The Detection and Partial Localisation of Heteroplasmic Mutations in the Mitochondrial Genome of Patients with Diabetic Retinopathy


Abstract
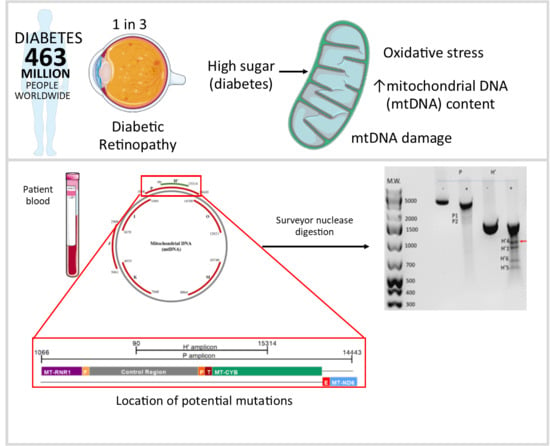
:
1. Introduction
2. Results
2.1. Characteristics of Patients Used in This Study
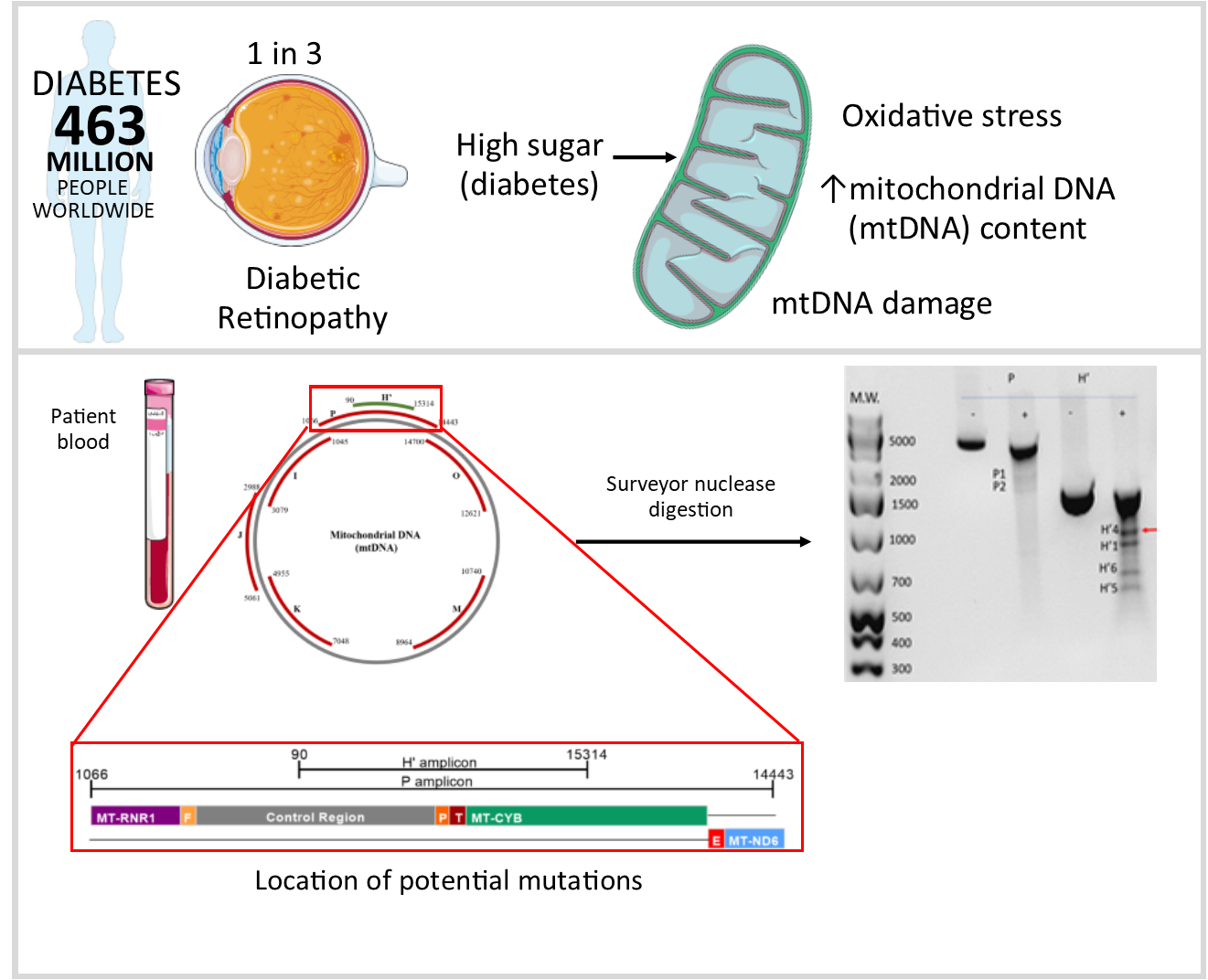
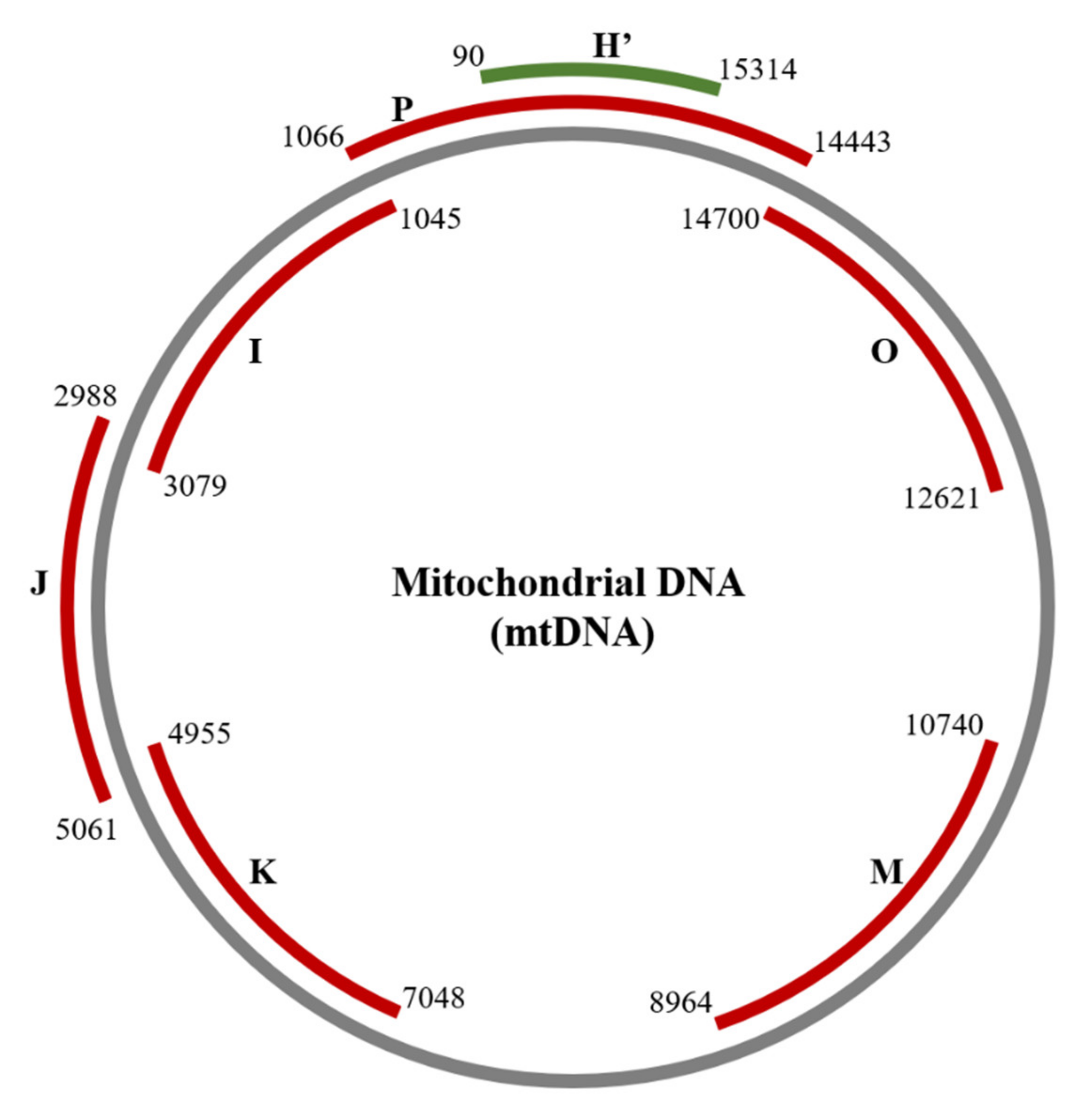
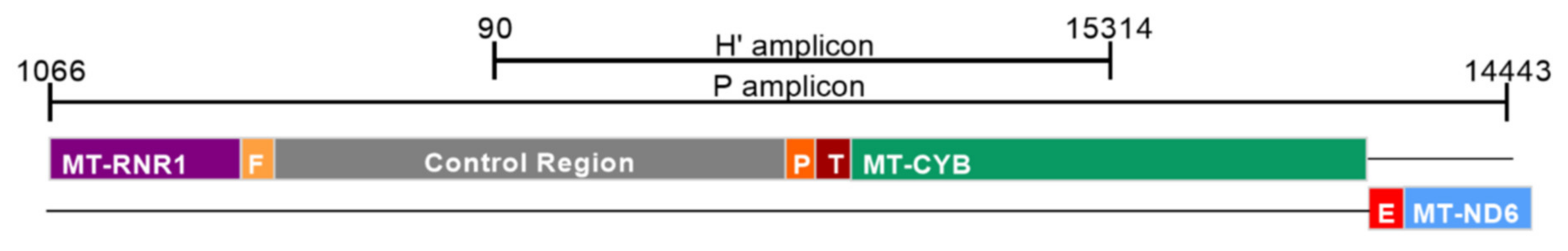
2.2. Detection of Damage in 6 Overlapping Regions of the Mitochondrial Genome: Pilot Study
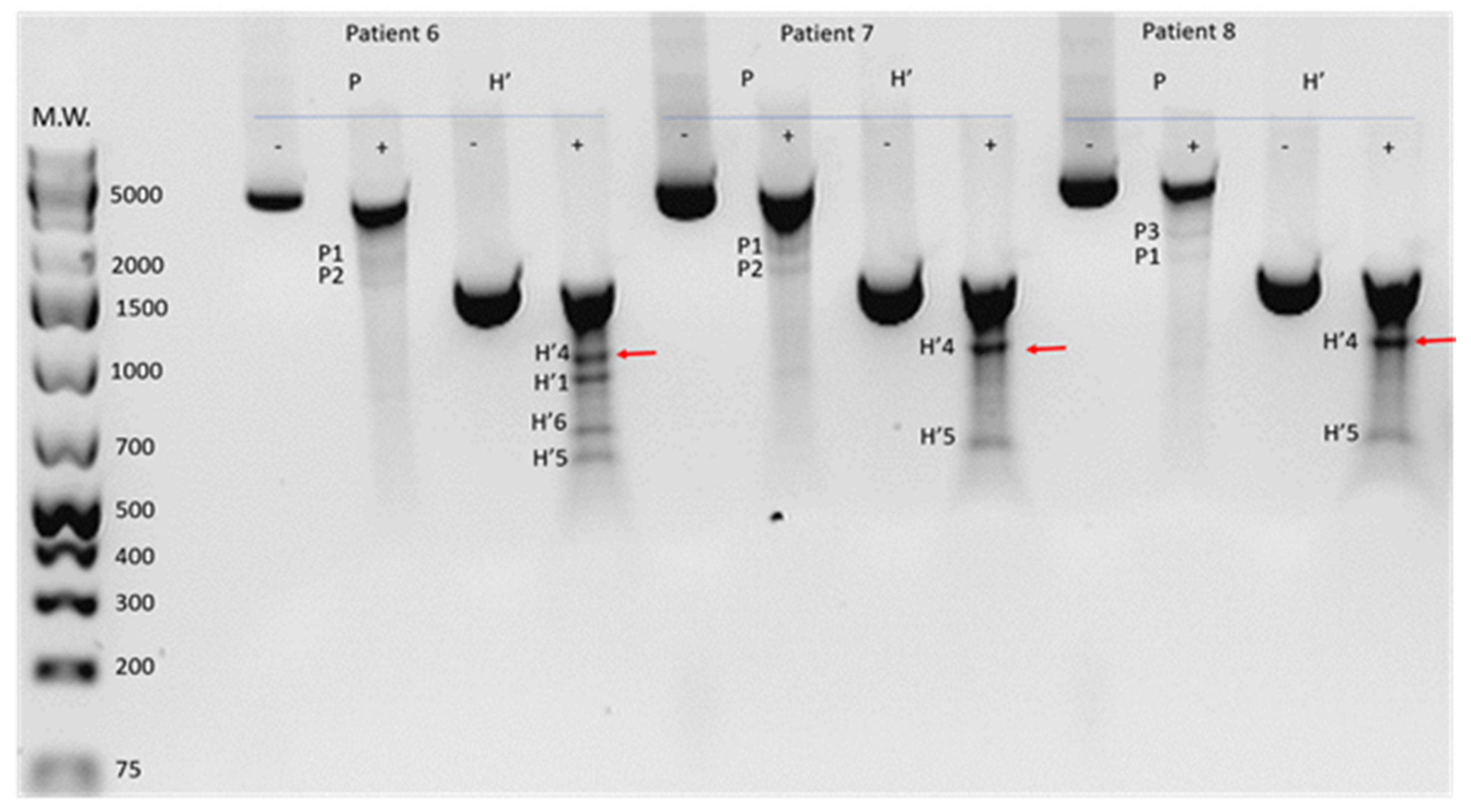
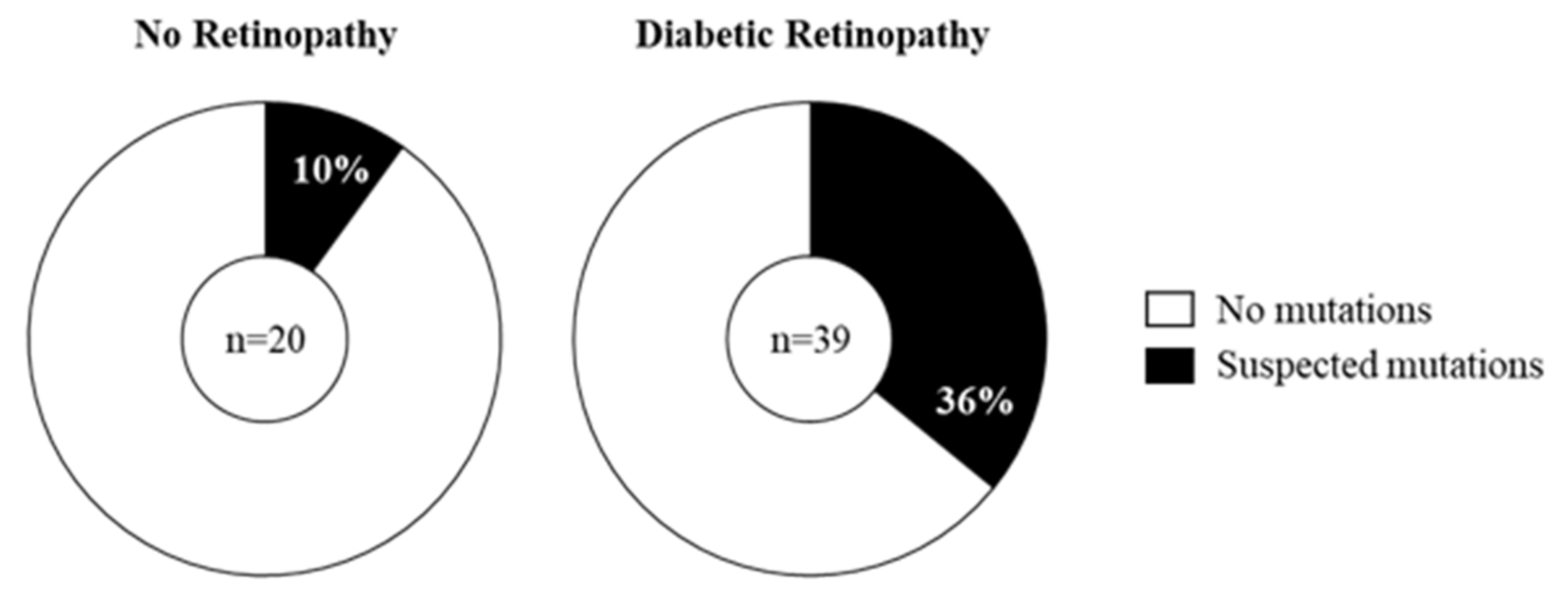
2.3. Prevalence of Mitochondrial DNA Mutations in Amplicon P in the Study Cohort
2.4. Does the Prevalence of Mutations Change with Severity of DR?
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Determination of Circulating mtDNA Content
4.3. Detection of mtDNA Damage Using the Surveyor Nuclease Method
4.4. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DR | Diabetic retinopathy |
| DN | Diabetic nephropathy |
| ETDRS | Early Treatment Diabetic Retinopathy Study |
| NPDR | Nonproliferative diabetic retinopathy |
| PDR | Proliferative diabetic retinopathy |
| ROS | Reactive oxygen species |
| OXPHOS | Oxidative phosphorylation |
| PCR | Polymerase chain reaction |
| HbA1c | Glycated haemoglobin |
| BMI | Body mass index |
| eGFR | Estimated glomerular filtration rate |
| MDRD | Modification of Diet in Renal Disease |
| tRNA | Transfer RNA |
| rRNA | Ribosomal RNA |
| mtDNA | Mitochondrial DNA |
| NADH | Nicotinamide adenine dinucleotide hydrogen |
| LHON | Leber’s hereditary optic neuropathy |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amplicon | Size (bp) | Forward Primer (5′-3′) | Position | Reverse Primer (5′-3′) | Position |
|---|---|---|---|---|---|
| H’ | 1345 | GCAGCCCTAGCAACACTCC | nt 15314–15332 | CAATGCTATCGCGTGCATACC | nt 90–70 |
| P | 3192 | CTCCTCAATAGCCATCGCTG | nt 14443–14462 | GGGTCTTAGCTATTGTGTGTTC | nt 1066–1045 |
| I | 2034 | GAACACACAATAGCTAAGACCC | nt 1045–1066 | CGGTCTGAACTCAGATCACGTA | nt 3079–3058 |
| J | 2073 | CGATGTTGGATCAGGACATCC | nt 2988–3008 | GGTTGTACGGTAGAACTGCTA | nt 5061–5041 |
