1. Introduction
Obesity and type 2 diabetes mellitus are metabolic conditions that are frequently associated with an increased risk of cardiovascular disease (CVD) [
1]. The functional characteristics of high-density lipoprotein (HDL) are distorted in subjects with obesity and diabetes [
2], and the latter partially accounts for the increased CVD risk in obese subjects and patients with diabetes.
In vivo macrophage-specific reverse cholesterol transport (m-RCT) is considered one of the most important anti-atherogenic properties of HDL [
3]. HDL also possesses potent antioxidant properties [
4,
5].
Previous data support a relationship between the main atheroprotective properties of HDL and its apolipoprotein A-I (ApoA-I) content [
4,
5]. Several experimental strategies aimed at elevating ApoA-I levels or using mimetic peptides proved effective at improving the main HDL atheroprotective functions [
6]. Accordingly, ApoA-I overexpression is associated with favorable anti-atherogenic effects, as it reduces atherosclerosis in different mouse models [
7,
8] and in rabbits [
9,
10]. In addition to its atheroprotective actions, compelling evidence also shows a role for ApoA-I in preventing adiposity in mice [
11], whereas its deficiency promotes fat gain [
12]. The overexpression of human (h)ApoA-I also alleviates experimental fatty liver and endoplasmic reticulum (ER) stress [
13,
14], which are common manifestations of obesity and diabetes [
15,
16]. In particular, hepatic ER stress directly influences hepatic cholesterol metabolism [
17], thereby contributing to the dysregulation of HDL metabolism and function [
18,
19,
20].
Although the hypothesis that ApoA-I improves the atheroprotective properties of HDL [
21,
22,
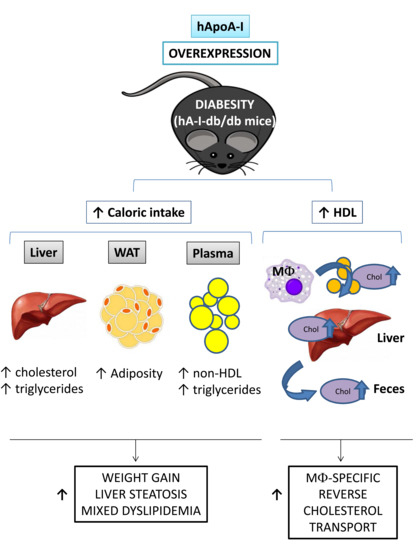
23] is well documented, the impact of its overexpression on these properties in an animal model of diabesity (db/db mice) with dysfunctional HDL is yet to be established. Thus, we addressed this question by testing the hypothesis that two of the most important anti-atherogenic properties attributed to HDL, which are compromised in db/db mice, would be improved following the overexpression of hApoA-I in db/db mice. Moreover, we also studied whether hApoA-I overexpression produced a concomitant amelioration of obesity-related complications observed in an experimental model of diabesity.
3. Discussion
For decades, overexpression of hApoA-I was reported to induce the main atheroprotective properties attributed to HDL, including m-RCT and antioxidant ability, and its clinical potential was assessed in animal and human models (reviewed in References [
3,
28]). To date, apoA-I was not reported to exert an effect on either body weight or food intake in mice with obesity induced by a high-fat diet [
