microRNAs Tune Oxidative Stress in Cancer Therapeutic Tolerance and Resistance


Burnett School of Biomedical Sciences, College of Medicine, University of Central Florida, 6900 Lake Nona Blvd, Orlando, FL 32827, USA
Int. J. Mol. Sci. 2019, 20(23), 6094; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236094
Submission received: 30 October 2019
/
Revised: 26 November 2019
/
Accepted: 27 November 2019
/
Published: 3 December 2019
(This article belongs to the Special Issue Crosstalk between MicroRNA and Oxidative Stress in Physiology and Pathology)
Abstract
:Relapsed disease following first-line therapy remains one of the central problems in cancer management, including chemotherapy, radiotherapy, growth factor receptor-based targeted therapy, and immune checkpoint-based immunotherapy. Cancer cells develop therapeutic resistance through both intrinsic and extrinsic mechanisms including cellular heterogeneity, drug tolerance, bypassing alternative signaling pathways, as well as the acquisition of new genetic mutations. Reactive oxygen species (ROSs) are byproducts originated from cellular oxidative metabolism. Recent discoveries have shown that a disabled antioxidant program leads to therapeutic resistance in several types of cancers. ROSs are finely tuned by dysregulated microRNAs, and vice versa. However, mechanisms of a crosstalk between ROSs and microRNAs in regulating therapeutic resistance are not clear. Here, we summarize how the microRNA–ROS network modulates cancer therapeutic tolerance and resistance and direct new vulnerable targets against drug tolerance and resistance for future applications.
1. Reactive Oxygen Species (ROSs)
There are many types of free radicals including oxygen- and nitrogen-based species. ROSs or reactive oxygen metabolites are free radicals containing oxygen metabolites such as single oxygen, the superoxide anion, hydrogen peroxide, and the hydroxyl radical [1]. ROSs are generated from cellular oxidative metabolism, including mitochondrial oxidative phosphorylation and electron transfer reactions, and optimal levels of ROSs play a pivotal role in many cellular functions [2]. At physiological levels, ROSs are considered signaling molecules or secondary messengers that participate in cell signal transduction, a process known as redox signaling [3]. In addition, the production of ROSs by phagocytic cells is recognized as an important part of innate immunity that kills invading pathogens [4].
The coordination between ROS generation and scavenging ensures that ROS levels are tightly controlled and fine-tuned so as to act as secondary messengers for cell signaling [5]. However, the aberrant production of ROSs, or the failure of the capacity to scavenge excessive ROSs, results in an imbalance in the redox environment of the cell [6]. High levels of ROSs have deleterious effects including nucleic acid (DNA and RNA), lipid, and protein oxidation, as well as membrane destruction by lipid peroxide formation, leading to the development of various diseases such as cancer [7]. Using antioxidant-based strategies [8] to decrease ROS levels or inhibit oxidative damage may prevent ROS-induced cell damage. For example, peroxisome proliferator-activated receptor-gamma coactivator 1 alpha upregulates expression levels of superoxide dismutase enzymes (SOD2/SOD3) and catalase to protect cells from oxidative damage via detoxification and DNA repair [9].
Aberrantly regulated metabolic pathways lead to tumorigenesis [10] and preferential survival of tumor cells [11]. Accumulating evidence suggests that tumorigenesis is dependent on mitochondrial metabolism [12], especially the tricarboxylic acid (TCA) cycle [13]. The TCA cycle is a central pathway in the metabolism of sugars, lipids, and amino acids [14]. Dysregulation of the TCA cycle can induce oncogenesis by activating pseudohypoxia responses, which result in the expression of hypoxia-associated proteins irrespective of oxygen status [15]. For example, succinate accumulation caused by functional loss of the TCA cycle enzyme succinate dehydrogenase complex stabilizes hypoxia-inducible factor (HIF)-1α via inhibition of prolyl hydroxylase (PHD) [16]. In addition, loss of function of the von Hippel–Lindau (VHL) protein [17] also induces pseudohypoxia responses through decreased ubiquitination and proteasomal degradation of HIF-1α [18]. Among the 1158 mitochondrial genes discovered in MitoCarta2.0 (Broad Institute) [19,20], the succinate dehydrogenase complex [21] inclusive of succinate dehydrogenase A [22], succinate dehydrogenase B [23], succinate dehydrogenase C [24], and succinate dehydrogenase D [25], as well as glycine decarboxylase [26,27,28,29] and glutaminase [30], is especially critical for tumorigenesis. Hypoxia, acting through HIF-1α, results in a low production of ROSs and high antioxidant defense in cancers such as leukemia [31]. It suggests that targeting key enzymes of hypoxia metabolism pathways might provide a new way to eradicate tumor formation [32].
2. microRNAs (miRNAs)
miRNAs are important regulators of mRNA expression [33] and play critical roles in regulating tumor initiation and progression [34]. Importantly, single miRNAs have been shown to regulate entire cell signaling networks in a cell-context dependent manner [35] and may also be utilized as biomarkers [36,37,38] for both invasive [39,40] and non-invasive [41,42,43] detection. Dysregulated expressions of miRNAs may function as oncogenes (oncomiRs) [44] such as miR-21 [45], miR-31 [46], miR-155 [47,48], and miR-10b [49] or as tumor suppressors such as let-7 [50] and miR-34 [51,52] in many cancers.
ROSs are finely tuned by dysregulated miRNAs, and vice versa. Many studies are focused on regulatory interactions between miRNAs and ROSs attributing to oxidative stress-related tissue [53]. It is important for a well-regulated cellular ROS level, and miRNAs fill in the role of maintaining this homeostasis. A dysregulation of normal physiological miRNA levels can thus lead to oxidative damage and the development of diseases such as cancer. For example, oncogenic miR-21 enhances both KRAS [54] and epidermal growth factor receptor (EGFR) signaling [55] and promotes tumorigenesis through stimulation of mitogen-activated protein kinase (MAPK)-mediated ROS production by downregulation of SOD2/SOD3 [56]. On the other hand, oxidative stress can alter the expression level of many miRNAs [57,58,59]. For instance, oxidative stress such as hydrogen peroxide elevates miR-34a with concomitant reduction of sirtuin-1 and sirtuin-6 in bronchial epithelial cells [60], which is associated with chronic obstructive pulmonary disease and tumorigenesis [61]. However, oxidative stress decreases expression levels of the let-7 family [62] in a p53-dependent manner in a variety of tumor cells [63]. These findings suggest that ROSs may exert a pivotal role in the regulation of microRNA expression in a cell-context-dependent manner.
miRNA-based monotherapy has not been developed well in clinical settings [64,65,66]. For example, a first-in-man, phase 1 clinical trial of miR-16-loaded nanoparticles as a treatment for recurrent malignant pleural mesothelioma patients has been completed [67]. Delivery of tumor suppressive miR-16 in 22 patients led to 5% objective response, 68% stable disease, and 27% progressive disease. Possible mechanisms of low objective response include miRNA sequestration through leaky cancer blood vessels as well as endocytosis by cancer cells [68]. Nevertheless, miR-16 expression levels in patients should be detected prior to receiving miR-16 treatment in future clinical trials [69]. Furthermore, miRNA-based treatment may combine with other current or potential therapeutics in combating cancer [70,71]. In addition, increasing evidence has revealed that miRNAs can be directly linked to therapeutic resistance in some cancers. For instance, overexpressing miR-205 sensitizes radioresistant breast cancer cells to radiation in a xenograft model [72]. Similarly, administration of miR-24 sensitizes radioresistant nasopharyngeal carcinoma cells to radiation in vitro [73]. miRNA-mediated regulation of signaling pathways involved in tumorigenesis as well as therapeutic tolerance and resistance is summarized in Table 1. It is revealed that miRNAs may serve both as drug targets and as therapeutic agents to eradicate cancer cells and sensitize therapeutic resistant cells [74].
3. Therapeutic Tolerance and Resistance
The discovery of genetic mutation on tyrosine kinase, such as EGFR mutations including exon 19 deletion (Del19) and exon 21 Leu858Arg substitution (L858R), that confer sensitivity to EGFR-targeted tyrosine kinase inhibitors in lung adenocarcinomas heralded the beginning of the era of precision medicine for lung cancer [91,92]. However, the success of EGFR-based therapy was compromised by therapeutic resistance following initial treatment response in most cancer patients [93]. Exon 20 Thr790Met substitution (T790M), affecting the ATP binding pocket of the EGFR kinase domain, accounts for approximately half of all lung cancer cases with acquired resistance to the current first generation EGFR tyrosine kinase inhibitors, erlotinib and gefitinib [94]. In erlotinib- and gefitinib-resistant lung tumors with EGFRT790M, rociletinib and osimertinib are highly active [95]. However, resistance to the third generation EGFR tyrosine kinase inhibitor osimertinib is now emerging clinically [96]. In addition to genetic mutations, intratumor heterogeneity also drives neoplastic progression and therapeutic resistance [97]. Recently, it has been found that EGFRT790M-positive drug-resistant cells are derived from EGFRT790M-negative drug-tolerant persister cells that survive initial EGFR tyrosine kinase inhibitors treatment [98,99]. It is therefore crucial to identify molecular changes that drive drug tolerance.
Consistently, Zhang et al. have revealed that lung tumor cells protect themselves with a drug-tolerance mechanism when the cells are treated with osimertinib [76]. These findings align with previous data showing that tumor cells enter into a tolerant state when they are treated with tyrosine kinase inhibitors in lung and other cancers [100,101,102]. These tolerant persister cells precede and evolve into resistant cells over time by acquiring EGFR-resistant mutations [98,99]. These tolerant cells are slow cycling and are enriched in the expression of stem-associated genes in the WNT/planar cell polarity signaling pathway, such as WNT5A, FZD2, and FZD7. These findings are conceptually similar to a recent report that post-drug transition to stable resistance consists of dedifferentiation [103].