| K | 2093 | CATAGCAGGCAGTTGAGGTG | nt 4955–4974 | GATAGGACATAGTGGAAGTGG | nt 7048–7028 |
| M | 1776 | CATCAGCCTACTCATTCAACC | nt 8964–8984 | GTACGTAGTCTAGGCCATATG | nt 10740–10720 |
| O | 2079 | CGTTACATGGTCCATCATAGAA | nt 12621–12642 | GTCGTGGTTGTAGTCCGTGC | nt 14700–14681 |
References
- Flaxman, S.R.; Bourne, R.R.A.; Resnikoff, S.; Ackland, P.; Braithwaite, T.; Cicinelli, M.V.; Das, A.; Jonas, J.B.; Keeffe, J.; Kempen, J.H.; et al. Global causes of blindness and distance vision impairment 1990–2020: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e1221–e1234. [Google Scholar] [CrossRef] [Green Version]
- Yau, J.W.; Rogers, S.L.; Kawasaki, R.; Lamoureux, E.L.; Kowalski, J.W.; Bek, T.; Chen, S.J.; Dekker, J.M.; Fletcher, A.; Grauslund, J.; et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 2012, 35, 556–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero-Aroca, P.; Navarro-Gil, R.; Valls-Mateu, A.; Sagarra-Alamo, R.; Moreno-Ribas, A.; Soler, N. Differences in incidence of diabetic retinopathy between type 1 and 2 diabetes mellitus: A nine-year follow-up study. Br. J. Ophthalmol. 2017, 101, 1346–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.; Klein, B.E.; Moss, S.E.; Davis, M.D.; DeMets, D.L. The Wisconsin epidemiologic study of diabetic retinopathy. III. Prevalence and risk of diabetic retinopathy when age at diagnosis is 30 or more years. Arch. Ophthalmol. 1984, 102, 527–532. [Google Scholar] [CrossRef]
- International Diabetes Federation. IDF Diabetes Atlas, 9th ed.; IDF: Brussels, Belgium, 2019; Available online: http://www.diabetesatlas.org (accessed on 5 December 2018).
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Zhu, X.; Hu, Y.; Li, T.; Gao, Y.; Shi, Y.; Tang, S. Mitochondrial DNA oxidative damage triggering mitochondrial dysfunction and apoptosis in high glucose-induced HRECs. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4203–4209. [Google Scholar] [CrossRef]
- Kanwar, M.; Chan, P.S.; Kern, T.S.; Kowluru, R.A. Oxidative damage in the retinal mitochondria of diabetic mice: possible protection by superoxide dismutase. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3805–3811. [Google Scholar]
