The Puzzling Role of Neuron-Specific PMCA Isoforms in the Aging Process


1
Department of Molecular Neurochemistry, Medical University, 92-215 Lodz, Poland
2
Department of Ophthalmology, Stanford University School of Medicine, Palo Alto, CA 94304, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(24), 6338; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246338
Submission received: 21 November 2019
/
Revised: 10 December 2019
/
Accepted: 13 December 2019
/
Published: 16 December 2019
(This article belongs to the Special Issue 8th ECS Workshop 'Calcium Signaling in Aging and Neurodegenerative Diseases')
Abstract
:The aging process is a physiological phenomenon associated with progressive changes in metabolism, genes expression, and cellular resistance to stress. In neurons, one of the hallmarks of senescence is a disturbance of calcium homeostasis that may have far-reaching detrimental consequences on neuronal physiology and function. Among several proteins involved in calcium handling, plasma membrane Ca2+-ATPase (PMCA) is the most sensitive calcium detector controlling calcium homeostasis. PMCA exists in four main isoforms and PMCA2 and PMCA3 are highly expressed in the brain. The overall effects of impaired calcium extrusion due to age-dependent decline of PMCA function seem to accumulate with age, increasing the susceptibility to neurotoxic insults. To analyze the PMCA role in neuronal cells, we have developed stable transfected differentiated PC12 lines with down-regulated PMCA2 or PMCA3 isoforms to mimic age-related changes. The resting Ca2+ increased in both PMCA-deficient lines affecting the expression of several Ca2+-associated proteins, i.e., sarco/endoplasmic Ca2+-ATPase (SERCA), calmodulin, calcineurin, GAP43, CCR5, IP3Rs, and certain types of voltage-gated Ca2+ channels (VGCCs). Functional studies also demonstrated profound changes in intracellular pH regulation and mitochondrial metabolism. Moreover, modification of PMCAs membrane composition triggered some adaptive processes to counterbalance calcium overload, but the reduction of PMCA2 appeared to be more detrimental to the cells than PMCA3.
1. Introduction
The aging process is a physiological phenomenon affecting all living organisms and is associated with perpetual changes in metabolic control, the expression and stability of genes, and cellular resistance to stress. Aging itself is not a disease, but can predispose to many diseases. This process is particularly noticeable in the central nervous system (CNS), as mature neurons have no or very limited capacity for renewal. The most difficult in studies on brain aging is to separate the effects from causes, because they frequently overlap [1]. The evident hallmarks of many age-related events are calcium dyshomeostasis, mitochondrial dysfunction, oxidative stress, and impaired proteostasis [2,3,4,5]. Senescence in neurons is related to protracted elevations in cytosolic Ca2+ ([Ca2+]c) levels and prolonged Ca2+ signaling following excitatory stimulation [6,7,8]. Physiologically, intracellular Ca2+ ([Ca2+]i) is regulated by balancing calcium influx through voltage-gated Ca2+ channels (VGCC), receptor-operated calcium channels (ROCC), and store-operated calcium entry (SOCE), with active efflux by plasma membrane Ca2+-ATPase (PMCA), sodium-calcium exchanger (NCX), and reuptake into the endoplasmic reticulum by sarco/endoplasmic Ca2+-ATPase (SERCA) [9,10,11].
Due to high buffering capacity of mitochondria, they are also an important component for maintaining calcium homeostasis and regulation of intracellular Ca2+ signaling [12,13]. The key role of mitochondria is production of ATP through oxidative phosphorylation, and the major energy source in the brain is glucose. The energy produced in mitochondria is vital to meet neuronal high basal energy requirements, including maintenance of the membrane potential [14]. It is well-documented that energy metabolism decreases during aging due to overproduction of reactive oxygen (ROS) and nitrogen species (RNS), as well as deterioration of protective and repair systems [15]. In oxidative stress conditions, overproduction of ROS is expected to profoundly alter cellular metabolism and its regulation [16]. According to the free radical theory of aging, which is currently the most accepted one to explain age-associated degeneration, the products of nucleic acids, proteins, lipids, and carbohydrates oxidation are all implicated in the aging process [17]. This theory proposed by Harman [18] implicates mitochondrial membrane potential loss and concomitant decrease in ATP production as a part of a negative feedback loop toward age-associated deregulation of [Ca2+]i.
The brain is especially vulnerable to oxidative damage but due to its high complexity, it is difficult to define precisely all the factors that trigger the aging process. It is now well established that deficits in ATP supply, which is critical for reestablishment of ion balance by the plasma membrane Ca2+-ATPase, can produce diverse defects in brain function. This posits disturbed calcium homeostasis in the center of several age-associated brain pathologies and should be considered as an important risk factor for the development of neurodegenerative diseases.
2. Plasma Membrane Ca2+-ATPase in Neuronal Cells
Despite the discovery of hundreds of proteins capable of calcium buffering, one of the main roles in controlling intracellular Ca2+ concentration is attributed to the plasma membrane Ca2+-ATPase, the most sensitive cellular calcium detector. There are four main isoforms of PMCA (PMCA1–PMCA4), that are similar in structure, despite low homology at the amino acid level [19,20]. Depending on the cell and tissue type, there may be nearly 30 different PMCA variants created by alternative splicing of the original transcripts, which generates enzymes of different structural and biochemical properties, such as Ca2+ affinity, velocity of Ca2+ transport, regulatory mechanisms, or ability to interact with different signaling and regulatory proteins [21,22,23]. PMCA1 and PMCA4 are thought to be the constituent forms and are expressed in large numbers of cells [19,24].
The presence of PMCA2 and PMCA3 is limited almost exclusively to the excitable cells, and because of their prominent expression in the brain, they are referred to as neuro-specific [19,24,25,26]. PMCA2 and PMCA3 possess a high basal activity and belong to so-called “fast” isoforms, whereas PMCA1 and PMCA4 are much slower. PMCA, by interacting locally with a number of protein partners, can be incorporated into intracellular signaling pathways [27]. It suggests that PMCA may play a role that is far beyond its classically attributed function of a calcium transporter. In the brain, the expression profile of PMCAs changes significantly during development, confirming the unique role of each isoform [26,28,29]. Even though PMCA is primarily localized at multisynaptic endings and in a close proximity to neurotransmitter release sites (active zone) of the presynaptic terminals, particular PMCA isoforms are distributed in the brain in a region-dependent manner. It is now well documented that PMCA activity and the amount of decline with age, and concomitant elevated calcium can lead to apoptosis or necrosis, depending on the extent of metabolic stress and cytosolic Ca2+ overload [2,30,31,32]. Although the half-life of PMCA in the whole brain homogenate was estimated to be 12 ± 1 days, there is no available data for different PMCA isoforms and variants turnover rates [33]. Moreover, accumulation of age-dependent modifications of PMCA protein structure, despite the relatively long half-life, may alter the enzyme efficiency in Ca2+ extrusion [34,35,36].
Interesting data related to neurospecific PMCAs function were obtained after discovering that some mutations in PMCA2 are linked to hereditary deafness [37,38] and mutation of PMCA3 was found in cerebellar ataxia [39], but in each case, the total efficiency of Ca2+ extrusion was reduced. It may suggest that impaired PMCA2 or PMCA3 function cannot be simply substituted for other isoforms. Using several models, including the adult and aged mouse brain, Alzheimer’s disease-affected human brain, SH-SY5Y human neuroblastoma cells, as well as purified synaptosomal PMCA from pig cerebrum, Mata’s group discovered that PMCA activity was inhibited by tau protein and amyloid-β peptides, and PMCA4b appeared to be the most sensitive target for that inhibition [40,41,42,43]. Moreover, CaM exhibited a protective action against functional impairment of PMCA by tau and Aβ peptide. Thus, although PMCA2 was shown to be more resistant to amyloid-β inhibition, its downregulation in the aging brain, together with lowered PMCA4 activity and the CaM amount, could be responsible for progressive development of AD symptoms.
One of the widely used in vitro model of sympathetic neurons are differentiated PC12 cells. This line was isolated from the pheochromocytoma tumor of rat adrenal glands that have an embryonic origin from the neural crest, which gives PC12 cells unique ability to acquire a number of features characteristic for mature neurons [44]. Upon treatment with nerve growth factor (NGF), they cease proliferation, start to extend neurite-like protrusions, express functional neuronal membrane channel, develop action potentials, and respond to acetylcholine treatment. Similar effects can be triggered by dibutyryl cyclic adenosine-3′, 5′-monophosphate (db-cAMP) [45,46]. Whereas in undifferentiated PC12 cells, all main PMCA isoforms have been detected, with PMCA4b constituting a major one, the differentiation process stimulates the expression of additional splice variants (i.e. 1c, 2a, 2c, 4a) although with unknown physiological function [47,48].
To evaluate the possible contribution of neuro-specific PMCA isoforms to the aging process, we developed PC12 lines deficient in PMCA2 (PC12_2) or PMCA3 (PC12_3). Our extensive functional and molecular analyses gave new insights into downstream cellular processes that can be modified by altered PMCA composition.
3. Differentiated PC12 Cells as a Model of Aging Neuron
The knockdown of PMCA isoforms was done using eukaryotic vectors containing antisense sequences designed to either PMCA2 or PMCA3, and in stably transfected PC12 lines the protein level of each isoform was reduced by almost 50% [49]. Staining of PMCA-deficient cells with the marker protein of growth cones showed the absence of a central structure surrounded by F-actin-rich filopodia and lamellipodia and strong condensation of this protein in neuronal terminals, suggesting that growth cones can be in the retraction phase. Reduced growing potency and neurite retraction are characteristic for aged neurons and frequently observed in response to injury or disease [50,51].
A direct functional consequence of a partial loss of each isoform was a moderate, but sustained, increase in resting Ca2+ concentration by 30–40 nM. In addition, PMCA2 deficiency increased the percentage of cells in the late apoptotic phase, which was detected by AnnexinV/propidium iodide assay and confirmed by the observed DNA fragmentation. Similar effects were reported for rat primary neurons and human SH-SY5Y neuroblastoma cells transfected with PMCA2 siRNA [52]. Beside higher basal Ca2+ level and impaired Ca2+ clearance following stimulation, these authors also demonstrated more intensive cell death upon exposure to excitotoxic concentrations of agents rising intracellular Ca2+. The correlation between PMCA2 down-regulation and disturbances of cell function, including the augmented cell death, was reported in neurons, which strongly implicated a protective PMCA2 role [53,54,55,56,57].
Although reduction of “fast” PMCA isoforms affected functionality of the cells, it did not change the total amount of PMCA protein which could be due to up-regulation of PMCA1 isoform in both PMCA-deficient lines, and PMCA4 that increased only in response to PMCA3 silencing [49]. Nevertheless, elevated cytoplasmic Ca2+ concentration indicated that these adaptive changes were insufficient to fully substitute even for the partial loss of PMCA2 or PMCA3 function. However, in the PC12_3 line, compensatory up-regulation of PMCA4 seemed to counteract Ca2+-dependent induction of apoptosis more effectively, suggesting that PMCA3 deficit could be less detrimental for intracellular calcium homeostasis.
Another protective mechanism aimed against Ca2+ cytotoxic effects was up-regulation of SERCA2 and SERCA3 isoforms in both PMCA-deficient lines [49]. It correlated with higher accumulation of Ca2+ in the endoplasmic reticulum. SERCA family includes three members (SERCA1–3), and two splice variants of SERCA2. SERCA2b is ubiquitously expressed, while SERCA2a and SERCA3 exhibit more discrete localization [58,59]. Since SERCA’s half-life (10–14 days) has been suggested to be similar to PMCA [2], and energetic cost of calcium transport by SERCA is lower than that of PMCA (2Ca2+/ATP vs. 1Ca2+/ATP, respectively) [60], this compensatory effect observed in PC12 cell lines could provide long-term multifaceted protection against calcium cytotoxicity. However, the molecular mechanism(s) of the interplay between these calcium pumps remains a mystery.
Further analysis of the effects of reduced PMCA2 or PMCA3 expression on the kinetics of Ca2+ extrusion showed a significantly increased influx and longer half time (t1/2) of Ca2+ clearance after KCl-induced depolarization [49]. In excitable cells, membrane potential-dependent [Ca2+]c raises are generated mainly by the voltage-gated Ca2+ channels (VGCCs), of which four types: N, L, P/Q, and T are present in PC12 cells [61,62]. The profiling of VGCC expression showed elevated transcript level of P/Q and L types in both PMCA-deficient lines and the T-type solely in PMCA2-deficient cells. Functional analysis with specific pharmacological inhibitors revealed that higher expression of these VGCCs corresponded to their increased contribution to the total Ca2+ load induced by KCl.
It has been widely documented that during aging, ion channel dysfunctions participate in the generation of ionic dyshomeostasis, alteration of membrane potential as well as signal transduction pathways, which can eventually modify cellular physiology [63]. Earlier studies have showed that the aging-related increase in Ca2+-mediated responses depended on greater activity of L-VGCC [64,65]. Moreover, a growing body of evidence indicated an increase in the expression and function of L-type Ca2+ channel with aging [66,67,68,69], and higher L-VGCC density in the hippocampus was positively correlated with cognitive impairment in aged animals [62]. An interesting observation was an up-regulation of T-type VGCC in response to PMCA2 depletion and its increased contribution to Ca2+ influx. Physiologically, T-type channels regulate neuronal excitability, differentiation, growth, and proliferation [70]. An important link was shown between T-type calcium channels and long-term potentiation (LTP) [71]. Downregulation of the T-type VGCC was demonstrated in the mouse model of Alzheimer’s disease [72]. It was also reported that a closing time in some populations of these channels is significantly longer than the other VGCC [72]. Therefore, one can hypothesise that together with a reduced Ca2+ extrusion, elevated presence of T-type channels in PMCA2-deficient line might contribute to sustained increase in [Ca2+]i.
Based on this data, it can be assumed that higher amplitude of [Ca2+]c rises during depolarization reflects the presence of more functional calcium channels in the plasma membrane. Activation of individual channels and coupling their functions with downstream cellular signaling pathways, can induce a different response e.g., by modulation of specific genes expression. In addition, changed VGCC profile in response to manipulations in PMCA expression strongly suggests a transcriptional link between these two opposing systems of Ca2+ transport.
The changes in main components of calcium handling systems described above revealed some differences in the cell response, which seemed to be driven by the presence of particular PMCA isoform (Table 1). Interestingly, knockdown of PMCA2 reproduced a wider spectrum of morphological and molecular abnormalities observed during physiological brain aging.
4. The Interplay between PMCA, Calcium, and Mitochondrial Function during Aging
Mitochondria are central hubs in neuronal pathology integrating energy production, Ca2+ homeostasis, cell signaling, and controlled cell death [73]. Preservation of these functions is especially important in the brain as neurons contain a large number of mitochondria to fulfill energy needs for synaptic processes. The decrease in mitochondrial functionality in the brain, especially in the hippocampus, is associated with the normal aging process and progressive loss of synaptic function [74]. Changes in mitochondrial activity during aging have also been associated with increased mitochondrial ROS production, causing cellular damage of mitochondrial and nuclear DNA and advancing age-related diseases [75].
One of the most important regulators of mitochondrial function in aging is the second messenger Ca2+. Buffering of [Ca2+]i by mitochondria increases when cells age, as was demonstrated in peripheral sympathetic neurons and intestinal smooth muscle cells [76,77]. The regulation of [Ca2+]I by mitochondria seems to be especially relevant in neuronal cells, in which local Ca2+ uptake by neighboring mitochondria regulates the duration of cytosolic Ca2+ fluxes generated by the influx through plasma membrane calcium channels [78]. The possibility that changes in intracellular Ca2+ homeostasis might explain mitochondrial impairments in senescent cells arise from calcium hypothesis of neuronal aging [79]. According to this hypothesis, a change in any of the Ca2+ transporting systems over a long time period should affect [Ca2+]i. To confirm it, several research groups showed elevated resting [Ca2+]i and reductions in the amplitude of stimulation-induced [Ca2+]i signal in aged neurons [80,81]. A similar effect on Ca2+ homeostasis was observed by us in response to PMCA2 or PMCA3 knockdown, pointing out a slower calcium clearance following Ca2+ loads, which is a common hallmark of aging cells. In PMCA-deficient cells, higher resting [Ca2+]i may directly reflect a partial loss of calcium clearing potency and sensitize mitochondria to buffer cytosolic calcium transients even in a condition of moderate increases in [Ca2+]i.
