Identification of Early Salinity Stress-Responsive Proteins in Dunaliella salina by isobaric tags for relative and absolute quantitation (iTRAQ)-Based Quantitative Proteomic Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
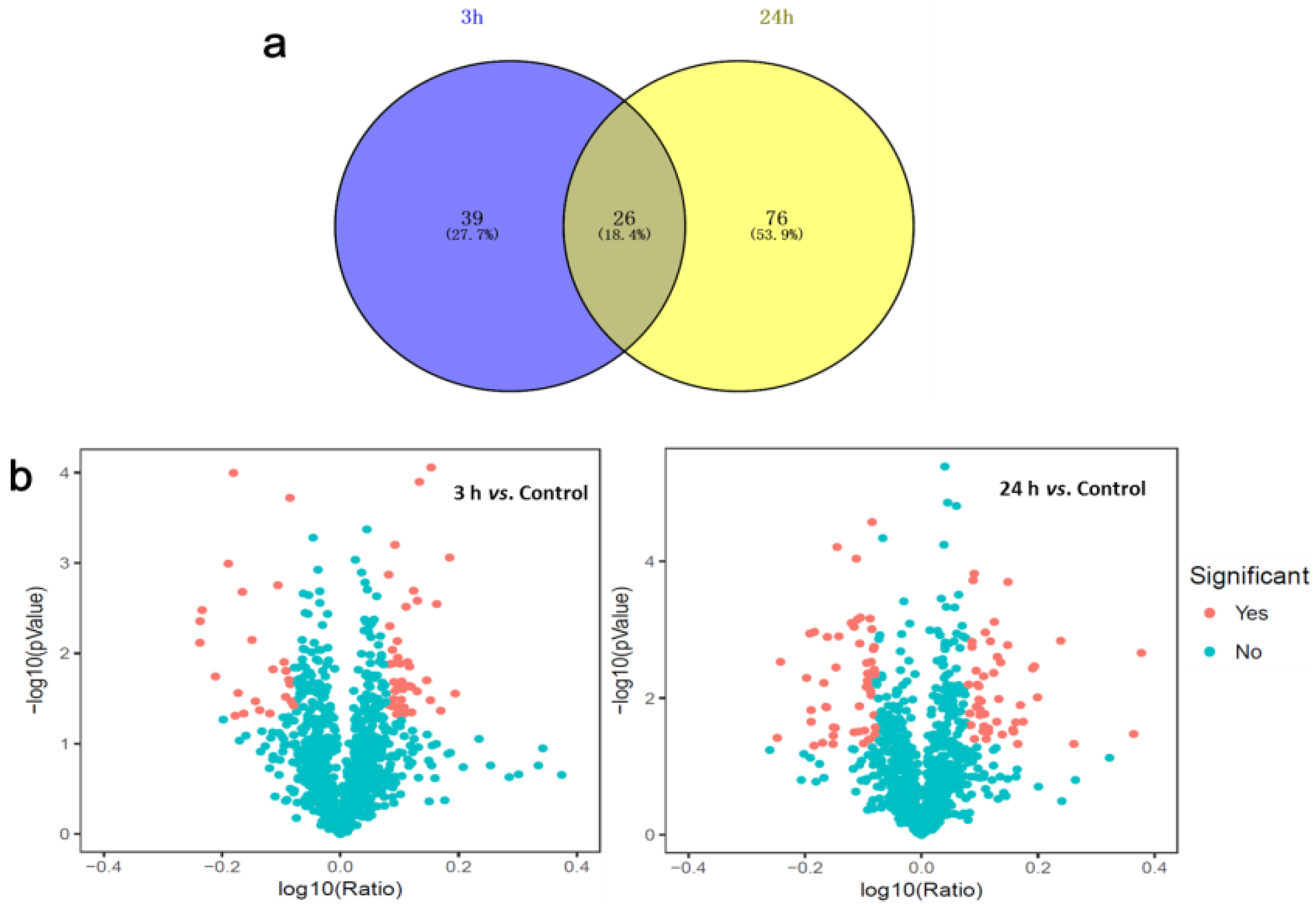
2.1. Identification of Dunaliella Salina Differentially Abundant Proteins (DAPs) Using Isobaric Tags for Relative and Absolute Quantitation (iTRAQ)
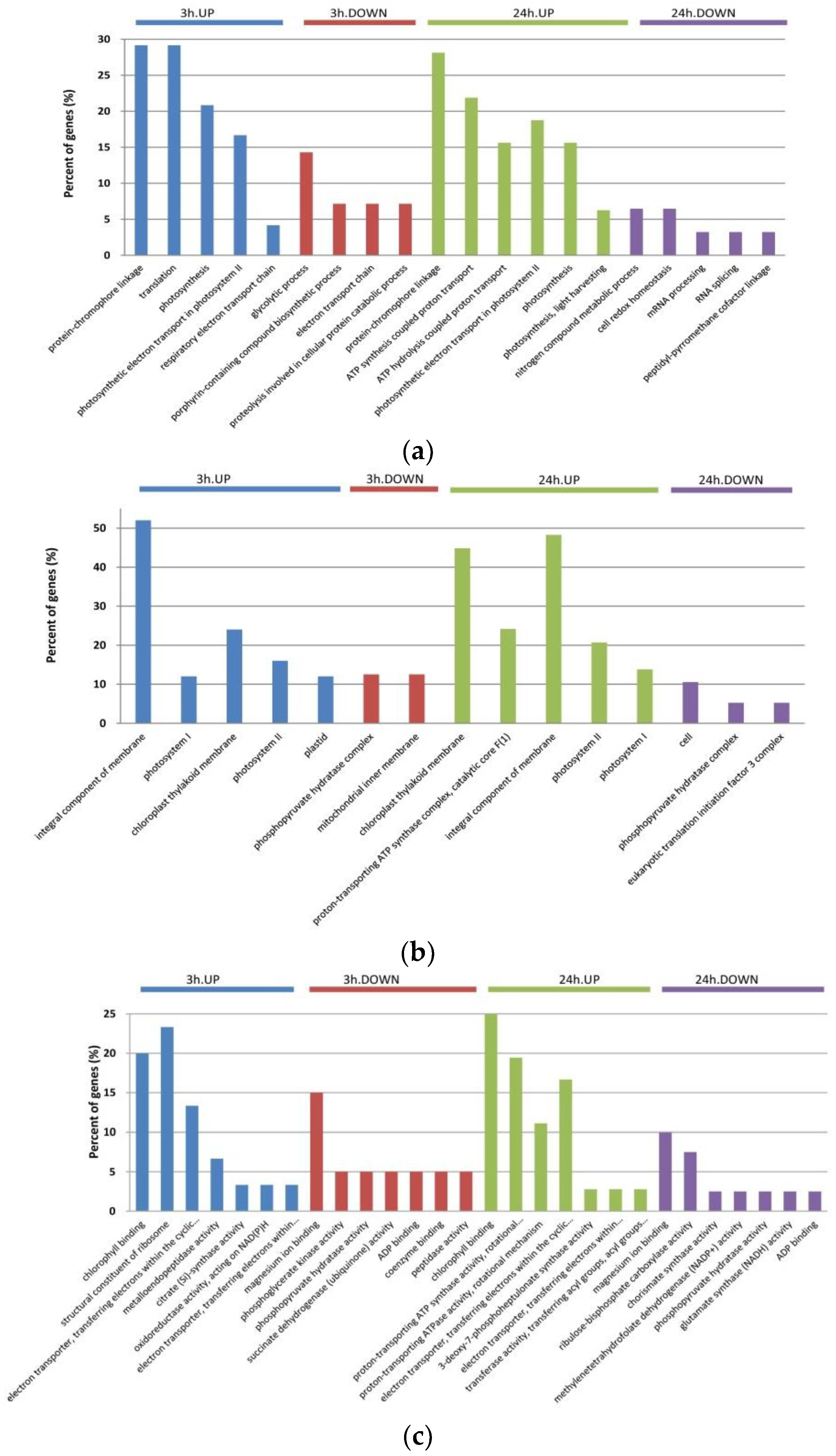
2.2. Gene Ontology (GO) Annotation of DAPs in Dunaliella Salina
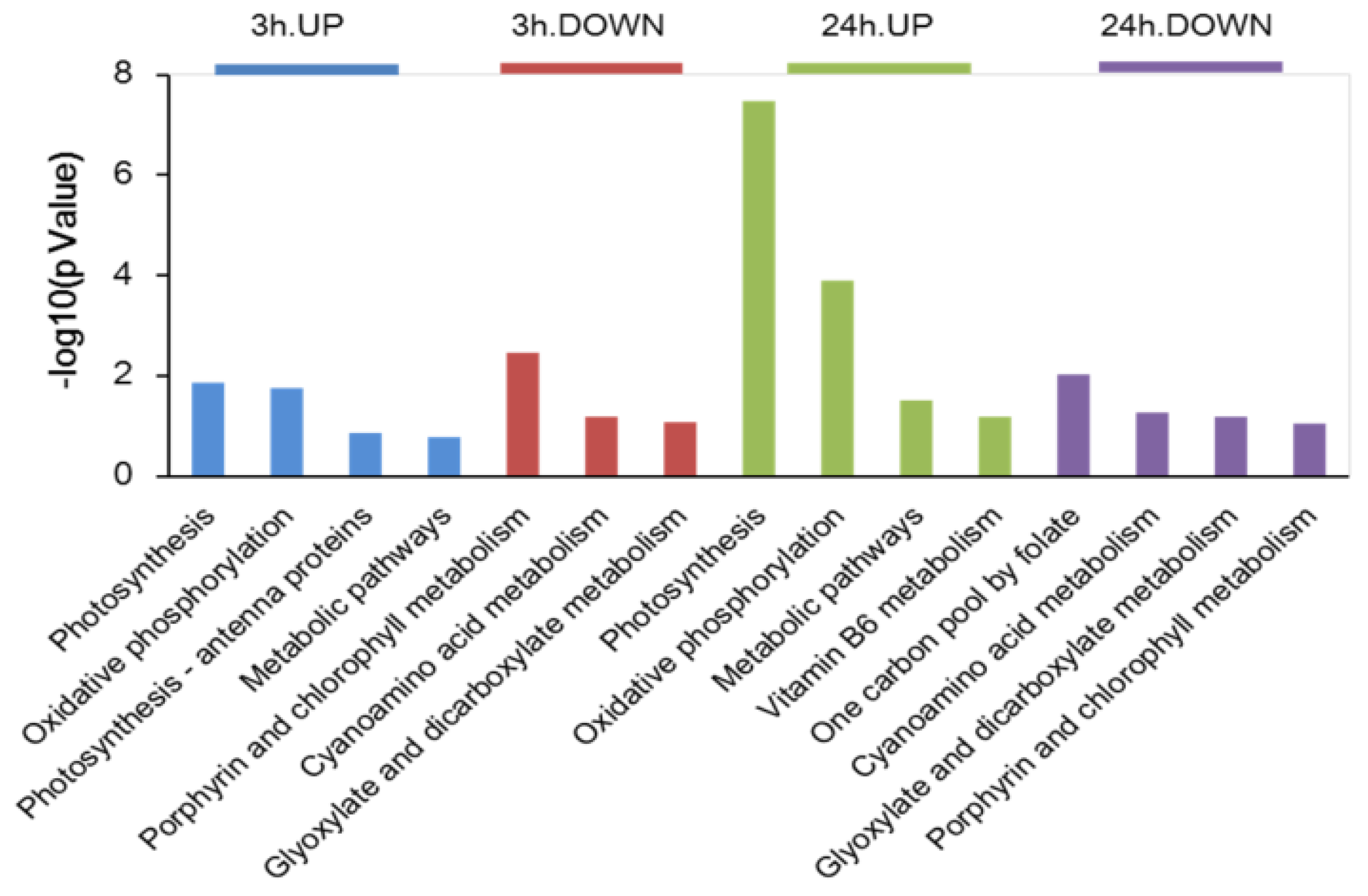
2.3. Kyoto Encyclopedia of Genes and Genomes (KEGG) Analysis of DAPs in Dunaliella Salina
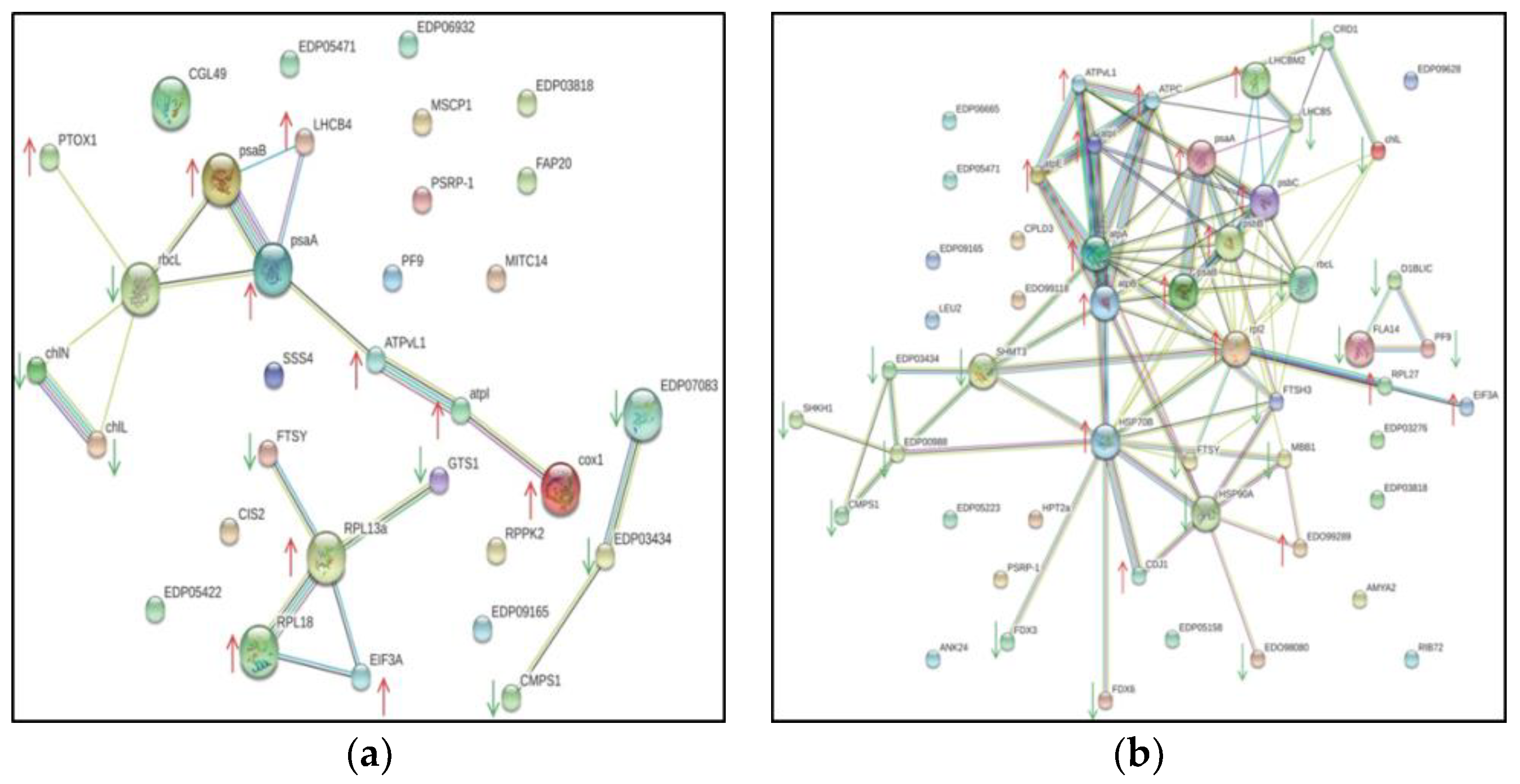
2.4. Search Tool for the Retrieval of Interacting Genes (STRING)-Based Protein-Protein Interaction (PPI) Analysis
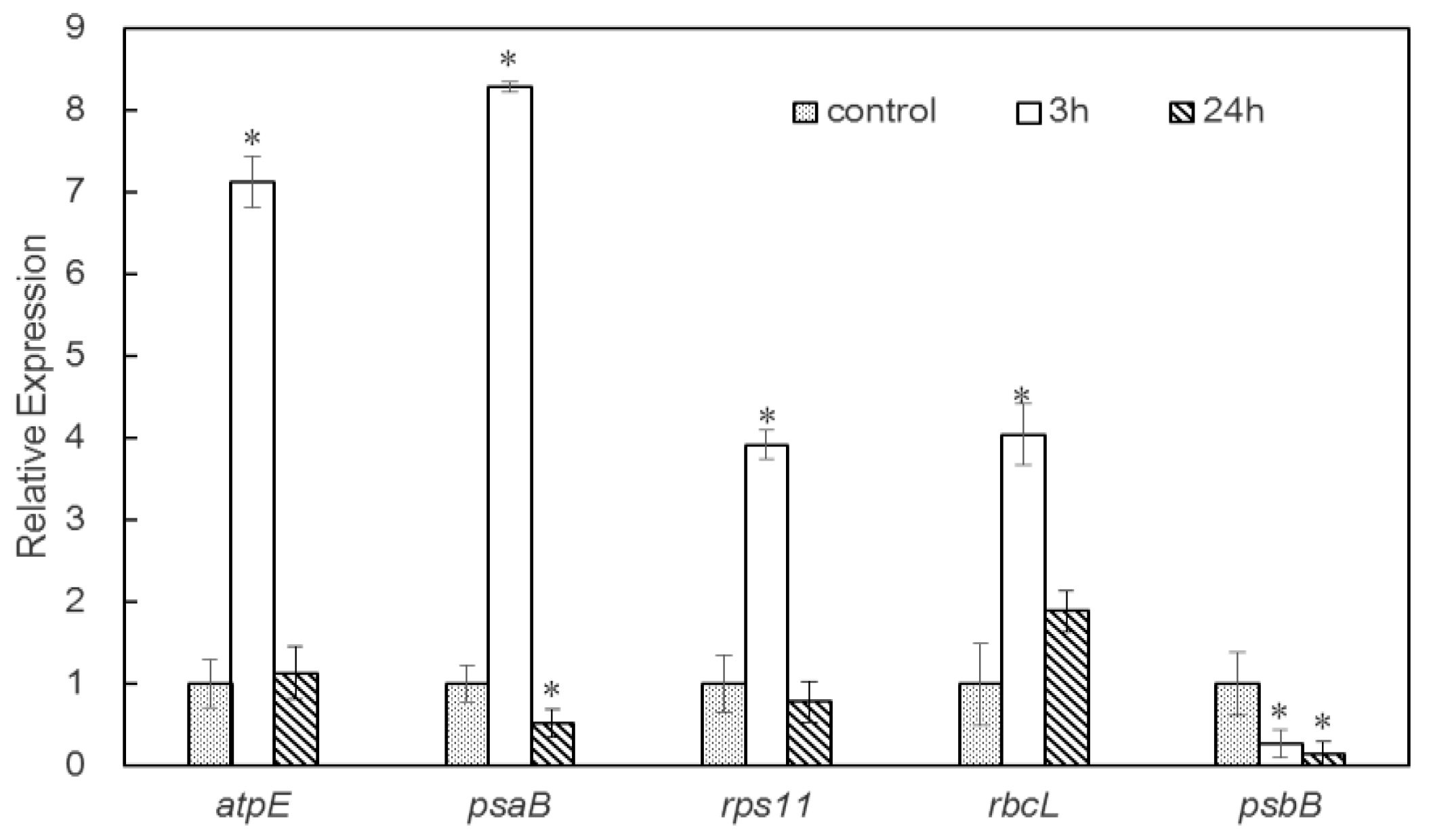
2.5. Analysis of Transcripts Encoding Selected DAPs
3. Discussion
4. Materials and Methods
4.1. Algae Culture
4.2. Protein Extraction and Quantification
4.3. Protein Digestionand iTRAQ Labeling and Fractionation by Strong Cationic Exchange (SCX)
4.4. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS) Analysis
4.5. Analysis of Differentially Abundant Proteins
4.6. Go, KEGG and STRING Enrichment Analyses
4.7. RNA Extraction and qRT-PCR
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| iTRAQ | Isobaric Tags for Relative and Absolute Quantitation |
| DAPs | differentially abundant proteins |
| BP | biological process |
| MF | molecular function |
| CC | cellular components |
| GO | Gene Ontology |
| KEGG | The Kyoto Encyclopedia of Genes and Genomes |
| STRING | Search Tool for the Retrieval of Interacting Genes |
| PPI | predict protein interaction |