- Madsen-Bouterse, S.A.; Mohammad, G.; Kanwar, M.; Kowluru, R.A. Role of mitochondrial DNA damage in the development of diabetic retinopathy, and the metabolic memory phenomenon associated with its progression. Antioxid. Redox Signal. 2010, 13, 797–805. [Google Scholar] [CrossRef] [Green Version]
- Santos, J.M.; Tewari, S.; Goldberg, A.F.; Kowluru, R.A. Mitochondrial biogenesis and the development of diabetic retinopathy. Free Radic. Biol. Med. 2011, 51, 1849–1860. [Google Scholar] [CrossRef] [Green Version]
- Malik, A.N.; Parsade, C.K.; Ajaz, S.; Crosby-Nwaobi, R.; Gnudi, L.; Czajka, A.; Sivaprasad, S. Altered circulating mitochondrial DNA and increased inflammation in patients with diabetic retinopathy. Diabetes Res. Clin. Pract. 2015, 110, 257–265. [Google Scholar] [CrossRef]
- Czajka, A.; Ajaz, S.; Gnudi, L.; Parsade, C.K.; Jones, P.; Reid, F.; Malik, A.N. Altered Mitochondrial Function, Mitochondrial DNA and Reduced Metabolic Flexibility in Patients With Diabetic Nephropathy. EBioMedicine 2015, 2, 499–512. [Google Scholar] [CrossRef] [Green Version]
- Furda, A.; Santos, J.H.; Meyer, J.N.; Van Houten, B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 2014, 1105, 419–437. [Google Scholar]
- Bannwarth, S.; Procaccio, V.; Paquis-Flucklinger, V. Surveyor Nuclease: a new strategy for a rapid identification of heteroplasmic mitochondrial DNA mutations in patients with respiratory chain defects. Hum. Mutat. 2005, 25, 575–582. [Google Scholar] [CrossRef]
- Bannwarth, S.; Procaccio, V.; Paquis-Flucklinger, V. Rapid identification of unknown heteroplasmic mutations across the entire human mitochondrial genome with mismatch-specific Surveyor Nuclease. Nat. Protoc. 2006, 1, 2037–2047. [Google Scholar] [CrossRef]
- Malik, A.N.; Rosa, H.S. Potential Mechanisms of Mitochondrial DNA Mediated Acquired Mitochondrial Disease. In Mitochondrial Biology and Experimental Therapeutics; Oliveira, P.J., Ed.; Springer: Berlin/Heidelberg, Germany, 2018. [Google Scholar]