It is expected that accumulation of Ca2+ by mitochondria will stimulate Ca2+-dependent dehydrogenases of the TCA cycle, providing reducing equivalents to boost the activity of respiratory chain and thereby, ATP production via oxidative phosphorylation (OXPHOS) [82]. The ATP supply is then needed for restoration of ion gradient across the plasma membrane by PMCA or Ca2+ transport to the ER by the SERCA pump. However, in the case of mitochondrial overwhelming with Ca2+, permeability of the inner mitochondrial membrane increases dramatically, resulting in the dissipation of mitochondrial membrane potential, loss of mitochondrial respiration, and finally, initiation of cell death [83,84]. Large dissipation of mitochondrial gradient was observed in PMCA2-deficient cells during massive Ca2+ entry, an event that was blocked by cyclosporine A or bongkrekic acid, suggesting the involvement of mitochondrial permeability transition pore (mPTP). The mPTP is a nonselective channel located in the inner mitochondrial membrane that allows the release of excessive Ca2+ accumulated in mitochondria. However, overload of the mitochondrial matrix with Ca2+ or ROS may trigger mPTP prolonged opening and liberation of small molecules and pro-apoptotic factors, finally causing membrane potential loss, reduction in ATP level, and eventually, cell death [85,86]. Similar to the sequence of events demonstrated in PMCA2-deficient cells during depolarization, Scorrano and coworkers showed that in vitro depolarization induced mPTP opening when mitochondria were suitably loaded with Ca2+ [87]. The explanation of this phenomenon is likely to involve the enhancement of ROS formation, what can be suggested in these cells by a higher level of GSH as an adaptive response to free radicals overload [88].
An enhanced susceptibility to mPTP opening and the loss in PMCA activity in synaptic membrane are both reported in the aged brain [89]. In contrast to the catastrophic nature of the mPTP opening, the mitochondrial membrane was able to repolarize in PMCA2-deficient cells upon stimulus withdrawal, pointing out the ability of the electron transport chain (ETC) to rebuild the proton gradient and restore membrane potential. Because even partial loss of PMCA2 led to higher percentage of apoptotic cells [49], it is plausible that only partial or brief opening of mPTP was sufficient to drive cell death. Recent comparative studies showed a decreased threshold for Ca2+ concentration that induced mPTP opening in mitochondria isolated from old rats [90]. Therefore, it is possible that PMCA2 loss and concomitant changes in the cytolic/mitochondrial calcium circuit can increase the probability of mPTP opening at lower rises in [Ca2+]i. Whether mPTP is involved in the physiological aging process is still under debate, but the latest data demonstrated reduced mitochondrial calcium buffering capacity and increased sensitivity to mPTP formation in putamen of aged monkeys [91].
Neurons rely almost exclusively on the mitochondrial OXPHOS system to generate ATP and the primary role of mitochondrial calcium ([Ca2+]m) is to boost energy production. During respiration, electrons from reducing equivalents (NADH and FADH2) are transferred through ETC complexes (complex I-V) and H+ are pumped out to intermembrane space, creating an electrical (ΔΨm) and pH (ΔpH) gradients, both thermodynamically equivalent to drive ATP synthesis. Formation of ΔΨm is fundamental for Ca2+ uptake to mitochondria and adequate mitochondrial function, mainly for ATP synthesis by the F0F1ATP synthase [92]. Mitochondria also buffer cytosolic Ca2+ elevations caused by the influx through plasma membrane Ca2+ channels [93] or by depletion of the internal Ca2+ stores [94]. The ER together with the mitochondria located in regions so called mitochondria-associated ER membranes (MAMs) form highly sophisticated toolkits enabling Ca2+ trafficking between these organelles [95].
It has been demonstrated that PMCA2 and PMCA4 are tethered to specialized calcium microdomains located at the plasma membrane through the interaction with postsynaptic density protein 95 (PSD-95) [96]. PMCA/PSD-95 complexes further recruit N-methyl-D-aspartate (NMDA) receptor subunits R1 and R2A to form multiprotein aggregates [97]. By bringing PMCA to a close proximity of Ca2+ entry sites, formation of such structures can allow for rapid and effective response to local Ca2+ increases. Disrupted NMDA/PSD-95 interaction, observed in animal models of several neurological diseases [98,99], is expected to result in receptor hypersensitivity and excitotoxicity, ultimately leading to cell death. Many important studies suggest that Ca2+ uptake by the mitochondrial uniporter (mtCU) plays an essential role in NMDA receptor activity-driven excitotoxicity [100,101,102]. NMDA receptor stimulation was found to increase mitochondrial membrane depolarization in neurons overexpressing mtCU, whereas knockdown of endogenous mtCU reduced NMDA-induced increases in mitochondrial Ca2+, making cells resistant to excitotoxicity [103]. In the scenario of NMDA receptor-induced and Ca2+-mediated mitochondrial swelling referred to as “mitochondrial aging”, progressive loss of respiratory control is in fact due to abrupt increase in mPTP conductance. Increased permeability of mitochondrial membrane initiated at the level of individual mitochondria is spread across increasing fraction of the mitochondrial population (for comprehensive review see [104]), and usually leads to a massive cell death. Such mode of neuronal death triggered by overstimulation of NMDA receptors is associated with cerebral ischemia and neurodegenerative diseases [105,106,107].
The mtCU complex consists of pore-forming subunit (MCU) and several regulatory subunits (MICU1, MICU2, MICU3, MCUb, and EMRE) [108,109,110]. Mitochondrial calcium uptake 1 and 2 (MICU1 and MICU2) form a central semiautonomous assembly [111] to establish a threshold for MCU opening, keeping it closed at nanomolar [Ca2+]c, thus protecting mitochondria from Ca2+ overload [112]. Elevations of [Ca2+]c above 2–3 µM are expected to trigger channel opening and promote cooperative increase in Ca2+ uptake as described in earlier studies [113,114]. The recent studies on MICU1−/− mouse showed that in purified mitochondria, the MICU1 absence augmented calcium uptake at low [Ca2+]c but inhibited the uptake rate when [Ca2+]c increased [115]. Young MICU1−/− mice had underdeveloped cerebellum, abnormal persistence of the outer granular layer at postnatal day 12, and altered arborization of Purkinje cells. Interestingly, surviving MICU1−/− mice appeared to improve over time and previous histological abnormalities seen in cerebellum were resolved [115]. In addition, the differences in resting calcium, ATP level, and muscle lactate were no longer visible, although MICU1−/− mice did continue to have neurological and muscular defects. The authors suggested that such gradual improvement is due to age-related functional remodeling of mitochondria, which has been recently demonstrated in mouse with reduced MCU activity [116]. In the case of MICU1 deletion, this remodeling appears to involve alterations in the expression ratio of MCU to EMRE.
EMRE is important for Ca2+ uptake and bridges the calcium-sensing role of MICU1 and MICU2 with the calcium-conducting function of MCU [109]. It seems to act as scaffold for the correct stoichiometric assembly of the complex and in its absence, uniporter current is lost despite proper MCU expression and oligomerization. Liu and colleagues [115] observed reduced EMRE expression in older MICU1−/− mice, which correlated with phenotypic improvement. They were, however, unable to obtain MICU1−/− EMRE−/− mice due to compromised survival. Nonetheless, deletion of one allele of EMRE on the background of MICU1−/− resulted in reduced calcium uptake at both low and high extra-mitochondrial calcium levels. Consistent with that, matrix Ca2+ level in brain mitochondria was similar between wild type and MICU1−/− EMRE+/− mice. This genetic background also produced age-appropriate cerebellar morphology but when compared to MICU1−/−, MICU1−/− EMRE+/− mice exhibited phenotypic and behavioral improvement. In neurons, non-assembled EMRE is degraded by m-AAA protease, which ensures sufficient assembly of gatekeeper subunits with MCU [117]. Very recently, it has been demonstrated that accumulation of constitutively active MCU-EMRE channels lacking gatekeeper subunits due to loss in m-AAA protease led to mitochondrial Ca2+ overload, mPTP opening, and subsequent neuronal death [117].
The function of other mitochondrial uniporter subunits is currently under intensive investigation. When looking at the tissue-specific MCU complex composition, MICU3 is predominantly expressed in the central nervous systems, although its low levels were also detected in skeletal muscle [118,119]. It can form heterodimers with MICU1 but not MICU2, but the function is opposite to MICU2, as its expression promotes mitochondrial Ca2+ accumulation at high [Ca2+]c. Therefore, MICU3 is considered to act as a highly potent cooperative MCU activator with no gatekeeping function. Patron and colleagues [120] have recently performed the extended analysis of MICU3 function in neurons. They found that MICU1/MICU2 dimers ensured low Ca2+ cycling in the steady-state conditions whereas MICU1/MICU3 dimers activated MCU-dependent Ca2+ uptake even in the low [Ca2+]c. MICU3, when silenced, was also the only MICU isoform that effectively decreased mitochondrial Ca2+ transients evoked by synaptic activity [120].
In principle, aged neurons undergo considerable changes in Ca2+ store content, mtCU expression, and ER-mitochondria domains [121]. Increased mtCU over time is thought to respond to higher Ca2+ release from the ER, which is in line with the observation of increased mitochondrial-ER coupling in aging in the face of mitochondrial depolarization [121]. In view of that, silencing of MICU3 could potentially slow down age-associated mitochondrial deficits by decreasing the amplitude of [Ca2+]c rises, as shown by Patron et al. [120], while MICU2 knockdown is expected to exert the opposite effect.
Overall, the effect of higher [Ca2+]m is expected to coordinate upregulation of OXPHOS machinery resulting in faster respiration and ATP production (Figure 1).
The recent report [122] showed that the basal rate of mitochondrial respiration was increased in aged endothelial cells, indicating enhanced mitochondrial contribution to energy balance in steady-state conditions and age-related increase in ER-mitochondrial Ca2+ transfer. In our study, a positive correlation between oxygen consumption and the ATP level was observed in PMCA2-deficient cells which indicates higher energy demands. Indeed, the experiments with KCN and oligomycin showed that OXPHOS rather than anaerobic glycolysis is a main energy suppling pathway in these cells. This was further supported by higher basal glucose consumption and lower NAD(P)H level accompanied by increased activity of proton-pumping ETC complexes (I, III, and IV). These findings are in agreement with the boosting effect of Ca2+ on mitochondrial metabolism and suggest that the rate of OXPHOS in PMCA2-deficient cells may be controlled at the activity of ETC proteins. Interestingly, knockdown of PMCA3 isoform did not change the rate of OXPHOS. However, enhanced lactate release and greater sensitivity of the ATP level to glucose withdrawal in the presence of pyruvate suggests the reliance on anaerobic glycolysis. This brings up an intriguing question about preferential fueling of PMCA isoforms by different energy-generating pathways. It is tempting to hypothesize that the loss of fast acting PMCA2 will stimulate other ATP-dependent Ca2+ extruding proteins to restore [Ca2+]i, thereby increasing cellular demand for ATP. When PMCA3 was depleted, [Ca2+]c rise was only moderate, so the local synthesis of ATP by glycolysis may be sufficient to provide adequate amount of energy (Figure 1).
Whatever the explanation might be, increased [Ca2+]i as a result of PMCA2 or PMCA3 knockdown seems to be central for understanding the specific coupling between PMCA2 and mitochondrial metabolism of aging neurons. Our results of increased mitochondrial activity, coexisting with higher Ca2+ accumulation in the ER, as described in a previous chapter, are consistent with the recent hypothesis of enhanced ER-mitochondrial Ca2+ influx during aging and are further supported by in vitro studies based on rat hippocampal neurons [121]. Although the exact mechanism is not known, the mild ER stress due to increase in ER loading with Ca2+ observed in PMCA-deficient cells, which yet does not induce massive cell death, can eventually cause slow Ca2+ leak and activation of proteins involved in ER-mitochondrial tethering. This crosstalk might help to sustain elevated mitochondrial ATP production, creating a positive feedback loop temporarily prolonging ER function during aging. Based on the biphasic model of mitochondrial aging reported for primates and rodents, increased respiration and activity of the ETC complexes in PMCA2-deficient cells may reflect the early stage in which Ca2+-dependent increased mitochondrial activity boosts the generation of harmful ROS. As the aging progresses, ROS accumulation combined with a loss of scavenging mechanisms negatively affect the activity of ETC, leading to severe mitochondrial dysfunction [123], which has been widely reported as a hallmark of ageing. Strong experimental data seem to support this hypothesis, as increased expression of mitochondrial genes of complexes I, III, IV, and V was reported in 18-month old mice in the hippocampus, medial prefrontal cortex, and striatum [124], whereas later in aging, the expression of ETC proteins as well as the activity of complexes I and IV decreased in substantia nigra, hippocampal dentate gyrus, frontal cortex, and cerebellum [125,126].
The fundamental question in respect to higher mitochondrial activity in PMCA2-deficient cells is how the driving force for ATP synthesis is generated. The findings of Poburko et al. [127] showed that bursts of proton accumulation in the cytosol generated by the activity of PMCA were readily transmitted to mitochondria. A similar relationship between pHmito and pHcyto was previously demonstrated in MDCK cells and was attributed to the activity of mitochondrial proton antiporters [128]. The pHmito recordings suggested that rapid equilibration of mitochondrial matrix pH to cytosolic pH changes is accounted by Pi/H+ symport and K+/H+ exchanger. However, the data regarding the effect of agonist-induced [Ca2+]c elevations on mitochondrial pH are conflicting, as both acidification and alkalization of matrix were reported [129,130,131]. In their excellent study, Poburko et al. provided several lines of evidence that PMCA was the source of H+ during [Ca2+]c-dependent mitochondrial acidification. First, the acidification matched the kinetic property expected for Ca2+/H+ antiporter coupling Ca2+ extrusion to the proportional load of H+; second, the acidification was attributed to Ca2+ fluxes at the plasma membrane but not to Ca2+ release from intracellular stores; third, alkaline extracellular pH or La3+, both known to inhibit PMCA activity, prevented cytosolic acidification. The transmission of acid generated by PMCA to mitochondria may constitute a mechanism, protecting cells from calcium excess, as low pHmito is known to inhibit Ca2+-dependent mPTP opening and reduce ROS generation [132,133].
Our concurrent measurements of pHcyto and pHmito showed, however, alkalization of mitochondria matrix in steady-state conditions in response to PMCA2 or PMCA3 knockdown (7.78 ± 0.01, 7.62 ± 0.01, respectively, vs. 7.53 ± 0.02 in control). Because pHcyto was also higher in PMCA-deficient cells, but still lower than pHmito, which is consistent with chemiosmotic coupling hypothesis, pH gradient across the inner mitochondrial membrane (ΔpH= pHmito − pHcyto) was increased in these cells. Based on the PMCA/pH relationship, we anticipate that knockdown of neuro-specific PMCA isoforms, which are considered as fast reacting, dramatically limits the number of H+ entering cytosol leading to pHmito increase. In these circumstances, the rise in pHmito could result from compensation of Ca2+ charge by moving H+ down the ETC complexes. It is therefore attractive to propose that increased [Ca2+]c observed in PMCA2-deficient cells, and hence higher [Ca2+]m, is a main trigger for increased proton motive force to drive higher rate of ATP synthesis. To support this, application of FCCP which collapses the inner mitochondrial membrane potential, evoked significant rise in [Ca2+]c in PMCA-deficient lines suggesting higher ΔΨm-dependent mitochondrial Ca2+ loading, especially when PMCA2 was knockdown. The phenomenon of higher magnitude of [Ca2+]c rises in response to FCCP was also demonstrated by Behringer and colleagues in old microvascular endothelium, suggesting greater capacity of mitochondria for Ca2+ during aging [134]. However, respiration-dependent Ca2+ entry is expected to lower ΔΨm, as clearly observed in isolated mitochondria exposed to supraphysiological levels of extramitochondrial Ca2+ [135]. Under physiological conditions, Ca2+ uptake would not always alter ΔΨm, which was demonstrated in isolated cardiomyocytes or it can provoke short-lasting ΔΨm decreases that would not inhibit ATP synthesis [136]. Transient and small fluctuations in ΔΨm in response to cytosolic Ca2+ peaks were indeed observed in other systems [137], suggesting plausible cell-specific effect of Ca2+ on ΔΨm-driven ATP synthesis. In line with that, Jouaville and colleagues [138] in their key paper suggested that mitochondrial ATP production can be differentially modulated in response to different Ca2+ signals generated be various external stimuli. In HeLa cells, that are glycolytic similar to PC12 cells, they demonstrated large increases in [Ca2+]c and [ATP]m and concomitant stable rise in ΔΨm upon histamine stimulation. This suggested that increased [ATP]m reflected long-lasting elevation of the mitochondrial energy state. Similarly, increase in both ΔΨm and ΔpH components of the proton motive force was seen after stimulation with vasopressin [139], which is known to produce cytosolic [Ca2+]c transients that are relayed to mitochondria. The authors suggested that respiratory chain activity may be regulated by a long-lasting mitochondrial signal generated by [Ca2+]m transients and thus, partially by [Ca2+]c levels, which is coherent with our data from PMCA2-depleted line. These findings also fit the mitochondrial memory mechanism proposed by Jouaville et al. [138] stating that long-term priming of mitochondrial activity persists longer than the rise in [Ca2+]m. This allows mitochondria to meet the energy needs without the risk of organelle Ca2+ overload. Although, the mechanistic explanation of such “memory” is unknown, it plausibly involves the changes in ΔΨm, as observed by us and/or Ca2+-dependent activation of F0F1ATP synthase. Indeed, several in vitro and in vivo studies demonstrated that the rate of ATP synthesis can be controlled directly at the level of F0F1ATP synthase, independently from the respiratory rate and ΔΨm [140,141]. F0F1ATP synthase can bind Ca2+ directly [142] but its activity can be also regulated by phosphorylation of the γ-subunit, which in turn is driven by mitochondrial Ca2+ [143]. Moreover, a recent study has shown that S100A1 protein binds F0F1ATP synthase in Ca2+-dependent fashion, increasing cellular capacity for ATP synthesis [144], thus providing another way for ΔΨm-independent regulation of energy generation.