| LHC | light-harvesting complex |
| qRT-PCR | quantitative real-time polymerase chain reaction |
| SDS-PAGE | sodium dodecyl sulfate -polyacrylamide gel electrophoresis |
| SCX | strong cationic exchange |
| LC-MS/MS | liquid chromatography-tandem mass spectrometry |
References
- Zhang, H.; Han, B.; Wang, T.; Chen, S.; Li, H.; Zhang, Y.; Dai, S. Mechanisms of plant salt response: Insights from proteomics. J. Proteome Res. 2012, 11, 49–67. [Google Scholar] [CrossRef]
- Parihar, P.; Singh, S.; Singh, R.; Singh, V.P.; Prasad, S.M. Effect of salinity stress on plants and its tolerance strategies: A review. Environ. Sci. Pollut. Res. Int. 2015, 22, 4056–4075. [Google Scholar] [CrossRef]
- Katz, A.; Waridel, P.; Shevchenko, A.; Pick, U. Salt-induced changes in the plasma membrane proteome of the halotolerant alga Dunaliella salina as revealed by blue native gel electrophoresis and nano-LC-MS/MS analysis. Mol. Cell. Proteom. 2007, 6, 1459–1472. [Google Scholar] [CrossRef]
- Wei, S.; Bian, Y.; Zhao, Q.; Chen, S.; Mao, J.; Song, C.; Cheng, K.; Xiao, Z.; Zhang, C.; Ma, W.; et al. Salinity-induced palmella formation mechanism in halotolerant algae Dunaliella salina revealed by quantitative proteomics and phosphoproteomics. Front. Plant Sci. 2017, 8, 810. [Google Scholar] [CrossRef]
- Morowvat, M.H.; Ghasemi, Y. Culture medium optimization for enhanced β-carotene and biomass production by Dunaliella salina in mixotrophic culture. Biocatal. Agric. Biotechnol. 2016, 7, 217–223. [Google Scholar] [CrossRef]
- Gong, W.-F.; Zhao, L.-N.; Hu, B.; Chen, X.-W.; Zhang, F.; Zhu, Z.-M.; Chen, D.-F. Identifying novel salt-tolerant genes from Dunaliella salina using a Haematococcus pluvialis expression system. Plant Cell Tissue Organ 2014, 117, 113–124. [Google Scholar] [CrossRef]
