Genetic Programs Driving Oncogenic Transformation: Lessons from In Vitro Models


Abstract
:
1. Introduction
2. Results
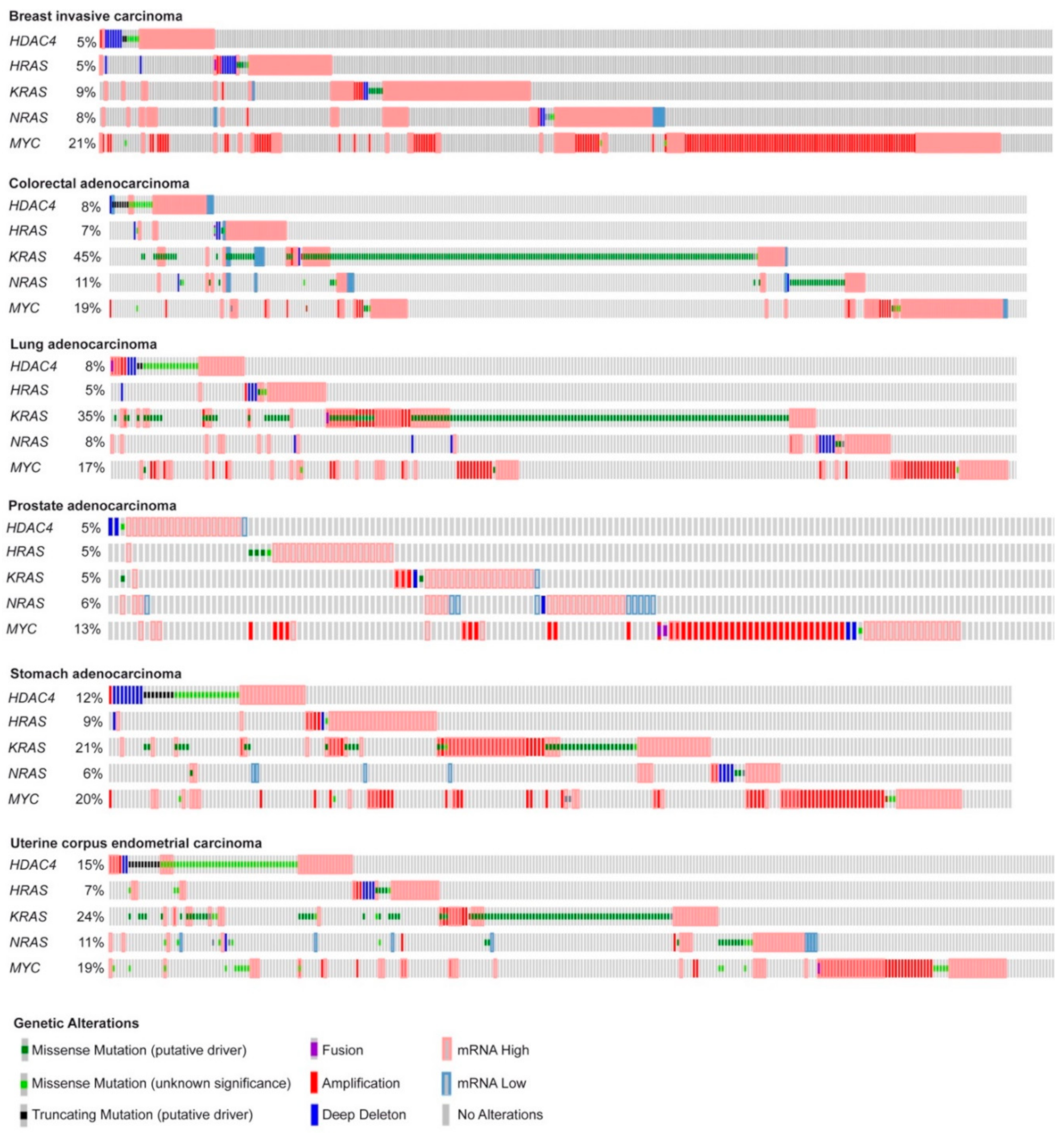
2.1. In Vitro Transformation of Human Fibroblasts Achieved by Different Oncogenes Leads to the Activation of Both Distinct and Common Genetic Programs
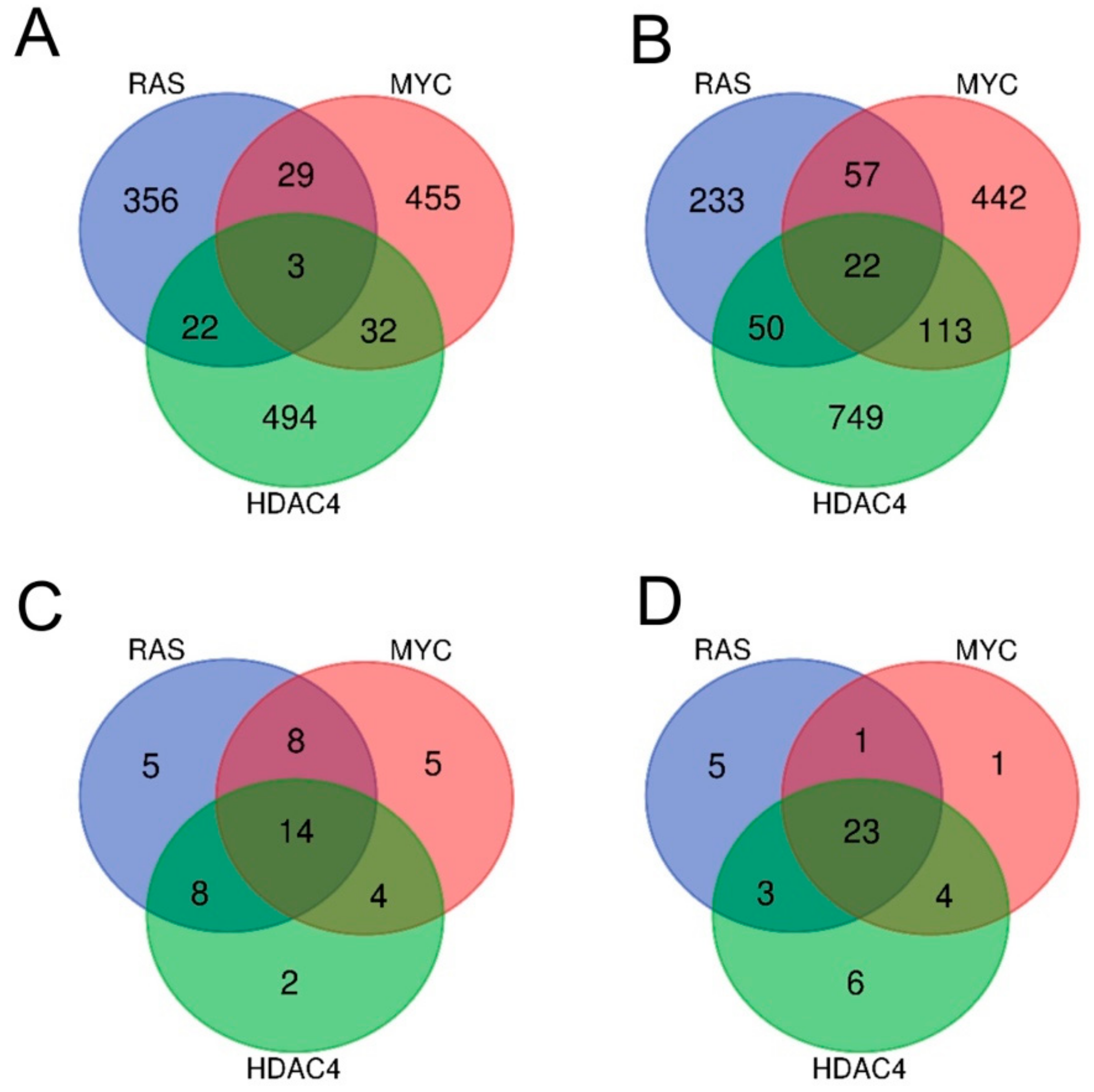
2.2. RAS, MYC, and HDAC4-Mediated Oncogenic Transformation Is Marked by the Dysregulation of Common Pathways Rather Than of Common Genes.
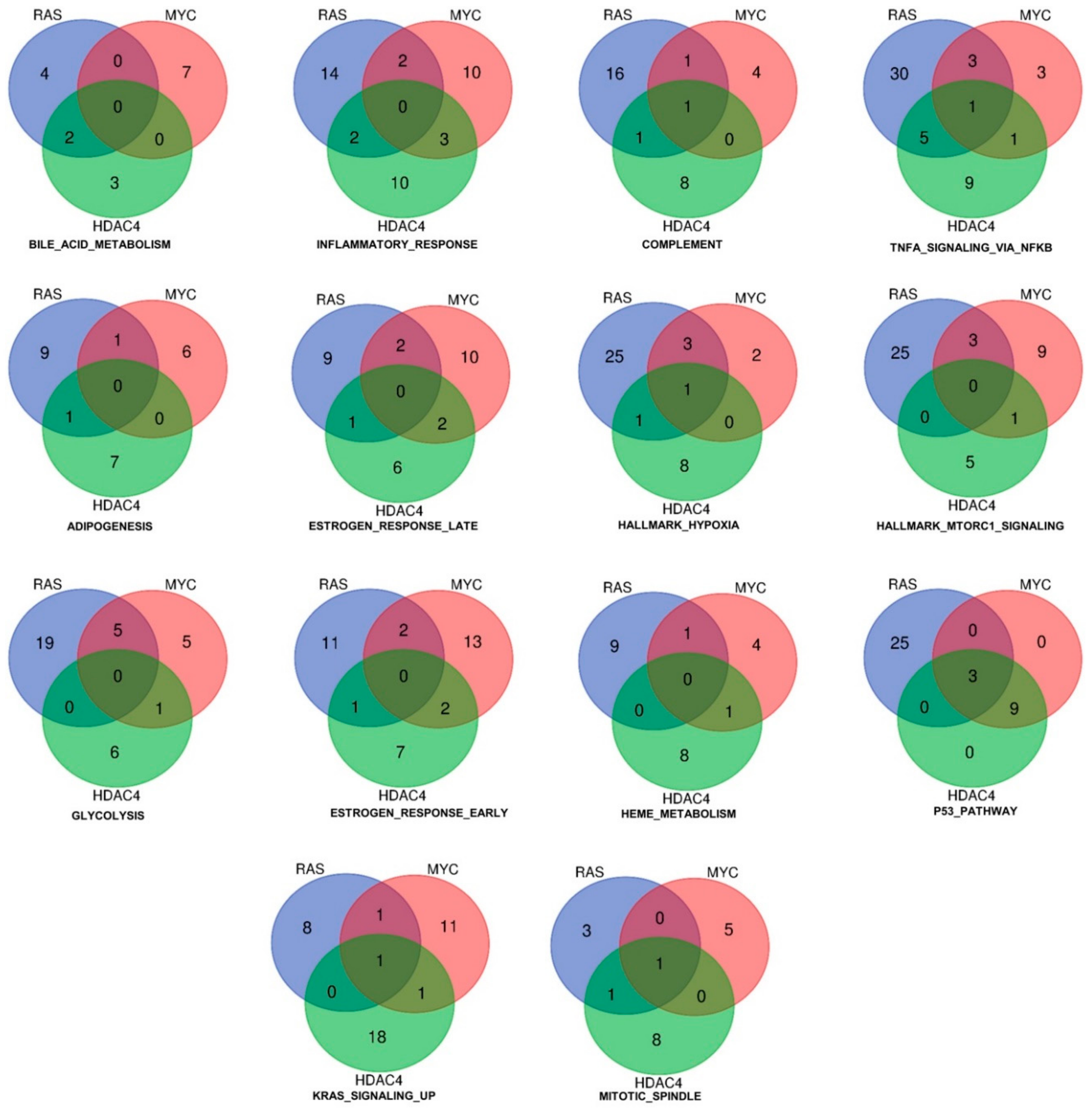
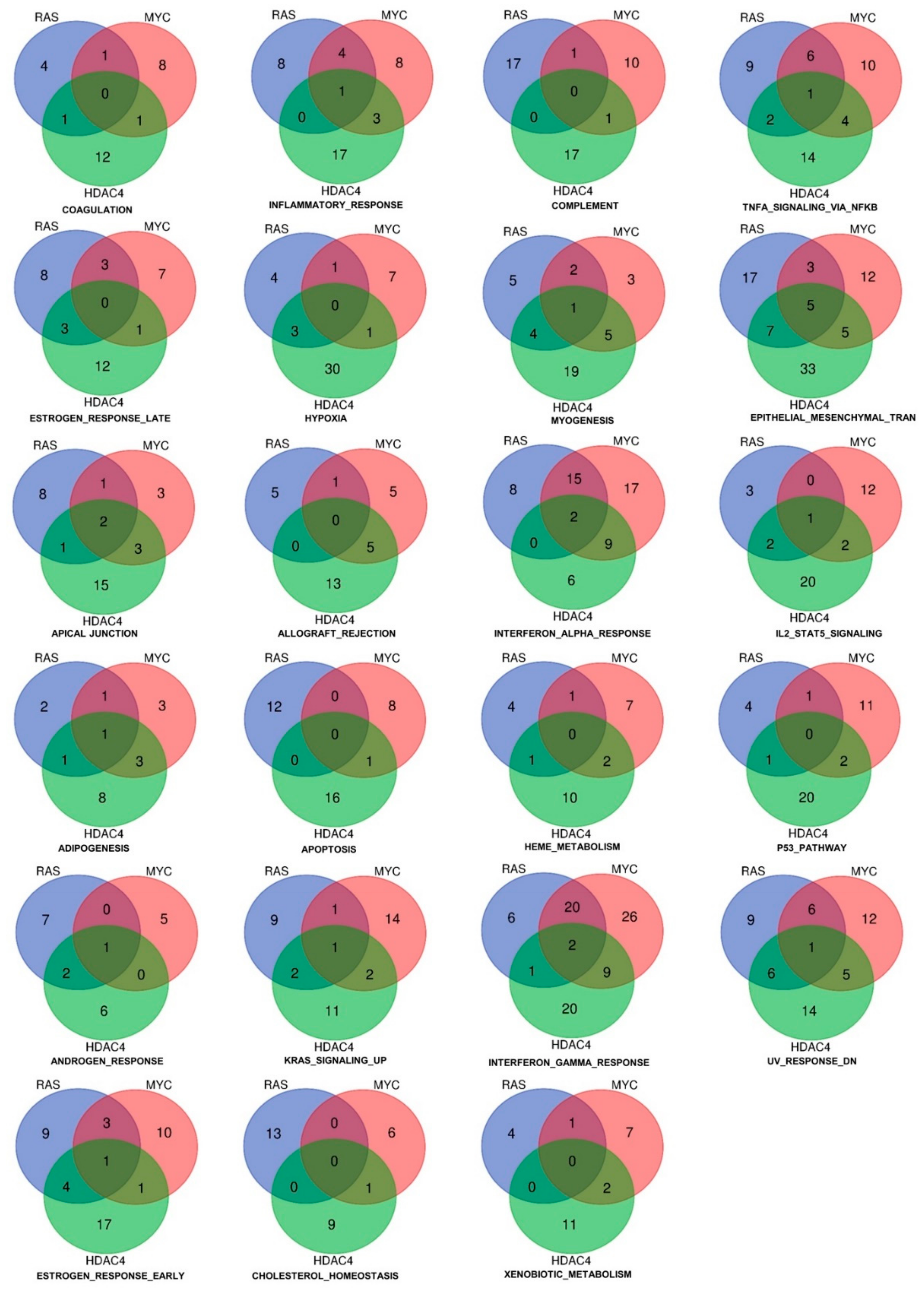
2.3. Identification of the Pathway Reprogramming Core that Defines the Common Trait of the in Vitro Transformation Process
2.4. Definition of the Minimal Signatures Regulated during in Vitro Transformation
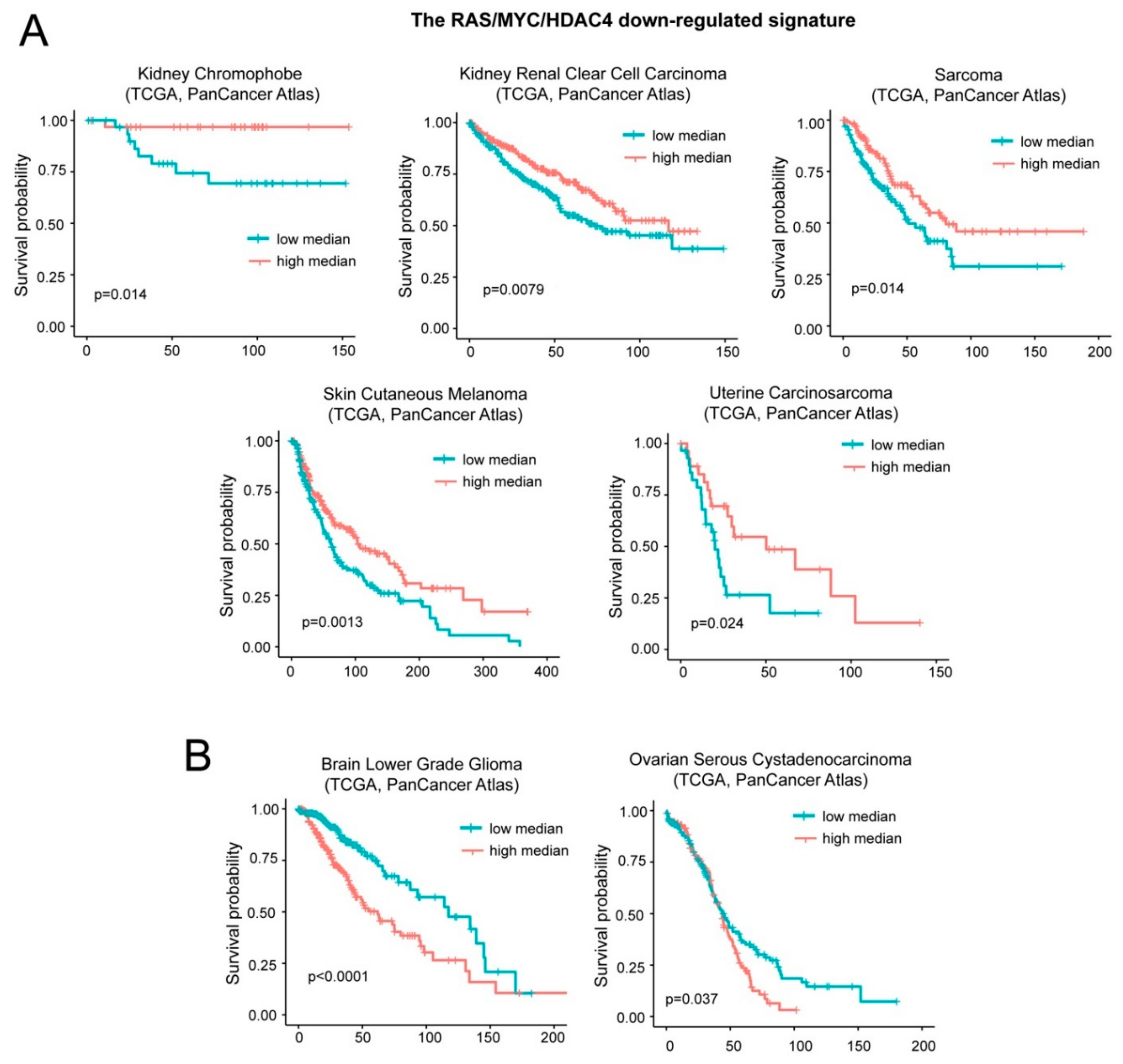
2.5. High mRNA Levels of the Upregulated Genes Correlate with Reduced Patients’ Survival in a Group of Different Tumors
2.6. High Levels of DOCK4, G0S2, and SRPX Expression Are Related to Worse Survival in Similar but also Different Tumor Types
2.7. A Group of Genes Repressed during in Vitro Transformation Predicts Patients’ Survival.
2.8. Inflammatory Genes Repressed during the in Vitro Transformation Process and Tissue-Infiltrating Immune Cells
2.9. Oncogene-Repressed Inflammatory Signature Provides an Additional Contribution to Overall Survival
3. Discussion
4. Materials and Methods
4.1. Data Retrieval and Analysis
4.2. Enrichment Analysis
4.3. Generation of the Signatures of Transformation
4.4. Analysis of Survival
4.5. Estimation of the Contribution/Perturbation of the Immune Infiltration to the Survival Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- You, J.S.; Jones, P.A. Cancer Genetics and Epigenetics: Two Sides of the Same Coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorsson, V.; Gibbs, D.L.; Brown, S.D.; Wolf, D.; Bortone, D.S.; Ou Yang, T.-H.; Porta-Pardo, E.; Gao, G.F.; Plaisier, C.L.; Eddy, J.A.; et al. The Immune Landscape of Cancer. Immunity 2018, 48, 812–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sompairac, N.; Nazarov, P.V.; Czerwinska, U.; Cantini, L.; Biton, A.; Molkenov, A.; Zhumadilov, Z.; Barillot, E.; Radvanyi, F.; Gorban, A.; et al. Independent Component Analysis for Unraveling the Complexity of Cancer Omics Datasets. Int. J. Mol. Sci. 2019, 20, 4414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laubenbacher, R.; Hower, V.; Jarrah, A.; Torti, S.V.; Shulaev, V.; Mendes, P.; Torti, F.M.; Akman, S. A systems biology view of cancer. Biochim. Biophys. Acta Rev. Cancer 2009, 1796, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Kuipers, J.; Jahn, K.; Beerenwinkel, N. Biochimica et Biophysica Acta Advances in understanding tumour evolution through single-cell sequencing. BBA Rev. Cancer 2017, 1867, 127–138. [Google Scholar]