Contrary, none of the PMCA-deficient lines showed a correlation between [Ca2+]c response to FCCP and mitochondrial membrane hyperpolarization. Instead, FCCP produced massive loss of ΔΨm and slight hyperpolarization of plasma membrane suggesting that the plasma membrane potential is not created by proton pump and the protonophore equilibrates pH across the plasma membrane by carrying H+ from cytoplasm to the extracellular milieu. It has been demonstrated that lack of protons in mitochondrial matrix under the excess of electron donors strongly promotes ROS generation [133]. Therefore, taking into account higher pHmito in PMCA-deficient cells, increased accumulation of Ca2+ by mitochondria consisted with the observation of over-activation of ETC complexes, our results point to a specific coupling between PMCA2 generated calcium fluxes and metabolic activity and may mirror specific cellular alterations observed in early aging.
5. PMCA and Ca2+-Regulated Proteins—CaM, GAP43, CaN in Ageing
The main function of PMCA is to maintain intracellular Ca2+ homeostasis, but it is now well documented that efficiency of this control is modulated by several independent regulatory mechanisms [145]. Among them, activation of PMCA by calmodulin (CaM) is the most prominent one, and PMCA is the only calcium pump directly regulated by CaM [146]. Both “fast” PMCA isoforms with a high basal activity exhibit 5 to 10-fold higher affinity for CaM compared to PMCA1 and PMCA4, but are weakly stimulated by CaM [20,147,148]. In rats, CaM is encoded by three non-allelic genes—Calm1, Calm2, and Calm3—all ultimately producing the identical CaM protein. The total CaM amount varies among tissues but the highest concentration (over 30 μM) can be detected in the brain [149]. Because CaM can regulate nearly 300 proteins, there is a high competition rate for CaM binding which is dictated by the affinity of its particular protein partners [150]. Such competition is thought to regulate frequently contradictory processes that are concurrently activated in response to [Ca2+]c rises. The prevalence in activation of certain CaM downstream signaling pathways may therefore sensitize neurons to aging-related physiological and pathological stimuli. Like PMCA, CaM shows functional defects in the aged brain, mainly due to intensive oxidation of multiple methionines [2]. Oxidatively modified CaM can still bind PMCA, but cannot stimulate pump activity. Moreover, it also prevents PMCA activation by unoxidized CaM. Although CaM half-life is approximately 18 ± 2 hours, these global structural CaM alterations provide a possible mechanism for the loss of proper calcium regulation during aging [32,151].
Early studies on PC12 cells suggested the inversed relationship between CaM level and neurite elongation in response to a differentiation signal [152]. In our studies, we observed decreased CaM immunoreactivity in both PMCA-deficient lines and proposed that the underlying mechanism involved differential regulation of CaM gene expression [153]. Interestingly, the expression of Calm1 and Calm2 was downregulated in both modified lines but PMCA2 depletion additionally reduced the expression of Calm3. Since PMCA2 and PMCA3 have high basal activity, but are less sensitive to CaM stimulation, limited CaM availability could result in decreased activation of other PMCA isoforms. As a consequence, impaired total PMCA extruding activity can lead to permanent rise in [Ca2+]i, more significantly in the PC12_2 line.
An additional physiological mechanism that can affect CaM function is a formation of complex with growth-associated protein 43 (GAP43, neuromodulin), which is also a marker of differentiating neurons [154,155]. A unique function of this multifunctional presynaptic protein is CaM binding, thereby it can control CaM availability in the cell. Especially high levels of GAP43 were reported at specific sites such as neuronal growth cones, where GAP43 may play a role of abundant CaM reservoir [156]. Since GAP43 half-life is about 2–3 days, this period may be sufficient for reprogramming of CaM-dependent signaling pathways in the cell. Critical for this process is the relationship between binding of CaM and protein kinase C (PKC)-mediated phosphorylation of GAP43. Dephosphorylated GAP43 binds CaM at low Ca2+ concentration, while phosphorylation by PKC at Ser 41 liberates CaM due to significant reduction in GAP43 affinity. Dephosphorylation by e.g., calcineurin (CaN), reverses this process and decreases available free CaM [156]. This mechanism directly controls the functional CaM level in the cell, and indirectly all Ca2+/CaM-dependent processes.
In neurons, GAP-43 is distributed throughout all cellular compartments with the highest density found in axon terminals and is implicated in synapse formation and neurite branching [154,157,158]. It has been proposed that the level of GAP-43 might determine the progress of neurogenesis, particularly in the hippocampus and associate cortex in the adult human brain [159]. Physiologically, GAP43 is linked with synaptic plasticity, regulation of neurotransmitter release, learning, and mnemonic function [156]. Behavioral studies demonstrated that reduced GAP-43 level led to some deficits, while increased GAP-43 level enhanced behavioral performance [160]. A study on rats indicated the contribution of this protein to the general age-dependent decline in brain plasticity [161]. During aging, less GAP43 mRNA was detected in the hippocampus, and the phosphorylation level decreased by nearly 50% [162]. Another study on rats reported a lowered GAP43 immunoreactivity in the dentate gyrus, cingulate cortex, and olfactory bulb [163,164,165]. In humans, GAP43 expression predominates during the first decade of life and remains nearly unchanged during further life [166]. However, in Alzheimer’s disease (AD) reduction of neuronal GAP-43 mRNA and GAP-43 protein has been linked to memory dysfunction [167,168].
Our studies showed increased GAP43 mRNA and protein level in both PMCA-deficient lines [169]. Interestingly, we detected a higher amount of dephosphorylated GAP43 protein, while its phosphorylation level (P-GAP43) was significantly decreased. Higher GAP43/P-GAP43 ratio indicates enhanced binding of CaM by dephosphorylated form of GAP43. Indeed, in PMCA-deficient lines, a much stronger fluorescent signal from GAP43 and calmodulin-specific antibodies was detected suggesting a more intensive formation of the GAP43/CaM complex whichwas further confirmed by co-immunoprecipitation. Both lowered CaM presence detected in these lines and stronger formation of CaM/GAP43 complex may additionally decrease CaM regulatory potency.
Further studies using cyclosporine A, an inhibitor of serine-threonine phosphatase—calcineurin (CaN), showed that formation of CaM/GAP43 is controlled by CaN activity [169]. CaN is the only reported phosphatase completely dependent on Ca2+/CaM, and it constitutes slightly more than 1% of all brain proteins [170]. In neurons, CaN can be detected in cytoplasm, but also in nearly all subcellular structures i.e., endoplasmic reticulum, Golgi apparatus, nucleus, synaptic vesicles, microsomes, outer mitochondrial membrane, and plasma membrane [30]. Distribution of CaN in the cell appears to be an important factor regulating its local activity. CaN controls numerous and diverse cellular processes mainly via activation of NFAT transcription factor (nuclear factor of activated T cells). Higher activity of calcineurin was observed in aging, as well as in rat and mice models of AD [171,172], and likewise other Ca2+/CaM -regulated enzymes, it was linked to an increase in [Ca2+]i. Some data suggested the relationship between CaN/NFAT-dependent regulation of PMCA and brain aging, along with the onset of neurodegenerative diseases [40,42,173].
To elucidate whether altered composition of PMCA isoforms could affect the expression and activity of CaN in PC12 cells, molecular and biochemical analyses were performed [169]. Using the real-time PCR technique, we detected up-regulation of CaN mRNA by approximately 60% and 85% in PMCA2 or PMCA3-deficient lines, respectively. These changes were also seen at the protein level and gave rise to higher CaN activity. Albeit, there was no direct interaction between GAP43 and PMCA, diminished calcium pumping activity considerably affected CaN and CaM functions.
One of the physiologically important CaN actions is an orchestration of a sequence of signaling events leading to dephosphorylation of conserved phosphoserine residues in the transcription factor NFAT, its translocation to the nucleus, and subsequent transcriptional activation of NFAT-regulated target genes [170]. However, besides the role of CaN in transcriptional signaling, increasing the number of studies reported the interaction of CaN with other substrates, targeting proteins and regulators of CaN activity. Among others, CaN was shown to interact directly with PMCA2 and PMCA4 isoforms, which resulted in the inhibition of CaN-regulated processes, including activation of NFAT [174,175,176,177]. On the other hand, the expression of PMCA4b in neurons was shown to be controlled by CaN [178]. In our model of differentiated PC12 cells, we detected strong interaction between PMCA2 and CaN. However, high-resolution confocal imaging supported by precipitation of PMCA/CaN immunocomplexes, demonstrated lower degree of colocalization and significantly decreased CaN binding to PMCA in PC12_2 cells. The same set of experiments showed lack of detectable changes in the ability of PMCA2/CaN complex formation between control and PC12_3 cells. Interestingly, in both PMCA-deficient lines, lower PMCA4/CaN immunoreactivity was present, suggesting that PMCA depletion may increase a pool of potentially active CaN. In this context, fast acting PMCA2 and PMCA3 isoforms may be seen as regulators of local CaN availability and function. Control of local CaN activity is beneficial for cell survival in the condition of calcium overload and together with reported protective role of PMCA4 [145,146,148] may be of paramount importance for effective anti-apoptotic protection in PMCA3-deficient line.
Taken together, changes in PC12 cell lines initiated by moderate, but prolonged increase in [Ca2+]c due to specific PMCAs depletion produced distinct cell responses, which clearly depended on PMCA isoforms composition and isoform-specific regulatory mechanisms. When induced changes are adaptive, the cells can effectively counteract Ca2+-dependent destructive processes and retain the ability to survive. However, these protective actions in PMCA2 down-regulated cells were found to be much less efficient. One can assume that not only changes in global [Ca2+]I concentration, but also spatially and temporally limited modifications in transmission of calcium signal played a decisive role in the cell response, and the detrimental effects over time were not sufficiently counterbalanced by these adaptive mechanisms.
6. PMCA-Deficient PC12 Cells as a Tool for Modeling Functional Adaptive Strategies in Neurons during Aging
As described above, aberrant PMCA-mediated Ca2+ clearance was associated with perturbation in mechanisms responsible for maintenance of calcium homeostasis. A summary of the detected changes in the expression of analyzed components in PC12_ and PC12_3 are presented in Table 1. As profound changes in CaM expression and availability demonstrated in PMCA-deficient cells could significantly alter CaN activation and CaN-downstream processes, we turned our attention toward a potential cross talk between PMCA, CaM, and CaN in shaping of the Ca2+ signal. One of the first basic question we asked was the role of the functional interaction between these proteins in the differential regulation of CaM transcripts and whether this may create a feedback loop for PMCA function.
Looking for a mechanism(s) that could underlie the diminished expression of CaM genes, a microarray screening of fundamental transcription factors was performed [153]. Among hundreds of analyzed genes, only the expression of Nfatc2, whose activity is associated with Ca2+, increased in both PC12-deficient lines, which led us to the hypothesis that NFATc2 may function as a transcriptional regulator of calmodulin genes. The NFAT family is composed of five proteins, of which NFATc1, NFATc2, NFATc3, and NFATc4 are activated by Ca2+ and their action is controlled by calcineurin [179,180]. In steady-state conditions, NFAT resides in the cytosol in the phosphorylated state, but increased [Ca2+]c activates CaN, which dephosphorylates NFAT causing its translocation to the nucleus, where it becomes transcriptionally active. A return of [Ca2+]c to the resting level initiates re-phosphorylation and rapid relocation of NFAT back to the cytosol. Our results showed that moderately increased basal [Ca2+]c in PMCA-deficient lines sufficiently induced its nuclear translocation and DNA binding activity. Moreover, we found that a sequence for NFATc2 binding is located in the promoter region of Calm2 and Calm3 genes, but not in Calm1. Chromatin immunoprecipitation assay revealed higher promoter occupancy by NFATc2 in PMCA-modified cell lines resulting in diminished transcription of both CaM genes. In addition, overexpression of the constitutively active form of CaN increased NFATc2 nuclear accumulation and its promoter activity, which potentiated Calm2 and Calm3 repression. This strongly indicated the NFATc2 repressive role toward CaM gene expression. Further experiments with NFATc2 silencing, using selective siRNA, showed a partial rescue of the expression of Calm2 in both lines and Calm3 in PC12_2 cells, and confirmed that the activation of the CaN/NFAT pathway may repress CaM genes, but to various extent in each of the PMCA-deficient lines.
The differences in PMCA isoform ratio could affect the regulation of the downstream events including CaN/NFAT-dependent regulation of Calm 2 and Calm3 genes. It was reported that CaN interacts with PMCA2 and PMCA4 which resulted in inhibition of its phosphatase activity [175]. In line with it, lowered PMCA2 amount could be partially responsible for diverse cell response. The second important player and limiting factor was the amount of CaM available for binding. This could profoundly interfere with CaN/NFAT activation in both PC12-deficient lines, further suggesting the existence of the feedback mechanism by which CaM could affect its own expression. This specific regulation seems to be a direct consequence of selective PMCA isoform silencing, because no similar effect was observed in the control PC12 cells. Finally, lowered CaM level may have potential consequences on Ca2+ extrusion by PMCA, as was reported in senescence neurons [30,32,181]. In addition, since aging was shown to be associated with excessive Ca2+ influx through L-type VGCC, which is inactivated by Ca2+/CaM complex and directly modulated by CaN [64,182,183], reduced CaM level may thus potentiate calcium influx and inhibit CaN activity as well. In the context of neuronal aging, these results shed new light on molecular basis of neurodegenerative diseases and demonstrated several lines of cellular protection from the negative effects of Ca2+ overload. In addition to the membrane components, the maintenance of calcium homeostasis is coupled with the multifunctional endoplasmic reticulum, which contains several Ca2+ sensitive transporters, including sarco/endoplasmic Ca2+-ATPase (SERCA), inositol 1,4,5-triphosphate receptors (IP3Rs), and ryanodine receptors (RyRs). Whereas SERCA decreases [Ca2+]c by the uptake into endoplasmic reticulum, IP3R and RyR act as channels releasing calcium from the ER following physiological stimulation. In PMCA-reduced cells, an increased level of SERCA2 and SERCA3 coexisted with higher Ca2+ accumulation in the ER, although the relationship between PMCA and SERCA expression has not been elucidated. More effective Ca2+ transport to the ER may decrease [Ca2+]c to its safe level, but also more Ca2+ could be released by activation of IP3R and RyR [184].
IP3 receptors are intracellular ubiquitously expressed Ca2+ channels that exist in three main isoforms: IP3R-1, IP3R-2, and IP3R-3. In the central nervous system, the presence of all isoforms, with the predominance of IP3R-1, was detected, although their subcellular compartmentalization varied in different brain regions [185,186,187]. In the rat brain, IP3R-1 was found in high amounts in Purkinje neurons in cerebellum and was localized to dendrites, dendritic spines, cell bodies, axons, and axonal terminals [188,189]. In the hippocampus, IP3R-1 is mostly expressed in the CA1 region, with substantially less expression in CA3 and only moderate levels in the granule cells of the dentate gyrus [185]. A particular role of IP3Rs in the hippocampus is related to learning and memory abilities, and changes in the IP3R isoform composition during aging may have an impact on increased deficits in these processes [190]. In other type of neurons, a high level of IP3R-1 was found in cell bodies and proximal dendrites. IP3R-2 was mostly detected in glia, whereas IP3R-3 was predominantly expressed in neuronal terminals in limbic and basal forebrain regions [191]. The expression of particular receptors during aging is differentially regulated, also in a brain region-specific manner [192,193]. Moreover, IP3Rs are dynamically regulated by the formation of homo- or heterotetrameric complexes, thus their relative expression together with other components will determine the final cell response [194,195]. It has been proposed that the level of IP3 receptors declines progressively during aging. However, due to the oxidative modifications that are found to increase IP3R function in the brain, IP3 downstream signaling may not be compromised which is thought to represent a compensation for an altered redox state [186]. Thus, diverse processing of each IP3R isoform may activate various cell signaling pathways that can initiate the adaptive response or lead to cell death [187,193]. Since IP3R-mediated Ca2+ release from the ER and mitochondrial Ca2+ homeostasis are physiologically coupled, their improper cooperation may significantly affect cell viability [196].