- Qi, H.; Casalena, G.; Shi, S.; Yu, L.; Ebefors, K.; Sun, Y.; Zhang, W.; D’Agati, V.; Schlondorff, D.; Haraldsson, B.; et al. Glomerular Endothelial Mitochondrial Dysfunction Is Essential and Characteristic of Diabetic Kidney Disease Susceptibility. Diabetes 2017, 66, 763–778. [Google Scholar] [CrossRef] [Green Version]
- Qiu, C.; Hevner, K.; Abetew, D.; Sedensky, M.; Morgan, P.; Enquobahrie, D.A.; Williams, M.A. Mitochondrial DNA copy number and oxidative DNA damage in placental tissues from gestational diabetes and control pregnancies: A pilot study. Clin. Lab. 2013, 59, 655–660. [Google Scholar] [CrossRef] [Green Version]
- Qiu, P.; Shandilya, H.; D’Alessio, J.M.; O’Connor, K.; Durocher, J.; Gerard, G.F. Mutation detection using Surveyor nuclease. Biotechniques 2004, 36, 702–707. [Google Scholar] [CrossRef]
- Tobe, S.S.; Kitchener, A.C.; Linacre, A.M. Reconstructing mammalian phylogenies: a detailed comparison of the cytochrome B and cytochrome oxidase subunit I mitochondrial genes. PLoS ONE 2010, 5, e14156. [Google Scholar] [CrossRef] [Green Version]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef]
- Blakely, E.L.; Mitchell, A.L.; Fisher, N.; Meunier, B.; Nijtmans, L.G.; Schaefer, A.M.; Jackson, M.J.; Turnbull, D.M.; Taylor, R.W. A mitochondrial cytochrome b mutation causing severe respiratory chain enzyme deficiency in humans and yeast. FEBS J. 2005, 272, 3583–3592. [Google Scholar] [CrossRef]
- Padma, G.; Ramchander, P.V.; Nandur, V.U.; Kumar, K.R.; Padma, T. Novel Mutations Affecting the Secondary Structure of MT-RNR1 Gene: A Causal Relationship with Profound Nonsyndromic Hearing Impairment. Genetic Test. Mol. Biomarkers 2012, 16, 1092–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, T.M.; Blakely, E.L.; Swalwell, H.; Carter, J.E.; Kartsounis, L.D.; O’Donovan, D.G.; Turnbull, D.M.; Taylor, R.W.; de Silva, R.N. Mitochondrial transfer RNA(Phe) mutation associated with a progressive neurodegenerative disorder characterized by psychiatric disturbance, dementia, and akinesia-rigidity. Arch. Neurol. 