Dual Roles of Astrocyte-Derived Factors in Regulation of Blood-Brain Barrier Function after Brain Damage

1
Laboratory of Pharmacology, Faculty of Pharmacy, Osaka Ohtani University, 3-11-1 Nishikiori-Kita, Tondabayashi, Osaka 584-8540, Japan
2
Laboratory of Pharmacology, Kobe Pharmaceutical University, 4-19-1 Motoyama-Kita Higashinada, Kobe 668-8558, Japan
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(3), 571; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030571
Submission received: 26 December 2018
/
Revised: 23 January 2019
/
Accepted: 27 January 2019
/
Published: 29 January 2019
(This article belongs to the Special Issue Astrocytes: Emerging Roles in the Pathogenesis and Treatment of CNS disorders)
Abstract
:The blood-brain barrier (BBB) is a major functional barrier in the central nervous system (CNS), and inhibits the extravasation of intravascular contents and transports various essential nutrients between the blood and the brain. After brain damage by traumatic brain injury, cerebral ischemia and several other CNS disorders, the functions of the BBB are disrupted, resulting in severe secondary damage including brain edema and inflammatory injury. Therefore, BBB protection and recovery are considered novel therapeutic strategies for reducing brain damage. Emerging evidence suggests key roles of astrocyte-derived factors in BBB disruption and recovery after brain damage. The astrocyte-derived vascular permeability factors include vascular endothelial growth factors, matrix metalloproteinases, nitric oxide, glutamate and endothelin-1, which enhance BBB permeability leading to BBB disruption. By contrast, the astrocyte-derived protective factors include angiopoietin-1, sonic hedgehog, glial-derived neurotrophic factor, retinoic acid and insulin-like growth factor-1 and apolipoprotein E which attenuate BBB permeability resulting in recovery of BBB function. In this review, the roles of these astrocyte-derived factors in BBB function are summarized, and their significance as therapeutic targets for BBB protection and recovery after brain damage are discussed.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
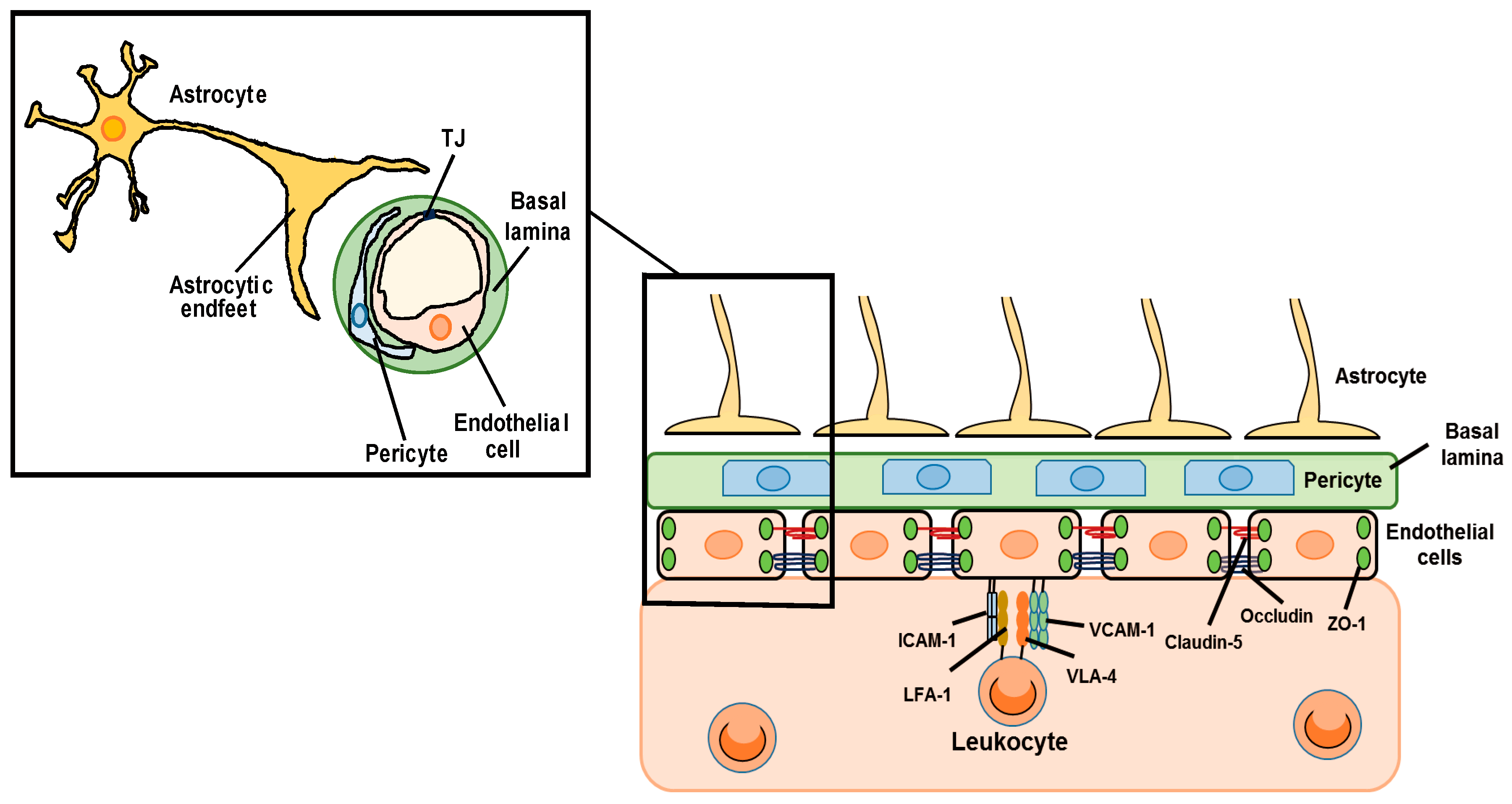
The blood–brain barrier (BBB) is a biological and functional barrier in the central nervous system (CNS), and comprises various types of cells including endothelial cells, pericytes and astrocytes (Figure 1). The BBB limits the influx of intravascular contents including serum proteins, blood cells and toxic substances into the cerebral parenchyma, and pumps out cerebral waste materials [1]. The BBB also expresses a range of transporters essential for movement of amino acids and glucose into the cerebral parenchyma to support the function and survival of brain cells. These static barrier functions and transportation systems of the BBB are regulated by endothelial cells, pericytes and astrocytes. Under physiological conditions, BBB permeability is strictly regulated by cell–cell interactions and cell-derived bioactive factors [2]. The static barrier function depends on endothelial tight junctions (TJs) and the basal lamina (Figure 1). The TJ is formed by TJ-related proteins including claudin (CLN), occludin (OCLN) and zonula occluden (ZO) [3]. The basal lamina is a layer of extracellular matrix known as the basement membrane, which consists of collagen, laminin and fibronectin. Astrocytes exist around cerebral microvessels and control BBB functions via astrocyte-derived factors and astrocytic terminal processes termed endfeet. Astrocytic endfeet express the potassium channel, Kir4.1, and aquaporin-4, which support the BBB function by controlling the ion and water balance [4]. BBB is also responsible for the regulation of leukocyte infiltration into the CNS (Figure 1). During the process of leukocyte infiltration, cell adhesion molecules (CAMs) on endothelial cells, including vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), have roles in leukocyte adhesion. Endothelial ICAM-1 and VCAM-1 interact with very late antigen-4 (VLA-4) and lymphocyte function-associated antigen 1 (LFA-1) in leukocytes, causing firm adhesion of endothelial cells and leukocytes. The expression of ICAM-1 and VCAM-1 in brain vascular endothelial cells is regulated by chemokines and inflammatory cytokines produced by astrocytes [5,6]. In this way, astrocytes can affect leukocyte infiltration into the CNS.
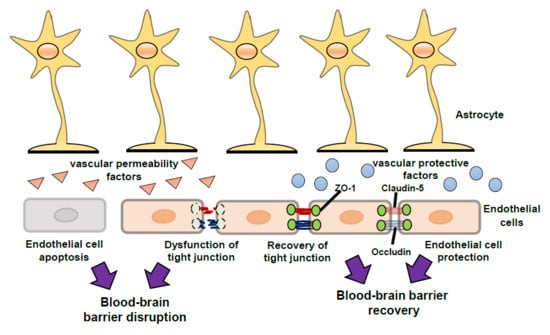
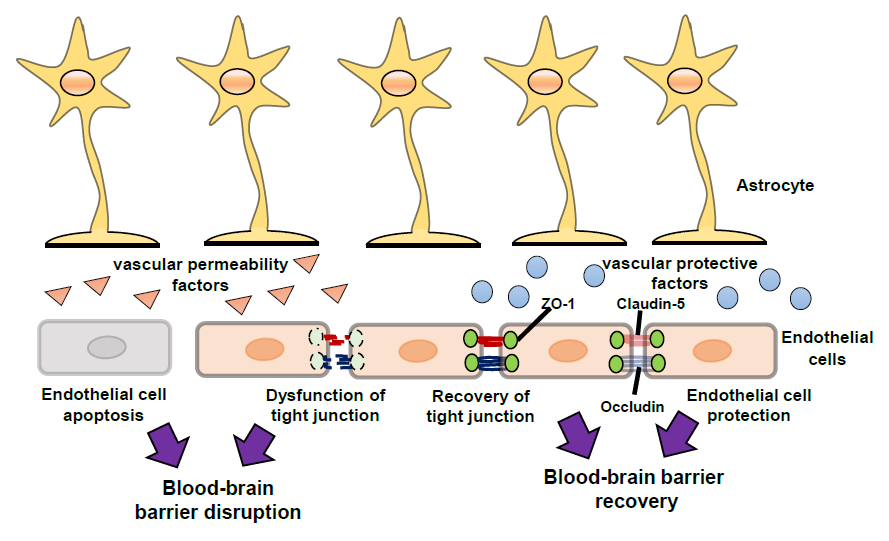
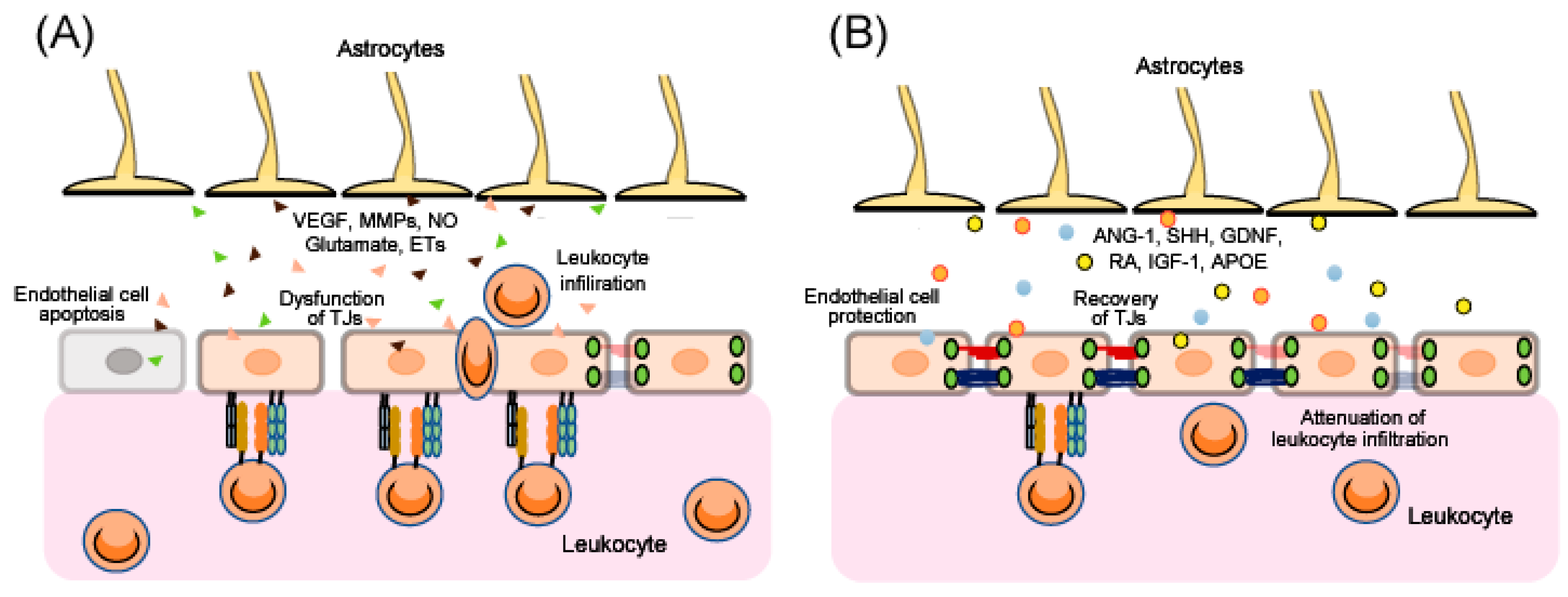
After traumatic brain injury (TBI), ischemia and various other CNS disorders, the functions of the BBB can be disrupted [7,8,9,10,11], and the resulting excessive BBB permeability causes secondary damage including brain edema and inflammatory injury. Therefore, BBB protection and recovery are essential for reducing the progression of brain damage. Apoptosis of endothelial cells and/or dysfunction of endothelial TJs results in disruption of BBB function (Figure 2). Upregulation of CAMs on endothelial cells accelerates leukocytes crossing the BBB (Figure 2). Further, after injury, astrocytes are converted from a resting form to a reactive form, and several astrocyte-derived factors induce endothelial cell apoptosis and decrease expression of endothelial TJ-related proteins, leading to aggravation of BBB disruption (Figure 2). By contrast, some astrocyte-derived factors can protect endothelial cells and enhance TJ reassembly, leading to BBB recovery (Figure 2). In addition, several astrocyte-derived factors also regulate CAMs on endothelial cells and control leukocyte crossing the BBB (Figure 2).
Therefore, the appropriate control of astrocyte-derived factors to reduce BBB damage and promote BBB recovery is becoming of increasing interest as a therapeutic strategy after brain damage. In this review, we describe several key astrocyte-derived factors involved in BBB function, and discuss the significance of these factors as novel therapeutic targets for BBB recovery after brain damage.
2. The Pathogenesis of BBB Disruption
BBB disruption causes extravasation of intravascular fluid and excessive infiltration of leukocytes including neutrophils, monocytes and lymphocytes into the cerebral parenchyma, resulting in brain edema and inflammatory injury, respectively. BBB disruption has been confirmed in patients with TBI and ischemic stroke [7,8], and is associated with the progression of various CNS disorders including Alzheimer’s disease, multiple sclerosis and Parkinson’s disease [9,10,11]. BBB disruption has also been reproduced in various models of brain disorders [12,13,14,15].