Excessive ROSs produced by damaged mitochondria can trigger mitophagy, a process that can scavenge impaired mitochondria and reduce ROS levels to maintain a stable mitochondrial function in cells [104]. Therefore, mitophagy helps maintain cellular homeostasis under oxidative stress. For example, protein kinase inhibitor sorafenib shows activities against many protein kinases, including vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), and rapidly accelerated fibrosarcoma (RAF) kinases [105]. Resistance to sorafenib in cancers such as hepatocellular carcinoma is frequent [106] partially due to antiangiogenic effects-mediated hypoxia [107]. Administration of tryptophan-derived metabolites such as melatonin [108] increased ROS production and mitophagy, resulting in increased sensitivity to sorafenib in hepatocellular carcinoma cells [109]. Additionally, melatonin downregulated the HIF-1α protein synthesis through inhibition of the mammalian target of rapamycin complex 1 (mTORC1)-mediated pathway [110]. Most recently, it was shown that drug-tolerant persister cancer cells were vulnerable to inhibition of the glutathione peroxidase 4, owing to a disabled antioxidant program [102]. It suggests that increasing ROS levels may re-sensitize therapeutic resistant cancer cells to current treatments.
4. miRNA–ROS Interaction Regulates Therapeutic Tolerance/Resistance at the Phenotypic Level
The miRNA–ROS network in a scenario of therapeutic tolerance/resistance is grouped at three levels including phenotype, signaling/metabolism, and genetics/epigenetics (Figure 1). Phenotypic changes include the enrichment of tumor-initiating cells, the histological transformation from EGFR-mutant non-small cell lung cancer to small cell lung cancer, and epithelial–mesenchymal transition resulting in therapeutic tolerance/resistance.
4.1. Enrichment of Tumor-Initiating Cells
Therapeutic resistance is frequent after primary and adjuvant cancer therapy, often evolving into a lethal relapse disease [111]. These observations may be attributed to the highly heterogeneous nature of tumors that contain distinct tumoral and microenvironment cells, all of which contribute in varying degrees toward self-renewal, drug resistance, and relapse [112]. The tumor-initiating cell or cancer stem cell model provides one explanation for the phenotypic and functional diversity among cancer cells in some tumors [113]. Tumor-initiating cells have been demonstrated to be more resistant to conventional therapeutic interventions [114] and are key drivers of relapse in many types of cancers including leukemia [115], lung cancer [116], breast cancer [117], brain cancer [118], colon cancer [119], and nasopharyngeal carcinoma [120]. There is, therefore, increasing interest in developing strategies that can specifically target tumor-initiating cells with novel and emerging therapeutic modalities, thereby halting cancer progression and improving disease outcome [121]. Tumor-initiating cells protect their genomes from ROS-mediated damage [122] via increased production of free radical scavengers [123] leading to low ROS levels [124]. Thus, heterogeneity of ROS levels in cancers such as glioma may influence the extent to which tumor-initiating cell-enriched populations are resistant to therapies such as ionizing radiation [125]. Tumor-initiating cells display heterogeneous phenotypes due to different genotypes in tumors [126]. Thus, the genetic backgrounds, such as mutant EGFR and RAS, need to be taken into consideration to better understand the association between tumor-initiating cells and therapeutic resistance in the future.
In non-small cell lung cancer, a panel of tumor-initiating cell-relevant miRNAs is enriched when assessed by a miRNA microarray [75]. Those top upregulated miRNAs include miR-1290 and miR-1246 (Table 1). The top downregulated miRNAs comprise miR-23a and let-7b/c/d/i. Further analysis showed that miR-1246 and miR-1290 regulate tumor-initiating cells via repressing cysteine-rich metal-binding proteins (metallothioneins) [75]. The reduced expression of metallothioneins has been implicated as biomarkers of low ROSs, which is consistent with the previous finding that pharmacological anti-oxidants such as N-acetyl cysteine or the knock-down of nuclear respiratory factor 2 (NRF2) prevented the induction of metallothionein-1 induced by tyrosine kinase inhibitor sorafenib [127]. Another direct target of miR-1290, glioma pathogenesis-related protein 1, promotes apoptosis through upregulating ROS production by activating the c-Jun-NH(2) kinase signaling cascade in cancer cells [128]. Other evidence has shown that extracellular miR-1246 could enhance radioresistance of lung cancer cells [129]. In addition, miR-21 is enriched in tumor-initiating cells in many types of cancers such as gastric and breast cancers [130]. Functional loss of miR-21 reduces a frequency of tumor-initiating cells, consistently with decreased capacity of therapeutic resistance against EGFR tyrosine kinase inhibitors [82] (Table 1). Whether these miRNAs regulate ROSs resulting in therapeutic tolerance and resistance still needs further study. Thus, targeting enriched tumor-initiating cells might overcome miRNA–ROS-mediated therapeutic tolerance/resistance.
4.2. Small Cell Lung Cancer Transformation
Small cell lung cancer is a highly aggressive disease that exhibits rapid growth and genetic instability including inactivated tumor suppressor retinoblastoma 1 (RB1) and amplified MYC proto-oncogene (MYC) [131]. Histologic transformation of EGFR mutant non-small cell lung cancer to small cell lung cancer is an important mechanism of resistance to EGFR tyrosine kinase inhibitors that occurs in approximately 3–10% of EGFR mutant non-small cell lung cancers [132]. Transformation to small cell lung cancer occurs in a subpopulation of EGFR mutant non-small cell lung cancer patients and is frequently associated with mutant RB1, TP53, and PIK3CA [133,134]. Future studies might help define which subsets of non-small cell lung cancer are most prone to small cell lung cancer transformation.
Frequent overexpression of the miR-17~92 cluster in small cell lung cancer [135] is a fine-tuner to reduce excessive ROS-induced DNA damage in RB1-inactivated small cell lung cancer cells [136]. Therefore, miR-17~92 may be excellent therapeutic target candidates to overcome small cell lung cancer transformation.
4.3. Epithelial–Mesenchymal Transition
An epithelial–mesenchymal transition is a biologic process that allows a polarized epithelial cell to undergo multiple biochemical changes that enable it to assume a mesenchymal cell phenotype, which includes increased resistance to apoptosis [137]. Epithelial–mesenchymal transition is tightly regulated by microRNAs. For example, downregulation of miR-200 family members is linked to enhanced epithelial–mesenchymal transition and tumor-initiating cell acquisition [138,139] in many cancers [140]. Reduced miR-200s directly increase p38α [141], leading to decreased levels of ROSs and subsequent inactivation of the NRF2 oxidative stress response pathway [142]. The decreased ROSs, in turn, inhibit expression of the miR-200s [143], thus establishing a miR-200s-activated stress signature, which strongly correlates with shorter patient survival caused by chemotherapeutic resistance. In addition, miR-30b/c and miR-222 mediate gefitinib-induced apoptosis and the epithelial–mesenchymal transition leading to therapeutic resistance in non-small cell lung cancer [87]. These discoveries collectively indicate potential roles of the miRNA family in the regulation of ROS homeostasis in tumor-initiating cells and therapeutic resistance.
5. miRNA–ROS Interaction Regulates Therapeutic Tolerance/Resistance at a Signaling/Metabolic Level
5.1. HIF-miR-210-ROS
Under hypoxic conditions, upregulated HIF-1α directly binds to a hypoxia-responsive element on the proximal miR-210 promoter and induces miR-210 expression in cancer cells [144]. miR-210 activates generation of ROSs [145] via suppressing iron–sulfur cluster assembly enzyme [146,147] and cytochrome c oxidase assembly protein [148] in the mitochondria electron transport chain and the TCA cycle. miR-210 knockdown decreased resistance to radiotherapy in hypoxic glioma stem cells and hepatoma cells [149,150]. These discoveries suggest that the HIF-miR-210-ROS [151] pathway might be a target to overcome therapeutic resistance (Figure 1).
5.2. EGFR-miR-147b-VHL-TCA Cycle
Increasing evidence suggests that the metabolic enzymes and the catalyzed metabolites, such as isocitrate dehydrogenase, succinate dehydrogenase, and succinate [16,152] in the TCA cycle, are involved in not only tumorigenesis but also therapeutic resistance. A hypoxia response is linked to tumor cell survival and drug-resistance in many cancers [153,154]. Dysregulated cancer metabolism has recently gained attention for its potential role in promoting therapeutic resistance by a therapeutic tolerance strategy in a novel manner [102]. Furthermore, Zhang et al. discovered that lung cancer cells adopt a tolerance strategy to protect from EGFR tyrosine kinase inhibitors by modulating miR-147b-dependent pseudohypoxia signaling pathways [76]. The study revealed that VHL [155] and succinate dehydrogenase play roles in tolerance-mediated cancer progression. Decreasing miR-147b and reactivation of the TCA cycle pathway provides a promising strategy to prevent therapeutic tolerance-mediated tumor relapse (Figure 1).
In addition, VHL regulates Akt activity [156], suggesting that miR-147b-VHL axis might confer therapeutic tolerance through activating Akt activity. In addition, other upstream transcription factors such as the inhibitor of DNA binding 2 might regulate VHL levels [157]. The interaction between miR-147b and other transcription factors controlling VHL needs to be investigated in the future.
Furthermore, the reciprocal changes of metabolites in the TCA cycle such as increased levels of succinate and 2-oxoglutarate (also known as α-ketoglutarate) [158] as well as decreased levels of malate and fumarate in osimertinib-tolerant cells indicate that silenced activity for succinate dehydrogenase is linked to therapeutic tolerance. In addition, small molecule inhibitor R59949 silencing succinate dehydrogenase activity enhances therapeutic tolerance, which is comparable to the function of miR-147b overexpression in tolerant persister cells. It is not surprising that accumulated succinate due to a loss of function of succinate dehydrogenase could activate the pseudohypoxia signaling pathway by repressing PHD2 as reported previously [16]. This is consistent with the findings that the miR-147b/succinate dehydrogenase axis could increase the gene expression for pseudohypoxia signaling pathways. In addition to inactivated VHL and succinate dehydrogenase, other factors such as reduced nicotinamide adenine dinucleotide (NAD+) and decreased glutathione [159] might also activate pseudohypoxia responses leading to therapeutic tolerance. In addition, these pseudohypoxia responses may further perturb the TCA cycle and cooperatively regulate therapeutic tolerance.
5.3. Myc-miR-23a/b-Glutaminase-ROS
Cancer cells depend on both glycolysis and glucose oxidation to support their growth [162,163] as well as glutaminolysis that catabolizes glutamine to generate ATP and lactate [164]. Oncogenic c-Myc represses miR-23a and miR-23b, resulting in increased levels of mitochondrial glutaminase in cancer cells [30]. Glutaminase converts glutamine to glutamate, which is further catabolized through the TCA cycle for the production of adenosine triphosphate (ATP) or serves as substrate for glutathione synthesis [165]. Glutamine withdrawal or glutaminase knockdown resulted in increased levels of ROSs. Thus, the Myc-miR-23-glutaminase axis provides a new mechanism for regulating ROS homeostasis in cancer cells. Considering that downregulated miR-23a is enriched in tumor-initiating cells [75], it is of great interest to explore a link between miR-23 and ROSs in therapeutic tolerance/resistance (Figure 1).