- Hahn, W.C.; Counter, C.M.; Lundberg, A.S.; Beijersbergen, R.L.; Brooks, M.W.; Weinberg, R.A. Creation of human tumour cells with defined genetic elements. Nature 1999, 400, 464–468. [Google Scholar] [CrossRef]
- Knudson, A.G. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 2001, 1, 637–641. [Google Scholar] [CrossRef]
- Rangarajan, A.; Weinberg, R.A. Comparative biology of mouse versus human cells: Modelling human cancer in mice. Nat Rev. Cancer 2003, 3, 952–959. [Google Scholar] [CrossRef]
- Boehm, J.S.; Hession, M.T.; Bulmer, S.E.; Hahn, W.C. Transformation of Human and Murine Fibroblasts without Viral Oncoproteins. Mol. Cell. Biol. 2005, 25, 6464–6474. [Google Scholar] [CrossRef] [Green Version]
- Drost, J.; van Boxtel, R.; Blokzijl, F.; Mizutani, T.; Sasaki, N.; Sasselli, V.; de Ligt, J.; Behjati, S.; Grolleman, J.E.; van Wezel, T.; et al. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 2017, 358, 234–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, W.C.; Weinberg, R.A. Rules for Making Human Tumor Cells. N. Engl. J. Med. 2002, 347, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Joshi, A.; Zhang, Y.; Jaeger, S.A.; Bulyk, M.; Tsichlis, P.N.; Shirley Liu, X.; Struhl, K. A Transcriptional Signature and Common Gene Networks Link Cancer with Lipid Metabolism and Diverse Human Diseases. Cancer Cell 2010, 17, 348–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malysheva, V.; Mendoza-parra, M.A.; Saleem, M.M.; Gronemeyer, H. Reconstruction of gene regulatory networks reveals chromatin remodelers and key transcription factors in tumorigenesis. Genome Med. 2016, 8, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paluvai, H.; Di Giorgio, E.; Brancolini, C. Unscheduled HDAC4 repressive activity in human fibroblasts triggers TP53-dependent senescence and favors cell transformation. Mol. Oncol. 2018, 12, 2165–2181. [Google Scholar] [CrossRef] [PubMed]
- Makridakis, N.M.; Caldas Ferraz, L.F.; Reichardt, J.K.V. Genomic analysis of cancer tissue reveals that somatic mutations commonly occur in a specific motif. Hum. Mutat. 2009, 30, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daly, M.J.; Patterson, N.; Mesirov, J.P.; Golub, T.R.; Tamayo, P.; Spiegelman, B. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Peruzzo, P.; Comelli, M.; Di Giorgio, E.; Franforte, E.; Mavelli, I.; Brancolini, C. Transformation by different oncogenes relies on speci fi c metabolic adaptations. Cell Cycle 2016, 15, 2656–2668. [Google Scholar] [CrossRef] [Green Version]
- Perucho, M.; Esteban, M. Inhibitory effect of interferon on the genetic and oncogenic transformation by viral and cellular genes. J. Virol. 1985, 54, 229–232. [Google Scholar]