The mechanisms underlying BBB disruption include direct injury to vascular endothelial cells in the core area and excessive BBB permeability in the peri-core area (Figure 2). The direct injury induces an irreversible BBB disruption due to the death of BBB cells. For example, endothelial cell apoptosis has been reported in ischemic animal models and following oxygen-glucose deprivation in vitro, resulting in a pathological increase in BBB permeability [16]. Brain endothelial cell apoptosis has also been reported in TBI model animals, including activation of the c-Jun N-terminal kinase, p38 mitogen-activated protein kinase and caspase-3 pathways [17].
In the peri-core area of brain injury, excessive BBB permeability can also result from increases in paracellular transport caused by dysfunction of endothelial TJs (Figure 2). For example, decreases in CLN-5, OCLN and ZO-1 were observed in ischemic stroke and TBI animal models [18,19,20]. Argaw et al. [21] and Wang et al. [22] have reported decreases in TJ-related proteins in animal models of CNS inflammation such as multiple sclerosis. Furthermore, phosphorylation of TJ-related proteins caused their detachment, leading to TJ dysfunction [3,23]. These observations suggest that protection of endothelial cells and promotion of recovery of endothelial TJ-related protein function are therapeutic targets for BBB disruption, which may reduce the pathogenesis of various CNS disorders and brain injuries.
The leukocytes that cross the BBB also accumulate in the damaged brain. The expression of VCAM-1 and ICAM-1 on endothelial cells was increased in experimental animals after brain damage [24,25,26], and the increased endothelial CAMs potentiated binding to adhesion molecules in leukocytes, such as VLA-4 and LFA-1. The interaction of these adhesion molecules is a key process for leukocytes crossing the BBB. The infiltration of neutrophils, monocytes and lymphocytes was observed around the injured core upon experimental brain damage [26,27,28,29]. Accumulation of leukocytes has also been shown in patients with TBI [30]. Moreover, astrocyte-derived chemokines, including monocyte chemotactic protein-1, and macrophage inflammatory proteins accelerate infiltration of leukocytes [31,32].
3. Regulation of BBB Function by Astrocyte-Derived Factors
Several studies suggest dual roles for astrocytes in the control of BBB function. Eilam et al. [33] revealed that loss of astroglial connections with blood vessels caused BBB disruption in an animal model of multiple sclerosis. By contrast, Begum et al. [13] showed that selective knock-out of the astrocytic Na+/H+ exchanger isoform 1 reduced astrogliosis after ischemic stroke in mice, with a resulting decrease in cerebral vessel damage and improved BBB function. Chiu et al. [14] also reported that ethyl-1-(4-(2,3,3-trichloroacrylamide)phenyl)-5-(trifluoromethyl)-1H-pyrazole-4-carboxylate decreased the pathological activation of astrocytes and reduced BBB destruction in intracerebral hemorrhage model rats. Overall, these studies imply that appropriate regulation of astrocyte function is required to attenuate BBB disruption and promote BBB function after brain injury. Astrocyte-derived factors are known to be responsible for both BBB disruption and repair (Figure 2). Below, we describe a range of astrocyte-derived factors and their roles in BBB disruption.
3.1. The Vascular Permeability Factors
3.1.1. Vascular Endothelial Growth Factor
Vascular endothelial growth factors (VEGFs) including VEGF-A, -B, -C, -D, -E and-F are known as an angiogenetic factor and exert angiogenetic functions via VEGF receptor-1 (VEGFR-1) and -2 (VEGFR-2), which are tyrosine kinase receptors expressed in endothelial cells. The activation of endothelial VEGF/VEGFR signal leads to endothelial proliferation and differentiation for angiogenesis [34]. On the other hands, VEGFs are also well-established to promote BBB permeability. For example, exogenous treatment with VEGF in animals and in cultured brain microvessel endothelial cells caused increased BBB permeability [35,36], while treatment with an anti-VEGF neutralizing antibody reduced BBB leakage (Evans blue staining) in cerebral ischemia/reperfusion [37] and focal TBI by fluid percussion injury (FPI) [12] animal models. Inhibition of VEGF signaling by SU5416, a VEGF receptor-2 inhibitor, and specific VEGF receptor-2 knockdown, also reduced BBB disruption after permanent ischemic damage by thrombosis [38]. Furthermore, VEGF was reported to protect against endothelial cell apoptosis under hypoglycemic conditions [39]. On the other hand, VEGF downregulated the expression of TJ-related proteins on brain endothelial cells [35,40]. Therefore, VEGF enhances BBB permeability by decreasing TJ-related proteins. In animal models of multiple sclerosis, normal expressions for VCAM-1 and ICAM-1 were displayed in the inactivation of astrocyte-specific VEGF-A mice, and the inactivation of astrocyte-specific VEGF-A reduced lymphocyte infiltration [40]. In human umbilical vascular endothelial cells (HUVECs), VEGF also induced ICAM-1 and VCAM-1 expressions, and induced leukocyte adhesion to HUVECs [41].
Following brain injury and in various CNS disorders, induction of VEGF was observed in reactive astrocytes although it is also produced in various types of cells in CNS. Several studies indicate the involvements of astrocytic VEGF for BBB disruption. Argaw et al. [40] reported that astrocytes expressed VEGF-A, while inactivation of a144strocyte-specific VEGF-A reduced BBB disruption in animal models of multiple sclerosis. Chapouly et al. [15] also reported VEGF-A expression on reactive astrocytes in human multiple sclerosis and experimental animal models, while blockade of VEGF-A by cavtratin, a selective inhibitor of VEGF-A signaling, protected against BBB disruption. Finally, we previously reported an increase in VEGF-A expression in astrocytes after brain damages in mice, and that blockade of VEGF-A using antibodies alleviated the BBB disruption [12]. In patients with brain damages including TBI and ischemic stroke, the increase of VEGF level was observed and suggested the relationships with degree of severity [42,43,44].
3.1.2. Matrix Metalloproteinases
MMPs are zinc-endopeptidases which degrade endothelial TJ-related proteins and extracellular matrix (ECM) molecules including collagen, laminin and fibronectin. The degradation of ECM and TJ-related proteins are essential processes for angiogenesis while accelerating BBB permeability. In patients with TBI, elevation of MMPs in cerebrospinal fluid and blood was indicated [43,45,46]. Chen et al. [47] found that overexpression of MMP-9 caused degradation of CLN-5 and OCLN, resulting in endothelial barrier disruption, while in experimental animals of cerebral ischemia/perfusion, the MMP-induced reduction of TJ-related proteins resulted in BBB disruption [48,49]. Guo et al. [50] also reported that MMP-9 activity was responsible for endothelial cell apoptosis following subarachnoid hemorrhage in rats. Moreover, the excessive activation of MMP-2 and MMP-9 led to cellular damage in cerebral endothelium after hypoxia-reoxygenation [51]. The beneficial effects of MMP inhibition on BBB disruption were also examined in experimental animal models. For example, blocking MMP activation or MMP-9 knock-out (KO) prevented degradation of CLN-5 and OCLN, and attenuated BBB disruption, in cerebral ischemia/reperfusion animal models [52,53]. In focal TBI animals by FPI, MMP-9 inhibition also reduced BBB disruption [12]. Moreover, blockade of MMP-9 activity by Ro32–3555, a broad spectrum MMP inhibitor reduced transmigration of neutrophils and monocytes in an in vitro model of CNS tuberculosis [54]. MMP inhibitors also regulated inflammatory cell migration by reducing ICAM-1 and VCAM-1 expression in lung tissues in asthma model animals [55]. Therefore, regulation of ICAM-1 and VCAM-1 expressions by MMP may be also involved in infiltration of leukocytes in CNS.
MMPs are produced in various types of cells in CNS. In experimental animal models of brain injury, the expression of MMPs was also observed in astrocytes. Jiang et al. [56] found that reactive astrocytes released MMP-2 and MMP-9, while in amyloid precursor protein/presenilin 1 transgenic mice, MMP-2 and MMP-9 immunoreactivities were selectively increased in activated astrocytes [57]. Astrocytic MMP-9 activation also compromised the BBB and exacerbated intracerebral hemorrhage in animal models [58]. Finally, we confirmed the induction of MMP-9 in astrocytes in TBI mice by FPI, and found that inhibition of MMP-9 attenuated the TBI-induced BBB disruption [12].
3.1.3. Nitric Oxide
Nitric oxide (NO) is a potent vasodilator and plays a role in neurovascular coupling by regulation of blood flow for neuronal activity [59]. NO is synthetized from L-arginine by NO synthase (NOS). There are three NOS isoforms, including neuronal NOS (NOS-1), inducible NOS (NOS-2) and endothelial NOS (NOS-3). NOS-1 and NOS-3 are constitutive and regulate endothelial cell functions under normal conditions, while NOS-2 is increased following injury to promote the inflammatory reaction. Various studies have also shown that astrocytes can produce NOS-2 in the CNS [60,61,62].
NO is known to induce BBB disruption. For example, blockade of NO production by Nomega-Nitro-l-arginine methyl ester, a non-specific NOS inhibitor, abolished BBB disruption following focal cerebral ischemia/perfusion in animal models [63,64]. However, the effects of NO on endothelial cell apoptosis are complicated. Shen et al. [65] showed that the anti-apoptotic effect of NO on endothelial cells was exerted through the cyclic guanosine monophosphate (cGMP) pathway, while NO induced apoptosis through cGMP-independent pathways. The effects of NO on TJ-related proteins are clearer, with a confirmed reduction in TJ-related proteins following NO production [66].
3.1.4. Glutamate
Glutamate is a major excitatory transmitter and play a key role in synaptic plasticity for learning and memory, which exerts its excitatory effects via glutamatergic receptors, including the N-methyl-D-aspartate (NMDA) receptor and the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor. Glutamate is not only released from neurons but also astrocytes, and astrocyte-derived glutamate acts as a gliotransmitter to nearby neurons to regulate synaptic plasticity and formation. NMDA receptors are also distributed in endothelial cells as well as neurons [67,68], and astrocyte-derived glutamate can induce vasodilatation that is dependent on NOS-3 and activation of endothelial NMDA receptors [69].
Although glutamate is essential for normal function of neurons and endothelial cells, excessive glutamate causes deleterious effects including neuronal death and BBB disruption. For example, perfusion of glutamate induced excessive vascular permeability via activation of NMDA receptors [70], while following permanent focal cerebral ischemia in rats, blockade of NMDA or AMPA receptors attenuated BBB disruption [71]. With respect to the effects of glutamate on endothelial TJ-related proteins, András et al. [68] suggested that treatment of glutamate decreased OCLN protein levels in brain endothelial cells. As excessive glutamate is released from astrocytes following brain injury, astrocyte-derived glutamate must be involved in BBB disruption via activation of endothelial glutamate receptors.
3.1.5. Endothelins
Endothelins (ETs) including ET-1, -2 and -3 are potent endogenous vasoconstrictors and exert various physiological actions other than vasoconstriction including regulation of endothelial function. There are two types of ET receptors including via endothelin receptor type A (ETA ) and type B (ETB ), and ETs exert bioactive functions via ETA and ETB receptors. In patients with brain damages including TBI and subarachnoid hemorrhage, ET-1 is increased in cerebrospinal fluid and associated with unfavorable outcomes [72,73]. The production of ET-1 is performed in various types of cells in CNS. In various experimental animal models, ET-1 production was also observed in astrocytes [74,75,76], while targeted overexpression of ET-1 in astrocytes led to a higher mortality, more severe neurological deficits and cerebral edema in subarachnoid hemorrhage and transient ischemia model mice [77,78]. Hung et al. [79] also reported that selective astrocytic ET-1 overexpression exacerbated cerebral edema, neurodegeneration, neuroinflammation, oxidative stress and memory deficits in transient cerebral ischemia mice.
The involvement of ET-1 in BBB disruption is supported by experimental models in vivo and in vitro. Repeated administration of ET-1 enhanced disruption of BBB permeability in dogs and rats [80]. Reijerkerk et al. [81] also reported that ET-1 contributed to the brain endothelial barrier passage of monocytes involved in BBB inflammation via ETB receptor signaling in brain endothelial cells. ET-1 also induced upregulation of ICAM-1 and VCAM-1 expression in human brain microvascular endothelial cells [82]. Further, astrocytic overexpression of ET-1 increased the severity of BBB breakdown in subarachnoid hemorrhage mice [78]. The effects of blockade of the ET system for BBB disruption have also been examined. For example, the selective ETA receptor antagonist S-0139 reduced BBB permeability, brain edema formation and infarct size after cerebral ischemia/reperfusion in rats [83], while Kim et al. [84,85] reported that the selective ETB receptor antagonist BQ788 blocked BBB disruption via inhibition of MMP-9 activation and ZO-1 protein degradation in experimental status epilepticus animals.
3.2. The Vascular Protective Factors
3.2.1. Angiopoietin-1
Angiopoietin-1 (ANG-1) is a glycoprotein with angiogenetic properties, which are exerted via Tie-2, a tyrosine kinase receptor expressed principally in endothelial cells. When ANG-1 binds Tie2, the cytoplasmic tyrosine residues of Tie2 is phosphorylated, resulting in activation of various intracellular signaling including Phosphoinositide 3-kinase /AKT, Ras and mitogen-activated protein kinase which are involved in the survival of endothelial cells and vascular remodeling and stability. A protective effect of ANG-1 via Tie-2 signaling in neurons after brain damage was also previously reported [86]. In CNS, endothelial cells produce ANG-1 while ANG-1 expression was also found in astrocytes in the cerebrum of experimental animals and in cultured cells [87,88,89,90,91]. A range of studies have found protective effects of ANG-1 on BBB function. Meng et al. [92] demonstrated that ANG-1 overexpression reduced BBB leakage, while exogenous ANG-1 or ANG-1 mimetic peptides suppressed BBB damage [93,94], in animal models of focal embolic cerebral ischemia. In subarachnoid hemorrhage rats, the administration of exogenous ANG-1 reduced BBB leakage [95]. In addition, blockade of Tie-2 activation exacerbated BBB disruption in TBI mice by controlled cortical impact (CCI) [96]. These observations suggest protective effects of ANG-1/Tie-2 against BBB damage. In patients with brain damages, alterations of ANG-1 level have been indicated. Plasma ANG-1 concentrations were low after ischemic stroke particularly in patients with poor stroke outcomes [97]. Sobrino et al. [98] suggested that high serum levels of ANG-1 were associated with good outcome in patients with intracerebral hemorrhage.
Interestingly, Nag et al. [99] found only minimal expression of caspase-3 after ANG-1 production by the endothelium following cortical cold injury in rats. Further, Zhao et al. [100] suggested that ANG-1 inhibited glycation end product-induced endothelial cell apoptosis. The functional effects of ANG-1 on endothelial TJ-related proteins have also been reported, with reversal of the decrease in TJ-related proteins with ANG-1 treatment following cerebral ischemia/perfusion in rats [101]. Further, Xia et al. [90] suggested that ANG-1 caused upregulation of ZO-1 and OCLN to repair TJs after permanent ischemic damage in rats. ANG-1 also suppressed VEGF-induced expression of ICAM-1 and VCAM-1, and reduced VEGF-induced leukocyte adhesion to HUVECs [41].