2010, 67, 1399–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Pozzo, P.; Cardaioli, E.; Malfatti, E.; Gallus, G.N.; Malandrini, A.; Gaudiano, C.; Berti, G.; Invernizzi, F.; Zeviani, M.; Federico, A. A novel mutation in the mitochondrial tRNA(Pro) gene associated with late-onset ataxia, retinitis pigmentosa, deafness, leukoencephalopathy and complex I deficiency. Eur. J. Hum. Genet. 2009, 17, 1092–1096. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Torroni, A.; Shoffner, J.M.; Wallace, D.C. Mitochondrial tRNA(Thr) mutations and lethal infantile mitochondrial myopathy. Am.J. Human Genet. 1992, 51, 446–447. [Google Scholar]
- Wang, M.; Liu, H.; Zheng, J.; Chen, B.; Zhou, M.; Fan, W.; Wang, H.; Liang, X.; Zhou, X.; Eriani, G.; et al. A deafness and diabetes associated tRNA mutation caused the deficient pseudouridinylation at position 55 in tRNAGlu and mitochondrial dysfunction. J. Biol. Chem. 2016. [Google Scholar] [CrossRef] [Green Version]
- Bronstein, O.; Kroh, A.; Haring, E. Mind the gap! The mitochondrial control region and its power as a phylogenetic marker in echinoids. BMC Evol. Biol. 2018, 18, 80. [Google Scholar] [CrossRef]
- Sharma, H.; Singh, A.; Sharma, C.; Jain, S.K.; Singh, N. Mutations in the mitochondrial DNA D-loop region are frequent in cervical cancer. Cancer Cell Int. 2005, 5, 34. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.-C.; Wang, C.-C.; Jiang, R.-S.; Wang, W.-Y.; Liu, S.-A. Impact of Somatic Mutations in the D-Loop of Mitochondrial DNA on the Survival of Oral Squamous Cell Carcinoma Patients. PLoS ONE 2015, 10, e0124322. [Google Scholar] [CrossRef]
- Miyazono, F.; Schneider, P.M.; Metzger, R.; Warnecke-Eberz, U.; Baldus, S.E.; Dienes, H.P.; Aikou, T.; Hoelscher, A.H. Mutations in the mitochondrial DNA D-Loop region occur frequently in adenocarcinoma in Barrett’s esophagus. Oncogene 2002, 21, 3780–3783. [Google Scholar] [CrossRef] [Green Version]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Yaribeygi, H.; Atkin, S.L.; Sahebkar, A. Mitochondrial dysfunction in diabetes and the regulatory roles of antidiabetic agents on the mitochondrial function. J. Cell. Physiol. 2019, 234, 8402–8410. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Kowluru, R.A. Retinal Mitochondrial DNA Mismatch Repair in the Development of Diabetic Retinopathy, and Its Continued Progression After Termination of Hyperglycemia. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6960–6967. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Lillvis, J.; Seyoum, B.; Kowluru, R.A. Peripheral Blood Mitochondrial DNA Damage as a Potential Noninvasive Biomarker of Diabetic Retinopathy. Investig. Ophthamol. Vis. Sci. 2016, 57, 4035–4044. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.-L.; Luo, W.-B.; Xie, L.; Mao, X.-B.; Wu, C.; You, Z.-P. Targeting human 8-oxoguanine DNA glycosylase to mitochondria protects cells from high glucose-induced apoptosis. Endocrine 2018, 60, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Shao, X.; Jia, S.; Qu, L.; Weng, C.; Shen, X.; Wang, Y.; Huang, H.; Wang, Y.; Wang, C.; et al. The Mitochondria-Targeted Metabolic Tubular Injury in Diabetic Kidney Disease. Cell Phys. Biochem. 2019, 52, 156–171. [Google Scholar]
- Garcia-Ramirez, M.; Francisco, G.; Garcia-Arumi, E.; Hernandez, C.; Martinez, R.; Andreu, A.L.; Simo, R. Mitochondrial DNA oxidation and manganese superoxide dismutase activity in peripheral blood mononuclear cells from type 2 diabetic patients. Diabetes Metab. 2008, 34, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Levey, A.S.; Bosch, J.P.; Lewis, J.B.; Greene, T.; Rogers, N.; Roth, D.; Modification of Diet in Renal Disease Study Group. A More Accurate Method To Estimate Glomerular Filtration Rate from Serum Creatinine: A New Prediction Equation. Ann. Int. Med. 1999, 130, 461–470. [Google Scholar] [CrossRef]
- Ajaz, S.; Czajka, A.; Malik, A. Accurate Measurement of Circulating Mitochondrial DNA Content from Human Blood Samples Using Real-Time Quantitative PCR. In Mitochondrial Medicine: Volume I, Probing Mitochondrial Function; Weissig, V., Edeas, M., Eds.; Springer: New York, NY, USA, 2015; pp. 117–131. [Google Scholar]




| Diabetes with No Retinopathy (DR-0) | Diabetic Retinopathy (DR) | p-Value | |
|---|---|---|---|
| Number of patients | 20 | 39 | |
| Age (years) | 66.1 ± 15.9 (31.0–85.0) | 58.8 ± 14.0 (30.0–82.0) | ns |
