Quercetin Suppresses the Progression of Atherosclerosis by Regulating MST1-Mediated Autophagy in ox-LDL-Induced RAW264.7 Macrophage Foam Cells

Abstract
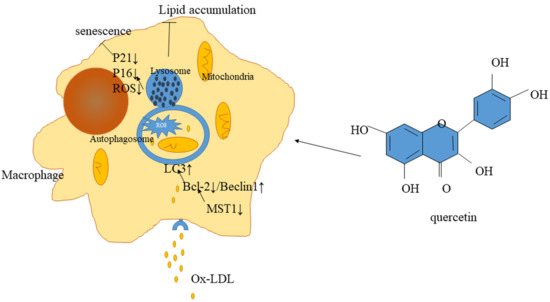
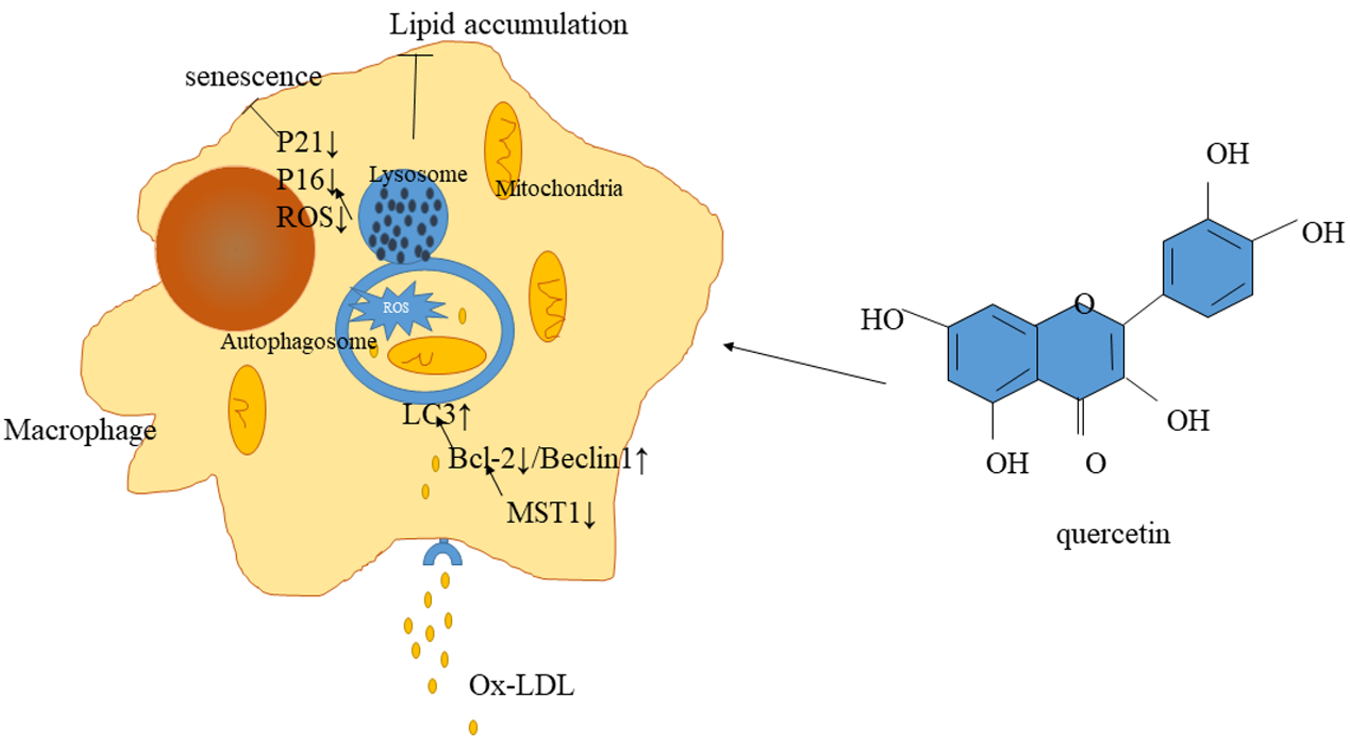
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
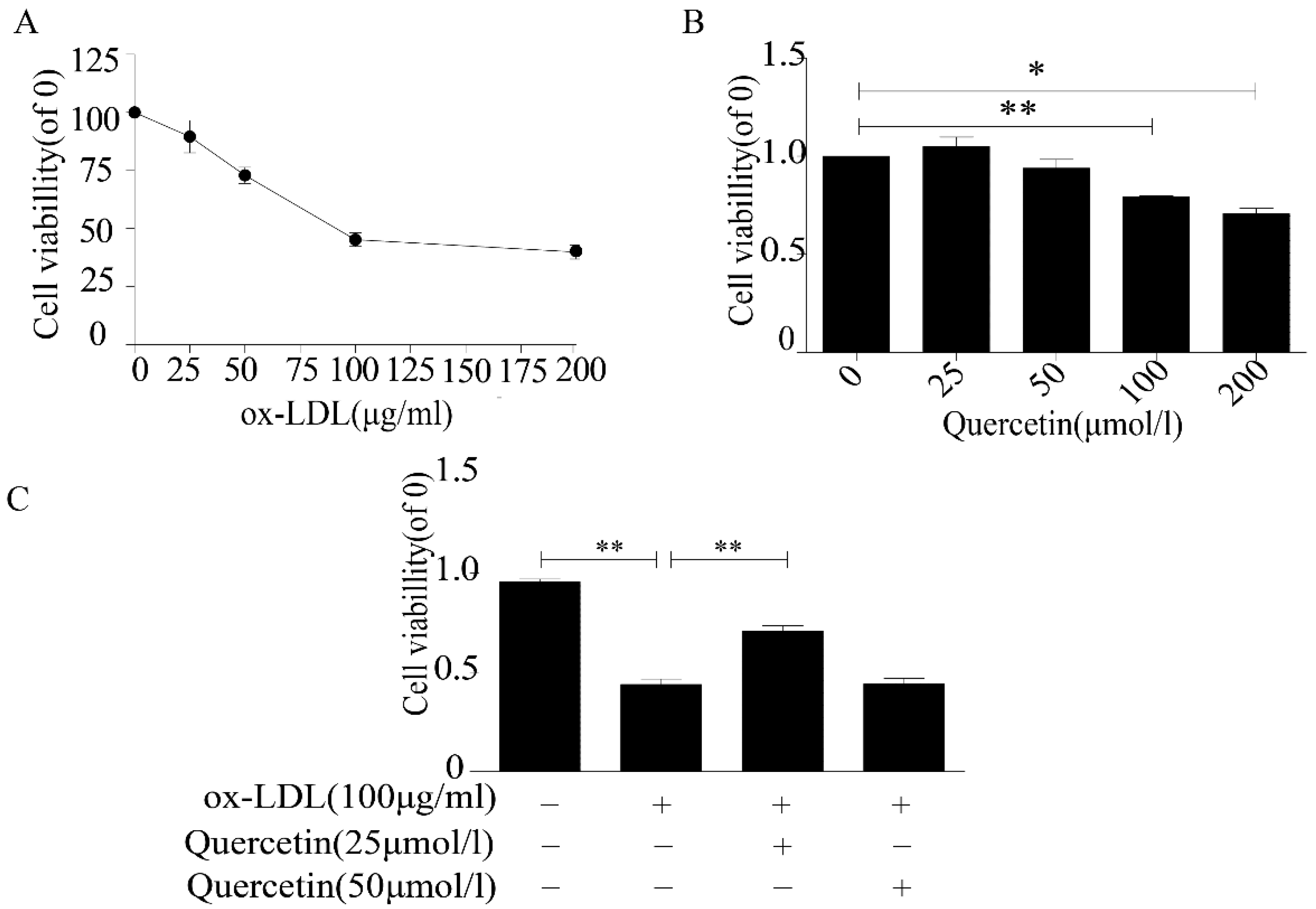
2.1. Quercetin Increased the Viability of RAW264.7 Cells
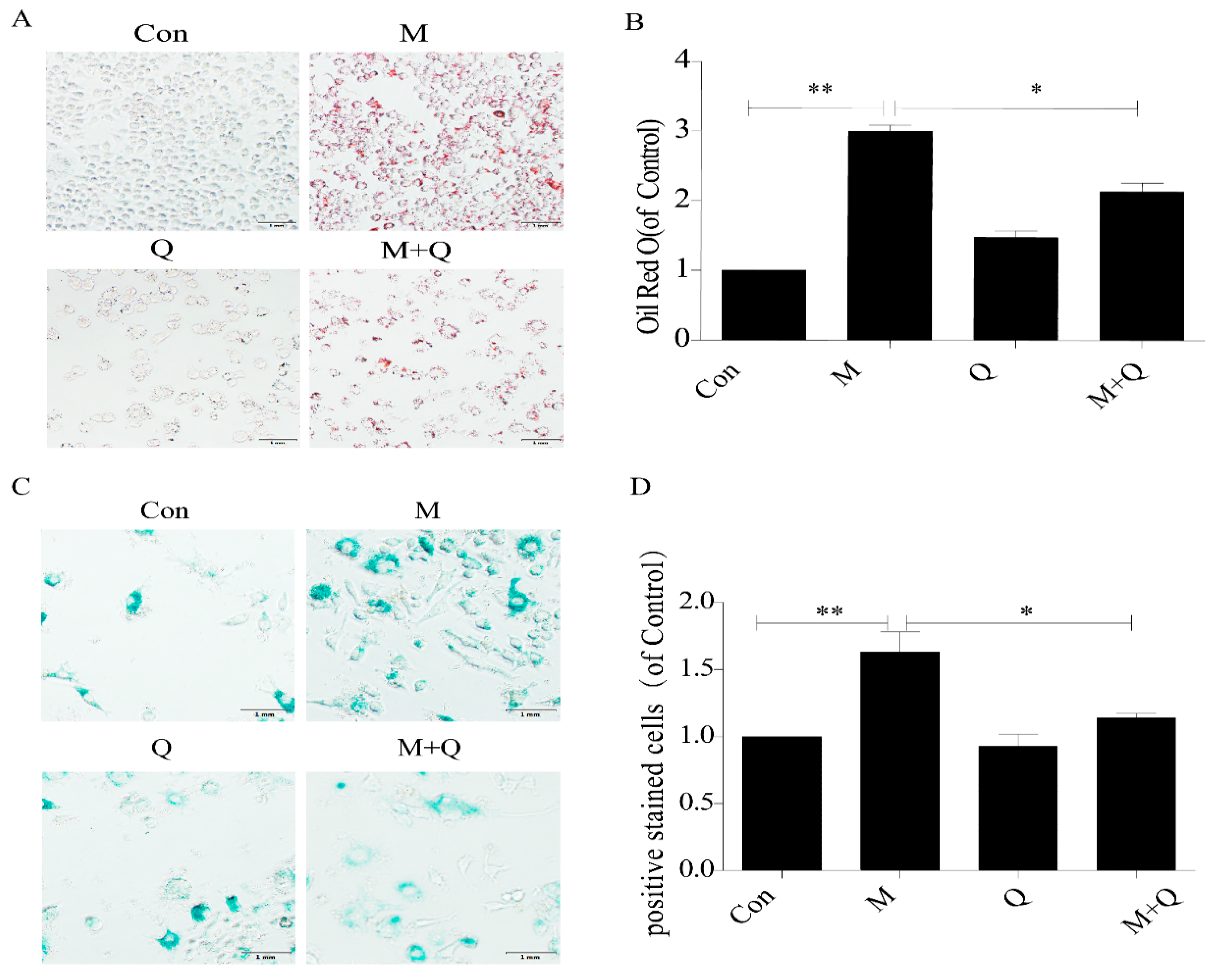
2.2. Quercetin Delayed Senescence and Reduced the Accumulation of Lipid in RAW264.7 Cells
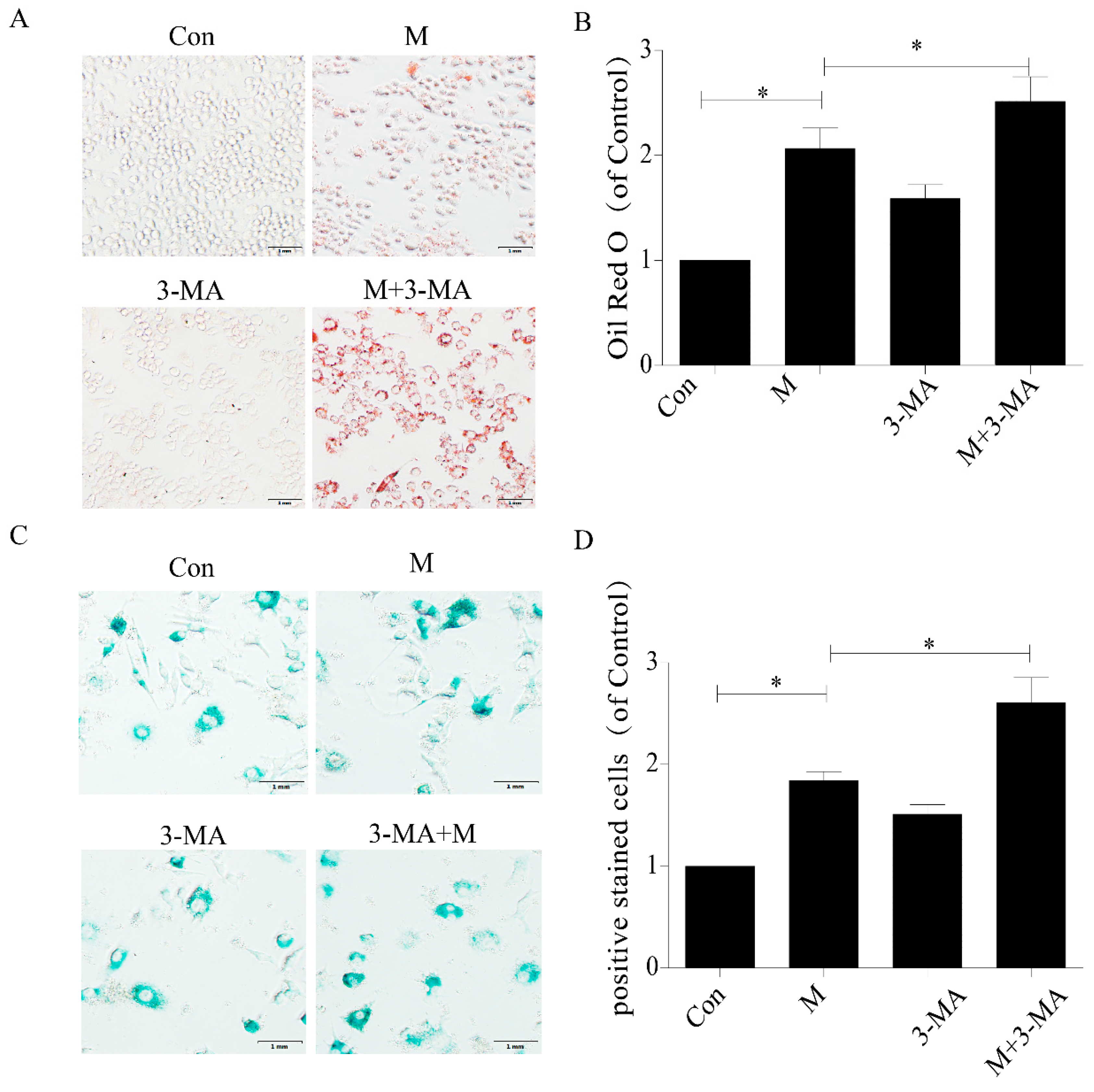
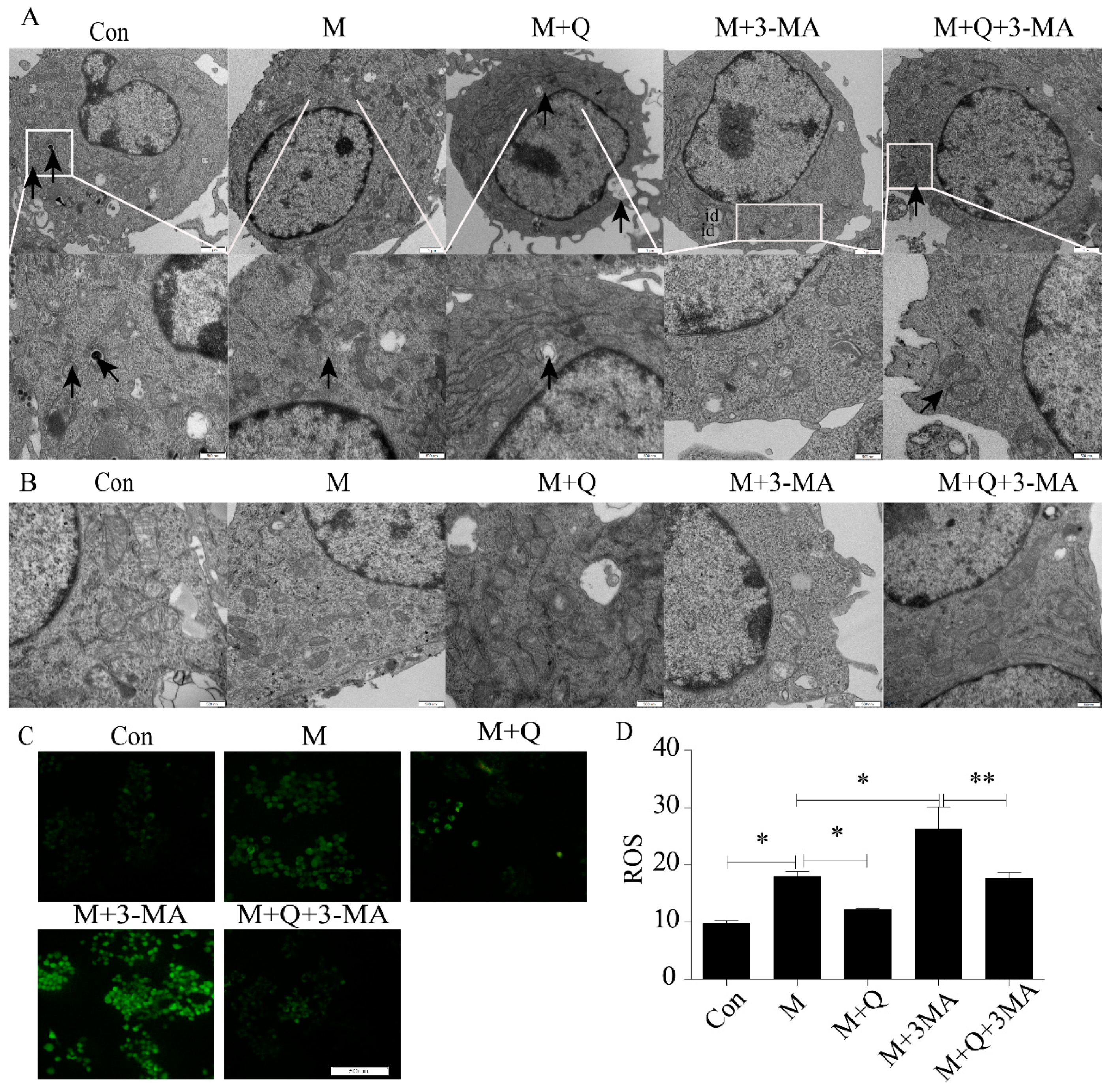
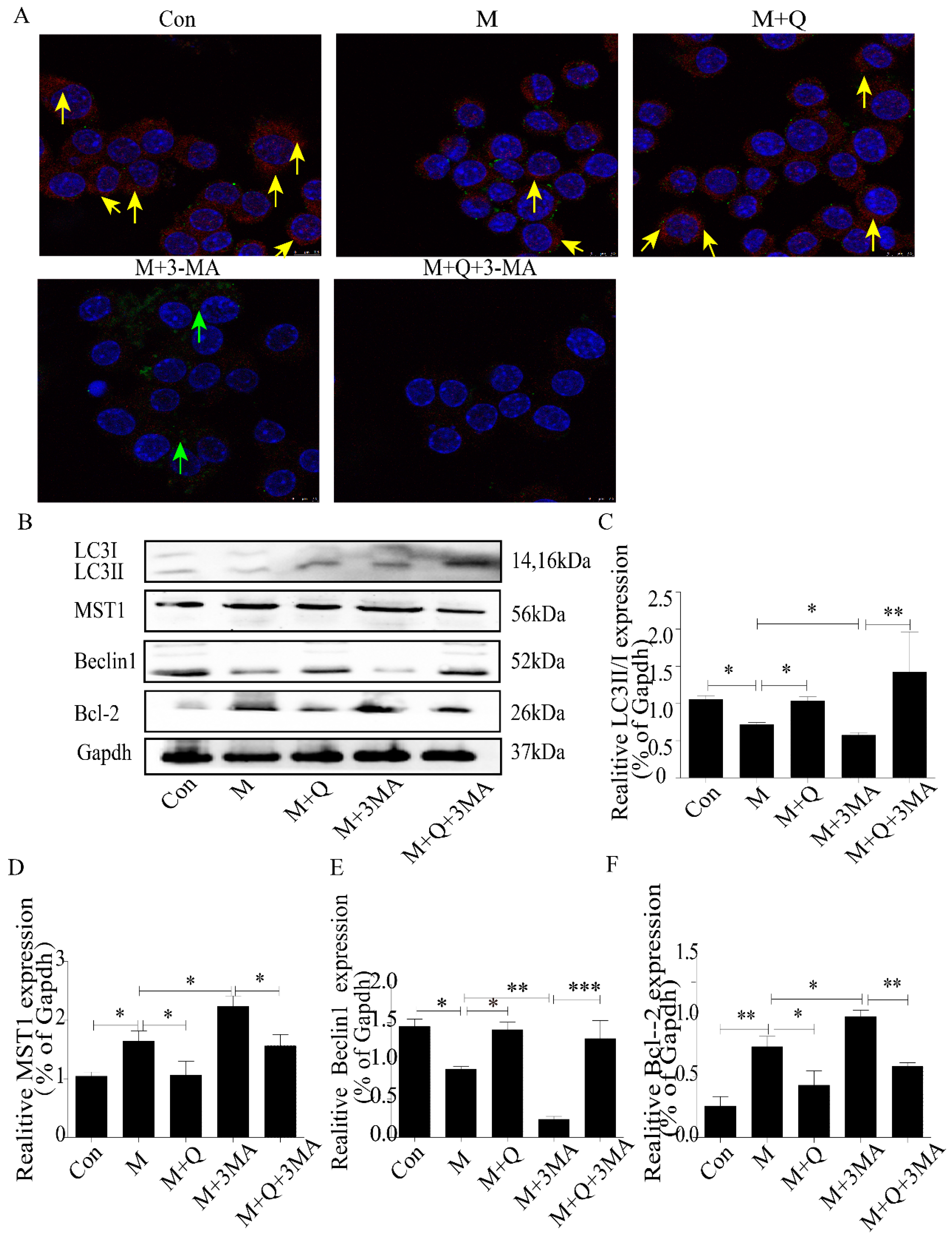
2.3. Inhibition of Autophagy Promoted the Lipid Accumulation and Senescence of RAW264.7 Cells
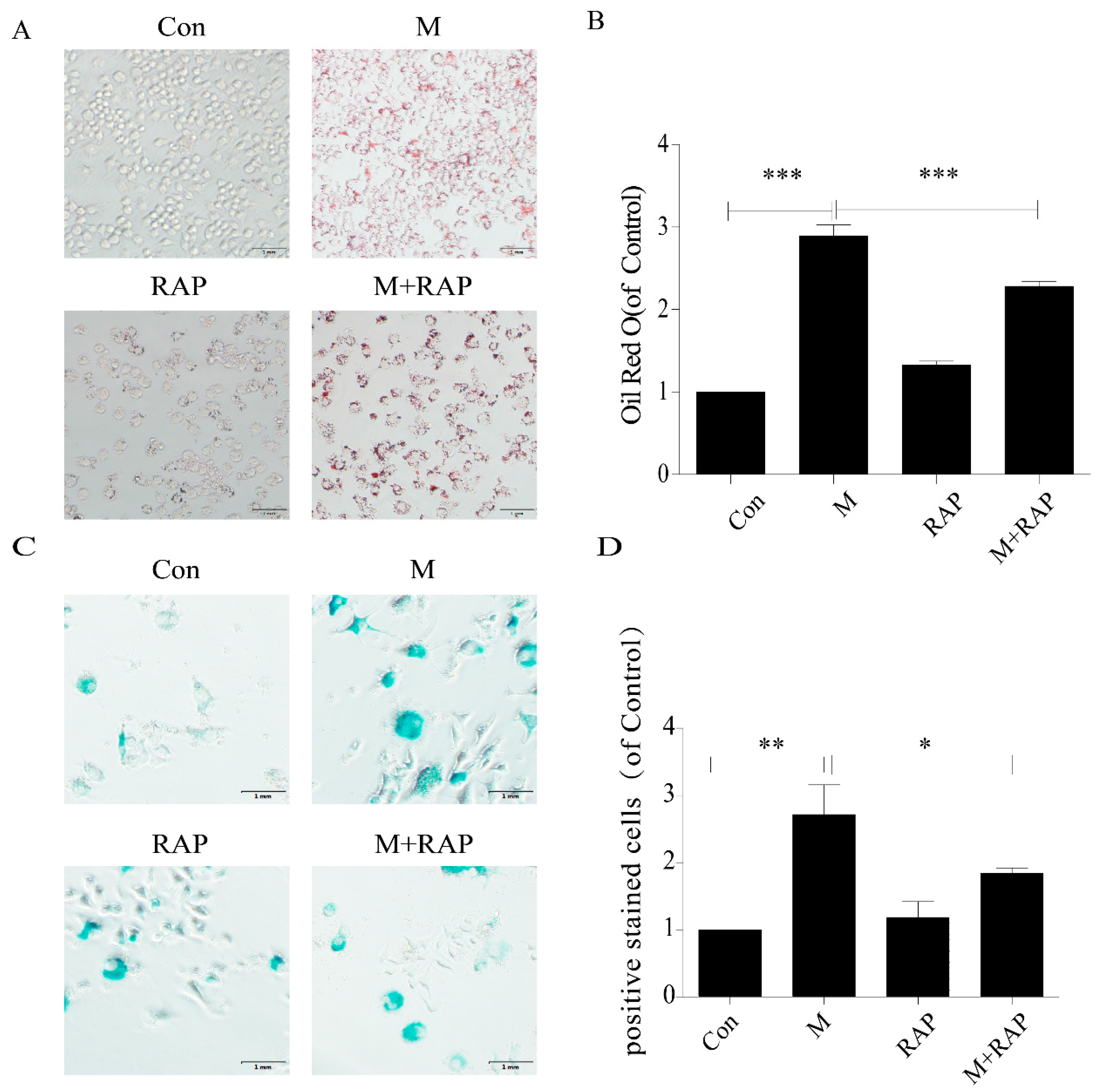
2.4. Promotion of Autophagy Inhibited the Lipid Accumulation and Senescence of RAW264.7 Cells
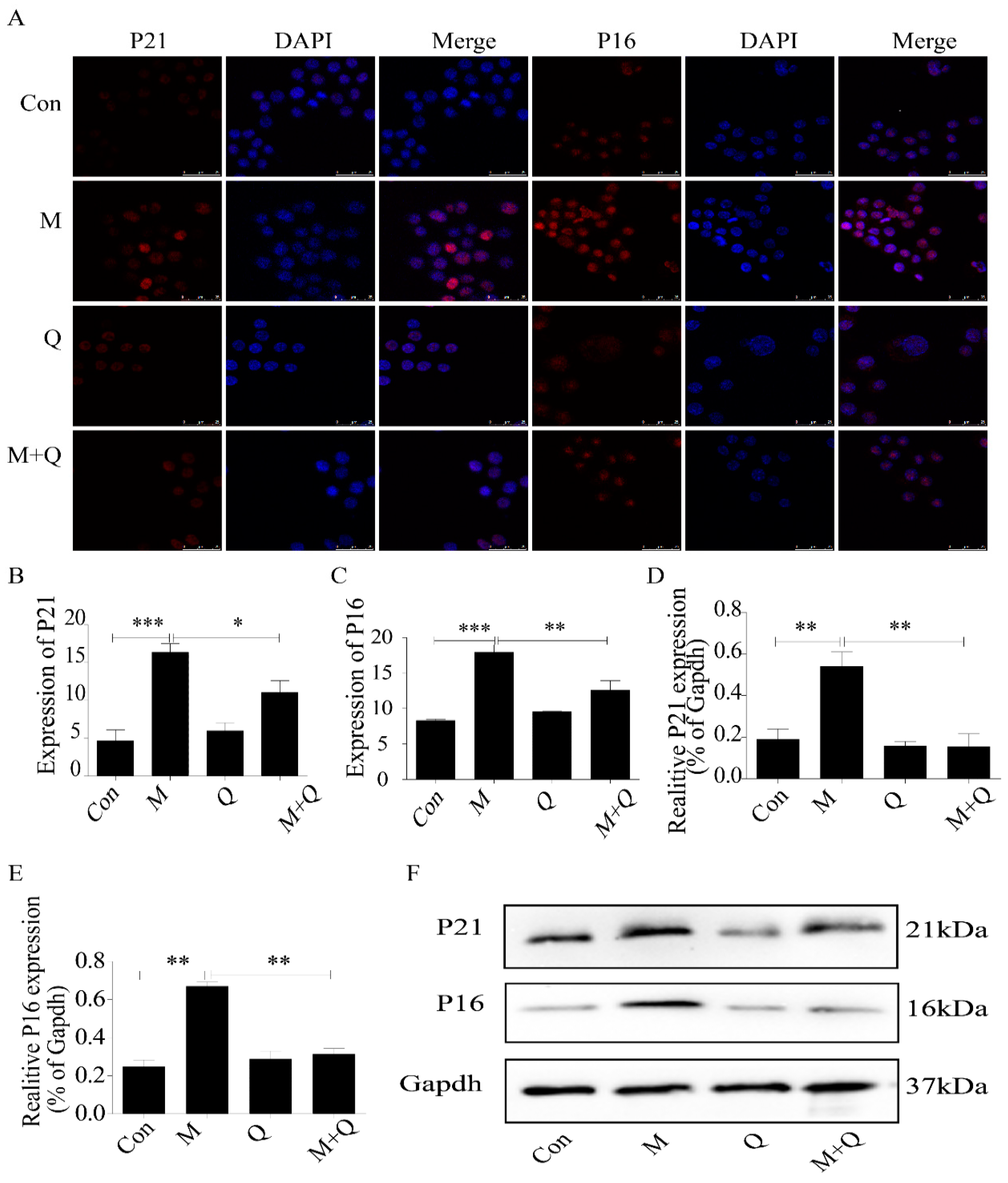
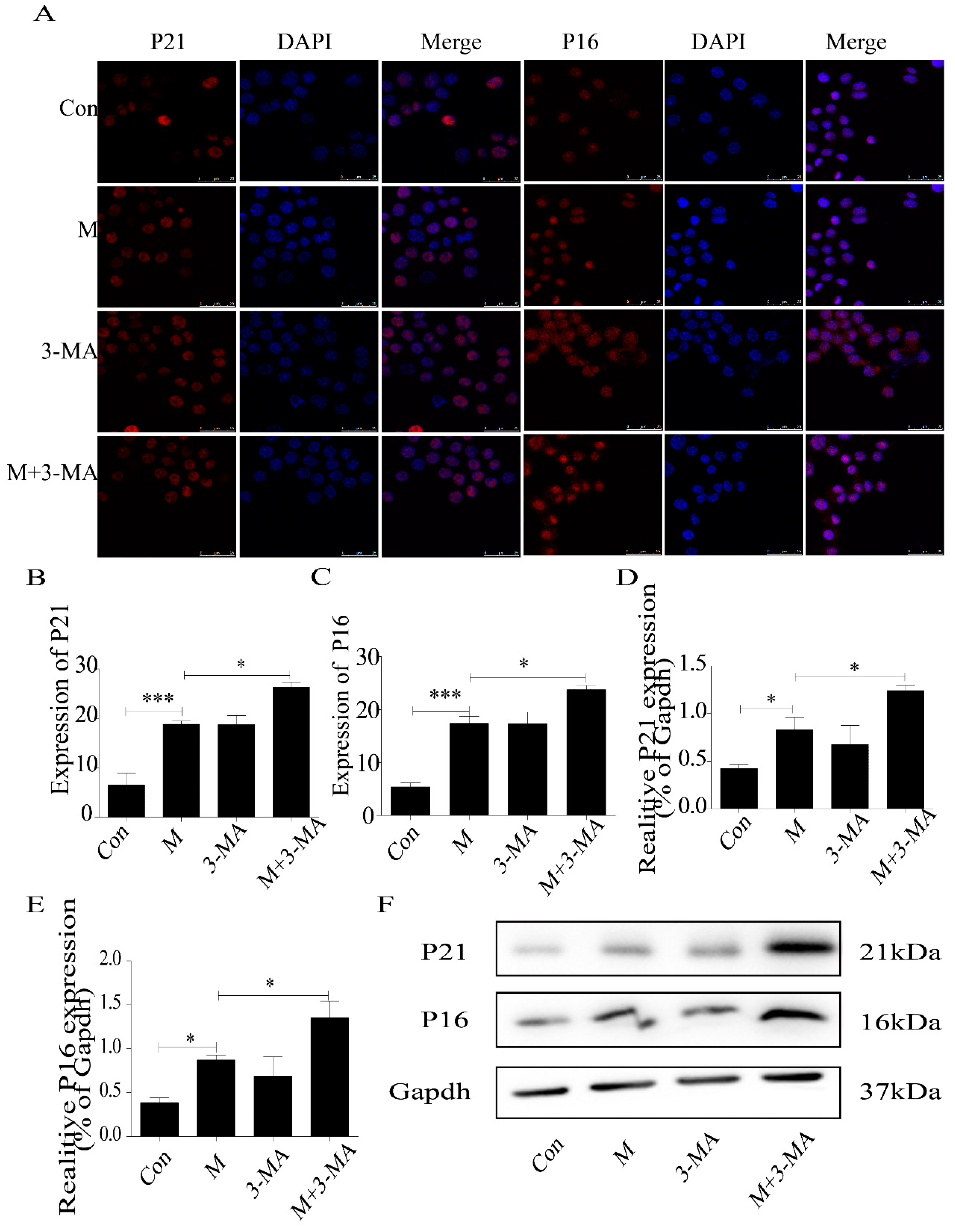
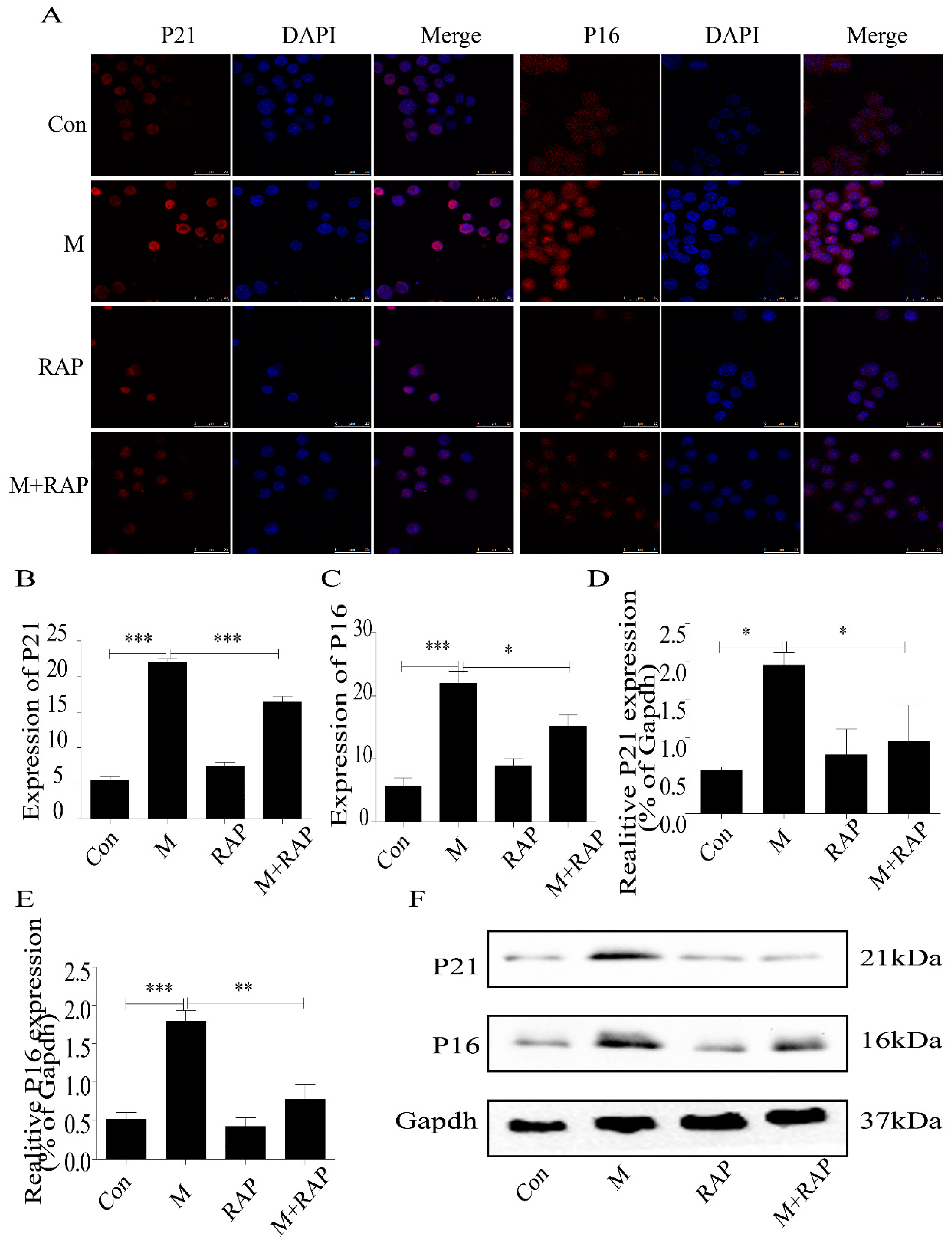
2.5. Quercetin Delayed Senescence, Which Was Associated with MST1-Mediated Autophagy
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. Cell Culture
4.3. Cell Proliferation Assay
4.4. Oil Red O Staining of Cells
4.5. SA-β-gal Staining of Cells
4.6. Transmission Electron Microscopy (TEM) Observation of Autophagosome and Mitochondria
4.7. Reactive Oxygen Species Assay of Cells
4.8. Immunofluorescence Detection of Protein Expression in Macrophages
4.9. Western Blot Detection of MST1, LC3II/I, Beclin1, Bcl-2, P21, and P16 Expression in Macrophages