3.2.2. Sonic Hedgehog
Sonic hedgehog (SHH) is a glycoprotein that belongs to the hedgehog family, and is essential for normal pattern formation and cellular differentiation in the developing CNS. The SHH signaling pathway is initiated by the binding of SHH to Patched-1 (PTCH1), which blocks the inhibitory action of the PTCH1 receptor to Smoothened, a membrane protein, resulting in activation of transcription factors [102]. In CNS, the production of SHH is observed in astrocytes, immune cells and endothelial cells [103]. In experimental animals and cultured cells, SHH production was predominantly observed in astrocytes [104,105,106,107,108], and astrocyte-derived SHH contributed to angiogenesis [106,107]. The beneficial effects of SHH for reducing BBB disruption have also been confirmed. Administration of recombinant SHH decreased BBB leakage in permanent ischemia model rats [90]. Furthermore, Alvarez et al. [105] showed that astrocyte-secreted SHH promoted BBB formation and integrity through endothelial hedgehog receptors.
Gao et al. [109] reported that downregulation of PTCH1 enhanced endothelial progenitor cell apoptosis induced by high glucose. Zhu et al. [110] also demonstrated that the SHH signaling pathway was protective against endothelial cells apoptosis. Therefore, SHH must exert anti-apoptotic effects via SHH signaling pathways in endothelial cells after brain damage. The effects of SHH on TJ-related proteins have also been reported. SHH or a SHH signaling agonist increased expression of CLN-5, OCLN and ZO-1 in brain endothelial cells, whereas a SHH signaling inhibitor blocked these effects [108]. Brilha et al. [54] also showed that treatment of exogenous SHH reduced the mycobacterium tuberculosis-induced BBB breakdown and reversed the decrease in CLN-5 in a co-culture BBB model consisting of brain microvascular endothelial cells and astrocytes. In permanent ischemia model rats, administration of SHH increased the expression of ZO-1 and OCLN [90]. Further, SHH reduced the levels of ICAM-1 expression in endothelial cells, and suppressed adhesion and transmigration of immune cells [105]. As a relationship of SHH for clinical disease, Drannik et al. [111] implied that SHH pathway may be compromised in ALS patients.
3.2.3. Glial-Derived Neurotrophic Factor
Glial-derived neurotrophic factor (GDNF) is a neurotrophic factor secreted from astrocytes and activates GDNF receptor alpha-1 and -2 expressed in neurons and endothelial cells, resulting in survival of neurons, axon guidance and synapse formation and control of endothelial functions. It was previously reported that GDNF can promote angiogenesis [112], and that GDNF is critical for normal postnatal development of the BBB [113]. Further, Igarashi et al. and Shimizu et al. [114,115] found that GDNF treatment increased CLN-5 expression and strengthened the barrier function in brain endothelial cells. Xiao et al. [116] also confirmed upregulation of OCLN and ZO-1 by GDNF. These results imply that GDNF exerts protective effects against BBB disruption by increasing TJ-related proteins in endothelial cells.
3.2.4. Retinoic Acid
Retinoic acid (RA) is an active metabolite of vitamin A, and is synthesized from retinol by retinaldehyde dehydrogenase (RALDH). RA acts as a ligand for nuclear RA receptors (RARs), which are important for growth and development in the CNS. RA are also associated with learning and memory behaviors by regulation of synaptic plasticity in the mature brain. The production of RA is observed in various types of cells including neurons and glial cells in CNS. RALDH2 is highly expressed in reactive astrocytes, which causes enhanced astrocytic RA synthesis [117].
Recent studies support a role for RA in the development and protection of the BBB. For example, Mizee et al. [118] suggested that RA is crucial for development of the brain endothelial cell barrier via RARβ signaling in the developing brain vasculature. During BBB differentiation, the inhibition of RAR activation caused leakage of serum proteins into the developing brain, and reduced the expression of BBB determinants [118]. The enhanced RA synthesis by increased expression of RALDH2 in reactive astrocytes also protected BBB function during inflammatory stimulation [117]. In addition, injection of RA increased expression of ZO-1 and vascular endothelial cadherin, which are crucial components of the BBB structure [119]. RA also reduced VCAM-1 expressions in cultured dermal microvascular endothelial cells during inflammatory conditions, and decreased VCAM-1-dependent T cell binding to microvascular endothelial cells [120]. Therefore, similar effects of RA may also exert in brain microvascular endothelial cells.
3.2.5. Insulin-Like Growth Factor-1
Insulin-like growth factor-1 (IGF-1) is a member of the insulin gene family, and exerts bioactive functions as a neurotrophic factor via activation of the IGF-1receptor. IGF-1 exerts multiple physiological roles including neurogenesis, prolonged neuronal survival, reduced cell death, resistance to injury, reparation and neuroplasticity in the adult brain [121]. Downregulation of the IGF-1 receptor promoted cellular apoptosis induced by advanced glycation end products in cultured vascular endothelial cells [122]. Therefore, anti-apoptotic effects of IGF-1 against brain endothelial cells are expected.
Astrocytes are one of product cells for IGF-1 although the production of IGF-1 is also observed in neurons, endothelial cells and other glial cells [123,124], and astrocyte-derived IGF-1 plays a key role in neuronal protection after brain damage. Astrocytic overexpression of IGF-1 also protected neurons against TBI by CCI [125], while astrocyte-IGF-1 gene transfer improved outcomes in rats following ischemia/perfusion [126].
Bake et al. [127] reported that IGF-1 reduced BBB permeability and decreased infarct volume in ischemia/perfusion rats. Further, in primary brain microvessel endothelial cells exposed to stroke-like conditions by oxygen-glucose deprivation, IGF-1 reversed the excessive dye transfer across the cell monolayer [128]. These results suggest that astrocyte-derived IGF-1 exerts protective effects against endothelial cell death, thus attenuating BBB disruption.
3.2.6. Apolipoprotein E
Apolipoprotein E (APOE) is a member of the apolipoprotein family which supports lipid transport and injury repair in the brain [129]. In experimental animals and humans, production of APOE is predominantly synthesized in and secreted from astrocytes in CNS [130,131,132].
Multiple studies indicate APOE is protective factor for BBB disruption in experimental animal models. In TBI mice by CCI, APOE-mimetic peptide COG1410 reduced Evans blue extravasation and suppressed the activity of MMP-9 [133]. On the other hand, the increased Evans blue extravasation was found in the brains of APOE KO mice after CCI compared with WT mice [134]. In addition, more activated MMP-9 was detected in APOE KO mice after CCI compared with WT mice while the expressions of OCLN and ZO-1 were decreased in APOE KO mice [134]. In animal models of CNS inflammation, Zheng et al. [135] suggested that APOE-deficient promoted BBB disruption, upregulated MMP-9 expression activity and decreased the expression of endothelial TJ-related proteins.
4. Astrocytic Molecules as Candidates for Therapeutic Strategies to Protect BBB
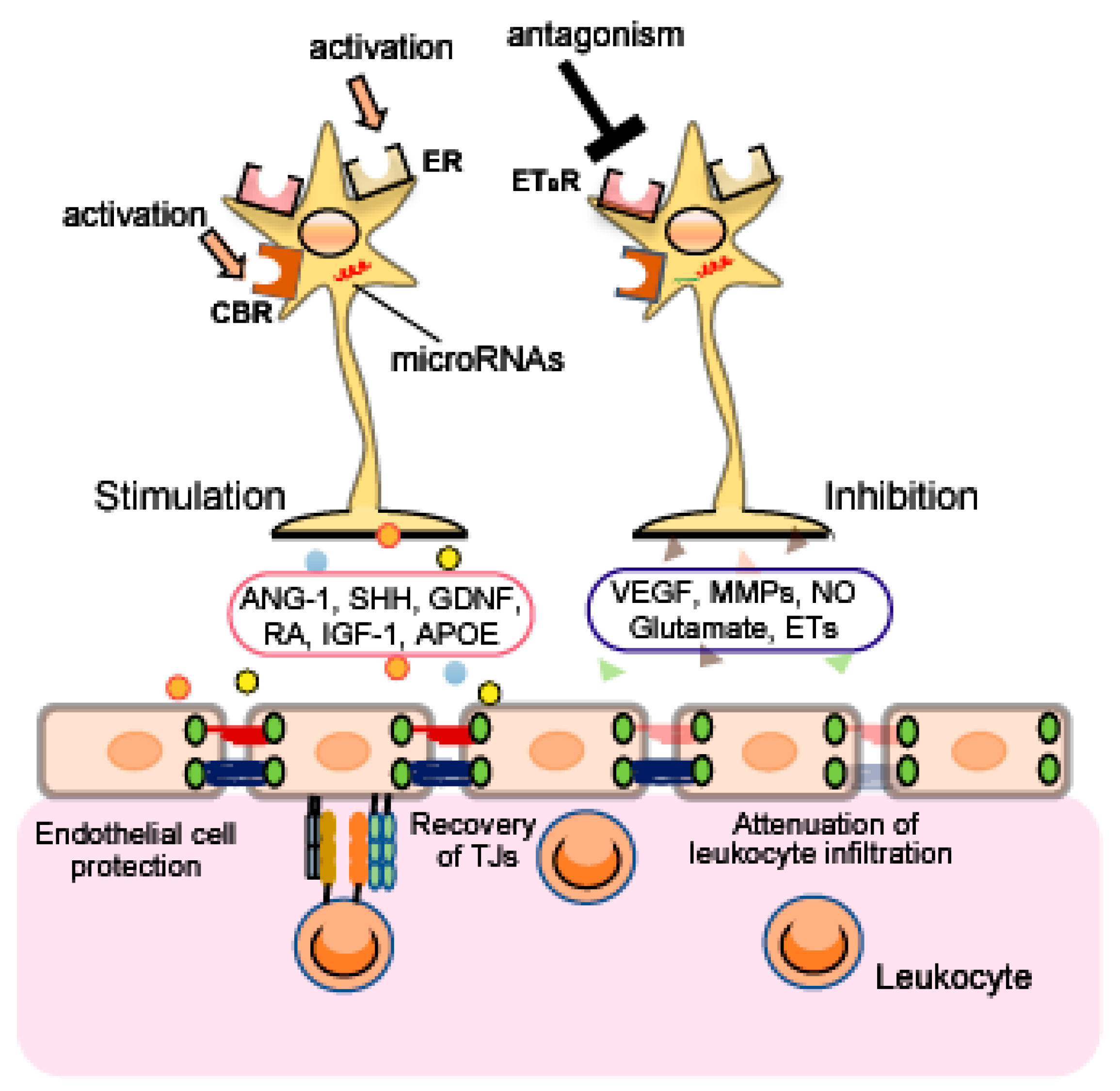
Therapeutic strategies to target astrocytes have been proposed in a range of neurodegenerative disorders [136,137,138], spinal cord injury [139], hyperalgesia [140], mental illnesses [141], TBI [142] and cerebral ischemia [143]. As astrocytes are involved in regulation of the BBB, targeting astrocytic function may protect against brain injury induced by BBB disruption. In this section, we describe several astrocytic molecules targeted for control of astrocyte function (Figure 3).
4.1. Estrogen Receptors
Estrogen and progesterone are known to control astrocyte functions and exert protective effects against brain damage. Arevalo et al. [144] and Acaz-Fonseca et al. [145] reported that the gonadal hormones suppressed astrogliosis and reduce neuroinflammation and brain edema after various types of CNS injury. In animal models of TBI by the Marmarou method and cerebral ischemia/perfusion, estradiol also attenuated BBB disruption [146,147,148,149]. Further, estradiol blocked the upregulation of MMPs after cerebral ischemia [150], and increased ANG-1 expression through ERα in the rat cerebrum [151]. Estradiol also inhibited induction of VCAM-1 and ICAM-1 expressions in cultured human endothelial cells during inflammatory conditions [152].
Numerous studies indicate that astrocytes express estrogen receptors (ERs) and that astrocytic ERs mediate the neuroprotective actions of estradiol [153,154,155,156]. The astrocytic ERs also regulate the production of several astrocyte-derived factors including neurotropic factors and chemokines [153,157,158]. These observations imply that activation of astrocytic ERs may be neuroprotective by alleviating BBB disruption.
4.2. Endothelin Receptor Type B
Although astrocytes can produce ET-1 (see Section 3.1.5.), astrocytes are also targets of ET-1. The predominant expression of ETB receptors in the brain is found in astrocytes [12,159,160]. Beneficial effects of ETB antagonist on BBB disruption have been reported in experimental animal models. For example, Kim et al. [84,85] found that the selective ETB antagonist BQ788 reduced BBB disruption in experimental status epilepticus. We also reported that BQ788 attenuated the BBB disruption and blocked the decrease in expression of TJ-related proteins after TBI in mice by FPI [12]. ETB antagonist may also ameliorate inflammatory damage because ET-1 increased in endothelial CAMs and contributed to the brain endothelial barrier passage of monocytes [81,82].
Astrocytic ETB receptors are known to control astrocyte functions. For example, activation of astrocytic ETB receptors causes astrocytes to transition from their resting form to a reactive form [12,159,160]. LeComte et al. [161] also showed that astrocyte-specific deletion of the ETB receptor causes a defect in reactive astrocyte proliferation after permanent cerebral ischemia. Further, we previously reported that activation of ETB receptors increased astrocytic MMP-9 and VEGF-A expression, and decreased astrocytic ANG-1 expression [89,162]. Therefore, blockade of astrocytic ETB receptors is an attractive candidate for repairing BBB disruption after brain injury.
4.3. Cannabinoid Receptors
The cannabinoid (CB) receptors, including CB1 and CB2, are a class of cell membrane G protein-coupled receptors that are activated by endocannabinoids or exogenous agonists. Numerous studies have shown a protective action of CB via CB receptors against BBB disruption and TJ-related proteins in experimental animals and cell models [163,164,165,166].
Astrocytes express CB receptors [167,168,169], and upregulation of CB receptors in reactive astrocytes was observed in animal models of amyotrophic lateral sclerosis and epilepsy [167,170]. Kozela et al. [168] also reported that increased glial fibrillary acidic protein, a marker of astrocyte activity, was suppressed by CB in various experimental animal models, suggesting that modulation of astrocytic CB receptors may have beneficial effects for treatment of brain disorders.