- Chai, X.; Liu, Y.; Wang, Y.; Wu, T.; Liu, L. Prokaryotic Expression, Purification and Cloning of MAPKK Kinase Gene DsMAPKKK from Dunaliella salina. Chin. Agric. Sci. Bull. 2015, 24, 1085–1091. [Google Scholar]
- Dani, V.; Simon, W.J.; Duranti, M.; Croy, R.R. Changes in the tobacco leaf apoplast proteome in response to salt stress. Proteomics 2005, 5, 737–745. [Google Scholar] [CrossRef]
- Liu, C.W.; Chang, T.S.; Hsu, Y.K.; Wang, A.Z.; Yen, H.C.; Wu, Y.P.; Wang, C.S.; Lai, C.C. Comparative proteomic analysis of early salt stress responsive proteins in roots and leaves of rice. Proteomics 2014, 14, 1759–1775. [Google Scholar] [CrossRef]
- Lv, D.W.; Subburaj, S.; Cao, M.; Yan, X.; Li, X.; Appels, R.; Sun, D.F.; Ma, W.; Yan, Y.M. Proteome and phosphoproteome characterization reveals new response and defense mechanisms of Brachypodium distachyon leaves under salt stress. Mol. Cell. Proteom. 2014, 13, 632–652. [Google Scholar] [CrossRef]
- McLoughlin, F.; Arisz, S.A.; Dekker, H.L.; Kramer, G.; de Koster, C.G.; Haring, M.A.; Munnik, T.; Testerink, C. Identification of novel candidate phosphatidic acid-binding proteins involved in the salt-stress response of Arabidopsis thaliana roots. Biochem. J. 2013, 450, 573–581. [Google Scholar] [CrossRef] [Green Version]
- Hossain, Z.; Khatoon, A.; Komatsu, S. Soybean proteomics for unraveling abiotic stress response mechanism. J. Proteome Res. 2013, 12, 4670–4684. [Google Scholar] [CrossRef]
- Ji, W.; Cong, R.; Li, S.; Li, R.; Qin, Z.; Li, Y.; Zhou, X.; Chen, S.; Li, J. Comparative proteomic analysis of soybean leaves and roots by iTRAQ provides insights into response mechanisms to short-term salt stress. Front Plant Sci. 2016, 7, 573. [Google Scholar] [CrossRef]