11,
29]. Instead, it increases energy expenditure and reduces the fat mass in hA-I mice [
11]. Consistent with the hApoA-I-mediated reduction in the fat mass, a number of independent studies also provided evidence for a protective role of hApoA-I in animal models of diet-induced fatty liver [
30,
31,
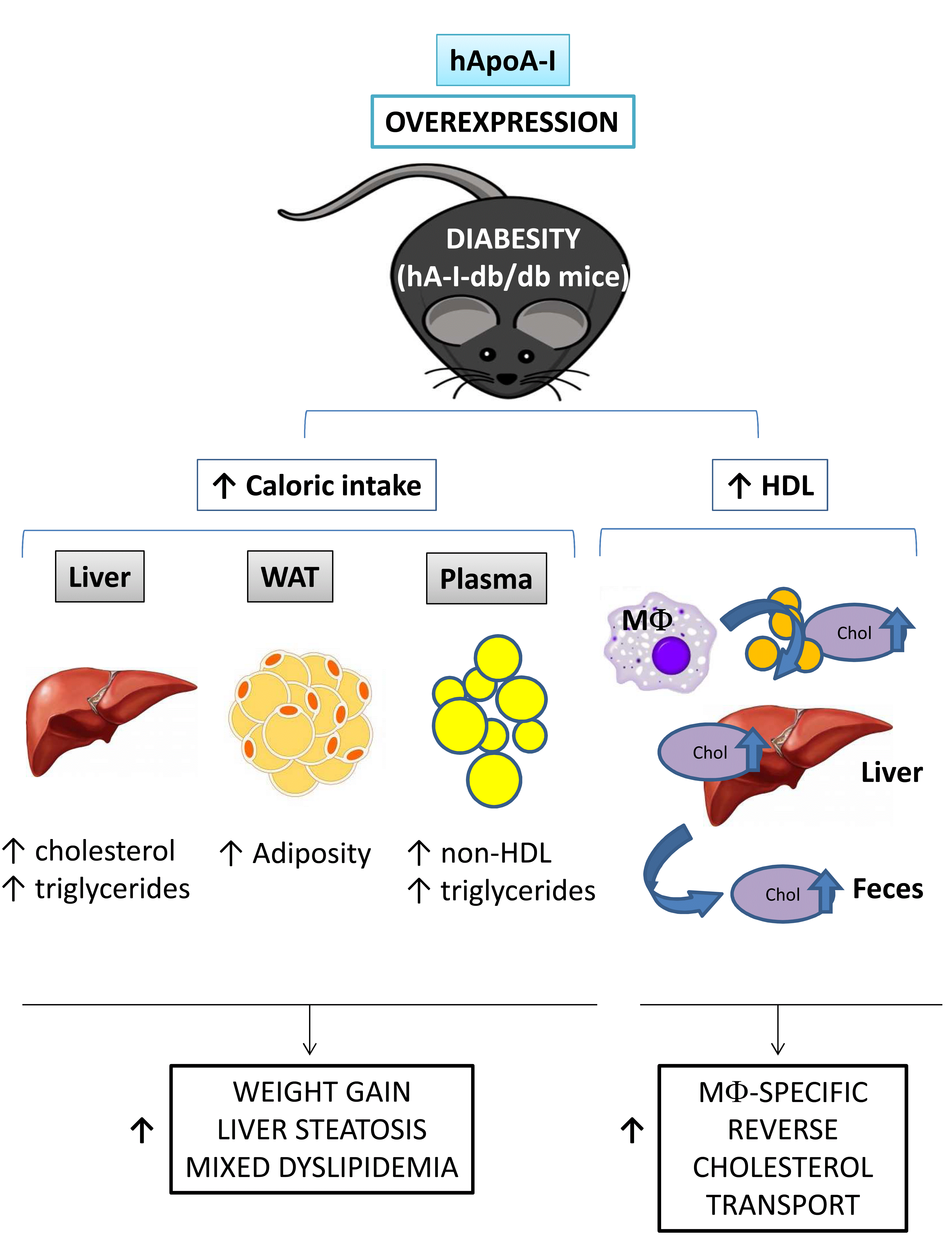
32]. However, to our knowledge, no published studies evaluated the impact of hApoA-I overexpression on the abnormal functions of HDL in an animal model of diabesity (db/db mice). In the present study, overexpression of hApoA-I in db/db mice unexpectedly promoted weight gain and exacerbated the fatty liver phenotype, despite the improvement in m-RCT.
The exacerbations of weight gain and fatty liver produced by the overexpression of hApoA-I in db/db mice were associated with an elevated daily caloric intake. Our study is not the first to show deleterious metabolic outcomes induced by long-term therapies designed to improve HDL concentrations or functions. Consistent with our findings, other experimental strategies (i.e., microRNA 33 (miR-33) antagonism and cholesteryl ester transfer protein (CETP) inhibition) aimed at increasing circulating HDL levels and inducing the m-RCT were also reported to exert long-term, adverse metabolic effects on obese mice [
33,
34,
35], including increased weight gain [
35], dyslipidemia [
34], and steatosis [
33,
34].
Overexpression of hApoA-I failed to prevent the fatty liver phenotype observed in db/db mice. The driving force underlying this failure was increased food intake, as steatosis was prevented by pair feeding with db/db mice. Fatty liver was mechanistically confirmed by a commensurate upregulation of lipogenic genes (i.e.,
Acac, and
Pparg) in the livers of db/db mice.
Pparg is generally activated in mouse models of diet-induced fatty liver [
36,
37]. Activation of
Pparg was revealed by the hepatic upregulation of
Cd36, a free fatty-acid uptake transporter and a known
Pparg-target gene [
38], in both db/db and hA-I-db/db mice. Consistent with this finding,
Cd36 upregulation was previously correlated with fatty liver development in different experimental settings and clinical studies [
39,
40,
41,
42]. Additional evidence for
Pparg induction was provided by the hepatic downregulation of the expression of the
APOA1 gene in hA-I-db/db mice. The expression of the
APOA1 gene is downregulated by a specific
Pparg agonist [
43]. Similar to
APOA1,
Apoa1 expression was also significantly downregulated in db/db mice compared with nonobese control mice. These findings were consistent with previous data showing similar transcriptional dysregulation of mouse ApoA-I in mice with defects in leptin signaling [
44].