- Samid, D.; Flessate, D.M.; Friedman, R.M. Interferon-induced revertants of ras-transformed cells: Resistance to transformation by specific oncogenes and retransformation by 5-azacytidine. Mol. Cell. Biol. 1987, 7, 2196–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.Y.; Lee, K.-H.; Chang, S.; Chung, C.; Lee, H.-W.; Yim, J.; Kim, T.K. Oncogenic Potential of a Dominant Negative Mutant of Interferon Regulatory Factor 3. J. Biol. Chem. 2003, 278, 15272–15278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Critchley-Thorne, R.J.; Yan, N.; Nacu, S.; Weber, J.; Holmes, S.P.; Lee, P.P. Down-Regulation of the Interferon Signaling Pathway in T Lymphocytes from Patients with Metastatic Melanoma. PLoS Med. 2007, 4, e176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katlinskaya, Y.V.; Katlinski, K.V.; Yu, Q.; Ortiz, A.; Beiting, D.P.; Brice, A.; Davar, D.; Sanders, C.; Kirkwood, J.M.; Rui, H.; et al. Suppression of Type I Interferon Signaling Overcomes Oncogene-Induced Senescence and Mediates Melanoma Development and Progression. Cell Rep. 2016, 15, 171–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manry, J.; Laval, G.; Patin, E.; Fornarino, S.; Itan, Y.; Fumagalli, M.; Sironi, M.; Tichit, M.; Bouchier, C.; Casanova, J.-L.; et al. Evolutionary genetic dissection of human interferons. J. Exp. Med. 2011, 208, 2747–2759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yajnik, V.; Paulding, C.; Sordella, R.; McClatchey, A.I.; Saito, M.; Wahrer, D.C.R.; Reynolds, P.; Bell, D.W.; Lake, R.; van den Heuvel, S.; et al. DOCK4, a GTPase activator, is disrupted during tumorigenesis. Cell 2003, 112, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Kawada, K.; Upadhyay, G.; Ferandon, S.; Janarthanan, S.; Hall, M.; Vilardaga, J.-P.; Yajnik, V. Cell Migration Is Regulated by Platelet-Derived Growth Factor Receptor Endocytosis. Mol. Cell. Biol. 2009, 29, 4508–4518. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Tai, Y.; Jin, Y.; Hammell, M.C.; Wilkinson, J.E.; Roe, J.; Vakoc, C.R.; Van Aelst, L. TGF-β/Smad signaling through DOCK4 facilitates lung adenocarcinoma metastasis. Genes Dev. 2015, 29, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Westbrook, J.A.; Wood, S.L.; Cairns, D.A.; McMahon, K.; Gahlaut, R.; Thygesen, H.; Shires, M.; Roberts, S.; Marshall, H.; Oliva, M.R.; et al. Identification and validation of DOCK4 as a potential biomarker for risk of bone metastasis development in patients with early breast cancer. J. Pathol. 2019, 247, 381–391. [Google Scholar] [CrossRef]