Release of Ca2+ from the ER by IP3 receptors requires binding of both, IP3—arising from stimulation of phospholipase C (PLC), and Ca2+. All isoforms exhibit specific characteristics: IP3R-1 possesses low Ca2+ affinity and moderate affinity for IP3, IP3R-2 represents the isoform with the highest affinity for both Ca2+ and IP3, and IP3R-3 is the most sensitive for modulation by Ca2+, but displays the lowest IP3 affinity [194,195,197]. Interestingly, cytosolic Ca2+ regulates IP3Rs in a biphasic way: low Ca2+ level stimulates the receptors, but the concentration above 300 nM inhibits their transport activity [197]. Our analysis showed that PC12 cells, like the majority of available cell lines, express all three main IP3Rs with IP3R-3 being the most prominent subtype [198], although IP3R-1 and IP3R-2 were also detected [193]. In PMCA-deficient cells IP3R-1 and IP3R-2 expression decreased, whereas IP3R-3 was higher (by 40%) than in the control, matching the changes at the protein level. Because this relationship occurred at the transcriptional level, our data could suggest the existence of Ca2+-dependent negative feedback loop for IP3R-1 and IP3R-2 expression. Western blot analysis using an antibody recognizing all IP3R isoforms revealed higher total protein level only in the PC12_3 line, pointing out a switch between IP3Rs ratio in response to PMCA depletion. Such changes, together with higher presence of PMCA4 and highly active PMCA2, may represent an additional cellular defence mechanism aimed in response to PMCA3 depletion to overcome potentially harmful consequences of calcium excitotoxicity. PMCA4 seems to be central for this defence as it can bind plasma membrane phosphatidylinositol 4,5-bisphosphate (PIP2), protecting it from hydrolysis by PLC, potentially decreasing IP3 production and, subsequently, Ca2+ release from the ER [199]. Some of the adaptive mechanisms revealed in our studies could be relevant to the physiological events occurring during aging, but long-lasting Ca2+ dyshomeostasis may promote a variety of potentially detrimental consequences including proteolytic processes triggering apoptosis and/or necrosis.
The wide spectrum of these processes can be propagated by pro-inflammatory chemokines [200] such as CCL5, which induces phospholipase C-mediated liberation of Ca2+ from the ER by IP3-gated channels. CCL5 can mobilize cytosolic Ca2+ after binding to three receptors—CCR1, CCR3, and CCR5, which are cell surface-associated, immune-regulatory G protein-coupled receptors (GPCRs) [201,202]. CCR5 activity has been implicated in normal cell development, but up-regulation of CCR5 was observed in a number of neurological disorders and models of CNS injury [203,204]. Over-activation of CCR5 with subsequent rise in cytosolic Ca2+ is known to affect chemotaxis, secretion, and gene expression and could lead to inflammatory and degenerative processes in the CNS [205]. Moreover, higher CCR5 expression in T cells during aging was demonstrated in humans and rodents at both mRNA and protein levels [206,207,208]. It should be noted that higher CCR5 activity may intensify not only CCL5-dependent events, but also the signaling induced by other known CCR5 ligands such as CCL3 and CCL4, of which expression is stimulated in the aging brain [209,210].
All three receptors were detected in our experimental model cells, but only CCR5 was localized in the plasma membrane which is required for CCL5 action. Interestingly, in each of the PMCA down-regulated PC12 lines, CCR5 expression increased by nearly 50%. Functional assays in Ca2+-free medium showed that binding of CCL5 to its membrane receptor mobilized Ca2+ from the ER more intensively in PMCA-deficient lines but the highest amplitude of [Ca2+]c rises was detected in PC12_2 cells. In addition, there were significant differences between lines with respect to the rate of Ca2+ clearance and the duration of the prolonged calcium signal upon chemokine stimulation. The return of Ca2+ to its new steady-state level was 3 times longer in PC12_2 and about 2 times longer in PC12_3 than in control cells. The differences in kinetic parameters could reflect actual rate of Ca2+ release from the ER and its extrusion to extracellular milieu. It is worth to emphasize that PMCA3-deficient cells still possess very active PMCA2, but also higher protein level of PMCA4, thus could operate more effectively to control [Ca2+]i. Despite that, the efficiency of this adaptation is still lower than in control PC12 cells with a full set of PMCA isoforms.
Structural and functional changes in PMCA-deficient PC12 cell lines were initiated by moderately increased cytosolic Ca2+, but downstream long-time effects seemed to be specifically linked to the function of particular isoforms. Some of the induced adaptive mechanisms were sufficient for protection of cell viability as observed in PMCA3-deficient cells (Figure 2) in comparison to less effective compensatory changes aimed in response to PMCA2 silencing (Figure 3). This underlines the unique role(s) of PMCA2 and PMCA3 in neuronal cells and indicates that long-lasting calcium dyshomeostasis due to diminished PMCA activity might be central to the aging process and variety of neuropathologies.
7. Conclusions
Aging, as a physiological process, involves the perpetual adaptation to varying internal and external conditions by generation of the complex protective mechanisms. This new adaptive state enables the cells to continuously make short-term adjustments for optimal functioning, but because of the multifactorial nature of aging, the efficiency of these adaptations depends on the intensity of senescent signaling [211,212]. Calcium dyshomeostasis is a factor with especially strong destructive potential. Less efficient Ca2+ extruding systems and aberrant regulation of downstream effectors could disturb the function of mitochondria and endoplasmic reticulum leading to cell death, as was widely documented in aging cells [122,213].
Studies performed on our PC12 cell model of senescent neurons revealed an apparent contribution of PMCA2 and PMCA3 isoforms to the aging process. A controlled deficiency of PMCA2 or PMCA3 and resulting permanent increase in intracellular Ca2+, allowed to track unique functions that each of these isoforms may potentially perform in aging. The most important findings demonstrate that the altered composition of PMCA isoforms is linked to:
- intracellular pH regulation and changes in the mitochondrial bioenergetic processes,
- adaptive functional changes in the VGCC and in Ca2+ transport systems to the ER,
- regulation of CaM availability by Ca2+/CaN-dependent complex formation with GAP43,
- reduced CaM level and repressive regulation of Calm2 and Calm3 genes by NFATc2,
- altered expression of CCL5-sensitive receptors—CCR1, CCR3, and CCR5,
- altered expression of IP3 receptors.
Comparison between PC12_2 and PC12_3 lines clearly indicates that down regulation of PMCA2, apart from several adaptive or compensatory processes, triggers more potentially damaging events that are also observed during neuronal aging, including higher sensitivity to inflammation. A declining potency to maintain cellular homeostasis as a potential contributor to age-dependent neuronal senescence markedly increases the risk for neurodegenerative processes. Although differentiated PC12 cells can only partially mimic the physiological processes in the functional neuron, the changes observed in our model might represent the age-related mechanisms common in functionally different cell types.
Author Contributions
T.B. and L.Z. conceived, designed the topic and wrote the manuscript. T.R. and B.F. Contributed to literature research and created the figures. All authors approved the submitted version.
Funding
This work was supported by the grants No. 503/6-086-02/503-61-001 from Medical University of Lodz, Poland and No. 2019/33/B/NZ4/00587 from National Science Centre, Poland.
Conflicts of Interest
The authors declare no conflict of interest
References
- Chandran, R.; Kumar, M.; Kesavan, L.; Jacob, R.S.; Gunasekaran, S.; Lakshmi, S.; Sadasivan, C.; Omkumar, R.V. Cellular calcium signaling in the aging brain. J. Chem. Neuroanat. 2019, 95, 95–114. [Google Scholar] [CrossRef] [PubMed]
- Squier, T.C.; Bigelow, D.J. Protein oxidation and age-dependent alterations in calcium homeostasis. Front. Biosci. 2000, 5, D504–D526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toescu, E.C.; Vreugdenhil, M. Calcium and normal brain ageing. Cell Calcium 2010, 47, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Gladyshev, V.N. Role of reactive oxygen species-mediated signaling in aging. Antioxid. Redox. Signal. 2013, 19, 1362–1372. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Yegla, B.; Foster, T.C. Redox Signaling in Neurotransmission and Cognition During Aging. Antioxid. Redox. Signal. 2018, 28, 1724–1745. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A. Calcium Signaling During Brain Aging and Its Influence on the Hippocampal Synaptic Plasticity. Adv. Exp. Med. Biol. 2020, 1131, 985–1012. [Google Scholar]
- Nikoletopoulou, V.; Tavernarakis, N. Calcium homeostasis in aging neurons. Front. Genet. 2012, 3, 200. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Bodhinathan, K.; Foster, T.C. Susceptibility to Calcium Dysregulation during Brain Aging. Front. Aging. Neurosci. 2009, 1, 2. [Google Scholar] [CrossRef] [Green Version]
- Kawamoto, E.M.; Vivar, C.; Camandola, S. Physiology and pathology of calcium signaling in the brain. Front. Pharmacol. 2012, 3, 61. [Google Scholar] [CrossRef] [Green Version]
- Lopreiato, R.; Giacomello, M.; Carafoli, E. The plasma membrane calcium pump: New ways to look at an old enzyme. J. Biol. Chem. 2014, 289, 10261–10268. [Google Scholar] [CrossRef] [Green Version]
- Stafford, N.; Wilson, C.; Oceandy, D.; Neyses, L.; Cartwright, E.J. The Plasma Membrane Calcium ATPases and Their Role as Major New Players in Human Disease. Physiol. Rev. 2017, 97, 1089–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunter, T.E.; Buntinas, L.; Sparagna, G.; Eliseev, R.; Gunter, K. Mitochondrial calcium transport: Mechanisms and functions. Cell Calcium 2000, 28, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Frazier, H.N.; Maimaiti, S.; Anderson, K.L.; Brewer, L.D.; Gant, J.C.; Porter, N.M.; Thibault, O. Calcium’s role as nuanced modulator of cellular physiology in the brain. Biochem. Biophys. Res. Commun. 2017, 483, 981–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, M.; Ahumada-Castro, U.; Sanhueza, M.; Gonzalez-Billault, C.; Court, F.A.; Cardenas, C. Mitochondria and Calcium Regulation as Basis of Neurodegeneration Associated With Aging. Front. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef] [PubMed]
- Akbar, M.; Essa, M.M.; Daradkeh, G.; Abdelmegeed, M.A.; Choi, Y.; Mahmood, L.; Song, B.J. Mitochondrial dysfunction and cell death in neurodegenerative diseases through nitroxidative stress. Brain Res. 2016, 1637, 34–55. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Wu, S.B.; Wu, Y.T.; Wei, Y.H. Oxidative stress response elicited by mitochondrial dysfunction: Implication in the pathophysiology of aging. Exp. Biol. Med. 2013, 238, 450–460. [Google Scholar] [CrossRef]
- Barja, G. Updating the mitochondrial free radical theory of aging: An integrated view, key aspects, and confounding concepts. Antioxid. Redox. Signal. 2013, 19, 1420–1445. [Google Scholar] [CrossRef] [Green Version]
- Harman, D. The Free Radical Theory of Aging: Effect of Age on Serum Copper Levels. J. Gerontol. 1965, 20, 151–153. [Google Scholar] [CrossRef]
- Strehler, E.E.; Zacharias, D.A. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol. Rev. 2001, 81, 21–50. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Carafoli, E. Calcium pumps in health and disease. Physiol. Rev. 2009, 89, 1341–1378. [Google Scholar] [CrossRef] [Green Version]
- Burette, A.; Rockwood, J.M.; Strehler, E.E.; Weinberg, R.J. Isoform-specific distribution of the plasma membrane Ca2+ ATPase in the rat brain. J. Comp. Neurol. 2003, 467, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, F.; Domi, T.; Fedrizzi, L.; Lim, D.; Carafoli, E. The plasma membrane Ca2+ ATPase of animal cells: Structure, function and regulation. Arch. Biochem. Biophys. 2008, 476, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Carafoli, E. The plasma membrane Ca2+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Cold Spring Harb. Perspect. Biol. 2011, 3, a004168. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, T.P.; Hilfiker, H.; Carafoli, E.; Strehler, E.E. Quantitative analysis of alternative splicing options of human plasma membrane calcium pump genes. J. Biol. Chem. 1993, 268, 25993–26003. [Google Scholar]
- Stauffer, T.P.; Guerini, D.; Carafoli, E. Tissue distribution of the four gene products of the plasma membrane Ca2+ pump. A study using specific antibodies. J. Biol. Chem. 1995, 270, 12184–12190. [Google Scholar] [CrossRef] [Green Version]
- Zacharias, D.A.; Kappen, C. Developmental expression of the four plasma membrane calcium ATPase (PMCA) genes in the mouse. Biochim. Biophys. Acta 1999, 1428, 397–405. [Google Scholar] [CrossRef]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life. Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef]
- Sepulveda, M.R.; Hidalgo-Sanchez, M.; Marcos, D.; Mata, A.M. Developmental distribution of plasma membrane Ca2+-ATPase isoforms in chick cerebellum. Dev. Dyn. 2007, 236, 1227–1236. [Google Scholar] [CrossRef]
- Kip, S.N.; Gray, N.W.; Burette, A.; Canbay, A.; Weinberg, R.J.; Strehler, E.E. Changes in the expression of plasma membrane calcium extrusion systems during the maturation of hippocampal neurons. Hippocampus 2006, 16, 20–34. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Bechtel, M.D.; Galeva, N.A.; Williams, T.D.; Michaelis, E.K.; Michaelis, M.L. Decreases in plasma membrane Ca2+-ATPase in brain synaptic membrane rafts from aged rats. J. Neurochem. 2012, 123, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, A.; Gao, J.; Squier, T.C.; Michaelis, M.L. Age-related decrease in brain synaptic membrane Ca2+-ATPase in F344/BNF1 rats. Neurobiol. Aging. 1998, 19, 487–495. [Google Scholar] [CrossRef]
- Michaelis, M.L.; Bigelow, D.J.; Schoneich, C.; Williams, T.D.; Ramonda, L.; Yin, D.; Huhmer, A.F.; Yao, Y.; Gao, J.; Squier, T.C. Decreased plasma membrane calcium transport activity in aging brain. Life Sci. 1996, 59, 405–412. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Chen, X.; Krainev, A.G.; Michaelis, E.K.; Bigelow, D.J. Protein half-lives of calmodulin and the plasma membrane Ca-ATPase in rat brain. Biochem. Biophys. Res. Commun. 1997, 237, 163–165. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A. Plasma membrane Ca-ATPases: Targets of oxidative stress in brain aging and neurodegeneration. World J. Biol. Chem. 2010, 1, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.; Michaelis, M.L. Effects of reactive oxygen species on brain synaptic plasma membrane Ca2+-ATPase. Free Radic. Biol. Med. 1999, 27, 810–821. [Google Scholar] [CrossRef]