ER stress was identified as a crucial mechanism of fatty liver [
45] that is commonly enhanced in the steatotic livers of obese mice deficient in leptin signaling, and it is induced by a high-fat diet [
46,
47,
48]. The unfolded protein response (UPR), which is regarded as a complementary adaptive defense mechanism against ER stress, is regulated by specific ER-resident chaperones that protect cells from activating ER-stress-induced apoptosis and cell death. The 78-kDa glucose-regulated protein (Grp78) (encoded by the
Hspa5 gene), which is one of the main representatives of these chaperone proteins, plays a crucial role in modulating the UPR [
49]. Consistent with previous observations [
25], the hepatic expression of
Hspa5 was consistently downregulated in db/db mice compared with nonobese mice. We did not determine the hepatic levels of this ER chaperone protein, but experimental overexpression of the
Hspa5 gene directly relates to protein function, and independent studies described the overexpression of this gene as a favorable protective mechanism against ER stress in hepatocytes [
49,
50]. Thus,
Hspa5 downregulation in the livers of db/db mice would also be consistent with the enhanced development of hepatic steatosis in these mice and was not prevented in our hA-I-db/db mice.
Our hA-I-db/db mice also developed a mixed type of dyslipidemia, characterized in part by elevated plasma levels of triglycerides and non-HDL cholesterol. The mechanism underlying the accumulation of non-HDL remains unknown, but non-HDL accumulation might be promoted by increased plasma levels of hApoA-I [
51]. HDL functions as apolipoprotein reservoir for the adequate maturation of triglyceride-rich lipoproteins [
52]. Therefore, the excess hApoA-I in HDL might contribute to hypertriglyceridemia by altering the HDL proteome, thereby abrogating triglyceride-rich lipoproteins. A similar mechanism was in fact also described in mice overexpressing the second major apolipoprotein of HDL, hApoA-II, to explain the altered triglyceride-rich lipoprotein metabolism observed in these transgenic mice [
53]. Alternatively, triglyceride-rich, non-HDL elevations might also be explained by an increased hepatic production. These mechanisms would likely be aggravated by the exacerbated hepatic steatosis and insulin resistance [
54] displayed in mice on the db/db background. Further research is warranted to elucidate the negative impact of hApoA-I overexpression on diabesity in the db/db mouse.
We also used our mouse model to evaluate the impact of hApoA-I overexpression on m-RCT, which is likely one of the main anti-atherogenic properties of HDL [
4]. Consistent with previous studies [
24], the relative fecal levels of the [
3H]-tracer (i.e., the last step in m-RCT) were reduced in db/db mice compared with nonobese control mice. As expected, overexpression of hApoA-I promoted the fecal excretion of the [
3H]-tracer in nonobese control mice. Notably, fecal excretion of the [
3H]-tracer was also stimulated in the hA-I-db/db mice and reached relative values similar to the nonobese control mice. The absence of changes in the analysis of the expression of the mRNAs encoding the main targets involved in cholesterol transport into the feces prompted us to hypothesize that the partial recovery of the relative fecal levels of the [
3H]-tracer in hA-I-db/db mice was indirectly attributed to an increased hApoA-I-related upload of cholesterol in the livers of these mice. Indeed, these data would be consistent with previous data showing that upregulation of hApoA-I induced by fenofibrate promotes the hepatic accumulation of [
3H]-cholesterol in male hA-I mice [
55]. This hepatic accumulation of cholesterol may be potentially explained, at least in part, by the downregulation of genes (i.e., ATP-binding cassette (ABC) transporters) involved in the trafficking of hepatic cholesterol to the bile and plasma. The Abcg5/g8 complex requires simultaneous expression of both cholesterol transporters [
56,
57]. Consistent with a previous report [
24], some liver X receptor (LXR) targets other than
Abcg5/g8, such as
Cyp7a1 and
Cyp27a1, were also downregulated in db/db mice, thereby suggesting the potential inhibition of LXR signaling in the livers of db/db mice [
24]. In the present study, similar results were also obtained in hA-I-db/db mice, thus suggesting that this hepatic signaling pathway was also defective.