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AS | Atherosclerosis |
| ABCA1 | ATP Binding Cassette Transporter A1 |
| CCK-8 | Cell Counting Kit-8 |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| FBS | Fetal Bovine Serum |
| LDL-C | Low Density Lipoprotein Cholesterol |
| mTOR | Mammalian rapamycin target protein |
| MST1 | Mammalian Ste20-like Kinase 1 |
| ox-LDL | Oxidized Low-Density Lipoprotein |
| PBS | Phosphate Buffer Saline |
| PBST | Phosphate Buffer Saline-Twen 20 |
| PMS | Phenylmethylsulphonyl Fluoride |
| QUE | Quercetin |
| ROS | Reactive Oxygen Species |
| RAP | Rapamycin |
| RIPA | Radio Immunoprecipitation |
| SA-β-gal | Senescence-Associated β-galactosidase |
| TEM | Transmission Electron Microscope |
| TLR | Toll-like Receptors |
| 3-MA | 3-Methyladenine |
References
- Cicero, A.F.G.; Fogacci, F.; Giovannini, M.; Grandi, E.; D’Addato, S.; Borghi, C. Interaction between low-density lipoprotein-cholesterolaemia, serum uric level and incident hypertension: Data from the Brisighella Heart Study. J. Hypertens. 2019, 37, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jiang, Y.; Song, X.; Guo, C.; Zhu, F.; Wang, X.; Wang, Q.; Shi, Y.; Wang, J.; Gao, F.; et al. Pdcd4 deficiency enhances macrophage lipoautophagy and attenuates foam cell formation and atherosclerosis in mice. Cell Death Dis. 2016, 7, e2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicero, A.F.G.; Kuwabara, M.; Johnson, R.; Bove, M.; Fogacci, F.; Rosticci, M.; Giovannini, M.; D’Addato, S.; Borghi, C. LDL-oxidation, serum uric acid, kidney function and pulse-wave velocity: Data from the Brisighella Heart Study cohort. Int. J. Cardiol. 