6. miRNA–ROS Interaction Regulates Therapeutic Tolerance/Resistance at a Genetic/Epigenetic Level
6.1. Mutant miRNAs
The whole genome sequencing analysis of lung adenocarcinomas showed noncoding somatic mutational hotspots near vacuolar membrane protein 1/MIR21 [166]. Samples harboring indels or single nucleotide variants in this locus demonstrated significantly higher levels of MIR21 expression. miR-21 high levels are linked to therapeutic resistance to several treatments, including EGFR tyrosine kinase inhibitors [167] and chemotherapeutic agents [168]. Thus, it is valuable to predict therapeutic response by detecting the sequence of miR-21 in biopsies from cancer patients before they receive treatments such as EGFR tyrosine kinase inhibitors (Figure 1).
6.2. RNA Editing
Adenosine deaminases acting on RNA (ADARs) convert adenosine to inosine in double-stranded RNA including both protein-coding [169] non-coding RNAs [170]. ADAR editase activation has been associated with progression of a broad array of malignancies including therapeutic resistance [171]. ADAR1 promotes tumor-initiating cell activity [172] and resistance to BCR-ABL1 inhibitor or janus kinase 2 inhibitor in chronic myeloid leukemia through inactivating biogenesis of the let-7 [173] or pri-miR-26a maturation [174]. In addition, most cancer patients either do not respond to the immune checkpoint blockade or develop resistance to it, often because of acquired mutations [175] that impair antigen presentation [176]. Loss of function of ADAR1 in tumor cells profoundly sensitizes tumors to immunotherapy and overcomes resistance to the programmed cell death protein 1 (PD-1) checkpoint blockade [177]. It is of interest to further study how the ADAR-miRNA axis regulates therapeutic tolerance/resistance through controlling potential genes encoding ROS scavengers [178] such as Drosophila homolog of the mammalian protein thioredoxin-1 and cytochrome P450 4g1 (Figure 1).
6.3. RNA m6A Modification
N6-methyladenosine (m6A) modification of mRNA (RNA m6A modification) is the most abundant RNA modification in eukaryotes and highly conserved among multiple species [179]. RNA m6A modification is emerging as an important regulator of gene expression that affects different developmental and biological processes [180], and altered m6A homeostasis is linked to cancer [181,182,183]. RNA m6A modification is catalyzed by the dynamic regulation of methyltransferases and demethylases. Methyltransferase include methyltransferase-like 3 (METTL3), METTL14, and Wilms’ tumor 1-associating protein, and the demethylases include fat mass- and obesity-associated protein and ALKB homolog 5 [184]. Upregulation of METTL3 is associated with poor prognosis in tumorigenesis and increased chemo- and radio-resistance in cancers such as glioblastomas [185] and pancreatic cancer [186]. Developing resistant phenotypes during tyrosine kinase inhibitor therapy is controlled by m6A modification [187]. Leukemia cells with mRNA m6A demethylation are more tolerant to tyrosine kinase inhibitor treatment. Recovery of m6A methylation re-sensitizes therapeutic resistant cells towards tyrosine kinase inhibitors. The findings identify a novel function for the m6A methylation in regulating reversible tyrosine kinase inhibitor-tolerance state, providing a mechanistic paradigm for drug resistance in cancer. In addition, METTL3 plays roles in the maturation process of miRNAs against ROSs in an m6A-dependent manner [188]. For example, METTL3-mediated miR-873 upregulation controls the kelch-like ECH associated protein 1 (KEAP1)-NRF2 [142] pathway against ROSs. These studies revealed that RNA m6A might regulate therapeutic tolerance/resistance through miRNA–ROS pathways (Figure 1).
7. Emerging Fields and Tools in Preventing and Overcoming Therapeutic Tolerance/Resistance
7.1. Artificial Intelligence (AI)
AI is an area of computer science that emphasizes the creation of intelligent machines that work and react like humans and that uses labeled big data along with markedly enhanced computing power and cloud storage [189]. The most common applications of AI in drug treatment have to do with matching patients to their optimal drug or combination of drugs, predicting drug–target or drug–drug interactions and optimizing treatment protocols [190]. AI-based models have been developed for predicting synergistic treatment combinations in many diseases such as infectious diseases [191] and cancers [192,193]. One challenge is determining how AI-based technology may design tools which improve identification of therapeutic tolerance and resistance and develop new treatment combinations against tolerant and resistant cancers. The success of this AI-based approach may provide earlier and targeted anticancer treatment, which would prevent therapeutic tolerance/resistance emerging and cure cancer patients more effectively (Figure 1).
7.2. Pathogens
Pathogens such as microbiomes and viruses are becoming increasingly recognized for their effects on tumorigenesis and therapeutic resistance to cancer treatment [194]. Bacterial dysbiosis accompanies carcinogenesis in several malignancies such as gastric [195], colon [196], liver [197], and pancreatic [198] cancers by affecting metabolism and impairing immune functions [199]. Additionally, fungi [200] and viruses [201] also induce carcinogenesis in several cancers. Furthermore, intratumoral bacteria induced therapeutic resistance through breakdown of chemotherapy gemcitabine into inactive metabolites via bacterial enzymes such as cytidine deaminase [202] and via impairing response to immune checkpoint blockade [198]. Gut microbiota plays a critical role in mediating colorectal cancer chemoresistance in response to chemotherapeutics via a selective target loss of miR-18a* (miR-18a-3p) and miR-4802, and via activation of the autophagy pathway [203]. In addition, miR-18a* is a tumor suppressor that inhibits KRAS expression [204]. Activating KRAS mutations confer both primary [205] and acquired [206] resistance to anti-EGFR cetuximab therapy in colorectal cancer. Thus, targeting intratumoral pathogens provide a new angle in cancer treatment to overcome therapeutic tolerance/resistance. Some intracellular pathogens interact directly with receptor tyrosine kinases, and this interaction is critical for pathogen entry [207]. This establishes that pathogen-encoded receptor tyrosine kinase-interacting epitopes represent promising candidates for the development of novel therapeutic and prophylactic vaccines and of small-molecule interaction disruptors [208]. It would be of great interest to investigate whether those pathogens will confer therapeutic tolerance/resistance in host tumor cells by regulating miRNA–ROS interaction (Figure 1).
8. Concluding Remarks and Future Directions
Therapeutic tolerance/resistance raise major problems for the successful treatment of cancer, including conventional therapy and recent molecular therapy. There is an increasing importance of studying the role of ROS-relevant miRNAs to identify more effective biomarkers and develop better therapeutic targets against therapeutic tolerance/resistance. The interaction between miRNAs and ROSs fits in with the opportunities and challenges of studying mechanisms by which cancer cells resist therapy and ways by which therapeutic tolerance/resistance can be overcome. New concepts and emerging research tools bring potential to overcome therapeutic tolerance/resistance. However, some major challenges should be addressed properly. First, cancer relapse is driven by a small subpopulation of drug-tolerant persister cells, known as minimal residual disease in clinic. Single cell-relevant technologies, such as single-cell sequencing [209] might be applied to track single tolerant persister cells to gain insights into drug tolerance dynamics and heterogeneity [210]. In addition, preventative strategies using potential agents targeting those therapeutic tolerant cells at early stages in combination with molecular therapeutics will help prevent therapeutic tolerance and the resulting therapeutic resistance [211]. Second, new ex vivo models such as the organoid have been widely applied in cancer treatment response and therapeutic tolerance/resistance [212,213]. One of the advantages of the three-dimensional organoid model compared to a conventional two-dimensional monolayer is that tumor microenvironments established in organoids are similar to those found in vivo. For example, cancer organoids show heterogeneous hypoxic regions and show their enriched tumor-initiating cells and relevant metabolism pathway [214]. The organoid model may be used for large-scale screening, especially when incorporated with AI-based technology, to optimize the best drug combinations and thus reduce therapeutic tolerance/resistance. However, lacking immune cells and other types of cells has challenged this model [215]. Thus, incorporating immune cells will help better understand tolerance and resistance to immunotherapy [216]. Third, applications of non-invasive biomarkers to predict drug response represents a future direction in clinical settings. For example, cell-free circulating miRNAs have been successfully combined with low dose computed tomography scanning for diagnoses of early-stage lung cancer patients [217]. It is reasonable to incorporate cell-free circulating miRNAs signature together with cell-free DNAs signature [218] to predict and track the emergence of therapeutic tolerance/resistance. However, microRNAs predicting therapeutic tolerance/resistance might be dependent on specific mutant driver genes. For instance, increased miR-147b is relevant to mutant EGFR [76], and downregulated miR-23a is relevant to mutant MYC [30]. Thus, genetic mutation background and specific treatment agents should be considered comprehensively. Ultimately, early intervention on genetic/epigenetic, signaling/metabolic, and phenotypic changes in the miRNA–ROS network should be considered comprehensively to prevent and overcome therapeutic tolerance/resistance.
Acknowledgments
This work is supported by the Burnett School of Biomedical Sciences, College of Medicine, University of Central Florida grant 25400714, the NIH-Yale SPORE in Lung Cancer Career Development Program Award, and NRSA grant 5T32HL007893 awarded to W.C.Z. We thank Joshua Roney for critical reading and comments. We apologize to all researches whose work could not be cited due to reference limitations.
Conflicts of Interest
The author declares no conflict of interest.