| Sex (male:female) | 7:13 | 22:17 | ns |
| Diabetes (type 1:type 2) | 5:15 | 9:30 | |
| Duration (years) | 13.0 ± 8.6 (5–43) | 17.7 ± 10.8 (1–50) | ns |
| BMI (kg/m2) | 28.7 ± 4.1 (21.9–34.7) | 31.0 ± 12.1 (20.0–82.7) | ns a |
| HbA1c (%) | 7.5 ± 1.2 (5.7–9.5) | 8.8 ± 2.1 (5.8–13.1) | p = 0.0127 |
| HbA1c (mmol/mol) | 58.2 ± 13.4 (38.8–80.3) | 72.6 ± 22.5 (39.9–119.7) | p = 0.0126 |
| eGFR (mL·min−1·1.73 m−2) | 61.1 ± 22.7 (17.0–98.0) | 77.3 ± 26.7 (10.0–161.0) | p = 0.0331 a |
| ACR (mg/mmol) | 2.9 ± 5.0 (0.1–20.8) | 7.2 ± 14.3 (0.2–51.9) | ns a |
| Systolic BP (mmHg) | 133.9 ± 19.0 (125.0–187.0) | 132.4 ± 16.3 (97.0–169.0) | ns |
| Diastolic BP (mmHg) | 75.5 ± 8.1 (60.0–97.0) | 74.8 ± 9.2 (56.0–96.0) | ns |
| Total cholesterol (mmol/L) | 4.3 ± 0.8 (2.9–5.8) | 4.3 ± 1.0 (2.3–7.4) | ns |
| mtDNA content (Mt/N) | 105.2 ± 54.5 (33.3–208.1) | 131.9 ± 117.3 (20.9–624.7) | ns |
| Category | Patient | P Amplicon | H’ Amplicon | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P3 | P4 | P5 | H’1 | H’2 | H’3 | H’4 | H’5 | H’6 | H’7 | ||
| DR-0 | 114 | + | + | - | - | - | + | - | + | + | + | - | - |
| 153 | + | + | - | - | - | + | - | + | + | - | - | - | |
| DR-m | 137 | + | + | - | - | - | + | + | - | - | - | - | - |
| 157 | + | + | - | - | - | + | + | - | - | - | - | - | |
| 151 | + | - | + | - | - | - | - | - | + | + | - | - | |
| 91 | + | + | - | - | + | + | - | - | + | + | + | - | |
| 136 | + | + | - | + | - | - | - | - | + | + | - | - | |
| 110 | + | - | + | + | - | - | - | - | + | + | - | - | |
| 58 | - | + | + | - | - | - | - | - | + | - | - | - | |
| DR-s | 161 | - | - | + | - | - | - | - | - | + | - | - | - |
| 65 | + | + | - | - | - | - | - | - | + | - | - | - | |
| 14 | + | + | - | - | - | - | - | - | + | + | - | + | |
| 16 | + | + | - | - | - | - | - | - | - | + | + | + | |
| 8 | + | - | + | - | - | - | - | - | + | - | + | + | |
| 9 | + | - | + | - | - | + | - | - | + | - | - | - | |
| 23 | + | + | - | - | - | n | n | n | n | n | n | n | |
| Mild Diabetic Retinopathy (DR-m) | Severe Diabetic Retinopathy (DR-s) | p Value | |
|---|---|---|---|
| N = | 20 | 19 | |