- Kip, S.N.; Strehler, E.E. Rapid downregulation of NCX and PMCA in hippocampal neurons following H2O2 oxidative stress. Ann. N. Y. Acad. Sci. 2007, 1099, 436–439. [Google Scholar] [CrossRef] [Green Version]
- Giacomello, M.; De Mario, A.; Lopreiato, R.; Primerano, S.; Campeol, M.; Brini, M.; Carafoli, E. Mutations in PMCA2 and hereditary deafness: A molecular analysis of the pump defect. Cell Calcium 2011, 50, 569–576. [Google Scholar] [CrossRef]
- Cali, T.; Lopreiato, R.; Shimony, J.; Vineyard, M.; Frizzarin, M.; Zanni, G.; Zanotti, G.; Brini, M.; Shinawi, M.; Carafoli, E. A Novel Mutation in Isoform 3 of the Plasma Membrane Ca2+ Pump Impairs Cellular Ca2+ Homeostasis in a Patient with Cerebellar Ataxia and Laminin Subunit 1α Mutations. J. Biol. Chem. 2015, 290, 16132–16141. [Google Scholar] [CrossRef] [Green Version]
- Zanni, G.; Cali, T.; Kalscheuer, V.M.; Ottolini, D.; Barresi, S.; Lebrun, N.; Montecchi-Palazzi, L.; Hu, H.; Chelly, J.; Bertini, E.; et al. Mutation of plasma membrane Ca2+ ATPase isoform 3 in a family with X-linked congenital cerebellar ataxia impairs Ca2+ homeostasis. Proc. Natl. Acad. Sci. USA 2012, 109, 14514–14519. [Google Scholar] [CrossRef] [Green Version]
- Berrocal, M.; Marcos, D.; Sepulveda, M.R.; Perez, M.; Avila, J.; Mata, A.M. Altered Ca2+ dependence of synaptosomal plasma membrane Ca2+-ATPase in human brain affected by Alzheimer’s disease. FASEB J. 2009, 23, 1826–1834. [Google Scholar] [CrossRef]
- Berrocal, M.; Sepulveda, M.R.; Vazquez-Hernandez, M.; Mata, A.M. Calmodulin antagonizes amyloid-β peptides-mediated inhibition of brain plasma membrane Ca2+-ATPase. Biochim. Biophys. Acta 2012, 1822, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Berrocal, M.; Corbacho, I.; Vazquez-Hernandez, M.; Avila, J.; Sepulveda, M.R.; Mata, A.M. Inhibition of PMCA activity by tau as a function of aging and Alzheimer’s neuropathology. Biochim. Biophys. Acta 2015, 1852, 1465–1476. [Google Scholar] [CrossRef] [Green Version]
- Berrocal, M.; Corbacho, I.; Sepulveda, M.R.; Gutierrez-Merino, C.; Mata, A.M. Phospholipids and calmodulin modulate the inhibition of PMCA activity by tau. Biochim. Biophys. Acta Mol. Cell. Res. 2017, 1864, 1028–1035. [Google Scholar] [CrossRef]
- Greene, L.A.; Tischler, A.S. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 1976, 73, 2424–2428. [Google Scholar] [CrossRef] [Green Version]
- Keller, D.; Grover, A.K. Nerve growth factor treatment alters Ca2+ pump levels in PC12 cells. Neuroreport 2000, 11, 65–68. [Google Scholar] [CrossRef]
- Lambeng, N.; Michel, P.P.; Agid, Y.; Ruberg, M. The relationship between differentiation and survival in PC12 cells treated with cyclic adenosine monophosphate in the presence of epidermal growth factor or nerve growth factor. Neurosci. Lett. 2001, 297, 133–136. [Google Scholar] [CrossRef]
- Hammes, A.; Oberdorf, S.; Strehler, E.E.; Stauffer, T.; Carafoli, E.; Vetter, H.; Neyses, L. Differentiation-specific isoform mRNA expression of the calmodulin-dependent plasma membrane Ca2+-ATPase. FASEB J. 1994, 8, 428–435. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.L.; Usachev, Y.M.; Thayer, S.A.; Strehler, E.E.; Windebank, A.J. Plasma membrane calcium ATPase plays a role in reducing Ca2+-mediated cytotoxicity in PC12 cells. J. Neurosci. Res. 2001, 64, 661–669. [Google Scholar] [CrossRef]
- Boczek, T.; Lisek, M.; Kowalski, A.; Pikula, S.; Niewiarowska, J.; Wiktorska, M.; Zylinska, L. Downregulation of PMCA2 or PMCA3 reorganizes Ca2+ handling systems in differentiating PC12 cells. Cell Calcium 2012, 52, 433–444. [Google Scholar] [CrossRef]
- Luo, L.; O’Leary, D.D. Axon retraction and degeneration in development and disease. Annu. Rev. Neurosci. 2005, 28, 127–156. [Google Scholar] [CrossRef] [Green Version]
- Baranov, S.V.; Baranova, O.V.; Yablonska, S.; Suofu, Y.; Vazquez, A.L.; Kozai, T.D.Y.; Cui, X.T.; Ferrando, L.M.; Larkin, T.M.; Tyurina, Y.Y.; et al. Mitochondria modulate programmed neuritic retraction. Proc. Natl. Acad. Sci. USA 2019, 116, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, D.; Zaidi, A.; Bean, J.; Hui, D.; Michaelis, M.L. RNAi—Induced silencing of the plasma membrane Ca2+-ATPase 2 in neuronal cells: Effects on Ca2+ homeostasis and cell viability. J. Neurochem. 2007, 102, 454–465. [Google Scholar] [CrossRef]
- Kurnellas, M.P.; Li, H.; Jain, M.R.; Giraud, S.N.; Nicot, A.B.; Ratnayake, A.; Heary, R.F.; Elkabes, S. Reduced expression of plasma membrane calcium ATPase 2 and collapsin response mediator protein 1 promotes death of spinal cord neurons. Cell. Death Differ. 2010, 17, 1501–1510. [Google Scholar] [CrossRef]
- Empson, R.M.; Turner, P.R.; Nagaraja, R.Y.; Beesley, P.W.; Knopfel, T. Reduced expression of the Ca2+ transporter protein PMCA2 slows Ca2+ dynamics in mouse cerebellar Purkinje neurones and alters the precision of motor coordination. J. Physiol. 2010, 588, 907–922. [Google Scholar] [CrossRef]
- Raza, M.; Deshpande, L.S.; Blair, R.E.; Carter, D.S.; Sombati, S.; DeLorenzo, R.J. Aging is associated with elevated intracellular calcium levels and altered calcium homeostatic mechanisms in hippocampal neurons. Neurosci. Lett. 2007, 418, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Mata, A.M.; Sepulveda, M.R. Plasma membrane Ca-ATPases in the nervous system during development and ageing. World J. Biol. Chem. 2010, 1, 229–234. [Google Scholar] [CrossRef]
- Strehler, E.E.; Thayer, S.A. Evidence for a role of plasma membrane calcium pumps in neurodegenerative disease: Recent developments. Neurosci. Lett. 2018, 663, 39–47. [Google Scholar] [CrossRef]
- Carafoli, E.; Brini, M. Calcium pumps: Structural basis for and mechanism of calcium transmembrane transport. Curr. Opin. Chem. Biol. 2000, 4, 152–161. [Google Scholar] [CrossRef]
- Mata, A.M.; Sepulveda, M.R. Calcium pumps in the central nervous system. Brain Res. Rev. 2005, 49, 398–405. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Rice, W.J.; Green, N.M. The mechanism of Ca2+ transport by sarco(endo)plasmic reticulum Ca2+-ATPases. J. Biol. Chem. 1997, 272, 28815–28818. [Google Scholar] [CrossRef] [Green Version]
- Janigro, D.; Maccaferri, G.; Meldolesi, J. Calcium channels in undifferentiated PC12 rat pheochromocytoma cells. FEBS Lett 1989, 255, 398–400. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Felix, R.; Gurnett, C.A.; De Waard, M.; Witcher, D.R.; Campbell, K.P. Expression and subunit interaction of voltage-dependent Ca2+ channels in PC12 cells. J. Neurosci. 1996, 16, 7557–7565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strickland, M.; Yacoubi-Loueslati, B.; Bouhaouala-Zahar, B.; Pender, S.L.F.; Larbi, A. Relationships between Ion Channels, Mitochondrial Functions and Inflammation in Human Aging. Front. Physiol. 2019, 10, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibault, O.; Landfield, P.W. Increase in single L-type calcium channels in hippocampal neurons during aging. Science 1996, 272, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.W.; Hao, S.Y.; Thibault, O.; Blalock, E.M.; Landfield, P.W. Aging changes in voltage-gated calcium currents in hippocampal CA1 neurons. J. Neurosci. 1996, 16, 6286–6295. [Google Scholar] [CrossRef]
- Brewer, L.D.; Thibault, O.; Staton, J.; Thibault, V.; Rogers, J.T.; Garcia-Ramos, G.; Kraner, S.; Landfield, P.W.; Porter, N.M. Increased vulnerability of hippocampal neurons with age in culture: Temporal association with increases in NMDA receptor current, NR2A subunit expression and recruitment of L-type calcium channels. Brain Res. 2007, 1151, 20–31. [Google Scholar] [CrossRef]
- Thibault, O.; Gant, J.C.; Landfield, P.W. Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: Minding the store. Aging Cell 2007, 6, 307–317. [Google Scholar] [CrossRef]
- Navakkode, S.; Liu, C.; Soong, T.W. Altered function of neuronal L-type calcium channels in ageing and neuroinflammation: Implications in age-related synaptic dysfunction and cognitive decline. Ageing Res. Rev. 2018, 42, 86–99. [Google Scholar] [CrossRef]
- Thibault, O.; Hadley, R.; Landfield, P.W. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: Relationship to impaired synaptic plasticity. J. Neurosci. 2001, 21, 9744–9756. [Google Scholar] [CrossRef] [Green Version]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [Green Version]
- Rice, R.A.; Berchtold, N.C.; Cotman, C.W.; Green, K.N. Age-related downregulation of the CaV3.1 T-type calcium channel as a mediator of amyloid beta production. Neurobiol. Aging 2014, 35, 1002–1011. [Google Scholar] [CrossRef] [Green Version]
- Del Toro, R.; Levitsky, K.L.; Lopez-Barneo, J.; Chiara, M.D. Induction of T-type calcium channel gene expression by chronic hypoxia. J. Biol. Chem. 2003, 278, 22316–22324. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Peleg, S. Biphasic Modeling of Mitochondrial Metabolism Dysregulation during Aging. Trends Biochem. Sci. 2017, 42, 702–711. [Google Scholar] [CrossRef] [Green Version]
- Bratic, A.; Larsson, N.G. The role of mitochondria in aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, D.V.; Wiley, C.D.; Velarde, M.C. Mitochondrial effectors of cellular senescence: Beyond the free radical theory of aging. Aging Cell 2015, 14, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Lopes, G.S.; Ferreira, A.T.; Oshiro, M.E.; Vladimirova, I.; Jurkiewicz, N.H.; Jurkiewicz, A.; Smaili, S.S. Aging-related changes of intracellular Ca2+ stores and contractile response of intestinal smooth muscle. Exp. Gerontol. 2006, 41, 55–62. [Google Scholar] [CrossRef]
- Buchholz, J.N.; Behringer, E.J.; Pottorf, W.J.; Pearce, W.J.; Vanterpool, C.K. Age-dependent changes in Ca2+ homeostasis in peripheral neurones: Implications for changes in function. Aging Cell 2007, 6, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Friel, D.D. Mitochondria as regulators of stimulus-evoked calcium signals in neurons. Cell Calcium 2000, 28, 307–316. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Toescu, E.C. Calcium and neuronal ageing. Trends Neurosci. 1998, 21, 2–7. [Google Scholar] [CrossRef]
- Martinez-Serrano, A.; Blanco, P.; Satrustegui, J. Calcium binding to the cytosol and calcium extrusion mechanisms in intact synaptosomes and their alterations with aging. J. Biol. Chem. 1992, 267, 4672–4679. [Google Scholar]
- Kirischuk, S.; Verkhratsky, A. Calcium homeostasis in aged neurones. Life Sci. 1996, 59, 451–459. [Google Scholar] [CrossRef]
- Denton, R.M.; Randle, P.J.; Bridges, B.J.; Cooper, R.H.; Kerbey, A.L.; Pask, H.T.; Severson, D.L.; Stansbie, D.; Whitehouse, S. Regulation of mammalian pyruvate dehydrogenase. Mol. Cell. Biochem. 1975, 9, 27–53. [Google Scholar] [CrossRef]
- McKenzie, M.; Lim, S.C.; Duchen, M.R. Simultaneous Measurement of Mitochondrial Calcium and Mitochondrial Membrane Potential in Live Cells by Fluorescent Microscopy. J. Vis. Exp. 2017, 119, e55166. [Google Scholar] [CrossRef] [Green Version]
- Madreiter-Sokolowski, C.T.; Sokolowski, A.A.; Graier, W.F. Dosis Facit Sanitatem—Concentration-Dependent Effects of Resveratrol on Mitochondria. Nutrients 2017, 9, 1117. [Google Scholar] [CrossRef] [Green Version]
- Gauba, E.; Guo, L.; Du, H. Cyclophilin D Promotes Brain Mitochondrial F1FO ATP Synthase Dysfunction in Aging Mice. J. Alzheimers Dis. 2017, 55, 1351–1362. [Google Scholar] [CrossRef] [Green Version]
- Panel, M.; Ghaleh, B.; Morin, D. Mitochondria and aging: A role for the mitochondrial transition pore? Aging Cell 2018, 17, e12793. [Google Scholar] [CrossRef]
- Scorrano, L.; Petronilli, V.; Bernardi, P. On the voltage dependence of the mitochondrial permeability transition pore. A critical appraisal. J. Biol. Chem. 1997, 272, 12295–12299. [Google Scholar] [CrossRef] [Green Version]
- Boczek, T.; Kozaczuk, A.; Taha, J.; Ferenc, B.; Zylinska, L. Adaptation of microsomal glutathione transferase 1 in PC12 cells with modified PMCA isoforms composition. Indian J. Biochem. Biophys. 2010, 47, 265–271. [Google Scholar]
- Marques-Aleixo, I.; Rocha-Rodrigues, S.; Santos-Alves, E.; Coxito, P.M.; Passos, E.; Oliveira, P.J.; Magalhaes, J.; Ascensao, A. In vitro salicylate does not further impair aging-induced brain mitochondrial dysfunction. Toxicology 2012, 302, 51–59. [Google Scholar] [CrossRef]
- Krestinina, O.; Azarashvili, T.; Baburina, Y.; Galvita, A.; Grachev, D.; Stricker, R.; Reiser, G. In aging, the vulnerability of rat brain mitochondria is enhanced due to reduced level of 2′,3′-cyclic nucleotide-3′-phosphodiesterase (CNP) and subsequently increased permeability transition in brain mitochondria in old animals. Neurochem. Int. 2015, 80, 41–50. [Google Scholar] [CrossRef]
- Pandya, J.D.; Grondin, R.; Yonutas, H.M.; Haghnazar, H.; Gash, D.M.; Zhang, Z.; Sullivan, P.G. Decreased mitochondrial bioenergetics and calcium buffering capacity in the basal ganglia correlates with motor deficits in a nonhuman primate model of aging. Neurobiol. Aging 2015, 36, 1903–1913. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Martino, P.L.; Capitanio, G.; Gaballo, A.; De Rasmo, D.; Signorile, A.; Petruzzella, V. The oxidative phosphorylation system in mammalian mitochondria. Adv. Exp. Med. Biol. 2012, 942, 3–37. [Google Scholar] [PubMed]
- Naghdi, S.; Waldeck-Weiermair, M.; Fertschai, I.; Poteser, M.; Graier, W.F.; Malli, R. Mitochondrial Ca2+ uptake and not mitochondrial motility is required for STIM1-Orai1-dependent store-operated Ca2+ entry. J. Cell Sci. 2010, 123, 2553–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waldeck-Weiermair, M.; Alam, M.R.; Khan, M.J.; Deak, A.T.; Vishnu, N.; Karsten, F.; Imamura, H.; Graier, W.F.; Malli, R. Spatiotemporal correlations between cytosolic and mitochondrial Ca2+ signals using a novel red-shifted mitochondrial targeted cameleon. PLoS ONE 2012, 7, e45917. [Google Scholar] [CrossRef] [PubMed]
- Raturi, A.; Simmen, T. Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim. Biophys. Acta 2013, 1833, 213–224. [Google Scholar] [CrossRef] [Green Version]
- Kruger, W.A.; Monteith, G.R.; Poronnik, P. The plasma membrane Ca2+-ATPase: Regulation by PSD-95/Dlg/Zo-1 scaffolds. Int. J. Biochem. Cell Biol. 2010, 42, 805–808. [Google Scholar] [CrossRef]
- Garside, M.L.; Turner, P.R.; Austen, B.; Strehler, E.E.; Beesley, P.W.; Empson, R.M. Molecular interactions of the plasma membrane calcium ATPase 2 at pre- and post-synaptic sites in rat cerebellum. Neuroscience 2009, 162, 383–395. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.; Ramsden, D.B. Huntington’s disease. Mol. Pathol. 2001, 54, 409–413. [Google Scholar] [CrossRef]
- Lisek, M.; Ferenc, B.; Studzian, M.; Pulaski, L.; Guo, F.; Zylinska, L.; Boczek, T. Glutamate Deregulation in Ketamine-Induced Psychosis-A Potential Role of PSD95, NMDA Receptor and PMCA Interaction. Front. Cell. Neurosci. 2017, 11, 181. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, D.G. Mitochondrial calcium function and dysfunction in the central nervous system. Biochim. Biophys. Acta 2009, 1787, 1416–1424. [Google Scholar] [CrossRef] [Green Version]