References
- Harman, S.M.; Liang, L.; Tsitouras, P.D.; Gucciardo, F.; Heward, C.B.; Reaven, P.D.; Ping, W.; Ahmed, A.; Cutler, R.G. Urinary excretion of three nucleic acid oxidation adducts and isoprostane F2α measured by liquid chromatography-mass spectrometry in smokers, ex-smokers, and nonsmokers. Free Radic. Biol Med. 2003, 1301, 35–1309. [Google Scholar] [CrossRef]
- Apel, K.; Hirt, H. Reactive oxygen species: Metabolism, oxidative stress, and signal transduction. Annu. Rev. Plant. Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011, 194, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Dupré-Crochet, S.; Erard, M.; Nüße, O. ROS production in phagocytes: Why, when, and where? J. Leukoc. Biol. 2013, 94, 657–670. [Google Scholar] [CrossRef]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in biology and medicine. React. Oxyg. Species 2016, 1, 9–21. [Google Scholar]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [Green Version]
- Yeung, A.W.K.; Tzvetkov, N.T.; El-Tawil, O.S.; Bungau, S.G.; Abdel-Daim, M.M.; Atanasov, A.G. Antioxidants: Scientific literature landscape analysis. Oxid. Med. Cell Longev. 2019, 8278, 2019454. [Google Scholar] [CrossRef]
- Valle, I.; Alvarez-Barrientos, A.; Arza, E.; Lamas, S.; Monsalve, M. Pgc-1α regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc. Res. 2005, 66, 562–573. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Thompson, C.B. Metabolic reprogramming: A cancer hallmark even Warburg did not anticipate. Cancer Cell 2012, 21, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Raimundo, N.; Baysal, B.E.; Shadel, G.S. Revisiting the TCA cycle: Signaling to tumor formation. Trends Mol. Med. 2011, 17, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Fernie, A.R.; Carrari, F.; Sweetlove, L.J. Respiratory metabolism: Glycolysis, the TCA cycle and mitochondrial electron transport. Curr. Opin. Plant Biol. 2004, 7, 254–261. [Google Scholar] [CrossRef]
- Dahia, P.L. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat. Rev. Cancer 2014, 14, 108–119. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G., Jr. Molecular basis of the VHL hereditary cancer syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef]
- Krieg, M.; Haas, R.; Brauch, H.; Acker, T.; Flamme, I.; Plate, K.H. Up-regulation of hypoxia-inducible factors HIF-1α and HIF-2α under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 2000, 19, 5435–5443. [Google Scholar] [CrossRef] [Green Version]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016, 44, D1251–D1257. [Google Scholar] [CrossRef]
- Gottlieb, E.; Tomlinson, I.P.M. Mitochondrial tumour suppressors: A genetic and biochemical update. Nat. Rev. Cancer 2005, 5, 857–866. [Google Scholar] [CrossRef]
- Wagner, A.J.; Remillard, S.P.; Zhang, Y.X.; Doyle, L.A.; George, S.; Hornick, J.L. Loss of expression of SDHA predicts SDHA mutations in gastrointestinal stromal tumors. Mod. Pathol. 2013, 26, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Astuti, D.; Latif, F.; Dallol, A.; Dahia, P.L.M.; Douglas, F.; George, E.; Skoldberg, F.; Husebye, E.S.; Eng, C.; Maher, E.R. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am. J. Hum. Genet. 2001, 69, 49–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemann, S.; Muller, U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nat. Genet. 2000, 26, 268–270. [Google Scholar] [CrossRef]
- Baysal, B.E.; Ferrell, R.E.; Willett-Brozick, J.E.; Lawrence, E.C.; Myssiorek, D.; Bosch, A.; van der Mey, A.; Taschner, P.E.M.; Rubinstein, W.S.; Myers, E.N.; et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 2000, 287, 848–851. [Google Scholar] [CrossRef]
- Zhang, W.C.; Shyh-Chang, N.; Yang, H.; Rai, A.; Umashankar, S.; Ma, S.; Soh, B.S.; Sun, L.L.; Tai, B.C.; Nga, M.E.; et al. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell 2012, 148, 259–272. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.C.; Lim, B. Targeting metabolic enzyme with locked nucleic acids in non-small cell lung cancer. Cancer Res. 2014, 74, 1438. [Google Scholar]
- Lim, B.; Zhang, W. Targeting Metabolic Enzymes in Human Cancer. U.S. Patent 9,297,813,B2, 11 November 2011. [Google Scholar]
- Go, M.K.; Zhang, C W.; Lim, B.; Yew, W.S. Glycine decarboxylase is an unusual amino acid decarboxylase involved in tumorigenesis. Biochemistry 2014, 53, 947–956. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. C-Myc suppression of miR-23a/B enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Testa, U.; Labbaye, C.; Castelli, G.; Pelosi, E. Oxidative stress and hypoxia in normal and leukemic stem cells. Exp. Hematol. 2016, 44, 540–560. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Kasinski, A.L.; Slack, F.J. MicroRNAs en route to the clinic: Progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer 2011, 11, 849–864. [Google Scholar] [CrossRef] [Green Version]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef]
- Fan, M.; Krutilina, R.; Sun, J.; Sethuraman, A.; Yang, H.C.; Wu, H.Z.; Yue, J.; Pfeffer, L.M. Comprehensive analysis of microRNA (miRNA) targets in breast cancer cells. J. Biol. Chem. 2013, 288, 27480–27493. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, A.E.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Nouraee, N.; Calin, G.A. MicroRNAs as cancer biomarkers. MicroRNA 2013, 2, 102–117. [Google Scholar] [CrossRef]
- Tentori, A.M.; Nagarajan, M.B.; Kim, J.J.; Zhang, W.C.; Slack, F.J.; Doyle, P.S. Quantitative and multiplex microRNA assays from unprocessed cells in isolated nanoliter well arrays. Lab Chip 2018, 18, 2410–2424. [Google Scholar] [CrossRef]
- Nagarajan, M.B.; Tentori, A.M.; Zhang, W.C.; Slack, F.J.; Doyle, P.S. Nonfouling, encoded hydrogel microparticles for multiplex microRNA profiling directly from formalin-fixed, paraffin-embedded tissue. Anal. Chem. 2018, 90, 10279–10285. [Google Scholar] [CrossRef]
- Anfossi, S.; Babayan, A.; Pantel, K.; Calin, G.A. Clinical utility of circulating non-coding RNAs—An update. Nat. Rev. Clin. Oncol. 2018, 15, 541–563. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Nishida, N.; Calin, G.A.; Pantel, K. clinical relevance of circulating cell-free microRNAs in cancer. Nat. Rev. Clin. Oncol. 2014, 11, 145–156. [Google Scholar] [CrossRef]
- Cortez, M.A.; Bueso-Ramos, C.; Ferdin, J.; Lopez-Berestein, G.; Sood, A.K.; Calin, G.A. MicroRNAs in body fluids—The mix of hormones and biomarkers. Nat. Rev. Clin. Oncol. 2011, 8, 467–477. [Google Scholar] [CrossRef] [Green Version]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs—MicroRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef]
- Medina, P.P.; Nolde, M.; Slack, F.J. Oncomir addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 2010, 467, 86–90. [Google Scholar] [CrossRef]
- Edmonds, M.D.; Boyd, K.L.; Moyo, T.; Mitra, R.; Duszynski, R.; Arrate, M.P.; Chen, X.; Zhao, Z.; Blackwell, T.S.; Andl, T.; et al. MicroRNA-31 initiates lung tumorigenesis and promotes mutant KRAS-driven lung cancer. J. Clin. Investig. 2016, 126, 349–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.J.; Bahal, R.; Babar, I.A.; Pincus, Z.; Barrera, F.; Liu, C.; Svoronos, A.; Braddock, D.T.; Glazer, P.M.; Engelman, D.M.; et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 2015, 518, 107–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babar, I.A.; Cheng, C.J.; Booth, C.J.; Liang, X.; Weidhaas, J.B.; Saltzman, W.M.; Slack, F.J. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, E1695–E1704. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Reinhardt, F.; Pan, E.; Soutschek, J.; Bhat, B.; Marcusson, E.G.; Teruya-Feldstein, J.; Bell, G.W.; Weinberg, R.A. Therapeutic silencing of miR-10b inhibits metastasis in a mouse mammary tumor model. Nat. Biotechnol. 2010, 28, 341–347. [Google Scholar] [CrossRef]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 microRNA family. Cell 2005, 120, 635–647. [Google Scholar] [CrossRef] [Green Version]
- Kasinski, A.L.; Slack, F.J. MiRNA-34 prevents cancer initiation and progression in a therapeutically resistant k-RAS and P53-induced mouse model of lung adenocarcinoma. Cancer Res. 2012, 72, 5576–5587. [Google Scholar] [CrossRef] [Green Version]
- Krzeszinski, J.Y.; Wei, W.; Huynh, H.; Jin, Z.; Wang, X.; Chang, T.C.; Xie, X.J.; He, L.; Mangala, L.S.; Lopez-Berestein, G.; et al. MiR-34a blocks osteoporosis and bone metastasis by inhibiting osteoclastogenesis and Tgif2. Nature 2014, 512, 431–435. [Google Scholar] [CrossRef]
- Banerjee, J.; Khanna, S.; Bhattacharya, A. MicroRNA regulation of oxidative stress. Oxid. Med. Cell Longev. 2017, 2872, 2017156. [Google Scholar] [CrossRef] [Green Version]
- Hatley, M.E.; Patrick, D.M.; Garcia, M.R.; Richardson, J.A.; Bassel-Duby, R.; van Rooij, E.; Olson, E.N. Modulation of K-Ras-dependent lung tumorigenesis by microRNA-21. Cancer Cell 2010, 18, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Seike, M.; Goto, A.; Okano, T.; Bowman, D.E.; Schetter, J.A.; Horikawa, I.; Mathe, A.E.; Jen, J.; Yang, P.; Sugimura, H.; et al. MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc. Natl. Acad. Sci. USA 2009, 106, 12085–12090. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Ng, W.L.; Wang, P.; Tian, L.; Werner, E.; Wang, H.; Doetsch, P.; Wang, Y. MicroRNA-21 modulates the levels of reactive oxygen species by targeting Sod3 Tnfα. Cancer Res. 2012, 72, 4707–4713. [Google Scholar] [CrossRef] [Green Version]
- Engedal, N.; Zerovnik, E.; Rudov, A.; Galli, F.; Olivieri, F.; Procopio, A.D.; Rippo, M.R.; Monsurro, V.; Betti, M.; Albertini, M.C. From oxidative stress damage to pathways, networks, and autophagy via microRNAs. Oxid. Med. Cell Longev. 2018, 2018, 4968321. [Google Scholar] [CrossRef]
- Poyil, P.; Son, Y.-O.; Divya, S.P.; Wang, L.; Zhang, Z.; Shi, H. Oncogenic transformation of human lung bronchial epithelial cells induced by arsenic involves ROS-dependent activation of STAT3-miR-21-PDCD4 mechanism. Sci. Rep. 2016, 6, 37227. [Google Scholar]