- Sobhanian, H.; Razavizadeh, R.; Nanjo, Y.; Ehsanpour, A.A.; Jazii, F.R.; Motamed, N.; Komatsu, S. Proteome analysis of soybean leaves, hypocotyls and roots under salt stress. Proteome Sci. 2010, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Luo, Q.; Wang, Q.; Zhang, X.; Qi, Z.; Xu, F.; Lei, X.; Cao, Y.; Chow, W.S.; Sun, G. Physiological and proteomic responses to salt stress in chloroplasts of diploid and tetraploid black locust (Robinia pseudoacacia L.). Sci. Rep. 2016, 6, 23098. [Google Scholar] [CrossRef]
- Jia, Y.L.; Chen, H.; Zhang, C.; Gao, L.J.; Wang, X.C.; Qiu, L.L.; Wu, J.F. Proteomic analysis of halotolerant proteins under high and low salt stress in Dunaliella salina using two-dimensional differential in-gel electrophoresis. Genet. Mol. Biol. 2016, 39, 239–247. [Google Scholar] [CrossRef]
- Liska, A.J.; Shevchenko, A.; Pick, U.; Katz, A. Enhanced photosynthesis and redox energy production contribute to salinity tolerance in Dunaliella as revealed by homology-based proteomics. Plant Physiol. 2004, 136, 2806–2817. [Google Scholar] [CrossRef]
- Nam, M.H.; Huh, S.M.; Kim, K.M.; Park, W.J.; Seo, J.B.; Cho, K.; Kim, D.Y.; Kim, B.G.; Yoon, I.S. Comparative proteomic analysis of early salt stress-responsive proteins in roots of SnRK2 transgenic rice. Proteome Sci. 2012, 10, 25. [Google Scholar] [CrossRef]
- Yin, X.; Komatsu, S. Comprehensive analysis of response and tolerant mechanisms in early-stage soybean at initial-flooding stress. J. Proteom. 2017, 169, 225–232. [Google Scholar] [CrossRef]