4.4. MicroRNAs
MicroRNAs (miRNAs) are small non-coding RNAs observed in the brains of humans and experimental animals, which regulate the expression of various genes under both normal and pathological conditions. The multifarious miRNAs are closely involved in both BBB disruption and protection in various experimental animal models [171,172,173,174,175]. Further, during neuroinflammation, expression of brain endothelial microRNA-125a-5p was suppressed, resulting in increased monocyte migration as a result of endothelial upregulation of ICAM-1 [176]. Recent studies suggest that astrocytes express various miRNAs, and these miRNAs control astrocytic functions [177,178,179,180,181,182]. Overexpression of miRNA-21 in astrocytes attenuated astrogliosis, while inhibition of miRNA-21 function enhanced astrocytic hypertrophy in spinal cord injury (SCI) animals [177]. Similarly, Wang et al. [183] showed that astrocyte-specific overexpression of miRNA-145 reduced astrogliosis in SCI rats. Therefore, astrocytic miRNAs are a potential therapeutic target for SCI by alleviating astrogliosis. Moreover, several studies have found that various miRNAs can regulate VEGF expression in endothelial cells in the cerebrum and in glioma cells [184,185,186]. The control of MMP expression by miRNAs was also shown following cerebral ischemia in rats, and in primary fetal astrocyte-enriched cell cultures and glioma cells [182,187,188]. As expression of these miRNAs is observed in astrocytes, a similar regulation of VEGF and MMPs may occur in astrocytes.
5. Conclusions
BBB disruption is commonly observed in TBI, cerebral ischemia and various CNS disorders including Alzheimer’s disease and multiple sclerosis, and results in severe secondary damage including brain edema and inflammatory changes. As current therapeutic strategies for various types of brain disorders do not sufficiently recover brain function, targeting BBB disruption is expected to be a novel therapeutic strategy for a wide range of brain disorders. The mechanisms of BBB disruption are complicated as they involve various types of cells and cell-derived factors. Numerous studies also suggest dual roles of astrocyte-derived factors for control of BBB function. Astrocyte-derived vascular permeability factors including VEGF, MMPs, NO, glutamate and ETs can increase BBB permeability, resulting in aggravation of BBB disruption. By contrast, astrocyte-derived protective factors including ANG-1, SHH, GDNF, RA, IGF-1 and APOE can attenuate the increase in BBB permeability leading to BBB protection. Because alterations of these factors are observed in TBI, cerebral ischemia and several CNS disorders in clinical practice, control of these factors may be significant. Astrocytes are a major therapeutic target for brain disorders, as numerous studies suggest that control of astrocytic functions can reduce brain injury in various experimental animal models. However, as described above, astrocyte-derived factors have both protective and detrimental actions against BBB disruption in brain disorders. Besides participation in formation of BBB, astrocyte is accepted to be a component of synapses, where astrocyte-derived factors regulate efficacy of neurotransmission. Because of these multiple functions, uncontrolled modulation of astrocytes may cause disturbance of brain functions including mentation and recognition. To avoid possible adverse actions in clinical use, selective stimulation of their beneficial actions without affecting the detrimental ones is required for the astrocyte-targeting therapy. Further investigation of mechanisms underlying astrocytic functions will lead to creation of more skillful methods for astrocytic control which can be applied to clinical use.
Author Contributions
S.M. and Y.K. contributed to the writing of this review.
Funding
This work was supported by a Grant-in-Aid for Scientific Research (C; Grant Number: 18K06695) and Grant-in-Aid for Young Scientists (B; Grant Number: 16K18890) from the Japan Society for the Promotion of Science (JSPS).
Acknowledgments
We thank Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| ANG-1 | angiopoietin-1 |
| BBB | blood-brain barrier |
| CAMs | cell adhesion molecules |
| CCI | controlled cortical impact |
| CB | cannabinoid |
| cGMP | cyclic guanosine monophosphate |
| CLN | claudin |
| CNS | central nervous system |
| ECM | extracellular matrix |
| ERs | estrogen receptors |
| ETs | endothelins |
| ETA | endothelin receptor type A |
| ETB | endothelin receptor type B |
| FPI | fluid percussion injury |
| GDNF | glial-derived neurotrophic factor |
| HUVECs | human umbilical vascular endothelial cells |
| ICAM-1 | intercellular adhesion molecule-1 |
| IGF-1 | insulin-like growth factor-1 |
| KO | knock-out |
| LFA-1 | lymphocyte function-associated antigen 1 |
| miRNAs | microRNAs |
| MMPs | matrix metalloproteinases |
| NMDA | N-methyl-D-aspartate |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| OCLN | occludin |
| PTCH1 | Patched-1 |
| RA | retinoic acid |
| RALDH | retinaldehyde dehydrogenase |
| RARs | retinoic acid receptors |
| SCI | spinal cord injury |
| SHH | sonic hedgehog |
| TBI | traumatic brain injury |
| TJ | tight junction |
| VCAM-1 | vascular cell adhesion molecule-1 |
| VEGF | vascular endothelial growth factor |
| VEGFR-1 | vascular endothelial growth factor receptor-1 |
| VEGFR-2 | vascular endothelial growth factor receptor-2 |
| VLA-4 | very late antigen-4 |
| ZO | zonula occluden |
References
- Wevers, N.R.; de Vries, H.E. Morphogens and blood-brain barrier function in health and disease. Tissue Barriers 2015, 4, e1090524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almutairi, M.M.; Gong, C.; Xu, Y.G.; Chang, Y.; Shi, H. Factors controlling permeability of the blood-brain barrier. Cell. Mol. Life Sci. 2016, 73, 57–77. [Google Scholar] [CrossRef] [PubMed]
- Luissint, A.C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.O. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Lien, C.F.; Mohanta, S.K.; Frontczak-Baniewicz, M.; Swinny, J.D.; Zablocka, B.; Gorecki, D.C. Absence of glial alpha-dystrobrevin causes abnormalities of the blood–brain barrier and progressive brain edema. J. Biol. Chem. 2012, 287, 41374–41385. [Google Scholar] [CrossRef]
- Takeshita, Y.; Ransohoff, R.M. Inflammatory cell trafficking across the blood-brain barrier: Chemokine regulation and in vitro models. Immunol. Rev. 2012, 248, 228–239. [Google Scholar] [CrossRef]
- Muller, W.A. How endothelial cells regulate transmigration of leukocytes in the inflammatory response. Am. J. Pathol. 2014, 184, 886–896. [Google Scholar] [CrossRef]
- Hay, J.R.; Johnson, V.E.; Young, A.M.; Smith, D.H.; Stewart, W. Blood-brain barrier disruption is an early event that may persist for many years after traumatic brain injury in humans. J. Neuropathol. Exp. Neurol. 2015, 74, 1147–1157. [Google Scholar]
- Arba, F.; Leigh, R.; Inzitari, D.; Warach, S.J.; Luby, M.; Lees, K.R. STIR/VISTA imaging collaboration. Blood-brain barrier leakage increases with small vessel disease in acute ischemic stroke. Neurology 2017, 89, 2143–2150. [Google Scholar] [CrossRef] [PubMed]
- Van de Haar, H.J.; Burgmans, S.; Jansen, J.F.; van Osch, M.J.; van Buchem, M.A.; Muller, M.; Hofman, P.A.; Verhey, F.R.; Backes, W.H. Blood-brain barrier leakage in patients with early Alzheimer Disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Cramer, S.P.; Simonsen, H.; Frederiksen, J.L.; Rostrup, E.; Larsson, H.B. Abnormal blood-brain barrier permeability in normal appearing white matter in multiple sclerosis investigated by MRI. Neuroimage Clin. 2013, 4, 182–189. [Google Scholar] [CrossRef]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef]
- Michinaga, S.; Kimura, A.; Hatanaka, S.; Minami, S.; Asano, A.; Ikushima, Y.; Matsui, S.; Toriyama, Y.; Fujii, M.; Koyama, Y. Delayed Administration of BQ788, an ETB antagonist, after experimental traumatic brain injury promotes recovery of blood-brain barrier function and a reduction of cerebral edema in mice. J. Neurotrauma 2018, 35, 1481–1494. [Google Scholar] [CrossRef] [PubMed]
- Begum, G.; Song, S.; Wang, S.; Zhao, H.; Bhuiyan, M.I.H.; Li, E.; Nepomuceno, R.; Ye, Q.; Sun, M.; Calderon, M.J.; Stolz, D.B.; et al. Selective knockout of astrocytic Na+ /H+ exchanger isoform 1 reduces astrogliosis, BBB damage, infarction, and improves neurological function after ischemic stroke. Glia 2018, 66, 126–144. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.D.; Yao, N.W.; Guo, J.H.; Shen, C.C.; Lee, H.T.; Chiu, Y.P.; Ji, H.R.; Chen, X.; Chen, C.C.; Chang, C. Inhibition of astrocytic activity alleviates sequela in acute stages of intracerebral hemorrhage. Oncotarget 2017, 8, 94850–94861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapouly, C.; Tadesse Argaw, A.; Horng, S.; Castro, K.; Zhang, J.; Asp, L.; Loo, H.; Laitman, B.M.; Mariani, J.N.; Straus Farber, R.; et al. Astrocytic TYMP and VEGFA drive blood-brain barrier opening in inflammatory central nervous system lesions. Brain 2015, 138, 1548–1567. [Google Scholar] [CrossRef] [Green Version]
- Sun, P.; Bu, F.; Min, J.W.; Munshi, Y.; Howe, M.D.; Liu, L.; Koellhoffer, E.C.; Qi, L.; McCullough, L.D.; Li, J. Inhibition of calcium/calmodulin-dependent protein kinase (CaMKK) exacerbates impairment of endothelial cell and blood-brain barrier after stroke. Eur. J. Neurosci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.X.; Jing, Y.; Liu, Y.L.; Xu, Z.M.; Yuan, F.; Wang, M.L.; Geng, Z.; Tian, H.L. Inhibition of transient receptor potential vanilloid 1 attenuates blood-brain barrier disruption after traumatic brain injury. in mice. J. Neurotrauma 2018. [Google Scholar] [CrossRef] [PubMed]
- Main, B.S.; Villapol, S.; Sloley, S.S.; Barton, D.J.; Parsadanian, M.; Agbaegbu, C.; Stefos, K.; McCann, M.S.; Washington, P.M.; Rodriguez, O.C.; et al. Apolipoprotein E4 impairs spontaneous blood brain barrier repair following traumatic brain injury. Mol. Neurodegener. 2018, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Fang, Z.; He, Q.W.; Li, Q.; Chen, X.L.; Baral, S.; Jin, H.J.; Zhu, Y.Y.; Li, M.; Xia, Y.P.; Mao, L.; et al. MicroRNA-150 regulates blood-brain barrier permeability via Tie-2 after permanent middle cerebral artery occlusion in rats. FASEB J. 2016, 30, 2097–2107. [Google Scholar] [CrossRef]
- Sun, J.; Yu, L.; Huang, S.; Lai, X.; Milner, R.; Li, L. Vascular expression of angiopoietin1, α5β1 integrin and tight junction proteins is tightly regulated during vascular remodeling in the post-ischemic brain. Neuroscience 2017, 362, 248–256. [Google Scholar] [CrossRef]
- Argaw, A.T.; Gurfein, B.T.; Zhang, Y.; Zameer, A.; John, G.R. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc. Natl. Acad. Sci. USA 2009, 106, 1977–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.S.; Fang, H.L.; Chen, Y.; Liang, S.S.; Zhu, Z.G.; Zeng, Q.Y.; Li, J.; Xu, H.Q.; Shao, B.; He, J.C.; et al. Idazoxan reduces blood-brain barrier damage during experimental autoimmune encephalomyelitis in mouse. Eur. J. Pharmacol. 2014, 736, 70–76. [Google Scholar] [CrossRef]
- González-Mariscal, L.; Tapia, R.; Chamorro, D. Crosstalk of tight junction components with signaling pathways. Biochim. Biophys. Acta 2008, 1778, 729–756. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.H.; Park, S.W.; Brooks, N.; Lang, B.T.; Vemuganti, R. PPARgamma agonist rosiglitazone is neuroprotective after traumatic brain injury via anti-inflammatory and anti-oxidative mechanisms. Brain Res. 2008, 1244, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Rancan, M.; Otto, V.; Hans, V.H.; Gerlach, I.; Jork, R.; Trentz, O.; Kossmann, T.; Morganti-Kossmann, M.C. Upregulation of ICAM-1 and MCP-1 but not of MIP-2 and sensorimotor deficit in response to traumatic axonal injury in rats. J. Neurosci. Res. 2001, 63, 438–446. [Google Scholar] [CrossRef]
- Williams, A.J.; Wei, H.H.; Dave, J.R.; Tortella, F.C. Acute and delayed neuroinflammatory response following experimental penetrating ballistic brain injury in the rat. J. Neuroinflamm. 2007, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Fee, D.; Crumbaugh, A.; Jacques, T.; Herdrich, B.; Sewell, D.; Auerbach, D.; Piaskowski, S.; Hart, M.N.; Sandor, M.; Fabry, Z. Activated/effector CD4+ T cells exacerbate acute damage in the central nervous system following traumatic injury. J. Neuroimmunol. 2003, 36, 54–66. [Google Scholar] [CrossRef]
- Harting, M.T.; Jimenez, F.; Adams, S.D.; Mercer, D.W.; Cox, C.S., Jr. Acute, regional inflammatory response after traumatic brain injury: Implications for cellular therapy. Surgery 2008, 144, 803–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenne, E.; Erlandsson, A.; Lindbom, L.; Hillered, L.; Clausen, F. Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. J. Neuroinflamm. 2012, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhind, S.G.; Crnko, N.T.; Baker, A.J.; Morrison, L.J.; Shek, P.N.; Scarpelini, S.; Rizoli, S.B. Prehospital resuscitation with hypertonic saline-dextran modulates inflammatory, coagulation and endothelial activation marker profiles in severe traumatic brain injured patients. J. Neuroinflamm. 2010, 7, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, J.M.; Downie, S.A.; Lyman, W.D.; Berman, J.W. Astrocyte-derived monocyte-chemoattractant protein-1 directs the transmigration of leukocytes across a model of the human blood-brain barrier. J. Immunol. 1998, 161, 6896–6903. [Google Scholar] [PubMed]