2018, 261, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Lee, A.S.; Lu, L.S.; Ke, L.Y.; Chen, W.Y.; Dong, J.W.; Lu, J.; Chen, Z.; Chu, C.S.; Chan, H.S.; et al. Human elecyronegative LDL induces mitochondrial dysfunction and premature senescence of vascular cells in vivo. Aging Cell 2018, 17, e12792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Tang, Y.; Cui, Y.; Zhang, H.; Zhang, D. Autophagy is associated with cell fate in the process of macrophage-derived foam cells formation and progress. J. Biomed. Sci. 2016, 23, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buljan, K.; Soldo, S.B.; Janculjak, D.; Kadojic, D.; Candrlic, M.; Bensic, M.; Buljan, V. Relationship between age and thickness of carotid arteries in a population without risk factors for atherosclerosis. Coll. Antropol. 2015, 39, 779–784. [Google Scholar]
- Huang, T.; Rivera-Pérez, J.A. Senescence-associated β-galactosidase activity marks the visceral endoderm of mouse embryos but is not indicative of senescence. Genesis 2014, 52, 300–308. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Liu, H.; Yang, M.; Zhang, Y.; Huang, B.; Zhou, Y. Disc cell senescence in intervertebral disc degeneration: Causes and molecular pathways. Cell Cycle 2016, 15, 1674–1684. [Google Scholar] [CrossRef] [Green Version]
- Dong, W.; Cheng, S.; Huang, F.; Fan, W.; Chen, Y.; Shi, H.; He, H. Mitochondrial dysfunction in long-term neuronal cultures mimics changes with aging. Med. Sci. Monit. 2011, 17, BR91–BR96. [Google Scholar] [CrossRef] [Green Version]