- Chen, T.; Zhang, L.; Shang, H.; Liu, S.; Peng, J.; Gong, W.; Shi, Y.; Zhang, S.; Li, J.; Gong, J.; et al. iTRAQ-based quantitative proteomic analysis of cotton roots and Leaves reveals pathways associated with salt stress. PLoS ONE 2016, 11, e0148487. [Google Scholar] [CrossRef]
- Lan, P.; Li, W.; Wen, T.N.; Shiau, J.Y.; Wu, Y.C.; Lin, W.; Schmidt, W. iTRAQ protein profile analysis of Arabidopsis roots reveals new aspects critical for iron homeostasis. Plant Physiol. 2011, 155, 821–834. [Google Scholar] [CrossRef]
- Long, R.; Gao, Y.; Sun, H.; Zhang, T.; Li, X.; Li, M.; Sun, Y.; Kang, J.; Wang, Z.; Ding, W.; et al. Quantitative proteomic analysis using iTRAQ to identify salt-responsive proteins during the germination stage of two Medicago species. Sci. Rep. 2018, 8, 9553. [Google Scholar] [CrossRef]
- Xia, F.; Yao, X.; Tang, W.; Xiao, C.; Yang, M.; Zhou, B. Isobaric tags for relative and absolute quantitation (iTRAQ)-based proteomic analysis of Hugan Qingzhi and its protective properties against free fatty acid-induced L02 hepatocyte Injury. Front. Pharmacol. 2017, 8, 99. [Google Scholar] [CrossRef]
- Vedeler, A.; Hollas, H.; Grindheim, A.K.; Raddum, A.M. Multiple roles of annexin A2 in post-transcriptional regulation of gene expression. Curr. Protein Pept. Sci. 2012, 13, 401–412. [Google Scholar] [CrossRef]
- Zhao, Y.; Hou, Y. Identification of NaHCO3 stress responsive proteins in Dunaliella salina HTBS using iTRAQ-based analysis. J. Proteom. Bioinform. 2016, 9, 137–143. [Google Scholar] [CrossRef]
- Ge, Y.; Ning, Z.; Wang, Y.; Zheng, Y.; Zhang, C.; Figeys, D. Quantitative proteomic analysis of Dunaliella salina upon acute arsenate exposure. Chemosphere 2016, 145, 112–118. [Google Scholar] [CrossRef]