- Guo, M.F.; Meng, J.; Li, Y.H.; Yu, J.Z.; Liu, C.Y.; Feng, L.; Yang, W.F.; Li, J.L.; Feng, Q.J.; Xiao, B.G.; et al. The inhibition of Rho kinase blocks cell migration and accumulation possibly by challenging inflammatory cytokines and chemokines on astrocytes. J. Neurol. Sci. 2014, 343, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Eilam, R.; Segal, M.; Malach, R.; Sela, M.; Arnon, R.; Aharoni, R. Astrocyte disruption of neurovascular communication is linked to cortical damage in an animal model of multiple sclerosis. Glia 2018, 66, 1098–1117. [Google Scholar] [CrossRef]
- Risau, W.; Esser, S.; Engelhardt, B. Differentiation of blood-brain barrier endothelial cells. Pathol. Biol. 1998, 46, 171–175. [Google Scholar] [CrossRef]
- Jiang, S.; Xia, R.; Jiang, Y.; Wang, L.; Gao, F. Vascular endothelial growth factors enhance the permeability of the mouse blood-brain barrier. PLoS ONE 2014, 9, e86407. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Ye, Z.; Pan, Y.; Li, X.; Fu, X.; Zhang, B.; Li, Y.; Lin, W.; Li, X.; Gao, Q. Vascular endothelial growth factor aggravates cerebral ischemia and reperfusion-induced blood-brain-barrier disruption through regulating LOC102640519/HOXC13/ZO-1 signaling. Exp. Cell Res. 2018, 369, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.T.; Zhang, P.; Gao, Y.; Li, C.L.; Wang, H.J.; Chen, L.C.; Feng, Y.; Li, R.Y.; Li, Y.L.; Jiang, C.L. Early VEGF inhibition attenuates blood-brain barrier disruption in ischemic rat brains by regulating the expression of MMPs. Mol. Med. Rep. 2017, 15, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Reeson, P.; Tennant, K.A.; Gerrow, K.; Wang, J.; Weiser Novak, S.; Thompson, K.; Lockhart, K.L.; Holmes, A.; Nahirney, P.C.; Brown, C.E. Delayed inhibition of VEGF signaling after stroke attenuates blood-brain barrier breakdown and improves functional recovery in a comorbidity-dependent manner. J. Neurosci. 2015, 35, 5128–5143. [Google Scholar] [CrossRef]
- Zhao, F.; Deng, J.; Yu, X.; Li, D.; Shi, H.; Zhao, Y. Protective effects of vascular endothelial growth factor in cultured brain endothelial cells against hypoglycemia. Metab. Brain Dis. 2015, 30, 999–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood–brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef]
- Kim, I.; Moon, S.O.; Park, S.K.; Chae, S.W.; Koh, G.Y. Angiopoietin-1 reduces VEGF-stimulated leukocyte adhesion to endothelial cells by reducing ICAM-1, VCAM-1, and E-selectin expression. Circ. Res. 2001, 89, 477–479. [Google Scholar] [CrossRef]
- Shore, P.M.; Jackson, E.K.; Wisniewski, S.R.; Clark, R.S.; Adelson, P.D.; Kochanek, P.M. Vascular endothelial growth factor is increased in cerebrospinal fluid after traumatic brain injury in infants and children. Neurosurgery 2004, 54, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Matsumoto, N.; Tasaki, O.; Nakamura, H.; Akagaki, F.; Shimazu, T. Delayed progression of edema formation around a hematoma expressing high levels of VEGF and mmp-9 in a patient with traumatic brain injury: Case report. Neurol. Med. Chir. 2013, 53, 609–612. [Google Scholar] [CrossRef]
- Matsuo, R.; Ago, T.; Kamouchi, M.; Kuroda, J.; Kuwashiro, T.; Hata, J.; Sugimori, H.; Fukuda, K.; Gotoh, S.; Makihara, N.; et al. Clinical significance of plasma VEGF value in ischemic stroke-research for biomarkers in ischemic stroke (REBIOS) study. BMC Neurol. 2013, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Grossetete, M.; Phelps, J.; Arko, L.; Yonas, H.; Rosenberg, G.A. Elevation of matrix metalloproteinases 3 and 9 in cerebrospinal fluid and blood in patients with severe traumatic brain injury. Neurosurgery 2009, 65, 702–708. [Google Scholar] [CrossRef]
- Liu, C.L.; Chen, C.C.; Lee, H.C.; Cho, D.Y. Matrix metalloproteinase-9 in the ventricular cerebrospinal fluid correlated with the prognosis of traumatic brain injury. Turk. Neurosurg. 2014, 24, 363–368. [Google Scholar]
- Chen, F.; Ohashi, N.; Li, W.; Eckman, C.; Nguyen, J.H. Disruptions of occludin and claudin-5 in brain endothelial cells in vitro and in brains of mice with acute liver failure. Hepatology 2009, 50, 1914–1923. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Estrada, E.Y.; Thompson, J.F.; Liu, W.; Rosenberg, G.A. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J. Cereb. Blood Flow Metab. 2007, 27, 697–709. [Google Scholar] [CrossRef]
- Zhang, S.; An, Q.; Wang, T.; Gao, S.; Zhou, G. Autophagy- and MMP-2/9-mediated reduction and redistribution of ZO-1 contribute to hyperglycemia-increased blood-brain barrier permeability during early reperfusion in stroke. Neuroscience 2018, 377, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Xu, L.; Wang, X.; Sun, X. MMP-9 expression and activity is concurrent with endothelial cell apoptosis in the basilar artery after subarachnoid hemorrhaging in rats. Neurol. Sci. 2015, 36, 1241–1245. [Google Scholar] [CrossRef]
- Lee, S.R.; Lo, E.H. Induction of caspase-mediated cell death by matrix metalloproteinases in cerebral endothelial cells after hypoxia-reoxygenation. J. Cereb. Blood Flow Metab. 2004, 24, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Rosenberg, G.A. MMP-mediated disruption of claudin-5 in the blood-brain barrier of rat brain after cerebral ischemia. Methods Mol. Biol. 2011, 762, 333–345. [Google Scholar]
- Asahi, M.; Wang, X.; Mori, T.; Sumii, T.; Jung, J.C.; Moskowitz, M.A.; Fini, M.E.; Lo, E.H. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood–brain barrier and white matter components after cerebral ischemia. J. Neurosci. 2001, 21, 7724–7732. [Google Scholar] [CrossRef] [PubMed]
- Brilha, S.; Ong, C.W.M.; Weksler, B.; Romero, N.; Couraud, P.O.; Friedland, J.S. Matrix metalloproteinase-9 activity and a downregulated Hedgehog pathway impair blood-brain barrier function in an in vitro model of CNS tuberculosis. Sci. Rep. 2017, 7, 16031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.S.; Jin, S.M.; Kim, H.J.; Lee, Y.C. Matrix metalloproteinase inhibitor regulates inflammatory cell migration by reducing ICAM-1 and VCAM-1 expression in a murine model of toluene diisocyanate-induced asthma. J. Allergy Clin. Immunol. 2003, 111, 1278–1284. [Google Scholar] [CrossRef]
- Jiang, L.; Pan, C.L.; Wang, C.Y.; Liu, B.Q.; Han, Y.; Hu, L.; Liu, L.; Yang, Y.; Qu, J.W.; Liu, W.T. Selective suppression of the JNK-MMP2/9 signal pathway by tetramethylpyrazine attenuates neuropathic pain in rats. J. Neuroinflamm. 2017, 14, 174. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.J.; Cirrito, J.R.; Yan, P.; Hu, X.; Xiao, Q.; Pan, X.; Bateman, R.; Song, H.; Hsu, F.F.; Turk, J.; et al. Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J. Neurosci. 2006, 26, 10939–10948. [Google Scholar] [CrossRef]
- Min, H.; Hong, J.; Cho, I.H.; Jang, Y.H.; Lee, H.; Kim, D.; Yu, S.W.; Lee, S.; Lee, S.J. TLR2-induced astrocyte MMP9 activation compromises the blood brain barrier and exacerbates intracerebral hemorrhage in animal models. Mol. Brain 2015, 8, 23. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, M.F.; Puebla, M.; Figueroa, X.F. Control of the neurovascular coupling by nitric oxide-dependent regulation of astrocytic Ca(2+) signaling. Front. Cell Neurosci. 2015, 9, 59. [Google Scholar] [CrossRef]
- Liu, B.; Neufeld, A.H. Expression of nitric oxide synthase-2 (NOS-2) in reactive astrocytes of the human glaucomatous optic nerve head. Glia 2000, 30, 178–186. [Google Scholar] [CrossRef]
- Buskila, Y.; Abu-Ghanem, Y.; Levi, Y.; Moran, A.; Grauer, E.; Amitai, Y. Enhanced astrocytic nitric oxide production and neuronal modifications in the neocortex of a NOS2 mutant mouse. PLoS ONE 2007, 2, e843. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.N.; Pahan, K. Signals for the induction of nitric oxide synthase in astrocytes. Neurochem. Int. 2006, 49, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Y.; Zheng, G.; Xu, M.; Li, Y.; Chen, X.; Zhu, W.; Tong, Y.; Chung, S.K.; Liu, K.J.; Shen, J. Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J. Neurochem. 2012, 120, 147–156. [Google Scholar] [CrossRef]
- Jiang, Z.; Li, C.; Arrick, D.M.; Yang, S.; Baluna, A.E.; Sun, H. Role of nitric oxide synthases in early blood-brain barrier disruption following transient focal cerebral ischemia. PLoS ONE 2014, 9, e93134. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.H.; Wang, X.L.; Wilcken, D.E. Nitric oxide induces and inhibits apoptosis through different pathways. FEBS Lett. 1998, 433, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Chen, Y.; Deng, X.; Jiang, W.; Li, B.; Fu, Z.; Du, M.; Ding, R. Hemoglobin-induced nitric oxide synthase overexpression and nitric oxide production contribute to blood-brain barrier disruption in the rat. J. Mol. Neurosci. 2013, 51, 352–363. [Google Scholar] [CrossRef] [PubMed]
- Sharp, C.D.; Hines, I.; Houghton, J.; Warren, A.; Jackson, T.H.; Jawahar, A.; Nanda, A.; Elrod, J.W.; Long, A.; Chi, A.; et al. Glutamate causes a loss in human cerebral endothelial barrier integrity through activation of NMDA receptor. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, 2592–2598. [Google Scholar] [CrossRef] [PubMed]
- András, I.E.; Deli, M.A.; Veszelka, S.; Hayashi, K.; Hennig, B.; Toborek, M. The NMDA and AMPA/KA receptors are involved in glutamate-induced alterations of occludin expression and phosphorylation in brain endothelial cells. J. Cereb. Blood Flow Metab. 2007, 27, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Hogan-Cann, A.D.; Globa, A.K.; Lu, P.; Nagy, J.I.; Bamji, S.X.; Anderson, C.M. Astrocytes drive cortical vasodilatory signaling by activating endothelial NMDA receptors. J. Cereb. Blood Flow Metab. 2017, 1. [Google Scholar] [CrossRef] [PubMed]
- Vazana, U.; Veksler, R.; Pell, G.S.; Prager, O.; Fassler, M.; Chassidim, Y.; Roth, Y.; Shahar, H.; Zangen, A.; Raccah, R.; et al. Glutamate-mediated blood-brain barrier opening: Implications for neuroprotection and drug delivery. J. Neurosci. 2016, 36, 7727–7739. [Google Scholar] [CrossRef]
- Liu, X.; Hunter, C.; Weiss, H.R.; Chi, O.Z. Effects of blockade of ionotropic glutamate receptors on blood-brain barrier disruption in focal cerebral ischemia. Neurol. Sci. 2010, 31, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Salonia, R.; Empey, P.E.; Poloyac, S.M.; Wisniewski, S.R.; Klamerus, M.; Ozawa, H.; Wagner, A.K.; Ruppel, R.; Bell, M.J.; Feldman, K.; et al. Endothelin-1 is increased in cerebrospinal fluid and associated with unfavorable outcomes in children after severe traumatic brain injury. J. Neurotrauma 2010, 27, 1819–1825. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.W.; Li, W.J.; Dou, X.J.; Jia, R.; Yang, H.; Liu, X.G.; Xu, C.B.; Liu, J.; Cao, Y.X.; Luo, G.G. Role of endothelin-1 and its receptors in cerebral vasospasm following subarachnoid hemorrhage. Mol. Med. Rep. 2018, 27, 5229–5236. [Google Scholar] [CrossRef] [PubMed]
- Petrov, T.; Steiner, J.; Braun, B.; Rafols, J.A. Sources of endothelin-1 in hippocampus and cortex following traumatic brain injury. Neuroscience 2002, 115, 275–283. [Google Scholar] [CrossRef]
- Hammond, T.R.; Gadea, A.; Dupree, J.; Kerninon, C.; Nait-Oumesmar, B.; Aguirre, A.; Gallo, V. Astrocyte-derived endothelin-1 inhibits remyelination through notch activation. Neuron 2014, 81, 588–602. [Google Scholar] [CrossRef]
- Ranno, E.; D’Antoni, S.; Spatuzza, M.; Berretta, A.; Laureanti, F.; Bonaccorso, C.M.; Pellitteri, R.; Longone, P.; Spalloni, A.; Iyer, A.M.; et al. Endothelin-1 is over-expressed in amyotrophic lateral sclerosis and induces motor neuron cell death. Neurobiol. Dis. 2014, 65, 160–171. [Google Scholar] [CrossRef]
- Yeung, P.K.; Shen, J.; Chung, S.S.; Chung, S.K. Targeted over-expression of endothelin-1 in astrocytes leads to more severe brain damage and vasospasm after subarachnoid hemorrhage. BMC Neurosci. 2013, 14, 131. [Google Scholar] [CrossRef]
- Lo, A.C.; Chen, A.Y.; Hung, V.K.; Yaw, L.P.; Fung, M.K.; Ho, M.C.; Tsang, M.C.; Chung, S.S.; Chung, S.K. Endothelin-1 overexpression leads to further water accumulation and brain edema after middle cerebral artery occlusion via aquaporin 4 expression in astrocytic end-feet. J. Cereb. Blood Flow Metab. 2005, 25, 998–1011. [Google Scholar] [CrossRef]
- Hung, V.K.; Yeung, P.K.; Lai, A.K.; Ho, M.C.; Lo, A.C.; Chan, K.C.; Wu, E.X.; Chung, S.S.; Cheung, C.W.; Chung, S.K. Selective astrocytic endothelin-1 overexpression contributes to dementia associated with ischemic stroke by exaggerating astrocyte-derived amyloid secretion. J. Cereb. Blood Flow Metab. 2015, 35, 1687–1696. [Google Scholar] [CrossRef]
- Narushima, I.; Kita, T.; Kubo, K.; Yonetani, Y.; Momochi, C.; Yoshikawa, I.; Ohno, N.; Nakashima, T. Highly enhanced permeability of blood-brain barrier induced by repeated administration of endothelin-1 in dogs and rats. Pharmacol. Toxicol. 2003, 92, 21–26. [Google Scholar] [CrossRef]
- Reijerkerk, A.; Lakeman, K.A.; Drexhage, J.A.; van Het Hof, B.; van Wijck, Y.; van der Pol, S.M.; Kooij, G.; Geerts, D.; de Vries, H.E. Brain endothelial barrier passage by monocytes is controlled by the endothelin system. J. Neurochem. 2012, 121, 730–737. [Google Scholar] [CrossRef] [PubMed]
- McCarron, R.M.; Wang, L.; Stanimirovic, D.B.; Spatz, M. Endothelin induction of adhesion molecule expression on human brain microvascular endothelial cells. Neurosci. Lett. 1993, 156, 31–34. [Google Scholar] [CrossRef]
- Matsuo, Y.; Mihara, S.; Ninomiya, M.; Fujimoto, M. Protective effect of endothelin type A receptor antagonist on brain edema and injury after transient middle cerebral artery occlusion in rats. Stroke 2001, 32, 2143–2148. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Ryu, H.J.; Kang, T.C. Status epilepticus induces vasogenic edema via tumor necrosis factor-α/endothelin-1-mediated two different pathways. PLoS ONE 2013, 8, e74458. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Ko, A.R.; Hyun, H.W.; Kang, T.C. ETB receptor-mediated MMP-9 activation induces vasogenic edema via ZO-1 protein degradation following status epilepticus. Neuroscience 2015, 304, 355–367. [Google Scholar] [CrossRef]
