik, M.C. Methods and applications of CRISPR-mediated base editing in eukaryotic genomes. Mol. Cell 2017, 68, 26–43. [Google Scholar] [CrossRef] [PubMed]
- Zafra, M.P.; Schatoff, E.M.; Katti, A.; Foronda, M.; Breinig, M.; Schweitzer, A.Y.; Simon, A.; Han, T.; Goswami, S.; Montgomery, E.; et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol. 2018, 36, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.H.; Miller, S.M.; Geurts, M.H.; Tang, W.; Chen, L.; Sun, N.; Zeina, C.M.; Gao, X.; Rees, H.A.; Lin, Z.; et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 2018, 556, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019. [Google Scholar] [CrossRef] [PubMed]
- Domanskyi, A.; Saarma, M.; Airavaara, M. Prospects of neurotrophic factors for Parkinson’s disease: Comparison of protein and gene therapy. Hum. Gene Ther. 2015, 26, 550–559. [Google Scholar] [CrossRef]
- Espay, A.J.; Brundin, P.; Lang, A.E. Precision medicine for disease modification in Parkinson disease. Nat. Rev. Neurol. 2017, 13, 119–126. [Google Scholar] [CrossRef]
- La Manno, G.; Gyllborg, D.; Codeluppi, S.; Nishimura, K.; Salto, C.; Zeisel, A.; Borm, L.E.; Stott, S.R.W.; Toledo, E.M.; Villaescusa, J.C.; et al. Molecular diversity of midbrain development in mouse, human, and stem cells. Cell 2016, 167, 566–580. [Google Scholar] [CrossRef] [Green Version]
- Creed, R.B.; Goldberg, M.S. New developments in genetic rat models of Parkinson’s disease. Mov. Disord. 2018, 33, 717–729. [Google Scholar] [CrossRef]
- Chesselet, M.F.; Richter, F. Modelling of Parkinson’s disease in mice. Lancet Neurol. 2011, 10, 1108–1118. [Google Scholar] [CrossRef]
- Blesa, J.; Przedborski, S. Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef] [Green Version]
- Berezikov, E.; Thuemmler, F.; van Laake, L.W.; Kondova, I.; Bontrop, R.; Cuppen, E.; Plasterk, R.H. Diversity of microRNAs in human and chimpanzee brain. Nat. Genet. 2006, 38, 1375–1377. [Google Scholar] [CrossRef] [PubMed]
- Londin, E.; Loher, P.; Telonis, A.G.; Quann, K.; Clark, P.; Jing, Y.; Hatzimichael, E.; Kirino, Y.; Honda, S.; Lally, M.; et al. Analysis of 13 cell types reveals evidence for the expression of numerous novel primate- and tissue-specific microRNAs. Proc. Natl. Acad. Sci. USA 2015, 112, E1106–E1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLoughlin, H.S.; Wan, J.; Spengler, R.M.; Xing, Y.; Davidson, B.L. Human-specific microRNA regulation of FOXO1: Implications for microRNA recognition element evolution. Hum. Mol. Genet. 2014, 23, 2593–2603. [Google Scholar] [CrossRef] [Green Version]
- Franca, G.S.; Hinske, L.C.; Galante, P.A.; Vibranovski, M.D. Unveiling the impact of the genomic architecture on the evolution of vertebrate microRNAs. Front. Genet. 2017, 8, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, N.; Zheng, J.; Chen, Z.; Liu, Y.; Dura, B.; Kwak, M.; Xavier-Ferrucio, J.; Lu, Y.C.; Zhang, M.; Roden, C.; et al. Single-cell microRNA-mRNA co-sequencing reveals non-genetic heterogeneity and mechanisms of microRNA regulation. Nat. Commun. 2019, 10, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engle, S.J.; Blaha, L.; Kleiman, R.J. Best practices for translational disease modeling using human iPSC-derived neurons. Neuron 2018, 100, 783–797. [Google Scholar] [CrossRef] [Green Version]
- Tolosa, E.; Botta-Orfila, T.; Morato, X.; Calatayud, C.; Ferrer-Lorente, R.; Marti, M.J.; Fernandez, M.; Gaig, C.; Raya, A.; Consiglio, A.; et al. MicroRNA alterations in iPSC-derived dopaminergic neurons from Parkinson disease patients. Neurobiol. Aging 2018, 69, 283–291. [Google Scholar] [CrossRef]
- Katerji, M.; Filippova, M.; Duerksen-Hughes, P. Approaches and methods to measure oxidative stress in clinical samples: Research applications in the cancer field. Oxid. Med. Cell Longev. 2019, 2019, 1279250. [Google Scholar] [CrossRef] [Green Version]