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duchen, M.R. Mitochondria, calcium-dependent neuronal death and neurodegenerative disease. Pflugers. Arch. 2012, 464, 111–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Tan, Y.W.; Hagenston, A.M.; Martel, M.A.; Kneisel, N.; Skehel, P.A.; Wyllie, D.J.; Bading, H.; Hardingham, G.E. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013, 4, 2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoratti, M.; Szabo, I. The mitochondrial permeability transition. Biochim. Biophys. Acta 1995, 1241, 139–176. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Li, V.; Wang, Y.T. Molecular mechanisms of NMDA receptor-mediated excitotoxicity: Implications for neuroprotective therapeutics for stroke. Neural Regen. Res. 2016, 11, 1752–1753. [Google Scholar]
- Zhou, Q.; Sheng, M. NMDA receptors in nervous system diseases. Neuropharmacology 2013, 74, 69–75. [Google Scholar] [CrossRef] [Green Version]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Markhard, A.L.; Kitami, T.; Kovacs-Bogdan, E.; Kamer, K.J.; Udeshi, N.D.; Carr, S.A.; Chaudhuri, D.; Clapham, D.E.; Li, A.A.; et al. EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 2013, 342, 1379–1382. [Google Scholar] [CrossRef] [Green Version]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell. Biol. 2015, 16, 545–553. [Google Scholar] [CrossRef]
- Kamer, K.J.; Jiang, W.; Kaushik, V.K.; Mootha, V.K.; Grabarek, Z. Crystal structure of MICU2 and comparison with MICU1 reveal insights into the uniporter gating mechanism. Proc. Natl. Acad. Sci. USA 2019, 116, 3546–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallilankaraman, K.; Doonan, P.; Cardenas, C.; Chandramoorthy, H.C.; Muller, M.; Miller, R.; Hoffman, N.E.; Gandhirajan, R.K.; Molgo, J.; Birnbaum, M.J.; et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell 2012, 151, 630–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csordas, G.; Golenar, T.; Seifert, E.L.; Kamer, K.J.; Sancak, Y.; Perocchi, F.; Moffat, C.; Weaver, D.; de la Fuente Perez, S.; Bogorad, R.; et al. MICU1 controls both the threshold and cooperative activation of the mitochondrial Ca2+ uniporter. Cell Metab. 2013, 17, 976–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patron, M.; Checchetto, V.; Raffaello, A.; Teardo, E.; Vecellio Reane, D.; Mantoan, M.; Granatiero, V.; Szabo, I.; De Stefani, D.; Rizzuto, R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell. 2014, 53, 726–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.C.; Liu, J.; Holmstrom, K.M.; Menazza, S.; Parks, R.J.; Fergusson, M.M.; Yu, Z.X.; Springer, D.A.; Halsey, C.; Liu, C.; et al. MICU1 Serves as a Molecular Gatekeeper to Prevent In Vivo Mitochondrial Calcium Overload. Cell Rep. 2016, 16, 1561–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, T.P.; Wu, Y.; Joiner, M.L.; Koval, O.M.; Wilson, N.R.; Luczak, E.D.; Wang, Q.; Chen, B.; Gao, Z.; Zhu, Z.; et al. Inhibition of MCU forces extramitochondrial adaptations governing physiological and pathological stress responses in heart. Proc. Natl. Acad. Sci. USA 2015, 112, 9129–9134. [Google Scholar] [CrossRef] [Green Version]
- Konig, T.; Troder, S.E.; Bakka, K.; Korwitz, A.; Richter-Dennerlein, R.; Lampe, P.A.; Patron, M.; Muhlmeister, M.; Guerrero-Castillo, S.; Brandt, U.; et al. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol. Cell. 2016, 64, 148–162. [Google Scholar] [CrossRef] [Green Version]
- Plovanich, M.; Bogorad, R.L.; Sancak, Y.; Kamer, K.J.; Strittmatter, L.; Li, A.A.; Girgis, H.S.; Kuchimanchi, S.; De Groot, J.; Speciner, L.; et al. MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS ONE 2013, 8, e55785. [Google Scholar] [CrossRef] [Green Version]
- Markus, N.M.; Hasel, P.; Qiu, J.; Bell, K.F.; Heron, S.; Kind, P.C.; Dando, O.; Simpson, T.I.; Hardingham, G.E. Expression of mRNA Encoding Mcu and Other Mitochondrial Calcium Regulatory Genes Depends on Cell Type, Neuronal Subtype, and Ca2+ Signaling. PLoS ONE 2016, 11, e0148164. [Google Scholar] [CrossRef]
- Patron, M.; Granatiero, V.; Espino, J.; Rizzuto, R.; De Stefani, D. MICU3 is a tissue-specific enhancer of mitochondrial calcium uptake. Cell Death Differ. 2019, 26, 179–195. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Rodriguez, M.; Garcia-Durillo, M.; Villalobos, C.; Nunez, L. In vitro aging promotes endoplasmic reticulum (ER)-mitochondria Ca2+ cross talk and loss of store-operated Ca2+ entry (SOCE) in rat hippocampal neurons. Biochim. Biophys. Acta 2016, 1863, 2637–2649. [Google Scholar] [CrossRef] [PubMed]
- Madreiter-Sokolowski, C.T.; Waldeck-Weiermair, M.; Bourguignon, M.P.; Villeneuve, N.; Gottschalk, B.; Klec, C.; Stryeck, S.; Radulovic, S.; Parichatikanond, W.; Frank, S.; et al. Enhanced inter-compartmental Ca2+ flux modulates mitochondrial metabolism and apoptotic threshold during aging. Redox Biol. 2019, 20, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Madreiter-Sokolowski, C.T.; Sokolowski, A.A.; Waldeck-Weiermair, M.; Malli, R.; Graier, W.F. Targeting Mitochondria to Counteract Age-Related Cellular Dysfunction. Genes 2018, 9, 165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manczak, M.; Jung, Y.; Park, B.S.; Partovi, D.; Reddy, P.H. Time-course of mitochondrial gene expressions in mice brains: Implications for mitochondrial dysfunction, oxidative damage, and cytochrome c in aging. J. Neurochem. 2005, 92, 494–504. [Google Scholar] [CrossRef]
- Itoh, K.; Weis, S.; Mehraein, P.; Muller-Hocker, J. Cytochrome c oxidase defects of the human substantia nigra in normal aging. Neurobiol. Aging 1996, 17, 843–848. [Google Scholar] [CrossRef]
- Bertoni-Freddari, C.; Fattoretti, P.; Giorgetti, B.; Solazzi, M.; Balietti, M.; Casoli, T.; Di Stefano, G. Cytochrome oxidase activity in hippocampal synaptic mitochondria during aging: A quantitative cytochemical investigation. Ann. N. Y. Acad. Sci. 2004, 1019, 33–36. [Google Scholar] [CrossRef]
- Poburko, D.; Santo-Domingo, J.; Demaurex, N. Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J. Biol. Chem. 2011, 286, 11672–11684. [Google Scholar] [CrossRef] [Green Version]
- Balut, C.; vandeVen, M.; Despa, S.; Lambrichts, I.; Ameloot, M.; Steels, P.; Smets, I. Measurement of cytosolic and mitochondrial pH in living cells during reversible metabolic inhibition. Kidney Int. 2008, 73, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Bolshakov, A.P.; Mikhailova, M.M.; Szabadkai, G.; Pinelis, V.G.; Brustovetsky, N.; Rizzuto, R.; Khodorov, B.I. Measurements of mitochondrial pH in cultured cortical neurons clarify contribution of mitochondrial pore to the mechanism of glutamate-induced delayed Ca2+ deregulation. Cell Calcium 2008, 43, 602–614. [Google Scholar] [CrossRef]
- Malli, R.; Frieden, M.; Osibow, K.; Zoratti, C.; Mayer, M.; Demaurex, N.; Graier, W.F. Sustained Ca2+ transfer across mitochondria is Essential for mitochondrial Ca2+ buffering, sore-operated Ca2+ entry, and Ca2+ store refilling. J. Biol. Chem. 2003, 278, 44769–44779. [Google Scholar] [CrossRef] [Green Version]
- Abad, M.F.; Di Benedetto, G.; Magalhaes, P.J.; Filippin, L.; Pozzan, T. Mitochondrial pH monitored by a new engineered green fluorescent protein mutant. J. Biol. Chem. 2004, 279, 11521–11529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petronilli, V.; Cola, C.; Bernardi, P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore. II. The minimal requirements for pore induction underscore a key role for transmembrane electrical potential, matrix pH, and matrix Ca2+. J. Biol. Chem. 1993, 268, 1011–1016. [Google Scholar] [PubMed]
- Selivanov, V.A.; Zeak, J.A.; Roca, J.; Cascante, M.; Trucco, M.; Votyakova, T.V. The role of external and matrix pH in mitochondrial reactive oxygen species generation. J. Biol. Chem. 2008, 283, 29292–29300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behringer, E.J.; Segal, S.S. Impact of Aging on Calcium Signaling and Membrane Potential in Endothelium of Resistance Arteries: A Role for Mitochondria. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1627–1637. [Google Scholar] [CrossRef]
- Gunter, T.E.; Pfeiffer, D.R. Mechanisms by which mitochondria transport calcium. Am. J. Physiol. 1990, 258, C755–C786. [Google Scholar] [CrossRef]
- Tarasov, A.I.; Griffiths, E.J.; Rutter, G.A. Regulation of ATP production by mitochondrial Ca2+. Cell Calcium 2012, 52, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Luciani, D.S.; Misler, S.; Polonsky, K.S. Ca2+ controls slow NAD(P)H oscillations in glucose-stimulated mouse pancreatic islets. J. Physiol. 2006, 572, 379–392. [Google Scholar] [CrossRef]
- Jouaville, L.S.; Pinton, P.; Bastianutto, C.; Rutter, G.A.; Rizzuto, R. Regulation of mitochondrial ATP synthesis by calcium: Evidence for a long-term metabolic priming. Proc. Natl. Acad. Sci. USA 1999, 96, 13807–13812. [Google Scholar] [CrossRef] [Green Version]
- Robb-Gaspers, L.D.; Burnett, P.; Rutter, G.A.; Denton, R.M.; Rizzuto, R.; Thomas, A.P. Integrating cytosolic calcium signals into mitochondrial metabolic responses. EMBO J. 1998, 17, 4987–5000. [Google Scholar] [CrossRef] [Green Version]
- Harris, D.A.; Das, A.M. Control of mitochondrial ATP synthesis in the heart. Biochem. J. 1991, 280, 561–573. [Google Scholar] [CrossRef] [Green Version]
- Scholz, T.D.; Balaban, R.S. Mitochondrial F1-ATPase activity of canine myocardium: Effects of hypoxia and stimulation. Am. J. Physiol. 1994, 266, H2396–H2403. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, M.J.; McHugh, N.J. Mitochondrial ATP synthase F1-β-subunit is a calcium-binding protein. FEBS Lett. 1996, 391, 323–329. [Google Scholar] [CrossRef] [Green Version]
- Hopper, R.K.; Carroll, S.; Aponte, A.M.; Johnson, D.T.; French, S.; Shen, R.F.; Witzmann, F.A.; Harris, R.A.; Balaban, R.S. Mitochondrial matrix phosphoproteome: Effect of extra mitochondrial calcium. Biochemistry 2006, 45, 2524–2536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boerries, M.; Most, P.; Gledhill, J.R.; Walker, J.E.; Katus, H.A.; Koch, W.J.; Aebi, U.; Schoenenberger, C.A. Ca2+ -dependent interaction of S100A1 with F1-ATPase leads to an increased ATP content in cardiomyocytes. Mol. Cell. Biol. 2007, 27, 4365–4373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padanyi, R.; Paszty, K.; Hegedus, L.; Varga, K.; Papp, B.; Penniston, J.T.; Enyedi, A. Multifaceted plasma membrane Ca2+ pumps: From structure to intracellular Ca2+ handling and cancer. Biochim. Biophys. Acta 2016, 1863, 1351–1363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cali, T.; Brini, M.; Carafoli, E. Regulation of Cell Calcium and Role of Plasma Membrane Calcium ATPases. Int. Rev. Cell. Mol. Biol. 2017, 332, 259–296. [Google Scholar]
- Elwess, N.L.; Filoteo, A.G.; Enyedi, A.; Penniston, J.T. Plasma membrane Ca2+ pump isoforms 2a and 2b are unusually responsive to calmodulin and Ca2+. J. Biol. Chem. 1997, 272, 17981–17986. [Google Scholar] [CrossRef] [Green Version]
- Strehler, E.E. Plasma membrane calcium ATPases: From generic Ca2+ sump pumps to versatile systems for fine-tuning cellular Ca2+. Biochem. Biophys. Res. Commun. 2015, 460, 26–33. [Google Scholar] [CrossRef]
- Toutenhoofd, S.L.; Strehler, E.E. The calmodulin multigene family as a unique case of genetic redundancy: Multiple levels of regulation to provide spatial and temporal control of calmodulin pools? Cell Calcium 2000, 28, 83–96. [Google Scholar] [CrossRef]
- Shen, X.; Valencia, C.A.; Szostak, J.W.; Dong, B.; Liu, R. Scanning the human proteome for calmodulin-binding proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 5969–5974. [Google Scholar] [CrossRef] [Green Version]
- Bigelow, D.J.; Squier, T.C. Redox modulation of cellular signaling and metabolism through reversible oxidation of methionine sensors in calcium regulatory proteins. Biochim. Biophys. Acta 2005, 1703, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Davidkova, G.; Zhang, S.P.; Nichols, R.A.; Weiss, B. Reduced level of calmodulin in PC12 cells induced by stable expression of calmodulin antisense RNA inhibits cell proliferation and induces neurite outgrowth. Neuroscience 1996, 75, 1003–1019. [Google Scholar] [CrossRef]
- Boczek, T.; Lisek, M.; Ferenc, B.; Zylinska, L. Cross talk among PMCA, calcineurin and NFAT transcription factors in control of calmodulin gene expression in differentiating PC12 cells. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 502–515. [Google Scholar] [CrossRef] [PubMed]
- Benowitz, L.I.; Routtenberg, A. GAP-43: An intrinsic determinant of neuronal development and plasticity. Trends Neurosci. 1997, 20, 84–91. [Google Scholar] [CrossRef]
- Denny, J.B. Molecular mechanisms, biological actions, and neuropharmacology of the growth-associated protein GAP-43. Curr. Neuropharmacol. 2006, 4, 293–304. [Google Scholar] [CrossRef] [Green Version]
- Holahan, M.R. A Shift from a Pivotal to Supporting Role for the Growth-Associated Protein (GAP-43) in the Coordination of Axonal Structural and Functional Plasticity. Front. Cell. Neurosci. 2017, 11, 266. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, C.J.; Park, M.; Spillane, M.; Yoo, S.; Pacheco, A.; Gomes, C.; Vuppalanchi, D.; McDonald, M.; Kim, H.H.; Merianda, T.T.; et al. Axonally synthesized β-actin and GAP-43 proteins support distinct modes of axonal growth. J. Neurosci. 2013, 33, 3311–3322. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.; Kim, H.H.; Kim, P.; Donnelly, C.J.; Kalinski, A.L.; Vuppalanchi, D.; Park, M.; Lee, S.J.; Merianda, T.T.; Perrone-Bizzozero, N.I.; et al. A HuD-ZBP1 ribonucleoprotein complex localizes GAP-43 mRNA into axons through its 3′ untranslated region AU-rich regulatory element. J. Neurochem. 2013, 126, 792–804. [Google Scholar] [CrossRef] [Green Version]
- Neve, R.L.; Finch, E.A.; Bird, E.D.; Benowitz, L.I. Growth-associated protein GAP-43 is expressed selectively in associative regions of the adult human brain. Proc. Natl. Acad. Sci. USA 1988, 85, 3638–3642. [Google Scholar] [CrossRef] [Green Version]
- Latchney, S.E.; Masiulis, I.; Zaccaria, K.J.; Lagace, D.C.; Powell, C.M.; McCasland, J.S.; Eisch, A.J. Developmental and adult GAP-43 deficiency in mice dynamically alters hippocampal neurogenesis and mossy fiber volume. Dev. Neurosci. 2014, 36, 44–63. [Google Scholar] [CrossRef] [Green Version]
- Schmoll, H.; Ramboiu, S.; Platt, D.; Herndon, J.G.; Kessler, C.; Popa-Wagner, A. Age influences the expression of GAP-43 in the rat hippocampus following seizure. Gerontology 2005, 51, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.A.; Mizumori, S.J.; Lovinger, D.M.; Sheu, F.S.; Murakami, K.; Chan, S.Y.; Linden, D.J.; Nelson, R.B.; Routtenberg, A. Selective decline in protein F1 phosphorylation in hippocampus of senescent rats. Neurobiol. Aging 1988, 9, 393–398. [Google Scholar] [CrossRef]
- Casoli, T.; Di Stefano, G.; Gracciotti, N.; Giovagnetti, S.; Fattoretti, P.; Solazzi, M.; Bertoni-Freddari, C. Cellular distribution of GAP-43 mRNA in hippocampus and cerebellum of adult rat brain by in situ RT-PCR. J. Histochem. Cytochem. 2001, 49, 1195–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casoli, T.; Spagna, C.; Fattoretti, P.; Gesuita, R.; Bertoni-Freddari, C. Neuronal plasticity in aging: A quantitative immunohistochemical study of GAP-43 distribution in discrete regions of the rat brain. Brain Res. 1996, 714, 111–117. [Google Scholar] [CrossRef]