- Debruyne, D.N.; Turchi, L.; Fareh, M.; Almairac, F.; Virolle, V. DOCK4 promotes loss of proliferation in glioblastoma progenitor cells through nuclear beta-catenin accumulation and subsequent miR-302-367 cluster expression. Nat. Publ. Gr. 2017, 37, 241–254. [Google Scholar] [CrossRef]
- Sundaravel, S.; Kuo, W.-L.; Jeong, J.J.; Choudhary, G.S.; Gordon-Mitchell, S.; Liu, H.; Bhagat, T.D.; McGraw, K.L.; Gurbuxani, S.; List, A.F.; et al. Loss of Function of DOCK4 in Myelodysplastic Syndromes Stem Cells is Restored by Inhibitors of DOCK4 Signaling Networks. Clin. Cancer Res. 2019, 25, 5638–5649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, R.A.; Hesselink, M.K. Recent advances in lipid droplet biology. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1129–1130. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Liu, C.; Cheng, C. REACTIN: Regulatory activity inference of transcription factors underlying human diseases with application to breast cancer. BMC Genom. 2013, 14, 504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, C.Y.; Sekula, D.J.; Hever-Jardine, M.P.; Liu, X.; Warzecha, J.M.; Tam, J.; Freemantle, S.J.; Dmitrovsky, E.; Spinella, M.J. G0S2 Suppresses Oncogenic Transformation by Repressing a MYC-Regulated Transcriptional Program. Cancer Res. 2016, 76, 1204–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, D.R.; Lerario, A.M.; Else, T.; Mukherjee, B.; Almeida, M.Q.; Vinco, M.; Rege, J.; Mariani, B.M.P.; Zerbini, M.C.N.; Mendonca, B.B.; et al. Targeted Assessment of G0S2 Methylation Identifies a Rapidly Recurrent, Routinely Fatal Molecular Subtype of Adrenocortical Carcinoma. Clin. Cancer Res. 2019, 25, 3276–3288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meindl, A.; Carvalho, M.R.; Herrmann, K.; Lorenz, B.; Achatz, H.; Lorenz, B.; Apfelstedt-Sylla, E.; Wittwer, B.; Ross, M.; Meitinger, T. A gene (SRPX) encoding a sushi-repeat-containing protein is deleted in patients with X-linked retinitis pigmentosa. Hum. Mol. Genet. 1995, 4, 2339–2346. [Google Scholar] [CrossRef]
- Jing, P.; Kazuyoshi, N.; Masuo, Y.; Hirokazu, I.; Qin, L.; Kiyomasa, O.; Naohisa, Y.; Akira, H. Isolation of a novel gene down-regulated by v-src. FEBS Lett. 1996, 383, 21–25. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, A.; Hakura, A.; Inoue, H. Suppression of anchorage-independent growth of human cancer cell lines by the drs gene. Oncogene 1999, 18, 4777–4787. [Google Scholar] [CrossRef] [Green Version]
- Shimakage, M.; Kawahara, K.; Kikkawa, N.; Sasagawa, T.; Yutsudo, M.; Inoue, H. Down-regulation of drs mRNA in human colon adenocarcinomas. Int. J. Cancer 2000, 87, 5–11. [Google Scholar] [CrossRef]
- Kim, C.J.; Shimakage, M.; Kushima, R.; Mukaisho, K.; Shinka, T.; Okada, Y.; Inoue, H. Down-regulation of drs mRNA in human prostate carcinomas. Hum. Pathol. 2003, 34, 654–657. [Google Scholar] [CrossRef]