- Tai, H.; Wang, Z.; Gong, H.; Han, X.; Zhou, J.; Wang, X.; Wei, X.; Ding, Y.; Huang, N.; Qin, J.; et al. Autophagy impairment with lysosomal and mitochondrial dysfunction is an important characteristic of oxidative stress-induced senescence. Autophagy 2017, 13, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Conway, O.; Akpinar, H.A.; Rogov, V.; Kirkin, V. Selective autophagy receptors in neuronal health and disease. J. Mol. Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.F.; Sebti, S.; Wei, Y.; Zou, Z.; Shi, M.; McMillan, K.L.; He, C.; Ting, T.; Liu, Y.; Chiang, W.C.; et al. Disruption of the beclin 1–BCL2 autophagy regulatory complex promotes longevity in mice. Nature 2018, 558, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Ogura, Y.; Koya, D. The protective role of Sirt1 in vascular tissue: Its relationship to vascular aging and atherosclerosis. Aging 2016, 8, 2290–2307. [Google Scholar] [CrossRef] [Green Version]
- Ouimet, M.; Ediriweera, H.; Afonso, M.S.; Bamkhelawon, B.; Singaravelu, R.; Liao, X.; Bandler, R.C.; Rahman, K.; Fisher, E.A.; Rayner, K.J.; et al. miR-33 regulates macrophage autophagy in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1058–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grootaert, M.O.; da Costa Martins, P.A.; Bitsch, N.; Pintelon, I.; De Meyer, G.R.; Martinet, W.; Schrijvers, D.W. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy 2015, 11, 2014–2032. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Sluimer, J.C.; Wang, Y.; Subramanian, M.; Brown, K.; Pattison, J.S.; Robbins, J.; Martinez, J.; Tabas, I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012, 15, 545–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.; Zhang, L.; Zhang, M.; Hu, J.; Wang, T.; Duan, Y.; Man, W.; Wu, B.; Feng, J.; Sun, L.; et al. Mst1 inhibits CMECs autophagy and participates in the development of diabetic coronary microvascular dysfunction. Sci. Rep. 2016, 6, 34199. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Zhang, L.; Hu, J.; Duan, Y.; Zhang, M.; Lin, J.; Man, W.; Pan, X.; Jiang, Z.; Zhang, G.; et al. Mst1 participates in the atherosclerosis progression through macrophage autopgahy inhibition and macrophage apoptosis enhancement. J. Mol. Cell. Cardiol. 2016, 98, 108–116. [Google Scholar] [CrossRef]
- Li, S.; Cao, H.; Shen, D.; Jia, Q.; Chen, C.; Xing, S.L. Quercetin protects against ox-LDL-induced injury via regulation of ABCAl, LXR-α and PCSK9 in RAW264.7 macrophages. Mol. Med. Rep. 2018, 18, 799–806. [Google Scholar] [CrossRef] [Green Version]
- Jia, Q.; Cao, H.; Shen, D.; Li, S.; Yan, L.; Chen, C.; Xing, S.; Dou, F. Quercetin protects against atherosclerosis by regulating the expression of PCSK9, CD36, PPARγ, LXRα and ABCA1. Int. J. Mol. Med. 2019, 44, 893–902. [Google Scholar] [CrossRef]
- Zhi, K.; Li, M.; Bai, J.; Wu, Y.; Zhou, S.; Zhang, X.; Qu, L. Quercitrin treatment protectsend othelial progenitor cells from oxidative damage via inducing autophagy through extracellular signal-regulated kinase. Angiogenesis 2016, 19, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Johnston, J.M.; Angyal, A.; Bauer, R.C.; Hanby, S.; Suvarna, S.K.; Baidzajevas, K.; Hegedus, Z.; Dear, T.N.; Turner, M.; Wilson, H.L.; et al. Myeloid Tribbles 1 induces early atherosclerosis via enhanced foam cell expansion. Sci. Adv. 2019, 5, eaax9183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhansali, S.; Khatri, S.; Dhawan, V. Terminalia Arjuna bark extract impedes foam cell formation and promotes apoptosis in ox-LDL-stimulated macrophages by enhancing UPR-CHOP pathway. Lipids Health Dis. 2019, 18, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, H.; Jia, Q.; Shen, D.; Yan, L.; Chen, C.; Xing, S. Quercetn has a protective effect on atherosclerosis via enhancement of autophagy in ApoE−/− mice. Exp. Ther. Med. 2019, 18, 2451–2458. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef] [Green Version]
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent insights into the cellular biology of atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef]
- Bhaskar, S.; Sudhakaran, P.R.; Helen, A. Quercetin attenuates atherosclerotic inflammation and adhesion molecule expression by modulating TLR-NF-kappaB signaling pathway. Cell. Immunol. 2016, 310, 131–140. [Google Scholar] [CrossRef]
- Cui, Y.; Hou, P.; Li, F.; Liu, Q.; Qin, S.; Zhou, G.; Xu, X.; Si, Y.; Guo, S. Quercetin improves macrophage reverse cholesterol transport in apolipoprotein E-deficient mice fed a high-fat diet. Lipids Health Dis. 2017, 16, 9. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Li, E.; Wang, F.; Wang, T.; Qin, Z.; Niu, S.; Qiu, C. Quercetin increases macrophage cholesterol efflux to inhibit foam cell formation through activating PPARγ-ABCA1 pathway. Int. J. Clin. Exp. Pathol. 2015, 8, 10854–10860. [Google Scholar]