- Sabirzhanov, B.; Faden, A.I.; Aubrecht, T.; Henry, R.; Glaser, E.; Stoica, B.A. MicroRNA-711-induced downregulation of angiopoietin-1 mediates neuronal cell death. J. Neurotrauma 2018, 35, 2462–2481. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, W.J.; Choi, Y.K.; Song, H.S.; Son, M.J.; Gelman, I.H.; Kim, Y.J.; Kim, K.W. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 2003, 9, 900–906. [Google Scholar] [CrossRef]
- Zacharek, A.; Chen, J.; Cui, X.; Li, A.; Li, Y.; Roberts, C.; Feng, Y.; Gao, Q.; Chopp, M. Angiopoietin1/Tie2 and VEGF/Flk1 induced by MSC treatment amplifies angiogenesis and vascular stabilization after stroke. J. Cereb. Blood Flow Metab. 2007, 27, 1684–1691. [Google Scholar] [CrossRef]
- Koyama, Y.; Maebara, Y.; Hayashi, M.; Nagae, R.; Tokuyama, S.; Michinaga, S. Endothelins reciprocally regulate VEGF-A and angiopoietin-1 production in cultured rat astrocytes: Implications on astrocytic proliferation. Glia 2012, 60, 1954–1963. [Google Scholar] [CrossRef]
- Xia, Y.P.; He, Q.W.; Li, Y.N.; Chen, S.C.; Huang, M.; Wang, Y.; Gao, Y.; Huang, Y.; Wang, M.D.; Mao, L.; Hu, B. Recombinant human sonic hedgehog protein regulates the expression of ZO-1 and occludin by activating angiopoietin-1 in stroke damage. PLoS ONE 2013, 8, e68891. [Google Scholar] [CrossRef]
- Kawamura, K.; Takahashi, T.; Kanazawa, M.; Igarashi, H.; Nakada, T.; Nishizawa, M.; Shimohata, T. Effects of angiopoietin-1 on hemorrhagic transformation and cerebral edema after tissue plasminogen activator treatment for ischemic stroke in rats. PLoS ONE 2014, 9, e98639. [Google Scholar] [CrossRef] [PubMed]
- Meng, Z.; Li, M.; He, Q.; Jiang, S.; Zhang, X.; Xiao, J.; Bai, Y. Ectopic expression of human angiopoietin-1 promotes functional recovery and neurogenesis after focal cerebral ischemia. Neuroscience 2014, 267, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, L.; Croll, S.D.; Chopp, M. Angiopoietin-1 reduces cerebral blood vessel leakage and ischemic lesion volume after focal cerebral embolic ischemia in mice. Neuroscience 2002, 113, 683–687. [Google Scholar] [CrossRef]
- Venkat, P.; Yan, T.; Chopp, M.; Zacharek, A.; Ning, R.; Van Slyke, P.; Dumont, D.; Landschoot-Ward, J.; Liang, L.; Chen, J. Angiopoietin-1 mimetic peptide promotes neuroprotection after stroke in type 1 diabetic rats. Cell Transplant. 2018, 20, 1744–1752. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Fei, Z.H.; Wang, Y.Q.; Yang, J.G.; Zhao, C.H.; Cai, Y.; Zhong, X.M. Angiopoietin-1 and angiopoietin-2 expression imbalance influence in early period after subarachnoid hemorrhage. Int. Neurourol. J. 2016, 20, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Brickler, T.R.; Hazy, A.; Guilhaume Correa, F.; Dai, R.; Kowalski, E.J.A.; Dickerson, R.; Chen, J.; Wang, X.; Morton, P.D.; Whittington, A.; et al. Angiopoietin/Tie2 axis regulates the age-at-injury cerebrovascular response to traumatic brain injury. J. Neurosci. 2018, 38, 9618–9634. [Google Scholar] [CrossRef]
- Golledge, J.; Clancy, P.; Maguire, J.; Lincz, L.; Koblar, S.; McEvoy, M.; Attia, J.; Levi, C.; Sturm, J.; Almeida, O.P.; et al. Plasma angiopoietin-1 is lower after ischemic stroke and associated with major disability but not stroke incidence. Stroke 2014, 45, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Sobrino, T.; Arias, S.; Rodríguez-González, R.; Brea, D.; Silva, Y.; de la Ossa, N.P.; Agulla, J.; Blanco, M.; Pumar, J.M.; Serena, J.; et al. High serum levels of growth factors are associated with good outcome in intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2009, 29, 1968–1974. [Google Scholar] [CrossRef]
- Nag, S.; Manias, J.L.; Kapadia, A.; Stewart, D.J. Molecular changes associated with the protective effects of angiopoietin-1 during blood-brain barrier breakdown post-injury. Mol. Neurobiol. 2017, 54, 4232–4242. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, L.; Shu, B.; Tang, J.; Zhang, L.; Xie, J.; Liu, X.; Xu, Y.; Qi, S. Angiopoietin-1 protects the endothelial cells against advanced glycation end product injury by strengthening cell junctions and inhibiting cell apoptosis. J. Cell. Physiol. 2015, 230, 1895–1905. [Google Scholar] [CrossRef]
- Yu, H.; Wang, P.; An, P.; Xue, Y. Recombinant human angiopoietin-1 ameliorates the expressions of ZO-1, occludin, VE-cadherin, and PKCα signaling after focal cerebral ischemia/reperfusion in rats. J. Mol. Neurosci. 2012, 46, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, Z.; Rikani, A.A.; Choudhry, A.M.; Tariq, S.; Zakaria, F.; Asghar, M.W.; Sarfraz, M.K.; Haider, K.; Shafiq, A.A.; Mobassarah, N.J. Sonic hedgehog signalling pathway: A complex network. Ann. Neurosci. 2014, 21, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Seifert, T.; Bauer, J.; Weissert, R.; Fazekas, F.; Storch, M.K. Differential expression of sonic hedgehog immunoreactivity during lesion evolution in autoimmune encephalomyelitis. J. Neuropathol. Exp. Neurol. 2005, 64, 404–411. [Google Scholar] [CrossRef]
- Amankulor, N.M.; Hambardzumyan, D.; Pyonteck, S.M.; Becher, O.J.; Joyce, J.A.; Holland, E.C. Sonic hedgehog pathway activation is induced by acute brain injury and regulated by injury-related inflammation. J. Neurosci. 2009, 29, 10299–10308. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef]
- He, Q.W.; Xia, Y.P.; Chen, S.C.; Wang, Y.; Huang, M.; Huang, Y.; Li, J.Y.; Li, Y.N.; Gao, Y.; Mao, L.; et al. Astrocyte-derived sonic hedgehog contributes to angiogenesis in brain microvascular endothelial cells via RhoA/ROCK pathway after oxygen-glucose deprivation. Mol. Neurobiol. 2013, 47, 976–987. [Google Scholar] [CrossRef]
- Li, Y.; Xia, Y.; Wang, Y.; Mao, L.; Gao, Y.; He, Q.; Huang, M.; Chen, S.; Hu, B. Sonic hedgehog (Shh) regulates the expression of angiogenic growth factors in oxygen-glucose-deprived astrocytes by mediating the nuclear receptor NR2F2. Mol. Neurobiol. 2013, 47, 967–975. [Google Scholar] [CrossRef]
- Wang, Y.; Jin, S.; Sonobe, Y.; Cheng, Y.; Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Mizuno, T.; Takeuchi, H.; Suzumura, A. Interleukin-1β induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS ONE 2014, 9, e110024. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Zhao, G.; Li, W.; Zhang, J.; Che, Y.; Song, M.; Gao, S.; Zeng, B.; Wang, Y. MiR-155 targets PTCH1 to mediate endothelial progenitor cell dysfunction caused by high glucose. Exp. Cell Res. 2018, 366, 55–62. [Google Scholar] [CrossRef]
- Zhu, S.L.; Luo, M.Q.; Peng, W.X.; Li, Q.X.; Feng, Z.Y.; Li, Z.X.; Wang, M.X.; Feng, X.X.; Liu, F.; Huang, J.L. Sonic hedgehog signalling pathway regulates apoptosis through Smo protein in human umbilical vein endothelial cells. Rheumatology 2015, 54, 1093–1102. [Google Scholar] [CrossRef] [PubMed]
- Drannik, A.; Martin, J.; Peterson, R.; Ma, X.; Jiang, F.; Turnbull, J. Cerebrospinal fluid from patients with amyotrophic lateral sclerosis inhibits sonic hedgehog function. PLoS ONE 2017, 12, e0171668. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Ba, H.; Lu, C.; Dai, J.; Sun, J. Glial cell line-derived neurotrophic factor (GDNF) promotes angiogenesis through the demethylation of the fibromodulin (FMOD) promoter in glioblastoma. Med. Sci. Monit. 2018, 24, 6137–6143. [Google Scholar] [CrossRef]
- Utsumi, H.; Chiba, H.; Kamimura, Y.; Osanai, M.; Igarashi, Y.; Tobioka, H.; Mori, M.; Sawada, N. Expression of GFRalpha-1, receptor for GDNF, in rat brain capillary during postnatal development of the BBB. Am. J. Physiol. Cell Physiol. 2000, 279, C361–C368. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Chiba, H.; Utsumi, H.; Miyajima, H.; Ishizaki, T.; Gotoh, T.; Kuwahara, K.; Tobioka, H.; Satoh, M.; Mori, M.; et al. Expression of receptors for glial cell line-derived neurotrophic factor (GDNF) and neurturin in the inner blood-retinal barrier of rats. Cell. Struct. Funct. 2000, 25, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, F.; Sano, Y.; Saito, K.; Abe, M.A.; Maeda, T.; Haruki, H.; Kanda, T. Pericyte-derived glial cell line-derived neurotrophic factor increase the expression of claudin-5 in the blood-brain barrier and the blood-nerve barrier. Neurochem. Res. 2012, 37, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Wang, W.; Chen, W.; Sun, L.; Li, X.; Zhang, C.; Yang, H. GDNF is involved in the barrier-inducing effect of enteric glial cells on intestinal epithelial cells under acute ischemia reperfusion stimulation. Mol. Neurobiol. 2014, 50, 274–289. [Google Scholar] [CrossRef]
- Mizee, M.R.; Nijland, P.G.; van der Pol, S.M.; Drexhage, J.A.; van Het Hof, B.; Mebius, R.; van der Valk, P.; van Horssen, J.; Reijerkerk, A.; de Vries, H.E. Astrocyte-derived retinoic acid: A novel regulator of blood-brain barrier function in multiple sclerosis. Acta Neuropathol. 2014, 128, 691–703. [Google Scholar] [CrossRef]
- Mizee, M.R.; Wooldrik, D.; Lakeman, K.A.; van het Hof, B.; Drexhage, J.A.; Geerts, D.; Bugiani, M.; Aronica, E.; Mebius, R.E.; Prat, A.; et al. Retinoic acid induces blood-brain barrier development. J. Neurosci. 2013, 33, 1660–1671. [Google Scholar] [CrossRef]
- Kong, L.; Wang, Y.; Wang, X.J.; Wang, X.T.; Zhao, Y.; Wang, L.M.; Chen, Z.Y. Retinoic acid ameliorates blood-brain barrier disruption following ischemic stroke in rats. Pharmacol. Res. 2015, 99, 125–136. [Google Scholar] [CrossRef]
- Gille, J.; Paxton, L.L.; Lawley, T.J.; Caughman, S.W.; Swerlick, R.A. Retinoic acid inhibits the regulated expression of vascular cell adhesion molecule-1 by cultured dermal microvascular endothelial cells. J. Clin. Investig. 1997, 99, 492–500. [Google Scholar] [CrossRef]
- Wrigley, S.; Arafa, D.; Tropea, D. Insulin-Like Growth Factor 1: At the Crossroads of Brain Development and Aging. Front. Cell Neurosci. 2017, 11, 14. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liang, H.; Liu, H.; Li, D.; Chen, X.; Li, L.; Zhang, C.Y.; Zen, K. Platelet-secreted microRNA-223 promotes endothelial cell apoptosis induced by advanced glycation end products via targeting the insulin-like growth factor 1 receptor. J. Immunol. 2014, 192, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Perez, A.I.; Borrajo, A.; Diaz-Ruiz, C.; Garrido-Gil, P.; Labandeira-Garcia, J.L. Crosstalk between insulin-like growth factor-1 and angiotensin-II in dopaminergic neurons and glial cells: Role in neuroinflammation and aging. Oncotarget 2016, 7, 30049–30067. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.; Wilcox, K.C.; Tortelli, V.; Diniz, L.P.; Oliveira, M.S.; Dobbins, C.; Yu, X.W.; Nandamuri, S.; Gomes, F.C.A.; DiNunno, N.; et al. Neuroprotective astrocyte-derived insulin/insulin-like growth factor 1 stimulates endocytic processing and extracellular release of neuron-bound Aβ oligomers. Mol. Biol. Cell. 2017, 28, 2623–2636. [Google Scholar] [CrossRef] [Green Version]
- Madathil, S.K.; Carlson, S.W.; Brelsfoard, J.M.; Ye, P.; D’Ercole, A.J.; Saatman, K.E. Astrocyte-specific overexpression of insulin-like growth factor-1 protects hippocampal neurons and reduces behavioral deficits following traumatic brain injury in mice. PLoS ONE 2013, 8, e67204. [Google Scholar] [CrossRef] [PubMed]
- Okoreeh, A.K.; Bake, S.; Sohrabji, F. Astrocyte-specific insulin-like growth factor-1 gene transfer in aging female rats improves stroke outcomes. Glia 2017, 65, 1043–1058. [Google Scholar] [CrossRef]
- Bake, S.; Okoreeh, A.K.; Alaniz, R.C.; Sohrabji, F. Insulin-like growth factor (IGF)-I modulates endothelial blood-brain barrier function in ischemic middle-aged female rats. Endocrinology 2016, 157, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Bake, S.; Okoreeh, A.; Khosravian, H.; Sohrabji, F. Insulin-like Growth Factor (IGF)-1 treatment stabilizes the microvascular cytoskeleton under ischemic conditions. Exp. Neurol. 2019, 311, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef]
- Morikawa, M.; Fryer, J.D.; Sullivan, P.M.; Christopher, E.A.; Wahrle, S.E.; DeMattos, R.B.; O’Dell, M.A.; Fagan, A.M.; Lashuel, H.A.; Walz, T.; et al. Production and characterization of astrocyte-derived human apolipoprotein E isoforms from immortalized astrocytes and their interactions with amyloid-beta. Neurobiol. Dis. 2005, 19, 66–76. [Google Scholar] [CrossRef]
- Ito, J.; Nagayasu, Y.; Miura, Y.; Yokoyama, S.; Michikawa, M. Astrocyte׳s endogenous apoE generates HDL-like lipoproteins using previously synthesized cholesterol through interaction with ABCA1. Brain Res. 2014, 1570, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Jiang, Y.; Wu, Y.; Zhong, J.; Liu, J.; Qin, X.; Chen, L.; Vitek, M.P.; Li, F.; Xu, L.; et al. Apolipoprotein E-Mimetic COG1410 Reduces Acute Vasogenic Edema following Traumatic Brain Injury. J. Neurotrauma 2016, 33, 175–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, Z.; Guo, Z.; Zhong, J.; Cheng, C.; Huang, Z.; Wu, Y.; Tang, S.; Luo, C.; Peng, X.; Wu, H.; et al. ApoE Influences the Blood-Brain Barrier Through the NF-κB/MMP-9 Pathway After Traumatic Brain Injury. Sci. Rep. 2017, 7, 6649. [Google Scholar] [CrossRef]
- Zheng, M.; Wei, J.; Tang, Y.; Yang, C.; Wei, Y.; Yin, X.; Liu, Q. ApoE-deficient promotes blood-brain barrier disruption in experimental autoimmune encephalomyelitis via alteration of MMP-9. J. Mol. Neurosci. 2014, 54, 282–290. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Therapeutic strategy of targeting astrocytes for neuroprotection in Parkinson’s Disease. Curr. Pharm. Des. 2017, 23, 4936–4947. [Google Scholar] [CrossRef]