- Shimakage, M.; Inoue, N.; Ohshima, K.; Kawahara, K.; Yamamoto, N.; Oka, T.; Tambe, Y.; Yasui, K.; Matsumoto, K.; Yutsudo, M.; et al. Downregulation of drs mRNA expression is associated with the progression of adult T-cell leukemia/lymphoma. Int. J. Oncol. 2007, 30, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Else, T.; Kim, A.C.; Sabolch, A.; Raymond, V.M.; Kandathil, A.; Caoili, E.M.; Jolly, S.; Miller, B.S.; Giordano, T.J.; Hammer, G.D. Adrenocortical Carcinoma. Endocr. Rev. 2014, 35, 282–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finotello, F.; Trajanoski, Z. Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immunol. Immunother. 2018, 67, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-puig, P.; Sautès-fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Li, Y.; Fan, X.; Zhang, C.; Wang, Y.; Jiang, T.; Liu, X. Molecular and clinical characterization of IDH associated immune signature in lower-grade gliomas. Oncoimmunology 2018, 7, e1434466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ischenko, I.; Zhi, J.; Moll, U.M.; Nemajerova, A.; Petrenko, O. Direct reprogramming by oncogenic Ras and Myc. Proc. Natl. Acad. Sci. USA 2013, 110, 3937–3942. [Google Scholar] [CrossRef] [Green Version]
- Di Giorgio, E.; Clocchiatti, A.; Piccinin, S.; Sgorbissa, A.; Viviani, G.; Peruzzo, P.; Romeo, S.; Rossi, S.; Dei Tos, A.P.; Maestro, R.; et al. MEF2 is a converging hub for histone deacetylase 4 and phosphatidylinositol 3-kinase/Akt-induced transformation. Mol. Cell. Biol. 2013, 33, 4473–4491. [Google Scholar] [CrossRef] [Green Version]
- Di Giorgio, E.; Franforte, E.; Cefalù, S.; Rossi, S.; Dei Tos, A.P.; Brenca, M.; Polano, M.; Maestro, R.; Paluvai, H.; Picco, R.; et al. The co-existence of transcriptional activator and transcriptional repressor MEF2 complexes influences tumor aggressiveness. PLoS Genet. 2017, 13, e1006752. [Google Scholar] [CrossRef]
- Di Giorgio, E.; Gagliostro, E.; Clocchiatti, A.; Brancolini, C. The Control Operated by the Cell Cycle Machinery on MEF2 Stability Contributes to the Downregulation of CDKN1A and Entry into S Phase. Mol. Cell. Biol. 2015, 35, 1633–1647. [Google Scholar] [CrossRef] [Green Version]
- Di Giorgio, E.; Brancolini, C. Regulation of class IIa HDAC activities: It is not only matter of subcellular localization. Epigenomics 2016, 8, 251–269. [Google Scholar] [CrossRef] [Green Version]