- Thulasingam, S.; Massilamany, C.; Gangaplara, A.; Dai, H.; Yarbaeva, S.; Subramaniam, S.; Riethoven, J.J.; Eudy, J.; Lou, M.; Reddy, J. MiR-27b*, an oxidative stress-responsive microRNA modulates nuclear factor-kB pathway in Raw 264.7 cells. Mol. Cell Biochem. 2011, 352, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Baker, J.R.; Vuppusetty, C.; Colley, T.; Papaioannou, A.I.; Fenwick, P.; Donnelly, L.; Ito, K.; Barnes, P.J. Oxidative stress dependent microRNA-34a activation via PI3Kα reduces the expression of sirtuin-1 and sirtuin-6 in epithelial cells. Sci. Rep. 2016, 6, 35871. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Fang, D. The roles of SIRT1 in cancer. Genes Cancer 2013, 4, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussing, I.; Slack, F.J.; Grosshans, H. Let-7 microRNAs in development, stem cells and cancer. Trends Mol. Med. 2008, 14, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Saleh, A.D.; Savage, J.E.; Cao, L.; Soule, B.P.; Ly, D.; DeGraff, W.; Harris, C.C.; Mitchell, J.B.; Simone, N.L. Cellular stress induced alterations in microRNA let-7a and let-7b expression are dependent on P53. PLoS ONE 2011, 6, e24429. [Google Scholar] [CrossRef] [PubMed]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef] [Green Version]
- Adams, B.D.; Parsons, C.; Walker, L.; Zhang, W.C.; Slack, F.J. Targeting noncoding RNAs in disease. J. Clin. Investig. 2017, 127, 761–771. [Google Scholar] [CrossRef]
- Rupaimoole, R.; Slack, F.J. MicroRNA therapeutics: Towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 2017, 16, 203–222. [Google Scholar] [CrossRef]
- Van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Van Zandwijk, N.; McDiarmid, J.; Brahmbhatt, H.; Reid, G. Response to an innovative mesothelioma treatment based on miR-16 mimic loaded EGFR targeted minicells (TargomiRs). Transl. Lung Cancer Res. 2018, 7, S60–S61. [Google Scholar] [CrossRef] [Green Version]
- Fennell, D. MiR-16: Expanding the range of molecular targets in mesothelioma. Lancet Oncol. 2017, 18, 1296–1297. [Google Scholar] [CrossRef]
- Slack, F.J.; Chinnaiyan, A.M. The role of non-coding RNAs in oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wang, L.; Rodriguez-Aguayo, C.; Yuan, Y.; Debeb, B.G.; Chen, D.; Sun, Y.; You, M.J.; Liu, Y.; Dean, C.D.; et al. MiR-205 acts as a tumour radiosensitizer by targeting ZEB1 and Ubc13. Nat. Commun. 2014, 5, 5671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Zhang, R.; Claret, F.X.; Yang, H. Involvement of microRNA-24 and DNA methylation in resistance of nasopharyngeal carcinoma to ionizing radiation. Mol. Cancer Ther. 2014, 13, 3163–3174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babar, I.A.; Czochor, J.; Steinmetz, A.; Weidhaas, J.B.; Glazer, P.M.; Slack, F.J. Inhibition of hypoxia-induced miR-155 radiosensitizes hypoxic lung cancer cells. Cancer Biol. Ther. 2011, 12, 908–914. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.C.; Chin, T.M.; Yang, H.; Nga, M.E.; Lunny, D.P.; Lim, E.K.; Sun, L.L.; Pang, Y.H.; Leow, Y.N.; Malusay, S.R.; et al. Tumour-initiating cell-specific miR-1246 and miR-1290 expression converge to promote non-small cell lung cancer progression. Nat. Commun. 2016, 7, 11702. [Google Scholar] [CrossRef]
- Zhang, W.C.; Wells, J.M.; Chow, K.H.; Huang, H.; Yuan, M.; Saxena, T.; Melnick, M.A.; Politi, K.; Asara, J.M.; Costa, D.B.; et al. MiR-147b-mediated TCA cycle dysfunction and pseudohypoxia initiate drug tolerance to EGFR inhibitors in lung adenocarcinoma. Nat. Metab. 2019, 1, 460–474. [Google Scholar] [CrossRef]
- Costinean, S.; Zanesi, N.; Pekarsky, Y.; Tili, E.; Volinia, S.; Heerema, N.; Croce, M.C. Pre-B Cell Proliferation and Lymphoblastic Leukemia/High-Grade Lymphoma in E(Mu)-Mir155 Transgenic Mice. Proc. Natl. Acad. Sci. USA 2006, 103, 7024–7029. [Google Scholar] [CrossRef] [Green Version]
- Mikamori, M.; Yamada, D.; Eguchi, H.; Hasegawa, S.; Kishimoto, T.; Tomimaru, Y.; Asaoka, T.; Noda, T.; Wada, H.; Kawamoto, K.; et al. MicroRNA-155 controls exosome synthesis and promotes gemcitabine resistance in pancreatic ductal adenocarcinoma. Sci. Rep. 2017, 7, 42339. [Google Scholar] [CrossRef]
- Moriyama, T.; Ohuchida, K.; Mizumoto, K.; Yu, J.; Sato, N.; Nabae, T.; Takahata, S.; Toma, H.; Nagai, E.; Tanaka, M. MicroRNA-21 modulates biological functions of pancreatic cancer cells including their proliferation, invasion, and chemoresistance. Mol. Cancer Ther. 2009, 8, 1067–1074. [Google Scholar] [CrossRef] [Green Version]
- Giovannetti, E.; Funel, N.; Peters, J G.; Del Chiaro, M.; Erozenci, L.A.; Vasile, E.; Leon, L.G.; Pollina, L.E.; Groen, A.; Falcone, A.; et al. MicroRNA-21 in pancreatic cancer: correlation with clinical outcome and pharmacologic aspects underlying its role in the modulation of gemcitabine activity. Cancer Res. 2010, 70, 4528–4538. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Wang, R.; Yan, H.; Jin, L.; Dou, X.; Chen, D. MicroRNA-21 modulates radiation resistance through upregulation of hypoxia-inducible factor-1α-promoted glycolysis in non-small cell lung cancer cells. Mol. Med. Rep. 2016, 13, 4101–4107. [Google Scholar] [CrossRef]
- Zhang, W.C.; Slack, F.J. MicroRNA-21 mediates resistance to EGFR tyrosine kinase inhibitors in lung cancer. J. Thorac. Oncol. 2017, 12, S1536. [Google Scholar] [CrossRef]
- Yu, F.; Yao, H.; Zhu, P.; Zhang, X.; Pan, Q.; Gong, C.; Huang, Y.; Hu, X.; Su, F.; Lieberman, J.; et al. Let-7 regulates self renewal and tumorigenicity of breast cancer cells. Cell 2007, 131, 1109–1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.; Hu, W.; Pan, L.; Fu, W.; Dai, L.; Jiang, Z.; Zhang, F.; Zhao, J. Let-7 and miR-17 promote self-renewal and drive gefitinib resistance in non-small cell lung cancer. Oncol. Rep. 2019, 42, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Deng, H.; Yao, H.; Liu, Q.; Su, F.; Song, E. MiR-30 reduction maintains self-renewal and inhibits apoptosis in breast tumor-initiating cells. Oncogene 2010, 29, 4194–4204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.S.; Yu, F.; Zhong, X.M.; Lu, G.X.; Cong, X.L.; Xue, S.B.; Xie, W.T.; Hou, L.K.; Pang, L.J.; Wu, W.; et al. MiR-30 family reduction maintains self-renewal and promotes tumorigenesis in NSCLC-initiating cells by targeting oncogene TM4SF1. Mol. Ther. 2018, 26, 2751–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, M.; Romano, G.; Di Leva, G.; Nuovo, G.; Jeon, Y.J.; Ngankeu, A.; Sun, J.; Lovat, F.; Alder, H.; Condorelli, G.; et al. EGFR and MET receptor tyrosine kinase-altered microRNA expression induces tumorigenesis and gefitinib resistance in lung cancers. Nat. Med. 2012, 18, 74–82. [Google Scholar] [CrossRef]
- Du, X.; Liu, B.; Luan, X.; Cui, Q.; Li, L. MiR-30 decreases multidrug resistance in human gastric cancer cells by modulating cell autophagy. Exp. Ther. Med. 2018, 15, 599–605. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Hermeking, H. MiR-34a and miR-34b/c suppress intestinal tumorigenesis. Cancer Res. 2017, 77, 2746–2758. [Google Scholar] [CrossRef] [Green Version]
- Zenz, T.; Mohr, J.; Eldering, E.; Kater, A.P.; Bühler, A.; Kienle, D.; Winkler, D.; Dürig, J.; van Oers, M.H.; Mertens, D.; et al. MiR-34a as part of the resistance network in chronic lymphocytic leukemia. Blood 2009, 113, 3801–3808. [Google Scholar] [CrossRef] [Green Version]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [Green Version]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef]
- Politi, K.; Herbst, R.S. Lung cancer in the era of precision medicine. Clin. Cancer Res. 2015, 21, 2213–2220. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Janne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Politi, K.; Ayeni, D.; Lynch, T. The next wave of EGFR tyrosine kinase inhibitors enter the clinic. Cancer Cell 2015, 27, 751–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thress, K.S.; Paweletz, C.P.; Felip, E.; Cho, B.C.; Stetson, D.; Dougherty, B.; Lai, Z.; Markovets, A.; Vivancos, A.; Kuang, Y.; et al. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21, 560–562. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.; Krishnamurthy Radhakrishna, V.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]
- Ramirez, M.; Rajaram, S.; Steininger, R.J.; Osipchuk, D.; Roth, M.A.; Morinishi, L.S.; Evans, L.; Ji, W.; Hsu, C.H.; Thurley, K.; et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016, 7, 10690. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.P.; Brunton, H.; Rowling, E.J.; Ferguson, J.; Arozarena, I.; Miskolczi, Z.; Lee, J.L.; Girotti, M.R.; Marais, R.; Levesque, M.P.; et al. Inhibiting drivers of non-mutational drug tolerance is a salvage strategy for targeted melanoma therapy. Cancer Cell 2016, 29, 270–284. [Google Scholar] [CrossRef] [Green Version]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, J.M.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, L.S.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlides, S.; Vera, I.; Gandara, R.; Sneddon, S.; Pestell, R.G.; Mercier, I.; Martinez-Outschoorn, U.E.; Whitaker-Menezes, D.; Howell, A.; Sotgia, F.; et al. Warburg meets autophagy: Cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid. Redox Signal. 2012, 16, 1264–1284. [Google Scholar] [CrossRef] [Green Version]
- Sebolt-Leopold, J.S.; English, J.M. Mechanisms of drug inhibition of signalling molecules. Nature 2006, 441, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.-j.; Zheng, B.; Wang, H.-y.; Chen, L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol. Sin. 2017, 38, 614–622. [Google Scholar] [CrossRef] [Green Version]
- Mendez-Blanco, C.; Fondevila, F.; Garcia-Palomo, A.; Gonzalez-Gallego, J.; Mauriz, L.J. Sorafenib resistance in hepatocarcinoma: Role of hypoxia-inducible factors. Exp. Mol. Med. 2018, 50, 134. [Google Scholar] [CrossRef] [Green Version]