- Laurent, J.M.; Vogel, C.; Kwon, T.; Craig, S.A.; Boutz, D.R.; Huse, H.K.; Nozue, K.; Walia, H.; Whiteley, M.; Ronald, P.C. Protein abundances are more conserved than mRNA abundances across diverse taxa. Proteomics 2010, 10, 4209–4212. [Google Scholar] [CrossRef] [Green Version]
- Drummond, D.A.; Bloom, J.D.; Adami, C.; Wilke, C.O.; Arnold, F.H. Why highly expressed proteins evolve slowly. Proc. Natl. Acad. Sci. USA 2005, 102, 14338–14343. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227. [Google Scholar] [CrossRef]
- Zhao, P.; Cui, R.; Xu, P.; Wu, J.; Mao, J.L.; Chen, Y.; Zhou, C.Z.; Yu, L.H.; Xiang, C.B. ATHB17 enhances stress tolerance by coordinating photosynthesis associated nuclear gene and ATSIG5 expression in response to abiotic stress. Sci. Rep. 2017, 7, 45492. [Google Scholar] [CrossRef] [Green Version]
- Asrar, H.; Hussain, T.; Hadi, S.; Gul, B.; Nielsen, B.; Khan, M. Salinity induced changes in light harvesting and carbon assimilating complexes of Desmostachya bipinnata (L.) Staph. Environ. Exp. Bot. 2017, 135, 86–95. [Google Scholar] [CrossRef]
- Shu, S.; Yuan, Y.; Chen, J.; Sun, J.; Zhang, W.; Tang, Y.; Zhong, M.; Guo, S. The role of putrescine in the regulation of proteins and fatty acids of thylakoid membranes under salt stress. Sci. Rep. 2015, 5, 14390. [Google Scholar] [CrossRef] [Green Version]
- Caffarri, S.; Tibiletti, T.; Jennings, R.; Santabarbara, S. A comparison between plant Photosystem I and Photosystem II architecture and functioning. Curr. Protein Pept. Sci. 2014, 15, 296–331. [Google Scholar] [CrossRef]
- Natali, A.; Croce, R. Characterization of the major light-harvesting complexes (LHCBM) of the green alga Chlamydomonas reinhardtii. PLoS ONE 2015, 10, e0119211. [Google Scholar] [CrossRef]