- Assefa, B.T.; Gebre, A.K.; Altaye, B.M. Reactive astrocytes as drug target in Alzheimer’s Disease. Biomed. Res. Int. 2018, 2018, 4160247. [Google Scholar] [CrossRef]
- Ponath, G.; Park, C.; Pitt, D. The role of astrocytes in multiple sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef]
- Okada, S.; Hara, M.; Kobayakawa, K.; Matsumoto, Y.; Nakashima, Y. Astrocyte reactivity and astrogliosis after spinal cord injury. Neurosci. Res. 2018, 126, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.; Dubner, R. Neuron-glia crosstalk gets serious: Role in pain hypersensitivity. Curr. Opin. Anaesthesiol. 2008, 21, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Elsayed, M.; Magistretti, P.J. A new outlook on mental illnesses: Glial involvement beyond the glue. Front. Cell. Neurosci. 2015, 9, 468. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 3, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Wilhelmsson, U.; Tatlisumak, T.; Pekna, M. Astrocyte activation and reactive gliosis-a new target in stroke? Neurosci. Lett. 2018, 18, 30490–30497. [Google Scholar] [CrossRef]
- Arevalo, M.A.; Santos-Galindo, M.; Acaz-Fonseca, E.; Azcoitia, I.; Garcia-Segura, L.M. Gonadal hormones and the control of reactive gliosis. Horm. Behav. 2013, 63, 216–221. [Google Scholar] [CrossRef] [Green Version]
- Acaz-Fonseca, E.; Sanchez-Gonzalez, R.; Azcoitia, I.; Arevalo, M.A.; Garcia-Segura, L.M. Role of astrocytes in the neuroprotective actions of 17β-estradiol and selective estrogen receptor modulators. Mol. Cell. Endocrinol. 2014, 389, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Wen, Y.; Perez, E.; Wang, X.; Day, A.L.; Simpkins, J.W.; Yang, S.H. 17beta-estradiol attenuates blood-brain barrier disruption induced by cerebral ischemia-reperfusion injury in female rats. Brain Res. 2005, 1060, 55–61. [Google Scholar] [CrossRef]
- Xiao, H.; Deng, M.; Yang, B.; Hu, Z.; Tang, J. Pretreatment with 17β-estradiol attenuates cerebral ischemia-induced blood-brain barrier disruption in aged rats: Involvement of antioxidant signaling. Neuroendocrinology 2018, 106, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Asl, S.Z.; Khaksari, M.; Khachki, A.S.; Shahrokhi, N.; Nourizade, S. Contribution of estrogen receptors alpha and beta in the brain response to traumatic brain injury. J. Neurosurg. 2013, 119, 353–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaksari, M.; Hajializadeh, Z.; Shahrokhi, N.; Esmaeili-Mahani, S. Changes in the gene expression of estrogen receptors involved in the protective effect of estrogen in rat’s traumatic brain injury. Brain Res. 2015, 1618, 1–8. [Google Scholar] [CrossRef]
- Na, W.; Lee, J.Y.; Kim, W.S.; Yune, T.Y.; Ju, B.G. 17β-estradiol ameliorates tight junction disruption via repression of MMP transcription. Mol. Endocrinol. 2015, 29, 1347–1361. [Google Scholar] [CrossRef] [PubMed]
- Ardelt, A.A.; McCullough, L.D.; Korach, K.S.; Wang, M.M.; Munzenmaier, D.H.; Hurn, P.D. Estradiol regulates angiopoietin-1 mRNA expression through estrogen receptor-alpha in a rodent experimental stroke model. Stroke 2005, 36, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Pei, F. Estradiol Inhibits Cytokine-Induced Expression of VCAM-1 and ICAM-1 in Cultured Human Endothelial Cells Via AMPK/PPARα Activation. Cell Biochem. Biophys. 2015, 72, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Spence, R.D.; Wisdom, A.J.; Cao, Y.; Hill, H.M.; Mongerson, C.R.; Stapornkul, B.; Itoh, N.; Sofroniew, M.V.; Voskuhl, R.R. Estrogen mediates neuroprotection and anti-inflammatory effects during EAE through ERα signaling on astrocytes but not through ERβ signaling on astrocytes or neurons. J. Neurosci. 2013, 33, 10924–10933. [Google Scholar] [CrossRef]
- Ma, Y.L.; Qin, P.; Feng, D.Y.; Li, Y.; Zhang, L.X.; Liu, Z.Y.; Yin, A.Q.; Tang, W.H.; Dong, H.L.; Meng, L.Z.; et al. Estrogen regulates the expression of Ndrg2 in astrocytes. Brain Res. 2014, 1569, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Stary, C.M.; Xu, L.; Li, L.; Sun, X.; Ouyang, Y.B.; Xiong, X.; Zhao, J.; Giffard, R.G. Inhibition of miR-181a protects female mice from transient focal cerebral ischemia by targeting astrocyte estrogen receptor-α. Mol. Cell. Neurosci. 2017, 82, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Duckles, S.P.; Weiss, J.H.; Li, X.; Krause, D.N. 17β-estradiol prevents cell death and mitochondrial dysfunction by an estrogen receptor-dependent mechanism in astrocytes after oxygen-glucose deprivation/reperfusion. Free Radic. Biol. Med. 2012, 52, 2151–2160. [Google Scholar] [CrossRef]
- Dhandapani, K.M.; Wade, F.M.; Mahesh, V.B.; Brann, D.W. Astrocyte-derived transforming growth factor-{beta} mediates the neuroprotective effects of 17{beta}-estradiol: Involvement of nonclassical genomic signaling pathways. Endocrinology 2005, 146, 2749–2759. [Google Scholar] [CrossRef]
- Karki, P.; Smith, K.; Johnson, J., Jr.; Lee, E. Astrocyte-derived growth factors and estrogen neuroprotection: Role of transforming growth factor-α in estrogen-induced upregulation of glutamate transporters in astrocytes. Mol. Cell. Endocrinol. 2014, 389, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Rogers, S.D.; Peters, C.M.; Pomonis, J.D.; Hagiwara, H.; Ghilardi, J.R.; Mantyh, P.W. Endothelin B receptors are expressed by astrocytes and regulate astrocyte hypertrophy in the normal and injured CNS. Glia 2003, 41, 180–190. [Google Scholar] [CrossRef]
- Wilhelmsson, U.; Li, L.; Pekna, M.; Berthold, C.H.; Blom, S.; Eliasson, C.; Renner, O.; Bushong, E.; Ellisman, M.; Morgan, T.E.; et al. Absence of glial fibrillary acidic protein and vimentin prevents hypertrophy of astrocytic processes and improves post-traumatic regeneration. J. Neurosci. 2004, 24, 5016–5021. [Google Scholar] [CrossRef]
- LeComte, M.D.; Shimada, I.S.; Sherwin, C.; Spees, J.L. Notch1-STAT3-ETBR signaling axis controls reactive astrocyte proliferation after brain injury. Proc. Natl. Acad. Sci. USA 2015, 112, 8726–8731. [Google Scholar] [CrossRef] [PubMed]
- Koyama, Y.; Tanaka, K. Intracerebroventricular administration of an endothelin ET(B)-receptor agonist increases expression of matrix metalloproteinase-2 and -9 in rat brain. J. Pharmacol. Sci. 2010, 114, 433–443. [Google Scholar] [CrossRef]
- Hind, W.H.; Tufarelli, C.; Neophytou, M.; Anderson, S.I.; England, T.J.; O’Sullivan, S.E. Endocannabinoids modulate human blood-brain barrier permeability in vitro. Br. J. Pharmacol. 2015, 172, 3015–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Yun, D.; Zhang, Y.; Tao, Y.; Tan, Q.; Qiao, F.; Luo, B.; Liu, Y.; Fan, R.; Xian, J.; et al. A cannabinoid receptor 2 agonist reduces blood-brain barrier damage via induction of MKP-1 after intracerebral hemorrhage in rats. Brain Res. 2018, 1697, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.C.; Shin, W.H.; Baek, J.Y.; Cho, E.J.; Baik, H.H.; Kim, S.R.; Won, S.Y.; Jin, B.K. CB2 receptor activation prevents glial-derived neurotoxic mediator production, BBB leakage and peripheral immune cell infiltration and rescues dopamine neurons in the MPTP model of Parkinson’s disease. Exp. Mol. Med. 2016, 48, e205. [Google Scholar] [CrossRef] [PubMed]
- Katz, P.S.; Sulzer, J.K.; Impastato, R.A.; Teng, S.X.; Rogers, E.K.; Molina, P.E. Endocannabinoid degradation inhibition improves neurobehavioral function, blood-brain barrier integrity, and neuroinflammation following mild traumatic brain injury. J. Neurotrauma 2015, 32, 297–306. [Google Scholar] [CrossRef]
- Meng, X.D.; Wei, D.; Li, J.; Kang, J.J.; Wu, C.; Ma, L.; Yang, F.; Zhu, G.M.; Ou-Yang, T.P.; Liu, Y.Y.; et al. Astrocytic expression of cannabinoid type 1 receptor in rat and human sclerotic hippocampi. Int. J. Clin. Exp. Pathol. 2014, 7, 2825–2837. [Google Scholar]
- Kozela, E.; Juknat, A.; Vogel, Z. Modulation of astrocyte activity by cannabidiol, a nonpsychoactive cannabinoid. Int. J. Mol. Sci. 2017, 18, 1669. [Google Scholar] [CrossRef]
- Hegyi, Z.; Oláh, T.; Kőszeghy, Á.; Piscitelli, F.; Holló, K.; Pál, B.; Csernoch, L.; Di Marzo, V.; Antal, M. CB1 receptor activation induces intracellular Ca2+ mobilization and 2-arachidonoylglycerol release in rodent spinal cord astrocytes. Sci. Rep. 2018, 8, 10562. [Google Scholar] [CrossRef]
- Fernández-Trapero, M.; Espejo-Porras, F.; Rodríguez-Cueto, C.; Coates, J.R.; Pérez-Díaz, C.; de Lago, E.; Fernández-Ruiz, J. Upregulation of CB2 receptors in reactive astrocytes in canine degenerative myelopathy, a disease model of amyotrophic lateral sclerosis. Dis. Models Mech. 2017, 10, 551–558. [Google Scholar] [CrossRef]
- Ma, Q.; Dasgupta, C.; Li, Y.; Huang, L.; Zhang, L. MicroRNA-210 suppresses junction proteins and disrupts blood-brain barrier integrity in neonatal rat hypoxic-ischemic brain injury. Int. J. Mol. Sci. 2017, 18, 1356. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, M.D.; Xia, Y.P.; Gao, Y.; Zhu, Y.Y.; Chen, S.C.; Mao, L.; He, Q.W.; Yue, Z.Y.; Hu, B. MicroRNA-130a regulates cerebral ischemia-induced blood-brain barrier permeability by targeting Homeobox A5. FASEB J. 2018, 32, 935–944. [Google Scholar] [CrossRef]
- Wan, Y.; Jin, H.J.; Zhu, Y.Y.; Fang, Z.; Mao, L.; He, Q.; Xia, Y.P.; Li, M.; Li, Y.; Chen, X.; et al. MicroRNA-149-5p regulates blood-brain barrier permeability after transient middle cerebral artery occlusion in rats by targeting S1PR2 of pericytes. FASEB J. 2018, 32, 3133–3148. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, J.; Ma, Y.; Tang, G.; Liu, Y.; Chen, X.; Zhang, Z.; Zeng, L.; Wang, Y.; Ouyang, Y.B.; et al. MicroRNA-29b is a therapeutic target in cerebral ischemia associated with aquaporin 4. J. Cereb. Blood Flow Metab. 2015, 35, 1977–1984. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.B.; Sun, Z.L.; Feng, D.F. The role of microRNA in traumatic brain injury. Neuroscience 2017, 367, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Reijerkerk, A.; Lopez-Ramirez, M.A.; van Het Hof, B.; Drexhage, J.A.; Kamphuis, W.W.; Kooij, G.; Vos, J.B.; van der Pouw Kraan, T.C.; van Zonneveld, A.J.; Horrevoets, A.J.; et al. MicroRNAs regulate human brain endothelial cell-barrier function in inflammation: Implications for multiple sclerosis. J. Neurosci. 2013, 33, 6857–6863. [Google Scholar] [CrossRef]
- Bhalala, O.G.; Pan, L.; Sahni, V.; McGuire, T.L.; Gruner, K.; Tourtellotte, W.G.; Kessler, J.A. microRNA-21 regulates astrocytic response following spinal cord injury. J. Neurosci. 2012, 32, 17935–17947. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.B.; Xu, L.; Lu, Y.; Sun, X.; Yue, S.; Xiong, X.X.; Giffard, R.G. Astrocyte-enriched miR-29a targets PUMA and reduces neuronal vulnerability to forebrain ischemia. Glia 2013, 61, 1784–1794. [Google Scholar] [CrossRef] [Green Version]
- Hong, P.; Jiang, M.; Li, H. Functional requirement of dicer1 and miR-17-5p in reactive astrocyte proliferation after spinal cord injury in the mouse. Glia 2014, 62, 2044–2060. [Google Scholar] [CrossRef]
- Sison, S.L.; Patitucci, T.N.; Seminary, E.R.; Villalon, E.; Lorson, C.L.; Ebert, A.D. Astrocyte-produced miR-146a as a mediator of motor neuron loss in spinal muscular atrophy. Hum. Mol. Genet. 2017, 26, 3409–3420. [Google Scholar]
- Meares, G.P.; Rajbhandari, R.; Gerigk, M.; Tien, C.L.; Chang, C.; Fehling, S.C.; Rowse, A.; Mulhern, K.C.; Nair, S.; Gray, G.K.; et al. MicroRNA-31 is required for astrocyte specification. Glia 2018, 66, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Korotkov, A.; Broekaart, D.W.M.; van Scheppingen, J.; Anink, J.J.; Baayen, J.C.; Idema, S.; Gorter, J.A.; Aronica, E.; van Vliet, E.A. Increased expression of matrix metalloproteinase 3 can be attenuated by inhibition of microRNA-155 in cultured human astrocytes. J. Neuroinflamm. 2018, 15, 211. [Google Scholar] [CrossRef]
- Wang, C.Y.; Yang, S.H.; Tzeng, S.F. MicroRNA-145 as one negative regulator of astrogliosis. Glia 2015, 63, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.C.; Mei, P.J.; Chen, C.; Miao, F.A.; Zhang, H.; Li, Z.L. MiR-29c inhibits glioma cell proliferation, migration, invasion and angiogenesis. J. Neurooncol. 2013, 115, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; He, X.; Wang, Y.; Tang, Y.; Zheng, C.; Cai, H.; Liu, J.; Wang, Y.; Fu, Y.; Yang, G.Y. MicroRNA-210 overexpression induces angiogenesis and neurogenesis in the normal adult mouse brain. Gene Ther. 2014, 21, 37–43. [Google Scholar] [CrossRef] [PubMed]
- He, Q.W.; Li, Q.; Jin, H.J.; Zhi, F.; Suraj, B.; Zhu, Y.Y.; Xia, Y.P.; Mao, L.; Chen, X.L.; Hu, B. MiR-150 regulates poststroke cerebral angiogenesis via vascular endothelial growth factor in rats. CNS Neurosci. Ther. 2016, 22, 507–517. [Google Scholar] [CrossRef]
- Deng, X.; Zhong, Y.; Gu, L.; Shen, W.; Guo, J. MiR-21 involve in ERK-mediated upregulation of MMP9 in the rat hippocampus following cerebral ischemia. Brain Res. Bull. 2013, 94, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Chopp, M.; Lu, Y.; Buller, B.; Jiang, F. MiR-15b and miR-152 reduce glioma cell invasion and angiogenesis via NRP-2 and MMP-3. Cancer Lett. 2013, 329, 146–154. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
The BBB comprises endothelial cells, pericytes and astrocytes. The low permeability to serum components results from dense formation of TJs between brain microvascular endothelial cells. TJs comprise TJ-related proteins including claudin-5, occludin and ZO-1. Astrocytes produce several factors that modulate the expression of the TJ-related proteins and regulate paracellular transport across vascular endothelial cells. In addition, astrocyte-derived factors affect the expression of endothelial ICAM-1 and VCAM-1, which interact with VLA-4 and LFA-1 in leukocytes. Increased ICAM-1 and VCAM-1 expression promotes leukocyte infiltration into the CNS.