- Vanecek, J. Cellular mechanisms of melatonin action. Physiol. Rev. 1998, 78, 687–721. [Google Scholar] [CrossRef]
- Prieto-Domínguez, N.; Ordóñez, R.; Fernández, A.; Méndez-Blanco, C.; Baulies, A.; Garcia-Ruiz, C.; Fernández-Checa, J.C.; Mauriz, J.L.; González-Gallego, J. Melatonin-induced increase in sensitivity of human hepatocellular carcinoma cells to sorafenib is associated with reactive oxygen species production and mitophagy. J. Pineal Res. 2016, 61, 396–407. [Google Scholar] [CrossRef]
- Prieto-Dominguez, N.; Mendez-Blanco, C.; Carbajo-Pescador, S.; Fondevila, F.; Garcia-Palomo, A.; Gonzalez-Gallego, J.; Mauriz, J.L. Melatonin enhances sorafenib actions in human hepatocarcinoma cells by inhibiting mTORC1/p70S6K/HIF-1α and hypoxia-mediated mitophagy. Oncotarget 2017, 8, 91402–91414. [Google Scholar] [CrossRef]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Rev. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Nguyen, L.V.; Vanner, R.; Dirks, P.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Rev. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef]
- Tang, D.G. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. 2012, 22, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Zhang, W.C.; Yang, H.; Soh, B.S.; Sun, L.L.; Chin, T.M.; Lim, E.H.; Lim, B. Abstract 487: Evidence for tumor initiating stem cells in lung cancer. Cancer Res. 2011, 71, 487. [Google Scholar]
- Tam, W.L.; Lu, H.; Buikhuisen, J.; Soh, B.S.; Lim, E.; Reinhardt, F.; Wu, Z.J.; Krall, J.A.; Bierie, B.; Guo, W.; et al. Protein kinase C α is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 2013, 24, 347–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Hoe, S.L.L.; Tan, L.P.; Jamal, J.; Peh, S.C.; Ng, C.C.; Zhang, W.C.; Ahmad, M.; Khoo, A.S.B. Evaluation of stem-like side population cells in a recurrent nasopharyngeal carcinoma cell line. Cancer Cell Int. 2014, 14, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Dando, I.; Cordani, M.; Dalla Pozza, E.; Biondani, G.; Donadelli, M.; Palmieri, M. Antioxidant mechanisms and ROS-related microRNAs in cancer stem cells. Oxid. Med. Cell Longev. 2015, 4257, 201508. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Curtis, S.J.; Sinkevicius, K.W.; Li, D.; Lau, A.N.; Roach, R.R.; Zamponi, R.; Woolfenden, A.E.; Kirsch, D.G.; Wong, K.K.; Kim, C.F. Primary tumor genotype is an important determinant in identification of lung cancer propagating cells. Cell Stem Cell 2010, 7, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Houessinon, A.; Francois, C.; Sauzay, C.; Louandre, C.; Mongelard, G.; Godin, C.; Bodeau, S.; Takahashi, S.; Saidak, Z.; Gutierrez, L.; et al. Metallothionein-1 as a biomarker of altered redox metabolism in hepatocellular carcinoma cells exposed to sorafenib. Mol. Cancer 2016, 15, 38. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Abdel Fattah, E.; Cao, G.; Ren, C.; Yang, G.; Goltsov, A.A.; Chinault, A.C.; Cai, W.W.; Timme, T.L.; Thompson, T.C. Glioma pathogenesis-related protein 1 exerts tumor suppressor activities through proapoptotic reactive oxygen species-c-Jun-NH2 kinase signaling. Cancer Res. 2008, 68, 434–443. [Google Scholar] [CrossRef] [Green Version]
- Yuan, D.; Xu, J.; Wang, J.; Pan, Y.; Fu, J.; Bai, Y.; Zhang, J.; Shao, C. Extracellular miR-1246 promotes lung cancer cell proliferation and enhances radioresistance by directly targeting DR5. Oncotarget 2016, 7, 32707–32722. [Google Scholar] [CrossRef] [Green Version]
- Golestaneh, A.F.; Atashi, A.; Langroudi, L.; Shafiee, A.; Ghaemi, N.; Soleimani, M. MiRNAs expressed differently in cancer stem cells and cancer cells of human gastric cancer cell line MKN-45. Cell Biochem. Funct. 2012, 30, 411–418. [Google Scholar] [CrossRef]
- Jackman, D.M.; Johnson, B.E. Small-cell lung cancer. Lancet 2005, 366, 1385–1396. [Google Scholar] [CrossRef]
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [Green Version]
- Marcoux, N.; Gettinger, S.N.; O’Kane, G.; Arbour, K.C.; Neal, J.W.; Husain, H.; Evans, T.L.; Brahmer, J.R.; Muzikansky, A.; Bonomi, P.D.; et al. EGFR-mutant adenocarcinomas that transform to small-cell lung cancer and other neuroendocrine carcinomas: clinical outcomes. J. Clin. Oncol. 2018, 37, 278–285. [Google Scholar] [CrossRef]
- Hayashita, Y.; Osada, H.; Tatematsu, Y.; Yamada, H.; Yanagisawa, K.; Tomida, S.; Yatabe, Y.; Kawahara, K.; Sekido, Y.; Takahashi, T. A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 2005, 65, 9628–9632. [Google Scholar] [CrossRef] [Green Version]
- Ebi, H.; Sato, T.; Sugito, N.; Hosono, Y.; Yatabe, Y.; Matsuyama, Y.; Yamaguchi, T.; Osada, H.; Suzuki, M.; Takahashi, T. Counterbalance between RB inactivation and miR-17-92 overexpression in reactive oxygen species and DNA damage induction in lung cancers. Oncogene 2009, 28, 3371–3379. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Lindahl-Allen, M.; Polytarchou, C.; Hirsch, H.A.; Tsichlis, P.N.; Struhl, K. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol. Cell 2010, 39, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Mateescu, B.; Batista, L.; Cardon, M.; Gruosso, T.; de Feraudy, Y.; Mariani, O.; Nicolas, A.; Meyniel, J.-P.; Cottu, P.; Sastre-Garau, X.; et al. MiR-141 and miR-200a act on ovarian tumorigenesis by controlling oxidative stress response. Nat. Med. 2011, 17, 1627–1635. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Magenta, A.; Cencioni, C.; Fasanaro, P.; Zaccagnini, G.; Greco, S.; Sarra-Ferraris, G.; Antonini, A.; Martelli, F.; Capogrossi, M.C. MiR-200c is upregulated by oxidative stress and induces endothelial cell apoptosis and senescence via Zeb1 inhibition. Cell Death Differ. 2011, 18, 1628–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Ding, L.; Bennewith, K.L.; Tong, R.T.; Welford, S.M.; Ang, K.K.; Story, M.; Le, Q.T.; Giaccia, A.J. Hypoxia-inducible miR-210 regulates normoxic gene expression involved in tumor initiation. Mol. Cell 2009, 35, 856–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favaro, E.; Ramachandran, A.; McCormick, R.; Gee, H.; Blancher, C.; Crosby, M.; Devlin, C.; Blick, C.; Buffa, F.; Li, J.L.; et al. MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PLoS ONE 2010, 5, e10345. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Li, Y.; Zhang, H.; Huang, P.; Luthra, R. Hypoxia-regulated microRNA-210 modulates mitochondrial function and decreases ISCU and COX10 expression. Oncogene 2010, 29, 4362–4368. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Sun, T.; Cao, J.; Liu, F.; Tian, Y.; Zhu, W. Downregulation of miR-210 expression inhibits proliferation, induces apoptosis and enhances radiosensitivity in hypoxic human hepatoma cells in vitro. Exp. Cell Res. 2012, 318, 944–954. [Google Scholar] [CrossRef]
- Yang, W.; Wei, J.; Guo, T.; Shen, Y.; Liu, F. Knockdown of miR-210 decreases hypoxic glioma stem cells stemness and radioresistance. Exp. Cell Res. 2014, 326, 22–35. [Google Scholar] [CrossRef]
- Ivan, M.; Huang, X. MiR-210: Fine-tuning the hypoxic response. Adv. Exp. Med. Biol. 2014, 772, 205–227. [Google Scholar]
- Calvert, A.E.; Chalastanis, A.; Wu, Y.; Hurley, L.A.; Kouri, F.M.; Bi, Y.; Kachman, M.; May, J.L.; Bartom, E.; Hua, Y.; et al. Cancer-associated IDH1 promotes growth and resistance to targeted therapies in the absence of mutation. Cell Rep. 2017, 19, 1858–1873. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G., Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat. Rev. Cancer 2008, 8, 865–873. [Google Scholar] [CrossRef]
- Guo, J.; Chakraborty, A.A.; Liu, P.; Gan, W.; Zheng, X.; Inuzuka, H.; Wang, B.; Zhang, J.; Zhang, L.; Yuan, M.; et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 2016, 353, 929–932. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Frattini, V.; Bansal, M.; Castano, A.M.; Sherman, D.; Hutchinson, K.; Bruce, J.N.; Califano, A.; Liu, G.; Cardozo, T.; et al. An ID2-dependent mechanism for VHL inactivation in cancer. Nature 2016, 529, 172–177. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, E.D.; Selak, M.A.; Tennant, D.A.; Payne, L.J.; Crosby, S.; Frederiksen, C.M.; Watson, D.G.; Gottlieb, E. Cell-permeating α-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol. Cell Biol. 2007, 27, 3282–3289. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [Green Version]
- Wilson, T.R.; Fridlyand, J.; Yan, Y.; Penuel, E.; Burton, L.; Chan, E.; Peng, J.; Lin, E.; Wang, Y.; Sosman, J.; et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature 2012, 487, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Niederst, M.J.; Engelman, J.A. Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer. Sci. Signal. 2013, 6, re6. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic heterogeneity in human lung tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef] [Green Version]
- Imielinski, M.; Guo, G.; Meyerson, M. Insertions and deletions target lineage-defining genes in human cancers. Cell 2017, 168, 460.e14–472.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Ren, S.; Li, X.; Wang, Y.; Garfield, D.; Zhou, S.; Chen, X.; Su, C.; Chen, M.; Kuang, P.; et al. MiR-21 overexpression is associated with acquired resistance of EGFR-TKI in non-small cell lung cancer. Lung Cancer 2014, 83, 146–153. [Google Scholar] [CrossRef]
- Zheng, G.; Li, N.; Jia, X.; Peng, C.; Luo, L.; Deng, Y.; Yin, J.; Song, Y.; Liu, H.; Lu, M.; et al. MYCN-mediated miR-21 overexpression enhances chemo-resistance via targeting CADM1 in tongue cancer. J. Mol. Med. (Berl.) 2016, 94, 1129–1141. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Chendrimada, T.P.; Wang, Q.; Higuchi, M.; Seeburg, P.H.; Shiekhattar, R.; Nishikura, K. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006, 13, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Lazzari, E.; Crews, L.A.; Wu, C.; Leu, H.; Ali, S.; Chiaramonte, R.; Minden, M.; Costello, C.; Jamieson, C.H.M. Abstract 2414: ADAR1-dependent RNA editing is a mechanism of therapeutic resistance in human plasma cell malignancies. Cancer Res. 2016, 76, 2414. [Google Scholar]