- Grenier, K.; Kao, J.; Diamandis, P. Three-dimensional modeling of human neurodegeneration: Brain organoids coming of age. Mol. Psychiatry 2019. [Google Scholar] [CrossRef]
- Windrem, M.S.; Schanz, S.J.; Morrow, C.; Munir, J.; Chandler-Militello, D.; Wang, S.; Goldman, S.A. A competitive advantage by neonatally engrafted human glial progenitors yields mice whose brains are chimeric for human glia. J. Neurosci. 2014, 34, 16153–16161. [Google Scholar] [CrossRef]
- Cardoso, T.; Adler, A.F.; Mattsson, B.; Hoban, D.B.; Nolbrant, S.; Wahlestedt, J.N.; Kirkeby, A.; Grealish, S.; Bjorklund, A.; Parmar, M. Target-specific forebrain projections and appropriate synaptic inputs of hESC-derived dopamine neurons grafted to the midbrain of parkinsonian rats. J. Comp. Neurol. 2018, 526, 2133–2146. [Google Scholar] [CrossRef] [PubMed]
- Grealish, S.; Diguet, E.; Kirkeby, A.; Mattsson, B.; Heuer, A.; Bramoulle, Y.; Van Camp, N.; Perrier, A.L.; Hantraye, P.; Bjorklund, A.; et al. Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson’s disease. Cell Stem Cell 2014, 15, 653–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, A.F.; Cardoso, T.; Nolbrant, S.; Mattsson, B.; Hoban, D.B.; Jarl, U.; Wahlestedt, J.N.; Grealish, S.; Bjorklund, A.; Parmar, M. HESC-derived dopaminergic transplants integrate into basal ganglia circuitry in a preclinical model of Parkinson’s disease. Cell Rep. 2019, 28, 3462–3473. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | Associated microRNAs Involved in Oxidative Stress Regulation | References |
|---|---|---|
| Alzheimer’s disease | miR-107 | [72,73,78] |
| miR-125b | [97] | |
| miR-130b | [91] | |
| miR-132/212 | [87,88] | |
| miR-134 | [72] | |
| miR-145 | [72] | |
| miR-146b | [89] | |
| miR-153 | [81] | |
| miR-186 | [79,99] | |
| miR-193b | [91] | |
| miR-200a-3p | [85] | |
| miR-20a | [91] | |
| miR-210 | [72] | |
| miR-219 | [86] | |
| miR-26 | [90] | |
| miR-296 | [91] | |
| miR-329 | [91] | |
| miR-330a | [92] | |
| miR-342-5p | [80] | |
| miR-98-5p | [96] | |
| Parkinson’s disease | miR-106a | [135] |
| miR-124 | [124,144] | |
| miR-133b | [103] | |
| miR-137 | [106] | |
| miR-142-5p | [123] | |
| miR-144 | [123] | |
| miR-146a | [124] | |
| miR-153 | [123,133] | |
| miR-155 | [124,141] | |
| miR-16-1 | [137] | |
| miR-214 | [132] | |
| miR-224 | [135] | |
| miR-26b | [135] | |
| miR-27a/b | [110,111,123] | |
| miR-301b | [135] | |
| miR-320 | [136] | |
| miR-34b/c | [118,134] | |
| miR-373 | [135] | |
| miR-379 | [135] | |
| mir-4639-5p | [117] | |
| miR-491 | [106] | |
| miR-494 | [116] | |
| miR-7 | [121,122,131,133] | |
| ALS | miR-142-5p | [155,163] |
| miR-155 | [157] | |
| miR-27a | [158,165] | |
| miR-338-3p | [162] | |
| miR-34a | [116,154] | |
| Huntington’s disease | miR-124a | [172] |
| miR-128 | [176] | |
| miR-132 | [171,172,176] | |
| miR-29a/b/c | [171,172,176] | |
| miR-330 | [171] | |
| miR-9 | [172] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konovalova, J.; Gerasymchuk, D.; Parkkinen, I.; Chmielarz, P.; Domanskyi, A. Interplay between MicroRNAs and Oxidative Stress in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 6055. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236055
Konovalova J, Gerasymchuk D, Parkkinen I, Chmielarz P, Domanskyi A. Interplay between MicroRNAs and Oxidative Stress in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2019; 20(23):6055. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236055
Chicago/Turabian StyleKonovalova, Julia, Dmytro Gerasymchuk, Ilmari Parkkinen, Piotr Chmielarz, and Andrii Domanskyi. 2019. "Interplay between MicroRNAs and Oxidative Stress in Neurodegenerative Diseases" International Journal of Molecular Sciences 20, no. 23: 6055. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236055