| Age (years) | 54.7 ± 15.1 (30.0–82.0) | 63.1 ± 11.5 (43.0–82.0) | ns |
| Sex (male:female) | 10:10 | 12:7 | ns |
| Diabetes (type1: type 2) | 8:12 | 1:18 | |
| Duration (years) | 19.0 ± 12.4 (1–50) | 16.4 ± 8.9 (1–33) | ns |
| BMI (kg/m2) | 32.0 ± 10.4 (20.3–59.6) | 30.0 ± 14.0 (20.0–82.7) | ns a |
| HbA1c (%) | 8.2 ± 2.2 (5.8–12.1) | 9.4 ± 1.7 (6.3–13.1) | ns |
| HbA1c (mmol/mol) | 66.5 ± 23.6 (39.9–108.7) | 80.8 ± 18.6 (45.4–119.7) | ns |
| eGFR (mL·min−1·1.73 m−2) | 86.6 ± 26.4 (29.0–161.0) | 66.1 ± 22.9 (10.0–91.0) | p = 0.0297 a |
| ACR (mg/mmol) | 5.5 ± 13.2 (0.2–51.0) | 9.9 ± 16.3 (0.5–51.9) | ns a |
| Systolic BP (mmHg) | 127.8 ± 15.6 (97.0–169.0) | 137.5 ± 15.6 (108.0–169.0) | ns |
| Diastolic BP (mmHg) | 74.4 ± 8.2 (56.0–89.0) | 75.3 ± 40.5 (60.0–96.0) | ns |
| Total cholesterol (mmol/L) | 4.3 ± 1.2 (2.3–7.4) | 4.6 ± 1.3 (3.2–7.4) | ns |
| mtDNA content (Mt/N) | 183.0 ± 139.8 (20.8–624.7) | 78.2 ± 50.1 (33.1–183.0) | p = 0.0063 a |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malik, A.N.; Rosa, H.S.; S. de Menezes, E.; Tamang, P.; Hamid, Z.; Naik, A.; Parsade, C.K.; Sivaprasad, S. The Detection and Partial Localisation of Heteroplasmic Mutations in the Mitochondrial Genome of Patients with Diabetic Retinopathy. Int. J. Mol. Sci. 2019, 20, 6259. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246259
Malik AN, Rosa HS, S. de Menezes E, Tamang P, Hamid Z, Naik A, Parsade CK, Sivaprasad S. The Detection and Partial Localisation of Heteroplasmic Mutations in the Mitochondrial Genome of Patients with Diabetic Retinopathy. International Journal of Molecular Sciences. 2019; 20(24):6259. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246259
Chicago/Turabian StyleMalik, Afshan N., Hannah S. Rosa, Eliane S. de Menezes, Priyanka Tamang, Zaidi Hamid, Anita Naik, Chandani Kiran Parsade, and Sobha Sivaprasad. 2019. "The Detection and Partial Localisation of Heteroplasmic Mutations in the Mitochondrial Genome of Patients with Diabetic Retinopathy" International Journal of Molecular Sciences 20, no. 24: 6259. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246259