- Zhang, W.C.; Slack, F.J. ADARs edit microRNAs to promote leukemic stem cell activity. Cell Stem Cell 2016, 19, 141–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zipeto, M.A.; Court, A.C.; Sadarangani, A.; Delos Santos, N.P.; Balaian, L.; Chun, H.J.; Pineda, G.; Morris, S.R.; Mason, C.N.; Geron, I.; et al. ADAR1 activation drives leukemia stem cell self-renewal by impairing Let-7 biogenesis. Cell Stem Cell 2016, 19, 177–191. [Google Scholar] [CrossRef]
- Jiang, Q.; Isquith, J.; Zipeto, M.A.; Diep, R.H.; Pham, J.; Delos Santos, N.; Reynoso, E.; Chau, J.; Leu, H.; Lazzari, E.; et al. Hyper-editing of cell-cycle regulatory and tumor suppressor RNA promotes malignant progenitor propagation. Cancer Cell 2019, 35, 81.e7–94.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Ishizuka, J.J.; Manguso, R.T.; Cheruiyot, C.K.; Bi, K.; Panda, A.; Iracheta-Vellve, A.; Miller, B.C.; Du, P.P.; Yates, K.B.; Dubrot, J.; et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 2019, 565, 43–48. [Google Scholar] [CrossRef]
- Chen, L.; Rio, D.C.; Haddad, G.G.; Ma, E. Regulatory role of dADAR in ROS metabolism in drosophila CNS. Brain Res. Mol. Brain Res. 2004, 131, 93–100. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 2012, 485, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Geula, S.; Moshitch-Moshkovitz, S.; Dominissini, D.; Mansour, A.A.; Kol, N.; Salmon-Divon, M.; Hershkovitz, V.; Peer, E.; Mor, N.; Manor, Y.S.; et al. M6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science 2015, 347, 1002–1006. [Google Scholar] [CrossRef]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Choe, J.; Lin, S.; Zhang, W.; Liu, Q.; Wang, L.; Ramirez-Moya, J.; Du, P.; Kim, W.; Tang, S.; Sliz, P.; et al. MRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature 2018, 561, 556–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, Q.; Liu, P.Y.; Haase, J.; Bell, J.L.; Huttelmaier, S.; Liu, T. The critical role of RNA m6A methylation in cancer. Cancer Res. 2019, 79, 1285–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Wei, J.; He, C. Where, when, and how: Context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 2019, 74, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Visvanathan, A.; Patil, V.; Arora, A.; Hegde, S.A.; Arivazhagan, A.; Santosh, V.; Somasundaram, K. Essential Role of Mettl3-Mediated M6a Modification in Glioma Stem-Like Cells Maintenance and Radioresistance. Oncogene 2018, 37, 522–533. [Google Scholar] [CrossRef]
- Taketo, K.; Konno, M.; Asai, A.; Koseki, J.; Toratani, M.; Satoh, T.; Doki, Y.; Mori, M.; Ishii, H.; Ogawa, K. The Epitranscriptome M6a Writer Mettl3 Promotes Chemo- and Radioresistance in Pancreatic Cancer Cells. Int. J. Oncol. 2018, 52, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; Al-Kali, A.; Zhang, Z.; Liu, J.; Pang, J.; Zhao, N.; He, C.; Litzow, M.R.; Liu, S. A dynamic N 6-methyladenosine methylome regulates intrinsic and acquired resistance to tyrosine kinase inhibitors. Cell Res. 2018, 28, 1062–1076. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ishfaq, M.; Xu, L.; Xia, C.; Chen, C.; Li, J. METTL3/m6A/miRNA-873–5p attenuated oxidative stress and apoptosis in colistin-induced kidney injury by modulating Keap1/Nrf2 pathway. Front. Pharmacol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Topol, E.J. High-performance medicine: The convergence of human and artificial intelligence. Nat. Med. 2019, 25, 44–56. [Google Scholar] [CrossRef]
- Romm, E.L.; Tsigelny, I.F. Artificial intelligence in drug treatment. Annu. Rev. Pharmacol. Toxicol. 2020, 60. [Google Scholar] [CrossRef]
- Mason, D.J.; Eastman, R.T.; Lewis, R.P.I.; Stott, I.P.; Guha, R.; Bender, A. Using machine learning to predict synergistic antimalarial compound combinations with novel structures. Front. Pharmacol. 2018. [Google Scholar] [CrossRef]
- Li, X.; Xu, Y.; Cui, H.; Huang, T.; Wang, D.; Lian, B.; Li, W.; Qin, G.; Chen, L.; Xie, L. Prediction of synergistic anti-cancer drug combinations based on drug target network and drug induced gene expression profiles. Artif. Intell. Med. 2017, 83, 35–43. [Google Scholar] [CrossRef]
- Xia, F.; Shukla, M.; Brettin, T.; Garcia-Cardona, C.; Cohn, J.; Allen, J.E.; Maslov, S.; Holbeck, S.L.; Doroshow, J.H.; Evrard, Y.A.; et al. Predicting tumor cell line response to drug pairs with deep learning. BMC Bioinform. 2018, 19, 486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McQuade, J.L.; Daniel, C.R.; Helmink, B.A.; Wargo, J.A. Modulating the microbiome to improve therapeutic response in cancer. Lancet Oncol. 2019, 20, e77–e91. [Google Scholar] [CrossRef]
- Peek, R.M., Jr.; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef] [Green Version]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Sam, Q.H.; Chang, M.W.; Chai, L.Y. The fungal mycobiome and its interaction with gut bacteria in the host. Int. J. Mol. Sci. 2017, 18, 330. [Google Scholar] [CrossRef] [Green Version]
- Aykut, B.; Pushalkar, S.; Chen, R.; Li, Q.; Abengozar, R.; Kim, J.I.; Shadaloey, S.A.; Wu, D.; Preiss, P.; Verma, N.; et al. The fungal mycobiome promotes pancreatic oncogenesis via activation of MBL. Nature 2019, 574, 264–267. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef] [Green Version]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum promotes chemoresistance to colorectal cancer by modulating autophagy. Cell 2017, 170, 548.e16–563.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, W.P.; Kwok, T.T. The miR-18a* microRNA functions as a potential tumor suppressor by targeting on K-Ras. Carcinogenesis 2009, 30, 953–959. [Google Scholar] [CrossRef]
- Lievre, A.; Bachet, J.B.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.F.; Cote, J.F.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006, 66, 3992–3995. [Google Scholar] [CrossRef] [Green Version]
- Misale, S.; Yaeger, R.; Hobor, S.; Scala, E.; Janakiraman, M.; Liska, D.; Valtorta, E.; Schiavo, R.; Buscarino, M.; Siravegna, G.; et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012, 486, 532–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haqshenas, G.; Doerig, C. Targeting of host cell receptor tyrosine kinases by intracellular pathogens. Sci. Signal. 2019, 12, eaau9894. [Google Scholar] [CrossRef] [Green Version]
- Schor, S.; Einav, S. Combating intracellular pathogens with repurposed host-targeted drugs. ACS Infect. Dis. 2018, 4, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.C.; Gay, C.M.; Xi, Y.; Fujimoto, J.; Kalhor, N.; Hartsfield, P.M.; Tran, H.; Fernandez, L.; Lu, D.; Wang, Y.; et al. Abstract 2899: Single-cell analyses reveal increasing intratumoral heterogeneity as an essential component of treatment resistance in small cell lung cancer. Cancer Res. 2019, 79, 2899. [Google Scholar]
- Rambow, F.; Rogiers, A.; Marin-Bejar, O.; Aibar, S.; Femel, J.; Dewaele, M.; Karras, P.; Brown, D.; Chang, Y.H.; Debiec-Rychter, M.; et al. Toward minimal residual disease-directed therapy in melanoma. Cell 2018, 174, 843.e19–855.e19. [Google Scholar] [CrossRef] [Green Version]
- Bivona, T.G.; Doebele, R.C. A framework for understanding and targeting residual disease in oncogene-driven solid cancers. Nat. Med. 2016, 22, 472–478. [Google Scholar] [CrossRef] [Green Version]
- Ponz-Sarvise, M.; Corbo, V.; Tiriac, H.; Engle, D.D.; Frese, K.K.; Oni, T.E.; Hwang, C.I.; Ohlund, D.; Chio, I.I.C.; Baker, L.A.; et al. Identification of resistance pathways specific to malignancy using organoid models of pancreatic cancer. Clin. Cancer Res. 2019, 25, 6742–6755. [Google Scholar] [CrossRef] [Green Version]
- Usui, T.; Sakurai, M.; Umata, K.; Elbadawy, M.; Ohama, T.; Yamawaki, H.; Hazama, S.; Takenouchi, H.; Nakajima, M.; Tsunedomi, R.; et al. Hedgehog signals mediate anti-cancer drug resistance in three-dimensional primary colorectal cancer organoid culture. Int. J. Mol. Sci. 2018, 19, 1098. [Google Scholar] [CrossRef] [Green Version]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A E.; Rich, N.J. A three-dimensional organoid culture system derived from human glioblastomas recapitulates the hypoxic gradients and cancer stem cell heterogeneity of tumors found in vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakrabarti, J.; Holokai, L.; Syu, L.; Steele, N.; Chang, J.; Dlugosz, A.; Zavros, Y. Mouse-derived gastric organoid and immune cell co-culture for the study of the tumor microenvironment. Methods Mol. Biol. 2018, 1817, 157–168. [Google Scholar] [PubMed]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid modeling of the tumor immune microenvironment. Cell 2018, 175, 1972.e16–1988.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sozzi, G.; Boeri, M.; Rossi, M.; Verri, C.; Suatoni, P.; Bravi, F.; Roz, L.; Conte, D.; Grassi, M.; Sverzellati, N.; et al. Clinical utility of a plasma-based miRNA signature classifier within computed tomography lung cancer screening: A correlative mild trial study. J. Clin. Oncol. 2014, 32, 768–773. [Google Scholar] [CrossRef]
- Siravegna, G.; Bardelli, A. Genotyping cell-free tumor DNA in the blood to detect residual disease and drug resistance. Genome Biol. 2014, 15, 449. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
miRNA–ROS interaction regulates cancer therapeutic tolerance and resistance through heterogeneous mechanisms. The mechanisms at hierarchy levels include phenotypic, signaling/metabolic, and genetic/epigenetic changes. ROS: reactive oxygen species; HIF: hypoxia-inducible factor; EGFR: epidermal growth factor receptor; VHL: von Hippel–Lindau; TCA: tricarboxylic acid; ↑: upregulation; ↓: downregulation.