- Casoli, T.; Stefano, G.D.; Fattoretti, P.; Solazzi, M.; Delfino, A.; Biagini, G.; Bertoni-Freddari, C. GAP-43 mRNA detection by in situ hybridization, direct and indirect in situ RT-PCR in hippocampal and cerebellar tissue sections of adult rat brain. Micron 2003, 34, 415–422. [Google Scholar] [CrossRef]
- Webster, M.J.; Elashoff, M.; Weickert, C.S. Molecular evidence that cortical synaptic growth predominates during the first decade of life in humans. Int. J. Dev. Neurosci. 2011, 29, 225–236. [Google Scholar] [CrossRef]
- De la Monte, S.M.; Ng, S.C.; Hsu, D.W. Aberrant GAP-43 gene expression in Alzheimer’s disease. Am. J. Pathol. 1995, 147, 934–946. [Google Scholar]
- Rekart, J.L.; Quinn, B.; Mesulam, M.M.; Routtenberg, A. Subfield-specific increase in brain growth protein in postmortem hippocampus of Alzheimer’s patients. Neuroscience 2004, 126, 579–584. [Google Scholar] [CrossRef]
- Boczek, T.; Ferenc, B.; Lisek, M.; Zylinska, L. Regulation of GAP43/calmodulin complex formation via calcineurin-dependent mechanism in differentiated PC12 cells with altered PMCA isoforms composition. Mol. Cell. Biochem. 2015, 407, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Kipanyula, M.J.; Kimaro, W.H.; Seke Etet, P.F. The Emerging Roles of the Calcineurin-Nuclear Factor of Activated T-Lymphocytes Pathway in Nervous System Functions and Diseases. J. Aging Res. 2016, 2016, 5081021. [Google Scholar] [CrossRef] [Green Version]
- Abdul, H.M.; Furman, J.L.; Sama, M.A.; Mathis, D.M.; Norris, C.M. NFATs and Alzheimer’s Disease. Mol. Cell. Pharmacol. 2010, 2, 7–14. [Google Scholar] [PubMed]
- Norris, C.M.; Kadish, I.; Blalock, E.M.; Chen, K.C.; Thibault, V.; Porter, N.M.; Landfield, P.W.; Kraner, S.D. Calcineurin triggers reactive/inflammatory processes in astrocytes and is upregulated in aging and Alzheimer’s models. J. Neurosci. 2005, 25, 4649–4658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Rao, A.; Hogan, P.G. Interaction of calcineurin with substrates and targeting proteins. Trends Cell. Biol. 2011, 21, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curry, M.C.; Luk, N.A.; Kenny, P.A.; Roberts-Thomson, S.J.; Monteith, G.R. Distinct regulation of cytoplasmic calcium signals and cell death pathways by different plasma membrane calcium ATPase isoforms in MDA-MB-231 breast cancer cells. J. Biol. Chem. 2012, 287, 28598–28608. [Google Scholar] [CrossRef] [Green Version]
- Holton, M.; Yang, D.; Wang, W.; Mohamed, T.M.; Neyses, L.; Armesilla, A.L. The interaction between endogenous calcineurin and the plasma membrane calcium-dependent ATPase is isoform specific in breast cancer cells. FEBS Lett. 2007, 581, 4115–4119. [Google Scholar] [CrossRef]
- Wu, X.; Chang, B.; Blair, N.S.; Sargent, M.; York, A.J.; Robbins, J.; Shull, G.E.; Molkentin, J.D. Plasma membrane Ca2+-ATPase isoform 4 antagonizes cardiac hypertrophy in association with calcineurin inhibition in rodents. J. Clin. Investig. 2009, 119, 976–985. [Google Scholar] [CrossRef] [Green Version]
- Baggott, R.R.; Mohamed, T.M.; Oceandy, D.; Holton, M.; Blanc, M.C.; Roux-Soro, S.C.; Brown, S.; Brown, J.E.; Cartwright, E.J.; Wang, W.; et al. Disruption of the interaction between PMCA2 and calcineurin triggers apoptosis and enhances paclitaxel-induced cytotoxicity in breast cancer cells. Carcinogenesis 2012, 33, 2362–2368. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.C.; Sharrow, K.M.; Masse, J.R.; Norris, C.M.; Kumar, A. Calcineurin links Ca2+ dysregulation with brain aging. J. Neurosci. 2001, 21, 4066–4073. [Google Scholar] [CrossRef] [Green Version]
- Sommerer, C.; Meuer, S.; Zeier, M.; Giese, T. Calcineurin inhibitors and NFAT-regulated gene expression. Clin. Chim. Acta 2012, 413, 1379–1386. [Google Scholar] [CrossRef]
- Muller, M.R.; Rao, A. NFAT, immunity and cancer: A transcription factor comes of age. Nat. Rev. Immunol. 2010, 10, 645–656. [Google Scholar] [CrossRef]
- Teolato, S.; Calderini, G.; Bonetti, A.C.; Toffano, G. Calmodulin content in different brain areas of aging rats. Neurosci. Lett. 1983, 38, 57–60. [Google Scholar] [CrossRef]
- Saimi, Y.; Kung, C. Calmodulin as an ion channel subunit. Annu. Rev. Physiol. 2002, 64, 289–311. [Google Scholar] [CrossRef] [PubMed]
- Norris, C.M.; Blalock, E.M.; Chen, K.C.; Porter, N.M.; Landfield, P.W. Calcineurin enhances L-type Ca2+ channel activity in hippocampal neurons: Increased effect with age in culture. Neuroscience 2002, 110, 213–225. [Google Scholar] [CrossRef] [Green Version]
- Chemaly, E.R.; Troncone, L.; Lebeche, D. SERCA control of cell death and survival. Cell Calcium 2018, 69, 46–61. [Google Scholar] [CrossRef]
- Foskett, J.K.; White, C.; Cheung, K.H.; Mak, D.O. Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 2007, 87, 593–658. [Google Scholar] [CrossRef] [Green Version]
- Decuypere, J.P.; Monaco, G.; Missiaen, L.; De Smedt, H.; Parys, J.B.; Bultynck, G. IP3 Receptors, Mitochondria, and Ca Signaling: Implications for Aging. J. Aging Res. 2011, 2011, 920178. [Google Scholar] [CrossRef] [Green Version]
- Vervloessem, T.; Yule, D.I.; Bultynck, G.; Parys, J.B. The type 2 inositol 1,4,5-trisphosphate receptor, emerging functions for an intriguing Ca2+-release channel. Biochim. Biophys. Acta 2015, 1853, 1992–2005. [Google Scholar] [CrossRef] [Green Version]
- Dent, M.A.; Raisman, G.; Lai, F.A. Expression of type 1 inositol 1,4,5-trisphosphate receptor during axogenesis and synaptic contact in the central and peripheral nervous system of developing rat. Development 1996, 122, 1029–1039. [Google Scholar]
- Mikoshiba, K. IP3 receptor/Ca2+ channel: From discovery to new signaling concepts. J. Neurochem. 2007, 102, 1426–1446. [Google Scholar] [CrossRef]
- Kumar, A.; Gibbs, J.R.; Beilina, A.; Dillman, A.; Kumaran, R.; Trabzuni, D.; Ryten, M.; Walker, R.; Smith, C.; Traynor, B.J.; et al. Age-associated changes in gene expression in human brain and isolated neurons. Neurobiol. Aging 2013, 34, 1199–1209. [Google Scholar] [CrossRef] [Green Version]
- Sharp, A.H.; Nucifora, F.C., Jr.; Blondel, O.; Sheppard, C.A.; Zhang, C.; Snyder, S.H.; Russell, J.T.; Ryugo, D.K.; Ross, C.A. Differential cellular expression of isoforms of inositol 1,4,5-triphosphate receptors in neurons and glia in brain. J. Comp. Neurol. 1999, 406, 207–220. [Google Scholar] [CrossRef]
- Simonyi, A.; Xia, J.; Igbavboa, U.; Wood, W.G.; Sun, G.Y. Age differences in the expression of metabotropic glutamate receptor 1 and inositol 1,4,5-trisphosphate receptor in mouse cerebellum. Neurosci. Lett. 1998, 244, 29–32. [Google Scholar] [CrossRef]
- Ivanova, H.; Vervliet, T.; Missiaen, L.; Parys, J.B.; De Smedt, H.; Bultynck, G. Inositol 1,4,5-trisphosphate receptor-isoform diversity in cell death and survival. Biochim. Biophys. Acta 2014, 1843, 2164–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, M.; Michikawa, T.; Bosanac, I.; Ikura, M.; Mikoshiba, K. Molecular basis of the isoform-specific ligand-binding affinity of inositol 1,4,5-trisphosphate receptors. J. Biol. Chem. 2007, 282, 12755–12764. [Google Scholar] [CrossRef] [Green Version]
- Parys, J.B.; De Smedt, H. Inositol 1,4,5-trisphosphate and its receptors. Adv. Exp. Med. Biol. 2012, 740, 255–279. [Google Scholar]
- Marchi, S.; Patergnani, S.; Missiroli, S.; Morciano, G.; Rimessi, A.; Wieckowski, M.R.; Giorgi, C.; Pinton, P. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 2018, 69, 62–72. [Google Scholar] [CrossRef]
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef] [Green Version]
- Radzik, T.; Boczek, T.; Ferenc, B.; Studzian, M.; Pulaski, L.; Zylinska, L. Calcium Dyshomeostasis Alters CCL5 Signaling in Differentiated PC12 Cells. Biomed. Res. Int. 2019, 2019, 9616248. [Google Scholar] [CrossRef]
- Penniston, J.T.; Padanyi, R.; Paszty, K.; Varga, K.; Hegedus, L.; Enyedi, A. Apart from its known function, the plasma membrane Ca2+ ATPase can regulate Ca2+ signaling by controlling phosphatidylinositol 4,5-bisphosphate levels. J. Cell. Sci. 2014, 127, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Sama, D.M.; Norris, C.M. Calcium dysregulation and neuroinflammation: Discrete and integrated mechanisms for age-related synaptic dysfunction. Ageing Res. Rev. 2013, 12, 982–995. [Google Scholar] [CrossRef] [Green Version]
- Marques, R.E.; Guabiraba, R.; Russo, R.C.; Teixeira, M.M. Targeting CCL5 in inflammation. Expert Opin. Ther. Targets 2013, 17, 1439–1460. [Google Scholar] [CrossRef] [PubMed]
- Pranzatelli, M.R. Advances in Biomarker-Guided Therapy for Pediatric- and Adult-Onset Neuroinflammatory Disorders: Targeting Chemokines/Cytokines. Front. Immunol. 2018, 9, 557. [Google Scholar] [CrossRef] [PubMed]
- Gamo, K.; Kiryu-Seo, S.; Konishi, H.; Aoki, S.; Matsushima, K.; Wada, K.; Kiyama, H. G-protein-coupled receptor screen reveals a role for chemokine receptor CCR5 in suppressing microglial neurotoxicity. J. Neurosci. 2008, 28, 11980–11988. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, Y.; Ransohoff, R.M. Inflammatory cell trafficking across the blood-brain barrier: Chemokine regulation and in vitro models. Immunol. Rev. 2012, 248, 228–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balistreri, C.R.; Caruso, C.; Grimaldi, M.P.; Listi, F.; Vasto, S.; Orlando, V.; Campagna, A.M.; Lio, D.; Candore, G. CCR5 receptor: Biologic and genetic implications in age-related diseases. Ann. N. Y. Acad. Sci. 2007, 1100, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Mo, R.; Lescure, P.A.; Misek, D.E.; Hanash, S.; Rochford, R.; Hobbs, M.; Yung, R.L. Aging is associated with increased T-cell chemokine expression in C57BL/6 mice. J. Gerontol. A Biol. Sci. Med. Sci. 2003, 58, 975–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumova, E.; Ivanova, M.; Pawelec, G. Immunogenetics of ageing. Int. J. Immunogenet. 2011, 38, 373–381. [Google Scholar] [CrossRef]
- Yung, R.L.; Mo, R. Aging is associated with increased human T cell CC chemokine receptor gene expression. J. Interferon Cytokine Res. 2003, 23, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Tripathy, D.; Thirumangalakudi, L.; Grammas, P. Expression of macrophage inflammatory protein 1-α is elevated in Alzheimer’s vessels and is regulated by oxidative stress. J. Alzheimers Dis. 2007, 11, 447–455. [Google Scholar] [CrossRef]
- Zhu, M.; Allard, J.S.; Zhang, Y.; Perez, E.; Spangler, E.L.; Becker, K.G.; Rapp, P.R. Age-related brain expression and regulation of the chemokine CCL4/MIP-1beta in APP/PS1 double-transgenic mice. J. Neuropathol. Exp. Neurol. 2014, 73, 362–374. [Google Scholar] [CrossRef] [Green Version]
- Pomatto, L.C.D.; Davies, K.J.A. The role of declining adaptive homeostasis in ageing. J. Physiol. 2017, 595, 7275–7309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pomatto, L.C.D.; Sun, P.Y.; Davies, K.J.A. To adapt or not to adapt: Consequences of declining Adaptive Homeostasis and Proteostasis with age. Mech. Ageing Dev. 2019, 177, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Janikiewicz, J.; Szymanski, J.; Malinska, D.; Patalas-Krawczyk, P.; Michalska, B.; Duszynski, J.; Giorgi, C.; Bonora, M.; Dobrzyn, A.; Wieckowski, M.R. Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell. Death Dis. 2018, 9, 332. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Differences in mitochondrial function in PMCA-deficient PC12 cells. Higher resting [Ca2+]i that reflected a partial loss of calcium clearing potency might sensitize mitochondria to buffer cytosolic calcium transients. Because PMCA operates as a 1Ca2+/1H+ antiporter, the extrusion of Ca2+ introduces large quantities of H+ that are readily transported to mitochondria. Due to alkalization of mitochondrial matrix in steady-state conditions and because pHcyto was higher, but still lower than pHmito, in the cells increased pH gradient which could drive a higher rate of ATP synthesis, more significantly in PC12_2 cells. The loss of fast acting PMCA2 could stimulate other ATP-dependent Ca2+ extruding proteins to restore [Ca2+]c, thereby increasing cellular demand for ATP. Also, in this line, OXPHOS rather than anaerobic glycolysis was a main energy supplying pathway. Interestingly, knockdown of PMCA3 isoform did not change the rate of OXPHOS. However, enhanced lactate release and greater sensitivity of the ATP level to glucose withdrawal in the presence of pyruvate suggests the reliance on anaerobic glycolysis, so the local synthesis of ATP may be sufficient to provide an adequate amount of energy. Other details are in the text. PMCA - plasma membrane Ca2+-ATPase.
Figure 1.
Differences in mitochondrial function in PMCA-deficient PC12 cells. Higher resting [Ca2+]i that reflected a partial loss of calcium clearing potency might sensitize mitochondria to buffer cytosolic calcium transients. Because PMCA operates as a 1Ca2+/1H+ antiporter, the extrusion of Ca2+ introduces large quantities of H+ that are readily transported to mitochondria. Due to alkalization of mitochondrial matrix in steady-state conditions and because pHcyto was higher, but still lower than pHmito, in the cells increased pH gradient which could drive a higher rate of ATP synthesis, more significantly in PC12_2 cells. The loss of fast acting PMCA2 could stimulate other ATP-dependent Ca2+ extruding proteins to restore [Ca2+]c, thereby increasing cellular demand for ATP. Also, in this line, OXPHOS rather than anaerobic glycolysis was a main energy supplying pathway. Interestingly, knockdown of PMCA3 isoform did not change the rate of OXPHOS. However, enhanced lactate release and greater sensitivity of the ATP level to glucose withdrawal in the presence of pyruvate suggests the reliance on anaerobic glycolysis, so the local synthesis of ATP may be sufficient to provide an adequate amount of energy. Other details are in the text. PMCA - plasma membrane Ca2+-ATPase.

Figure 2.
Functional changes in PMCA3-deficient PC12 cells. PMCA3 silencing increased [Ca2+]i, although PMCA1 and PMCA4 were up-regulated as compensatory effect, but it did not change the total amount of PMCA protein. Increased expression of L- and P/Q types of voltage-gated Ca2+ channels (VGCCs) could also raise [Ca2+]i. The observed up-regulation of CaN affected transcriptional activity of NFATc2, which repressed CaM genes and finally decreased available CaM. Since CaN was also shown to interact directly with PMCA2 and PMCA4 isoforms, the noticed lower PMCA4/CaN complex formation (not shown) could further increase a pool of potentially active CaN. The amount of CCR5 increased in PC12_3 cells, which is linked to IP3-mediated Ca2+ release from endoplasmic reticulum (ER). The altered ratio of IP3Rs expression with the enlarged total quantity, could further increase [Ca2+]i. However, PMCA4 was shown to bind plasma membrane PIP2, potentially decreasing IP3 production and, subsequently, Ca2+ release from the ER. All these changes, together with higher presence of PMCA4 and highly active PMCA2, may overcome potentially harmful consequences of calcium excitotoxicity. Green up-arrows reflect the increase and red down-arrows reflect the decrease of the amount or activity. Other details are in the text.