Figure 1.
The BBB comprises endothelial cells, pericytes and astrocytes. The low permeability to serum components results from dense formation of TJs between brain microvascular endothelial cells. TJs comprise TJ-related proteins including claudin-5, occludin and ZO-1. Astrocytes produce several factors that modulate the expression of the TJ-related proteins and regulate paracellular transport across vascular endothelial cells. In addition, astrocyte-derived factors affect the expression of endothelial ICAM-1 and VCAM-1, which interact with VLA-4 and LFA-1 in leukocytes. Increased ICAM-1 and VCAM-1 expression promotes leukocyte infiltration into the CNS.
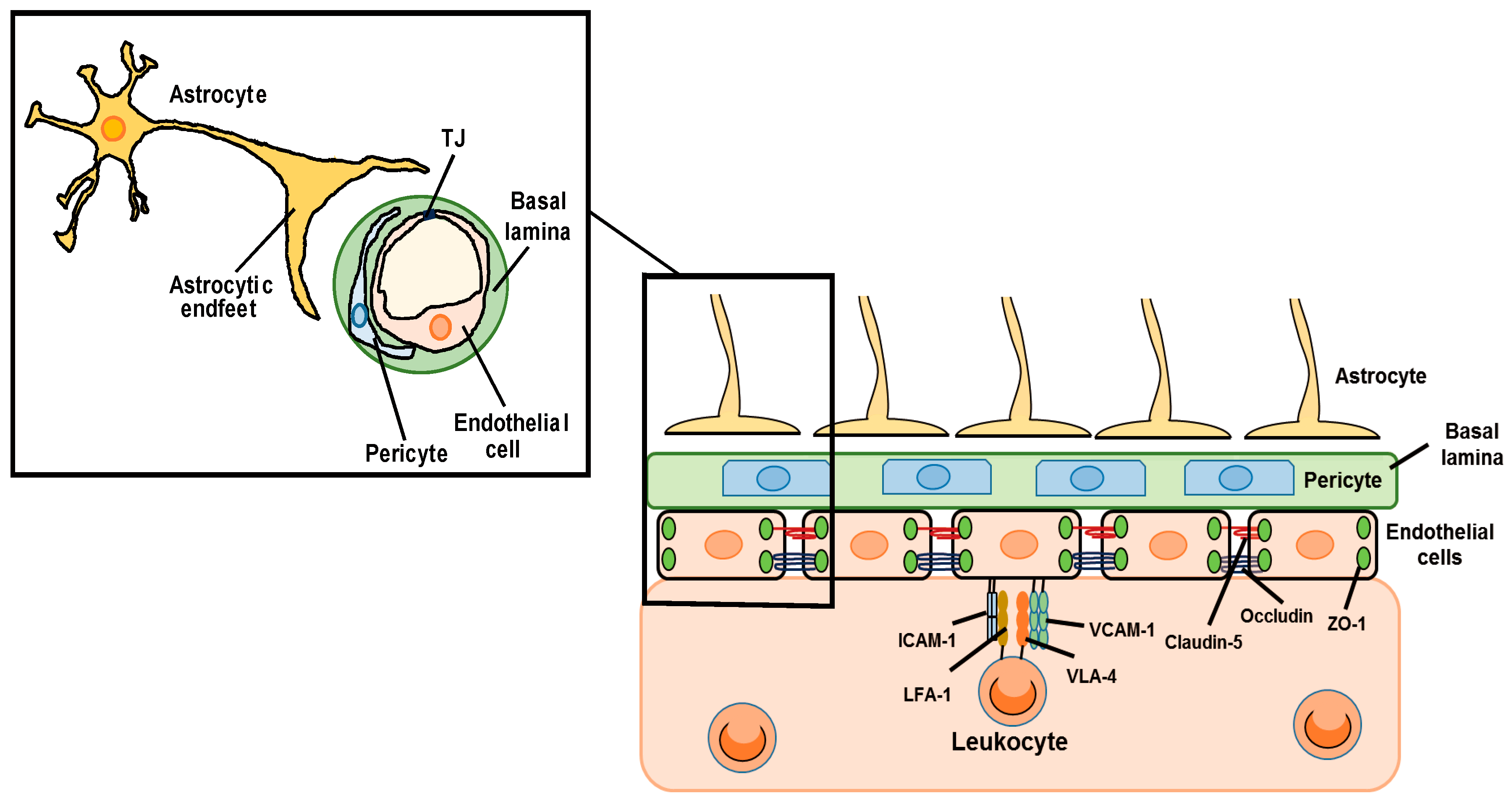
Figure 2.
Dual roles of astrocyte-derived factors in the regulation of BBB functions. In brain disorders, astrocytes release various kinds of extracellular signaling molecules. (A) Vascular permeability factors: Astrocyte-derived vascular endothelial growth factors (VEGFs), matrix metalloproteinases (MMPs), nitric oxide (NO), glutamate and endothelins (ETs) cause endothelial apoptosis and downregulation of TJ-related proteins, resulting in BBB disruption. Some of these factors also upregulate endothelial CAMs, which induce leukocyte transmigration. (B) Vascular protective factors: Astrocyte-derived angiopoietin-1 (ANG-1), sonic hedgehog (SHH), glial-derived neurotrophic factor (GDNF), retinoic acid (RA), insulin-like growth factor-1 (IGF-1) and apolipoprotein E (APOE) protect endothelial cells from apoptosis and promote recovery of TJ function. Some of these factors also decrease endothelial CAMs’ expression and reduce leukocyte infiltration.
Figure 2.
Dual roles of astrocyte-derived factors in the regulation of BBB functions. In brain disorders, astrocytes release various kinds of extracellular signaling molecules. (A) Vascular permeability factors: Astrocyte-derived vascular endothelial growth factors (VEGFs), matrix metalloproteinases (MMPs), nitric oxide (NO), glutamate and endothelins (ETs) cause endothelial apoptosis and downregulation of TJ-related proteins, resulting in BBB disruption. Some of these factors also upregulate endothelial CAMs, which induce leukocyte transmigration. (B) Vascular protective factors: Astrocyte-derived angiopoietin-1 (ANG-1), sonic hedgehog (SHH), glial-derived neurotrophic factor (GDNF), retinoic acid (RA), insulin-like growth factor-1 (IGF-1) and apolipoprotein E (APOE) protect endothelial cells from apoptosis and promote recovery of TJ function. Some of these factors also decrease endothelial CAMs’ expression and reduce leukocyte infiltration.
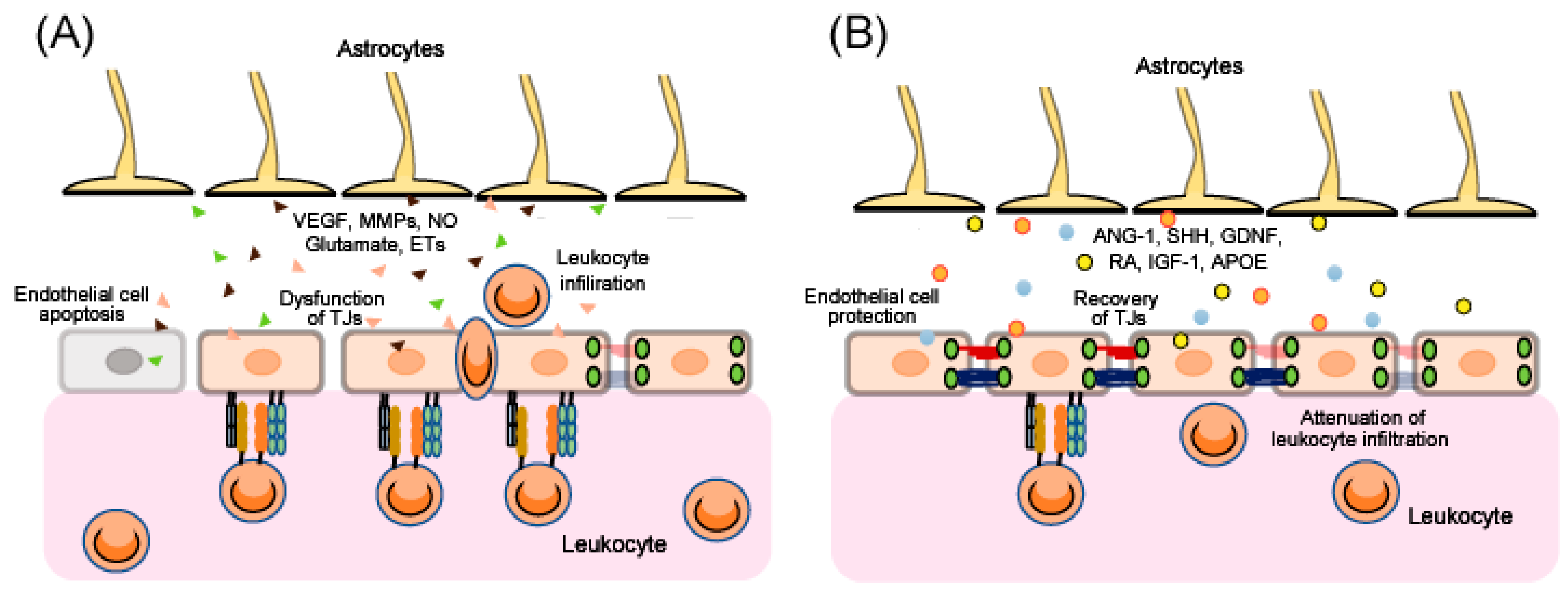
Figure 3.
Therapeutic strategy for BBB protection and recovery of BBB function by controlling the function of astrocyte-derived factors. Astrocytic estrogen receptor (ER), ETB receptor (ETBR), cannabinoid receptor (CBR) and microRNAs are involved in the regulation of astrocyte-derived factors production. Hence, these receptors and microRNAs are candidates for protection/recovery of BBB functions by modulating the actions of astrocyte-derived factors.
Figure 3.
Therapeutic strategy for BBB protection and recovery of BBB function by controlling the function of astrocyte-derived factors. Astrocytic estrogen receptor (ER), ETB receptor (ETBR), cannabinoid receptor (CBR) and microRNAs are involved in the regulation of astrocyte-derived factors production. Hence, these receptors and microRNAs are candidates for protection/recovery of BBB functions by modulating the actions of astrocyte-derived factors.

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Michinaga, S.; Koyama, Y. Dual Roles of Astrocyte-Derived Factors in Regulation of Blood-Brain Barrier Function after Brain Damage. Int. J. Mol. Sci. 2019, 20, 571. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030571
AMA Style
Michinaga S, Koyama Y. Dual Roles of Astrocyte-Derived Factors in Regulation of Blood-Brain Barrier Function after Brain Damage. International Journal of Molecular Sciences. 2019; 20(3):571. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030571
Chicago/Turabian StyleMichinaga, Shotaro, and Yutaka Koyama. 2019. "Dual Roles of Astrocyte-Derived Factors in Regulation of Blood-Brain Barrier Function after Brain Damage" International Journal of Molecular Sciences 20, no. 3: 571. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030571
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.