Figure 1.
miRNA–ROS interaction regulates cancer therapeutic tolerance and resistance through heterogeneous mechanisms. The mechanisms at hierarchy levels include phenotypic, signaling/metabolic, and genetic/epigenetic changes. ROS: reactive oxygen species; HIF: hypoxia-inducible factor; EGFR: epidermal growth factor receptor; VHL: von Hippel–Lindau; TCA: tricarboxylic acid; ↑: upregulation; ↓: downregulation.


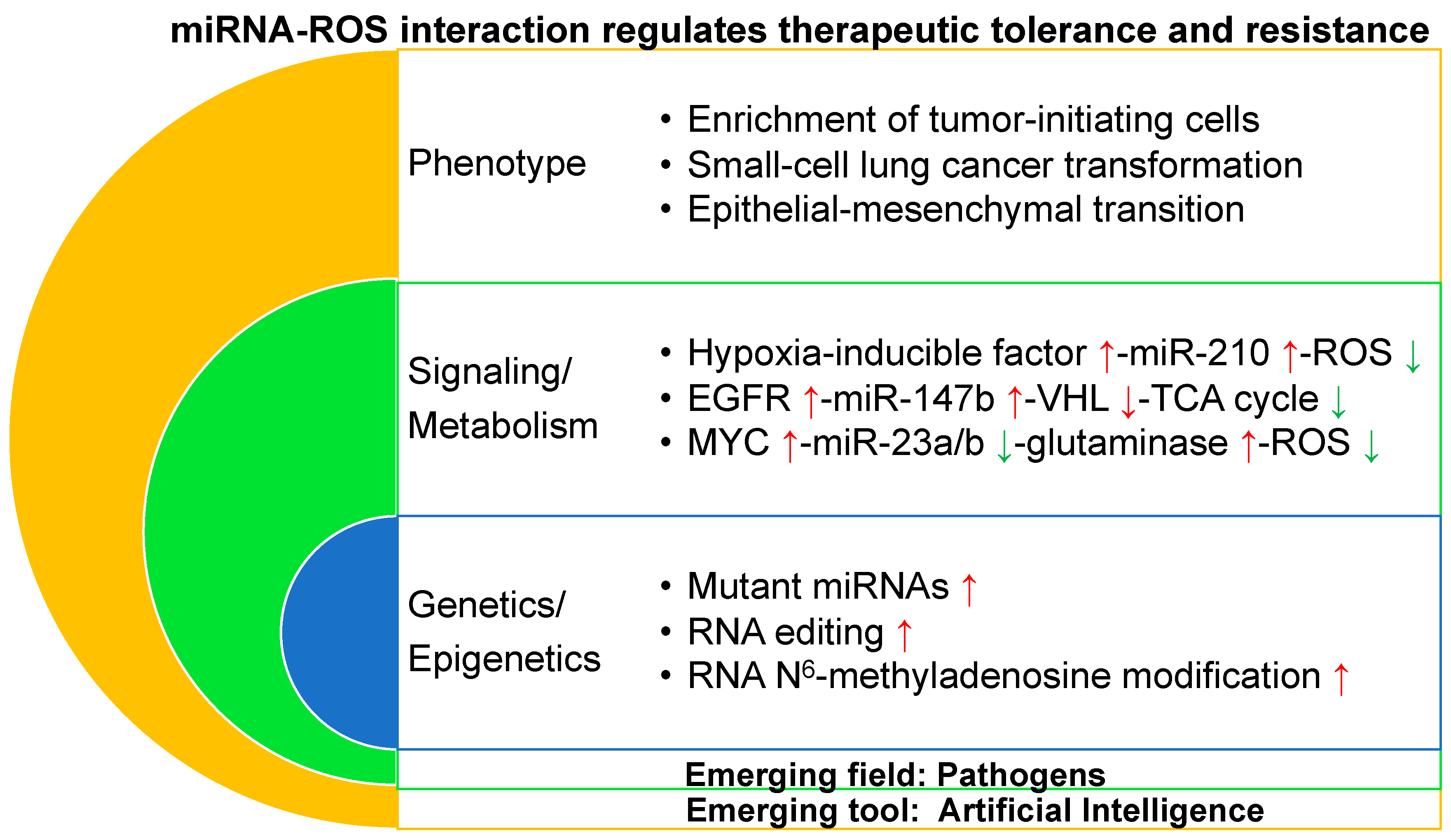{kind=link}
Table 1.
miRNA-mediated regulation of signaling pathways involved in tumorigenesis as well as therapeutic tolerance and resistance.
Table 1.
miRNA-mediated regulation of signaling pathways involved in tumorigenesis as well as therapeutic tolerance and resistance.
| miRNA | Signaling Involved in Tumorigenesis | Signaling Involved in Therapeutic Tolerance and Resistance |
|---|---|---|
| miR-1246 and miR-1290 ↑ | (+) tumorigenesis via repressing metallothioneins in human non-small cell lung cancer [75] | (+) resistance to EGFR tyrosine kinase inhibitor gefitinib via repressing metallothioneins in human non-small cell lung cancer [75] |
| miR-147b ↑ | N.A. | (+) tolerance to EGFR tyrosine kinase inhibitor osimertinib through activating pseudohypoxia signaling pathways via repressing VHL and succinate dehydrogenase in human non-small cell lung cancer [76] |
| miR-155 ↑ | (+) tumorigenesis in mouse miR155 transgenic B cell lymphomas [77] | (+) chemoresistance to gemcitabine through decreasing apoptosis in human pancreatic cancer [78] |
| miR-21 ↑ | (+) Ras/MEK/ERK signaling via repressing negative regulators of the Ras/MEK/ERK pathway and inhibition of apoptosis in mouse KRAS transgenic non-small cell lung cancer [54] | (+) chemoresistance to gemcitabine through decreasing apoptosis and activating Akt phosphorylation in human pancreatic cancer [79,80] (+) radioresistance through upregulation of hypoxia-inducible factor 1α in human non-small cell lung cancer [81] (+) resistance to EGFR tyrosine kinase inhibitors through activating PI3K-AKT signaling pathway in human non-small cell lung cancer [82] |
| miR-31 ↑ | (+) tumorigenesis through activating RAS/MAPK signaling via repressing negative regulators of RAS/MAPK signaling in mouse KRAS transgenic non-small cell lung cancer [46] | N.A. |
| let-7 family ↓ | (+) tumorigenesis in human breast cancer through repressing H-RAS and high mobility group AT-hook 2 [83] | (+) resistance to EGFR tyrosine kinase inhibitor gefitinib through upregulation of MYC in human non-small cell lung cancer [84] |
| miR-30 ↓ | ||
| miR-34a/b/c ↓ | (+) chemoresistance to fludarabine through p53 inactivation and apoptosis resistance in human chronic lymphocytic leukemia [90] |
EGFR: epidermal growth factor receptor; Akt: Akt Serine/Threonine Kinase; MAPK: mitogen-activated protein kinase; MEK: Mitogen-activated protein kinase kinase; ERK: extracellular-signal-regulated kinase; PI3K: phosphatidylinositol 3-kinase; AXL: AXL receptor tyrosine kinase; Apc: adenomatous polyposis coli; VHL: Von Hippel–Lindau; mTOR: mammalian target of rapamycin; ↑: upregulation; ↓: downregulation; (+): promotion; (−): repression; N.A.: not available.
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zhang, W.C. microRNAs Tune Oxidative Stress in Cancer Therapeutic Tolerance and Resistance. Int. J. Mol. Sci. 2019, 20, 6094. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236094
AMA Style
Zhang WC. microRNAs Tune Oxidative Stress in Cancer Therapeutic Tolerance and Resistance. International Journal of Molecular Sciences. 2019; 20(23):6094. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236094
Chicago/Turabian StyleZhang, Wen Cai. 2019. "microRNAs Tune Oxidative Stress in Cancer Therapeutic Tolerance and Resistance" International Journal of Molecular Sciences 20, no. 23: 6094. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236094
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.