- Huang, K.; Li, S.Q.; Wang, W.J.; Liu, L.S.; Jiang, Y.G.; Feng, P.N.; Wang, Y.Q.; Wang, S.M. Oral FTY720 administration induces immune tolerance and inhibits early development of atherosclerosis in apolipoprotein E-deficient mice. Int. J. Immunopathol. Pharmacol. 2012, 25, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Vion, A.C.; Kheloufi, M.; Hammoutene, A.; Poisson, J.; Lasselin, J.; Devue, C.; Pic, I.; Dupont, N.; Busse, J.; Stark, K.; et al. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow. Proc. Natl. Acad. Sci. USA 2017, 144, e8675–e8684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, B.Z.; Han, B.Z.; Zeng, Y.X.; Su, D.F.; Liu, C. The roles of macrophage autophagy in atherosclerosis. Acta Pharmacol. Sin. 2016, 37, 150–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, L.; Zhang, X.; Liu, M.; Liu, X.; Dong, M.; Cheng, J.; Zhang, X.; Zhai, C.; Song, Y.; Lu, H.; et al. Corrigendum: Ginsenoside Rb1 enhances atherosclerotic plaque stability by improving autophagy and lipid metabolism in macrophage foam cells. Front. Pharmacol. 2018, 8, 964. [Google Scholar] [CrossRef] [PubMed]
- Daum, B.; Walter, A.; Horst, A.; Osiewacz, H.D.; Kuhlbrandt, W. Age-dependent dissociation of ATP synthase dimers and loss of inner-membrane cristae in mitochondria. Proc. Natl. Acad. Sci. USA 2013, 110, 15301–15306. [Google Scholar] [CrossRef] [Green Version]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [Green Version]