Figure 2.
Functional changes in PMCA3-deficient PC12 cells. PMCA3 silencing increased [Ca2+]i, although PMCA1 and PMCA4 were up-regulated as compensatory effect, but it did not change the total amount of PMCA protein. Increased expression of L- and P/Q types of voltage-gated Ca2+ channels (VGCCs) could also raise [Ca2+]i. The observed up-regulation of CaN affected transcriptional activity of NFATc2, which repressed CaM genes and finally decreased available CaM. Since CaN was also shown to interact directly with PMCA2 and PMCA4 isoforms, the noticed lower PMCA4/CaN complex formation (not shown) could further increase a pool of potentially active CaN. The amount of CCR5 increased in PC12_3 cells, which is linked to IP3-mediated Ca2+ release from endoplasmic reticulum (ER). The altered ratio of IP3Rs expression with the enlarged total quantity, could further increase [Ca2+]i. However, PMCA4 was shown to bind plasma membrane PIP2, potentially decreasing IP3 production and, subsequently, Ca2+ release from the ER. All these changes, together with higher presence of PMCA4 and highly active PMCA2, may overcome potentially harmful consequences of calcium excitotoxicity. Green up-arrows reflect the increase and red down-arrows reflect the decrease of the amount or activity. Other details are in the text.

Figure 3.
Functional changes in PMCA2-deficient PC12 cells. Down-regulation of PMCA2 increased [Ca2+]i due to lowered calcium extrusion potency, despite compensatory up-regulation of PMCA1 and unchanged total PMCA protein level. Elevated expression of VGCC types, particularly T-type that exhibits slow deactivation rate, might contribute to the Ca2+ influx. Up-regulated CaN affected transcriptional activity of NFATc2, which repressed CaM genes and lowered CaM amount. Limited CaM availability could weaken its stimulatory effect on PMCA. The amount of CCR5 increased in PC12_2, also with subsequent IP3-mediated Ca2+ release from ER. Altered ratio of IP3Rs expression with higher in the presence of IP3R-3, the most sensitive for modulation by Ca2+, but with the lowest IP3 affinity, could further increase [Ca2+]i. PMCA2 depletion induced some adaptive changes to counteract Ca2+-dependent destructive processes, but because of the enlarged percentage of apoptotic cells, these protective actions appeared to be much less efficient. Green up-arrows reflect the increase and red down-arrows reflect the decrease of the amount or activity. Other details are in the text.
Figure 3.
Functional changes in PMCA2-deficient PC12 cells. Down-regulation of PMCA2 increased [Ca2+]i due to lowered calcium extrusion potency, despite compensatory up-regulation of PMCA1 and unchanged total PMCA protein level. Elevated expression of VGCC types, particularly T-type that exhibits slow deactivation rate, might contribute to the Ca2+ influx. Up-regulated CaN affected transcriptional activity of NFATc2, which repressed CaM genes and lowered CaM amount. Limited CaM availability could weaken its stimulatory effect on PMCA. The amount of CCR5 increased in PC12_2, also with subsequent IP3-mediated Ca2+ release from ER. Altered ratio of IP3Rs expression with higher in the presence of IP3R-3, the most sensitive for modulation by Ca2+, but with the lowest IP3 affinity, could further increase [Ca2+]i. PMCA2 depletion induced some adaptive changes to counteract Ca2+-dependent destructive processes, but because of the enlarged percentage of apoptotic cells, these protective actions appeared to be much less efficient. Green up-arrows reflect the increase and red down-arrows reflect the decrease of the amount or activity. Other details are in the text.


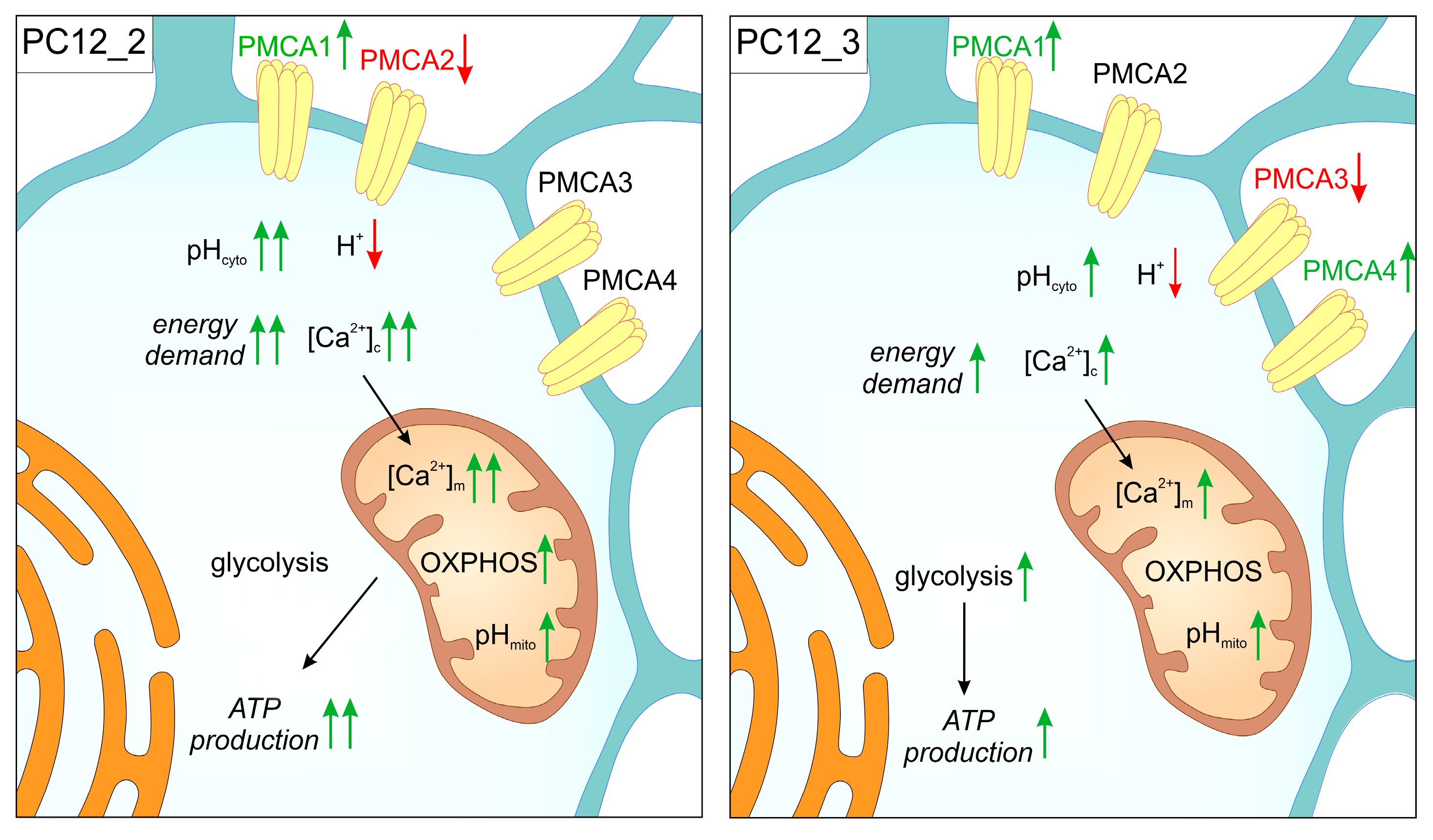{kind=link}
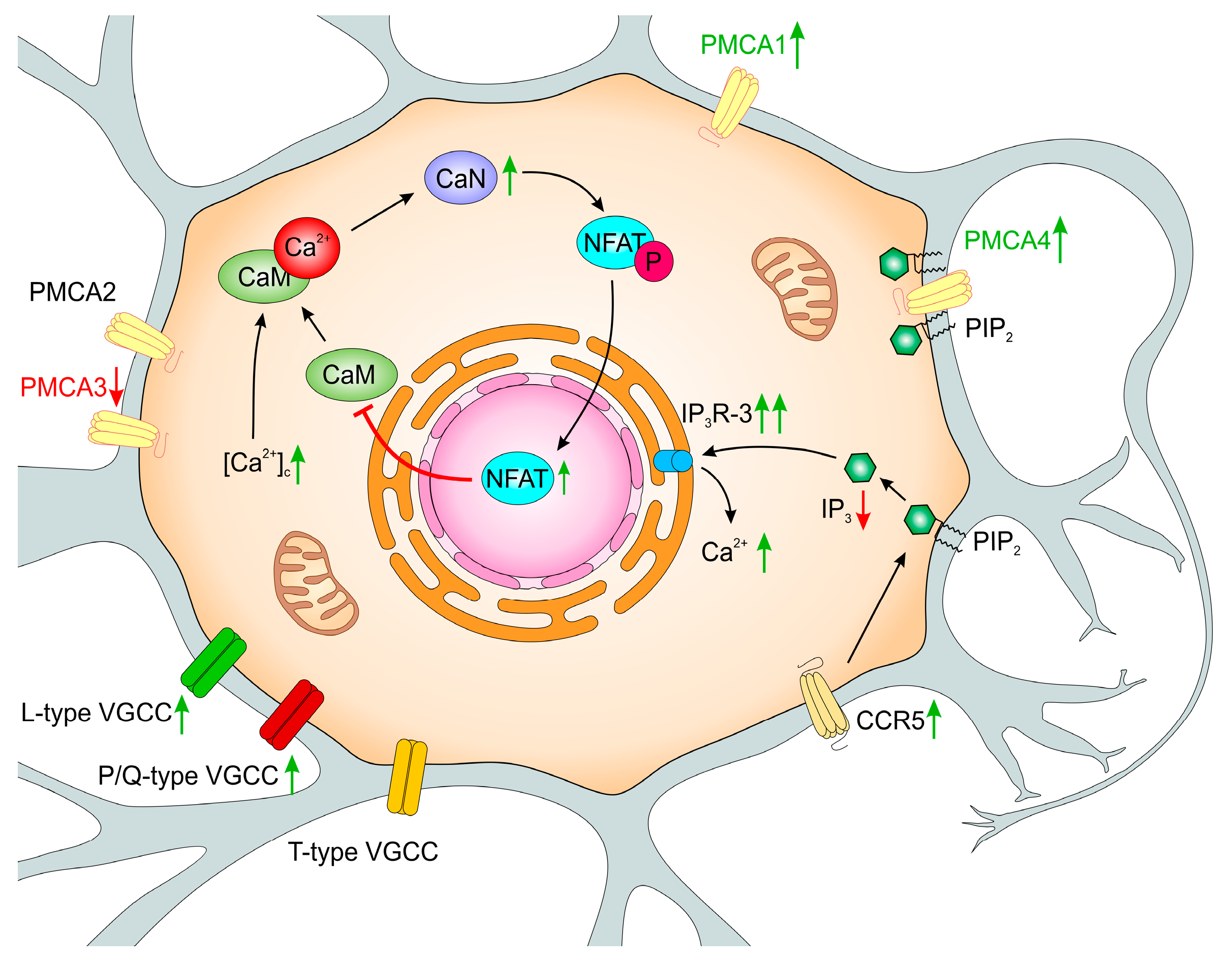{kind=link}
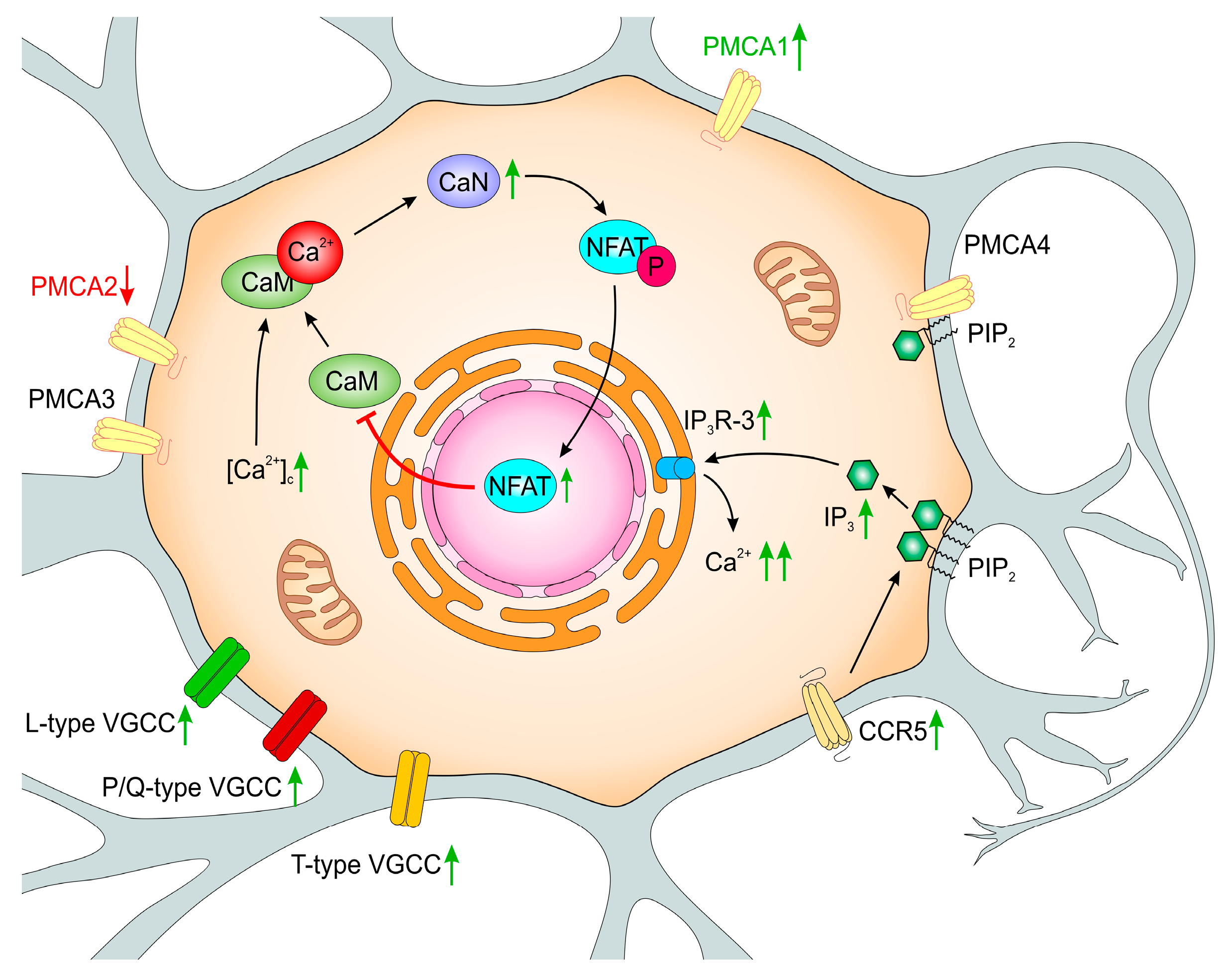{kind=link}
Table 1.
Summary of differences between PMCA2 and PMCA3 down regulation in PC12 cells.
| Protein or mRNA | PC12_2 | PC12_3 |
|---|---|---|
| PMCA1 | increased | increased |
| PMCA2 | decreased | unchanged |
| PMCA3 | unchanged | decreased |
| PMCA4 | unchanged | increased |
| Total PMCA | unchanged | unchanged |
| SERCA2 | increased | increased |
| SERCA3 | increased | increased |
| CaM | decreased | decreased |
| CaN | increased | increased |
| L-type VGCC | increased | increased |
| P/Q-type VGCC | increased | increased |
| T-type VGCC | increased | unchanged |
| CCR1 | increased | unchanged |
| CCR3 | unchanged | increased |
| CCR5 | increased | increased |
| IP3R1 | decreased | decreased |
| IP3R2 | decreased | decreased |
| IP3R3 | increased | increased |
| Total IP3R | unchanged | increased |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Boczek, T.; Radzik, T.; Ferenc, B.; Zylinska, L. The Puzzling Role of Neuron-Specific PMCA Isoforms in the Aging Process. Int. J. Mol. Sci. 2019, 20, 6338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246338
AMA Style
Boczek T, Radzik T, Ferenc B, Zylinska L. The Puzzling Role of Neuron-Specific PMCA Isoforms in the Aging Process. International Journal of Molecular Sciences. 2019; 20(24):6338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246338
Chicago/Turabian StyleBoczek, Tomasz, Tomasz Radzik, Bozena Ferenc, and Ludmila Zylinska. 2019. "The Puzzling Role of Neuron-Specific PMCA Isoforms in the Aging Process" International Journal of Molecular Sciences 20, no. 24: 6338. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246338
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.