- Zhang, H.; Li, L.; Ye, T.; Chen, R.; Gao, X.; Xu, Z. Molecular characterization, expression pattern and function analysis of the OsHSP90 family in rice. Biotechnol. Biotechnol. Equip. 2016, 30, 669–676. [Google Scholar] [CrossRef]
- Rutgers, M.; Muranaka, L.S.; Schulz-Raffelt, M.; Thoms, S.; Schurig, J.; Willmund, F.; Schroda, M. Not changes in membrane fluidity but proteotoxic stress triggers heat shock protein expression in Chlamydomonas reinhardtii. Plant Cell Environ. 2017, 40, 2987–3001. [Google Scholar] [CrossRef]
- Tang, T.; Yu, A.; Li, P.; Yang, H.; Liu, G.; Liu, L. Sequence analysis of the Hsp70 family in moss and evaluation of their functions in abiotic stress responses. Sci. Rep. 2016, 6, 33650. [Google Scholar] [CrossRef] [Green Version]
- Yokthongwattana, C.; Mahong, B.; Roytrakul, S.; Phaonaklop, N.; Narangajavana, J.; Yokthongwattana, K. Proteomic analysis of salinity-stressed Chlamydomonas reinhardtii revealed differential suppression and induction of a large number of important housekeeping proteins. Planta 2012, 235, 649–659. [Google Scholar] [CrossRef]
- Hahn, A.; Bublak, D.; Schleiff, E.; Scharf, K.D. Crosstalk between Hsp90 and Hsp70 chaperones and heat stress transcription factors in tomato. Plant Cell 2011, 23, 741–755. [Google Scholar] [CrossRef]
- Liu, A.; Xiao, Z.; Li, M.W.; Wong, F.L.; Yung, W.S.; Ku, Y.S.; Wang, Q.; Wang, X.; Xie, M.; Yim, A.K.; et al. Transcriptomic reprogramming in soybean seedlings under salt stress. Plant Cell Environ. 2018, 12, e0189159. [Google Scholar] [CrossRef]