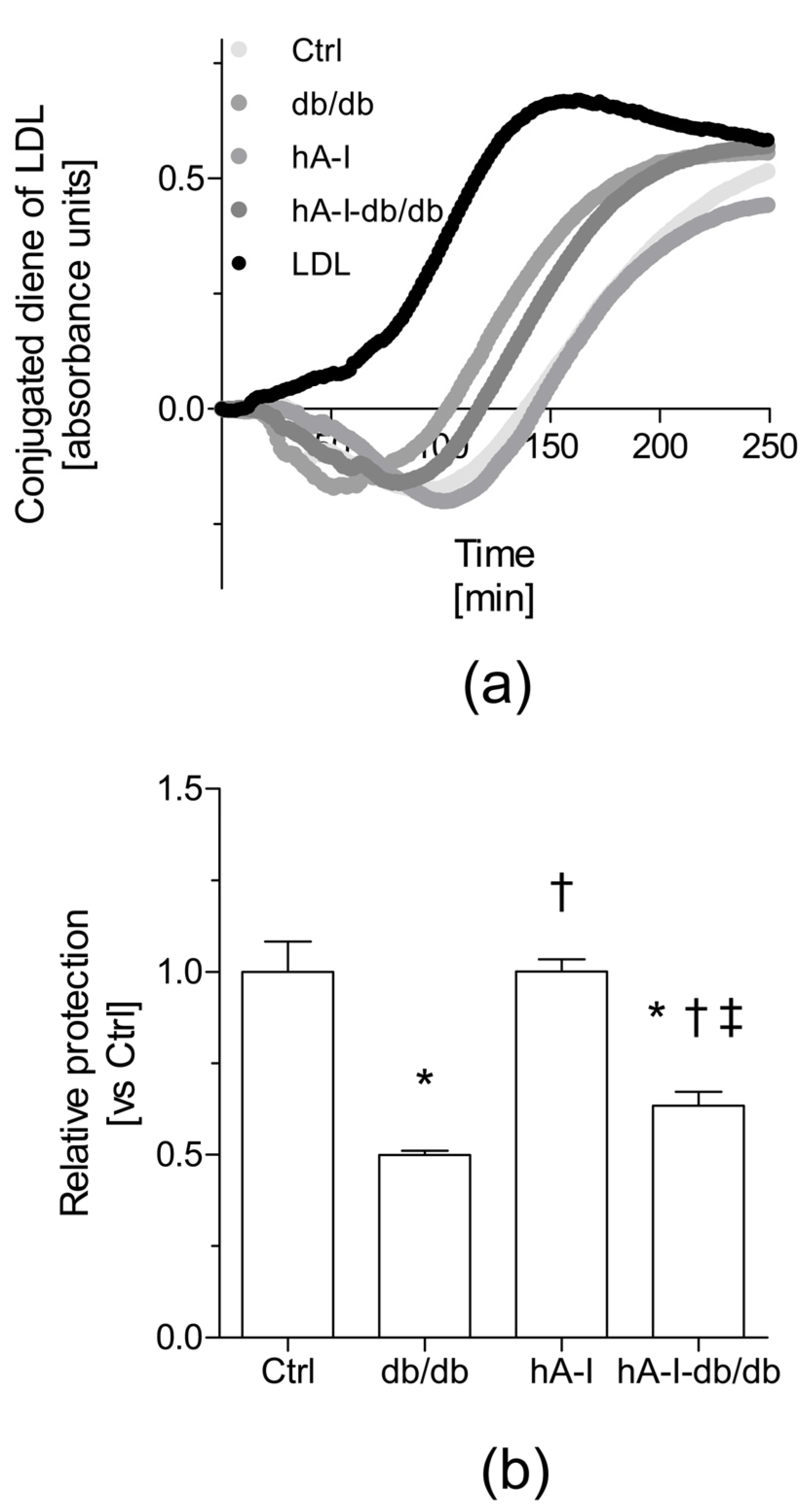
Another crucial atheroprotective action of HDL is to protect against low-density lipoprotein (LDL) oxidation, mainly by inhibiting the formation of toxic lipid hydroperoxides and promoting their removal from LDL [
23]. In our study, HDL isolated from the plasma of db/db mice failed to protect LDL from oxidation compared with the HDL from nonobese control mice. This finding was first related to a relative reduction in the levels of antioxidant HDL-associated proteins (i.e., PON1, Lp-PLA2, and ApoA-I) in db/db mice. The overexpression of hApoA-I in db/db mice improved the antioxidant ability of HDL by preventing LDL oxidation ex vivo compared the HDL from db/db mice. This effect was not explained by favorable changes in the plasma levels of HDL-associated Lp-PLA2 and PON-1 activities. In fact, the plasma levels of PON1 were decreased in the groups of mice overexpressing hApoA-I. Similar to mouse ApoA-I [
26,
27], hApoA-I might conceivably compete with PON1 and displaces it from the HDL particles in hA-I-db/db mice. Therefore, based on our data, the enhanced antioxidant properties of the HDL from hA-I-db/db mice might be attributed to an increase in the relative hApoA-I content.
The present study has several limitations. Firstly, the observed changes in mRNA expression do not necessarily reflect changes in the protein content and function. Additionally, an analysis of the levels of the abovementioned cholesterol transporter proteins is warranted to confirm and extend our present observations. Finally, the mechanisms underlying the decreased activity of the LXR pathway and the increase in hepatic cholesterol accumulation were not explored. Thus, further studies are warranted.

 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}