- Heckmann, B.L.; Zhang, X.; Xie, X.; Saarinen, A.; Lu, X.; Yang, X.; Liu, J. Defective Adipose Lipolysis and Altered Global Energy Metabolism in Mice with Adipose Overexpression of the Lipolytic Inhibitor G 0/G 1 Switch Gene 2 (G0S2). J. Biol. Chem. 2014, 289, 1905–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, C.Y.; Bikorimana, E.; Khan, E.; Warzecha, J.M.; Shin, L.; Rodriguez, J.; Dmitrovsky, E.; Freemantle, S.J.; Spinella, M.J. G0S2 represses PI3K/mTOR signaling and increases sensitivity to PI3K/mTOR pathway inhibitors in breast cancer. Cell Cycle 2017, 16, 2146–2155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukunaga, T.; Fujita, Y.; Kishima, H.; Yamashita, T. Methylation dependent down-regulation of G0S2 leads to suppression of invasion and improved prognosis of IDH1-mutant glioma. PLoS ONE 2018, 13, e0206552. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Kwon, Y.-J.; Ye, D.-J.; Baek, H.-S.; Kwon, T.-U.; Choi, H.-K.; Chun, Y.-J. G0/G1 Switch 2 Induces Cell Survival and Metastasis through Integrin-Mediated Signal Transduction in Human Invasive Breast Cancer Cells. Biomol. Ther. 2019, 12, 1–12. [Google Scholar] [CrossRef]
- Balachandran, S.; Porosnicu, M.; Barber, G.N. Oncolytic activity of vesicular stomatitis virus is effective against tumors exhibiting aberrant p53, Ras, or myc function and involves the induction of apoptosis. J. Virol. 2001, 75, 3474–3479. [Google Scholar] [CrossRef] [Green Version]
- Klampfer, L.; Huang, J.; Corner, G.; Mariadason, J.; Arango, D.; Sasazuki, T.; Shirasawa, S.; Augenlicht, L. Oncogenic Ki-Ras Inhibits the Expression of Interferon-responsive Genes through Inhibition of STAT1 and STAT2 Expression. J. Biol. Chem. 2003, 278, 46278–46287. [Google Scholar] [CrossRef] [Green Version]
- Battcock, S.M.; Collier, T.W.; Zu, D.; Hirasawa, K. Negative regulation of the alpha interferon-induced antiviral response by the Ras/Raf/MEK pathway. J. Virol. 2006, 80, 4422–4430. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, Y.; Christian, S.L.; Ho, N.; Pongnopparat, T.; Licursi, M.; Hirasawa, K. Oncogenic Ras inhibits IRF1 to promote viral oncolysis. Oncogene 2015, 34, 3985–3993. [Google Scholar] [CrossRef]
- Komatsu, Y.; Hirasawa, K.; Christian, S.L. Global gene analysis identifying genes commonly regulated by the Ras/Raf/MEK and type I IFN pathways. Genom. Data 2015, 4, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Jonak, G.J.; Knight, E. Selective reduction of c-myc mRNA in Daudi cells by human beta interferon. Proc. Natl. Acad. Sci. USA 1984, 81, 1747–1750. [Google Scholar] [CrossRef] [Green Version]
- Dani, C.; Mechti, N.; Piechaczyk, M.; Lebleu, B.; Jeanteur, P.; Blanchard, J.M. Increased rate of degradation of c-myc mRNA in interferon-treated Daudi cells. Proc. Natl. Acad. Sci. USA 1985, 82, 4896–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlee, M.; Hölzel, M.; Bernard, S.; Mailhammer, R.; Schuhmacher, M.; Reschke, J.; Eick, D.; Marinkovic, D.; Wirth, T.; Rosenwald, A.; et al. c-MYC activation impairs the NF-κB and the interferon response: Implications for the pathogenesis of Burkitt’s lymphoma. Int. J. Cancer 2007, 120, 1387–1395. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M.; Schuhmacher, M.; Hölzel, M.; Laux, G.; Bornkamm, G.W. c-MYC Impairs Immunogenicity of Human B Cells. Adv. Cancer Res. 2007, 97, 167–188. [Google Scholar] [PubMed]