- Katz, A.; Avron, M. Determination of intracellular osmotic volume and sodium concentration in dunaliella. Plant Physiol. 1985, 78, 817–820. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Polle, J.E.W.; Barry, K.; Cushman, J.; Schmutz, J.; Tran, D.; Hathwaik, L.T.; Yim, W.C.; Jenkins, J.; McKie-Krisberg, Z.; Prochnik, S.; et al. Draft Nuclear Genome Sequence of the Halophilic and Beta-Carotene-Accumulating Green Alga Dunaliella salina Strain CCAP19/18. Genome Announc. 2017, 5, e01105–e01117. [Google Scholar] [CrossRef]
- Vizcaino, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. Data, information, knowledge and principle: Back to metabolism in KEGG. Nucleic Acids Res. 2014, 42, D199–D205. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Cong, Y.; Wang, Y.; Guo, Z.; Yue, J.; Xing, Z.; Gao, X.; Chai, X. Identification of Early Salinity Stress-Responsive Proteins in Dunaliella salina by isobaric tags for relative and absolute quantitation (iTRAQ)-Based Quantitative Proteomic Analysis. Int. J. Mol. Sci. 2019, 20, 599. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030599
Wang Y, Cong Y, Wang Y, Guo Z, Yue J, Xing Z, Gao X, Chai X. Identification of Early Salinity Stress-Responsive Proteins in Dunaliella salina by isobaric tags for relative and absolute quantitation (iTRAQ)-Based Quantitative Proteomic Analysis. International Journal of Molecular Sciences. 2019; 20(3):599. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030599
Chicago/Turabian StyleWang, Yuan, Yuting Cong, Yonghua Wang, Zihu Guo, Jinrong Yue, Zhenyu Xing, Xiangnan Gao, and Xiaojie Chai. 2019. "Identification of Early Salinity Stress-Responsive Proteins in Dunaliella salina by isobaric tags for relative and absolute quantitation (iTRAQ)-Based Quantitative Proteomic Analysis" International Journal of Molecular Sciences 20, no. 3: 599. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20030599