- Maejima, Y.; Kyoi, S.; Zhai, P.; Liu, T.; Li, H.; Ivessa, A.; Sciarretta, S.; Dei Re, D.P.; Zablocki, D.K.; Hsu, C.P.; et al. MST1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 2013, 19, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Qin, F.; Tian, J.; Zhou, D. MST1 and MST2 kinase: Regulations and disease. Cell Biosci. 2013, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Wang, C.; Croce, C.M.; Guan, J.L. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014, 28, 1204–1216. [Google Scholar] [CrossRef] [Green Version]









© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, H.; Jia, Q.; Yan, L.; Chen, C.; Xing, S.; Shen, D. Quercetin Suppresses the Progression of Atherosclerosis by Regulating MST1-Mediated Autophagy in ox-LDL-Induced RAW264.7 Macrophage Foam Cells. Int. J. Mol. Sci. 2019, 20, 6093. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236093
Cao H, Jia Q, Yan L, Chen C, Xing S, Shen D. Quercetin Suppresses the Progression of Atherosclerosis by Regulating MST1-Mediated Autophagy in ox-LDL-Induced RAW264.7 Macrophage Foam Cells. International Journal of Molecular Sciences. 2019; 20(23):6093. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236093
Chicago/Turabian StyleCao, Hui, Qingling Jia, Li Yan, Chuan Chen, Sanli Xing, and Dingzhu Shen. 2019. "Quercetin Suppresses the Progression of Atherosclerosis by Regulating MST1-Mediated Autophagy in ox-LDL-Induced RAW264.7 Macrophage Foam Cells" International Journal of Molecular Sciences 20, no. 23: 6093. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236093