- Polioudakis, D.; Bhinge, A.A.; Killion, P.J.; Lee, B.; Abell, N.S.; Iyer, V.R. A Myc–microRNA network promotes exit from quiescence by suppressing the interferon response and cell-cycle arrest genes. Nucleic Acids Res. 2013, 41, 2239–2254. [Google Scholar] [CrossRef]
- Wee, Z.N.; Li, Z.; Lee, P.L.; Lee, S.T.; Lim, Y.P.; Yu, Q. EZH2-Mediated Inactivation of IFN-γ-JAK-STAT1 Signaling Is an Effective Therapeutic Target in MYC-Driven Prostate Cancer. Cell Rep. 2014, 8, 204–216. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.W.; Hong, S.; Lin, Y.; Murat, E.; Joo, H.; Kim, T.; Pascual, V.; Liu, Y. Transcriptional Repression of IFN Regulatory Factor 7 by MYC Is Critical for Type I IFN Production in Human Plasmacytoid Dendritic Cells. J. Immunol. 2016, 197, 3348–3359. [Google Scholar] [CrossRef] [Green Version]
- Layer, J.P.; Kronmüller, M.T.; Quast, T.; Boorn-Konijnenberg, D.V.D.; Effern, M.; Hinze, D.; Althoff, K.; Schramm, A.; Westermann, F.; Peifer, M.; et al. Amplification of N-Myc is associated with a T-cell-poor microenvironment in metastatic neuroblastoma restraining interferon pathway activity and chemokine expression. Oncoimmunology 2017, 6, e1320626. [Google Scholar] [CrossRef] [Green Version]
- Zou, J.; Zhuang, M.; Yu, X.; Li, N.; Mao, R.; Wang, Z.; Wang, J.; Wang, X.; Zhou, H.; Zhang, L.; et al. MYC inhibition increases PD-L1 expression induced by IFN-γ in hepatocellular carcinoma cells. Mol. Immunol. 2018, 101, 203–209. [Google Scholar] [CrossRef]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; DeStefano Shields, C.E.; Niknafs, N.; Yen, R.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017, 171, 1284–1300. [Google Scholar] [CrossRef] [Green Version]
- Rathi, A.V.; Cantalupo, P.G.; Sarkar, S.N.; Pipas, J.M. Induction of interferon-stimulated genes by Simian virus 40 T antigens. Virology 2010, 406, 202–211. [Google Scholar] [CrossRef] [Green Version]
- Nozawa, H.; Oda, E.; Nakao, K.; Ishihara, M.; Ueda, S.; Yokochi, T.; Ogasawara, K.; Nakatsuru, Y.; Shimizu, S.; Ohira, Y.; et al. Loss of transcription factor IRF-1 affects tumor susceptibility in mice carrying the Ha-ras transgene or nullizygosity for p53. Genes Dev. 1999, 13, 1240–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sica, A.; Allavena, P.; Mantovani, A. Cancer related inflammation: The macrophage connection. Cancer Lett. 2008, 267, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, M.T.; Luster, A.D. Chemokines in Cancer. Cancer Immunol. Res. 2014, 2, 1125–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spranger, S.; Gajewski, T.F. Impact of oncogenic pathways on evasion of antitumour immune responses. Nat. Rev. Cancer 2018, 18, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; MacDonald, M.E.; Leßmann, V.; Humbert, S.; et al. Huntingtin Controls Neurotrophic Support and Survival of Neurons by Enhancing BDNF Vesicular Transport along Microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Dai, M. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005, 33, e175. [Google Scholar] [CrossRef] [Green Version]
- Irizarry, R.A. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 2003, 4, 249–264. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Jacobsen, A. Cgdsr: R-Based API for Accessing the MSKCC Cancer Genomics Data Server (CGDS). Version 1.3.0. Available online: http://CRAN.R-project.org/package=cgdsr (accessed on 30 October 2019).
- Therneau, T. A package for survival analysis. In S. Version 2.44-1.1. Available online: https://CRAN.R-project.org/package=survivaL (accessed on 30 October 2019).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENE | SIGNATURE | HALLMARK |
|---|---|---|
| DOCK4 | Upregulated | Complement/Spindle |
| G0S2 | Upregulated | TNF-NFKB/KRAS |
| SRPX | Upregulated | Hypoxia |
| CDH11 | Downregulated A | EMT/APICAL |
| DKK1 | Downregulated A | EMT |
| GREM1 | Downregulated A | EMT |
| MYLK | Downregulated A | Adipogenesis/Myogenesis/EMT |
| SPRY2 | Downregulated A | KRAS |
| ARID5B | Downregulated B | Androgen/IFNγ |
| DUSP4 | Downregulated B | TNFα-NFKB |
| ELF1 | Downregulated B | IFNα/ESTROGEN |
| LPAR1 | Downregulated B | Inflammation/UV |
| MX1 | Downregulated B | IFNα/IFNγ |
| SOCS2 | Downregulated B | IL2-STAT5 |
| TNFRSF11B | Downregulated B | EMT/APICAL |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Giorgio, E.; Paluvai, H.; Picco, R.; Brancolini, C. Genetic Programs Driving Oncogenic Transformation: Lessons from In Vitro Models. Int. J. Mol. Sci. 2019, 20, 6283. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246283
Di Giorgio E, Paluvai H, Picco R, Brancolini C. Genetic Programs Driving Oncogenic Transformation: Lessons from In Vitro Models. International Journal of Molecular Sciences. 2019; 20(24):6283. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246283
Chicago/Turabian StyleDi Giorgio, Eros, Harikrishnareddy Paluvai, Raffaella Picco, and Claudio Brancolini. 2019. "Genetic Programs Driving Oncogenic Transformation: Lessons from In Vitro Models" International Journal of Molecular Sciences 20, no. 24: 6283. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246283