Synthesis, Characterization, and Bacterial Fouling-Resistance Properties of Polyethylene Glycol-Grafted Polyurethane Elastomers

 and
and
Abstract
:
1. Introduction
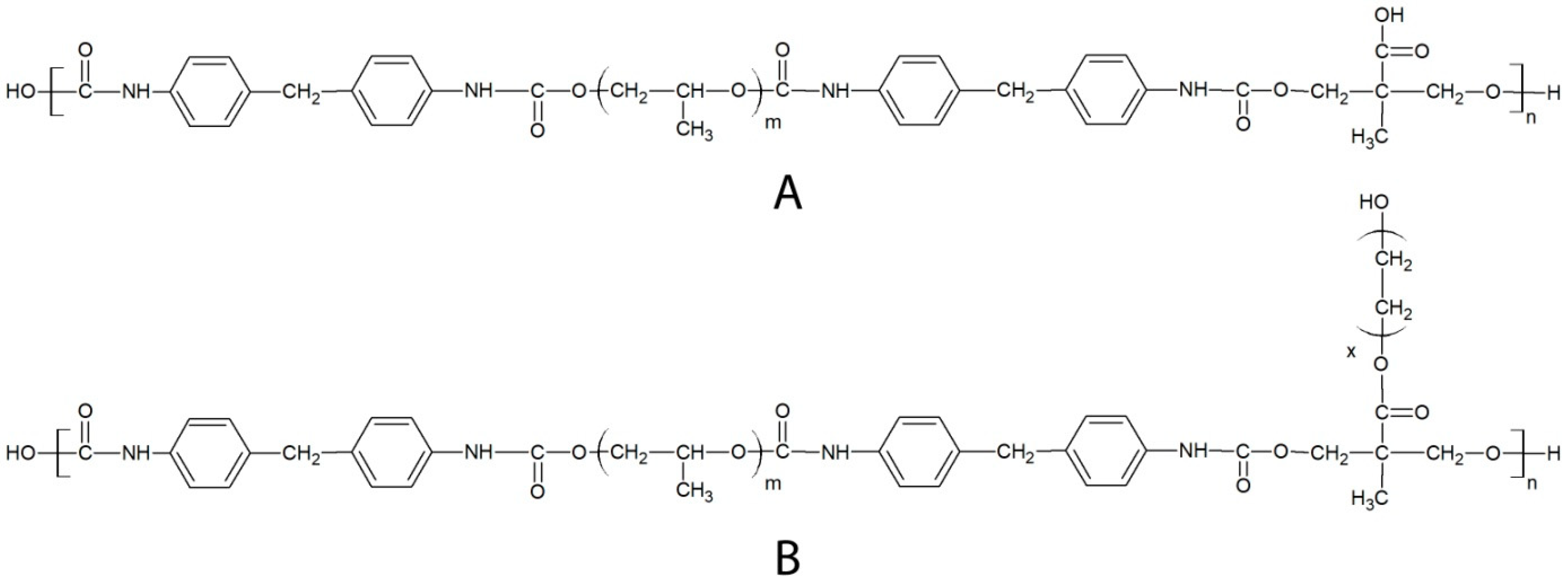
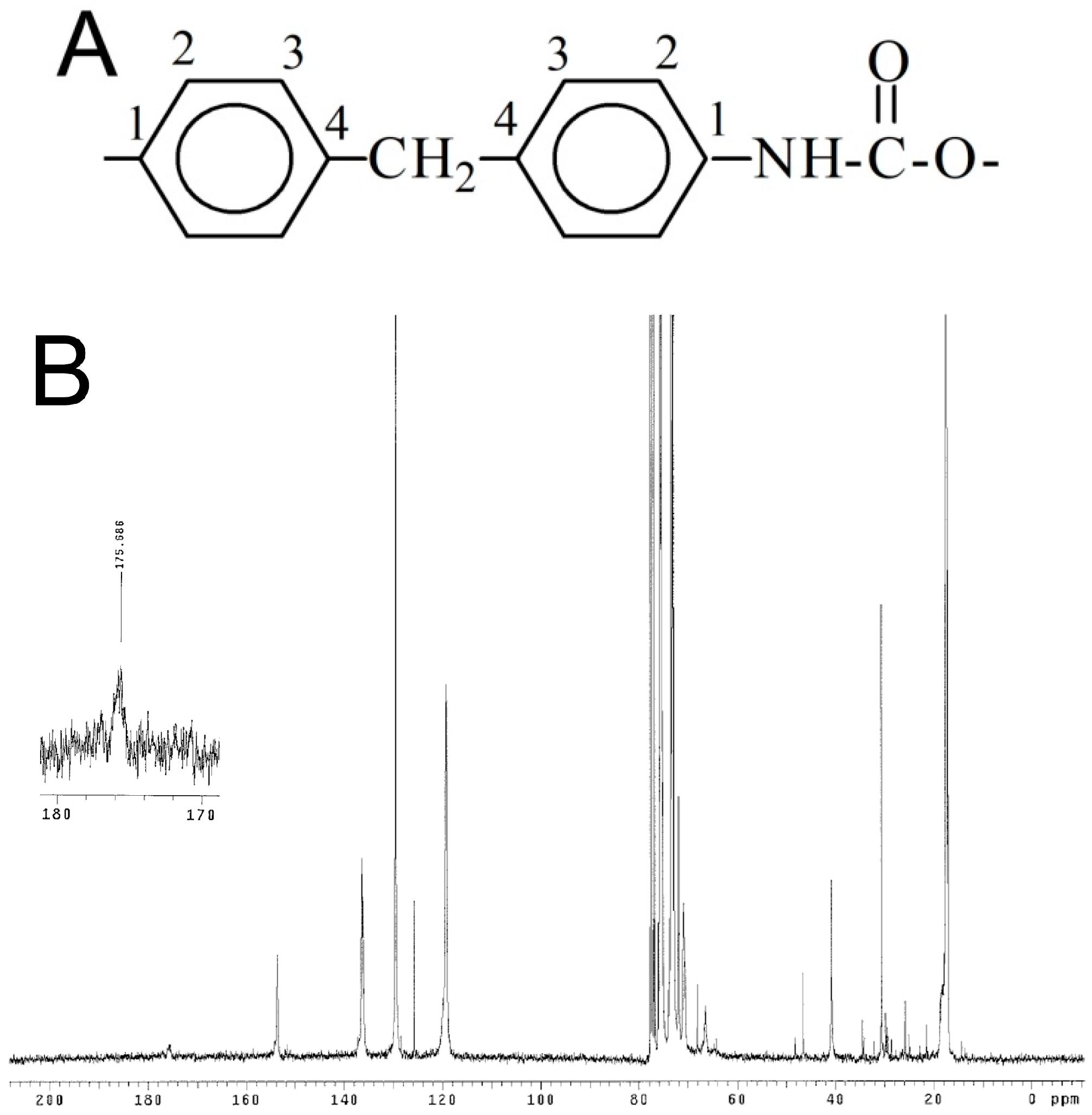
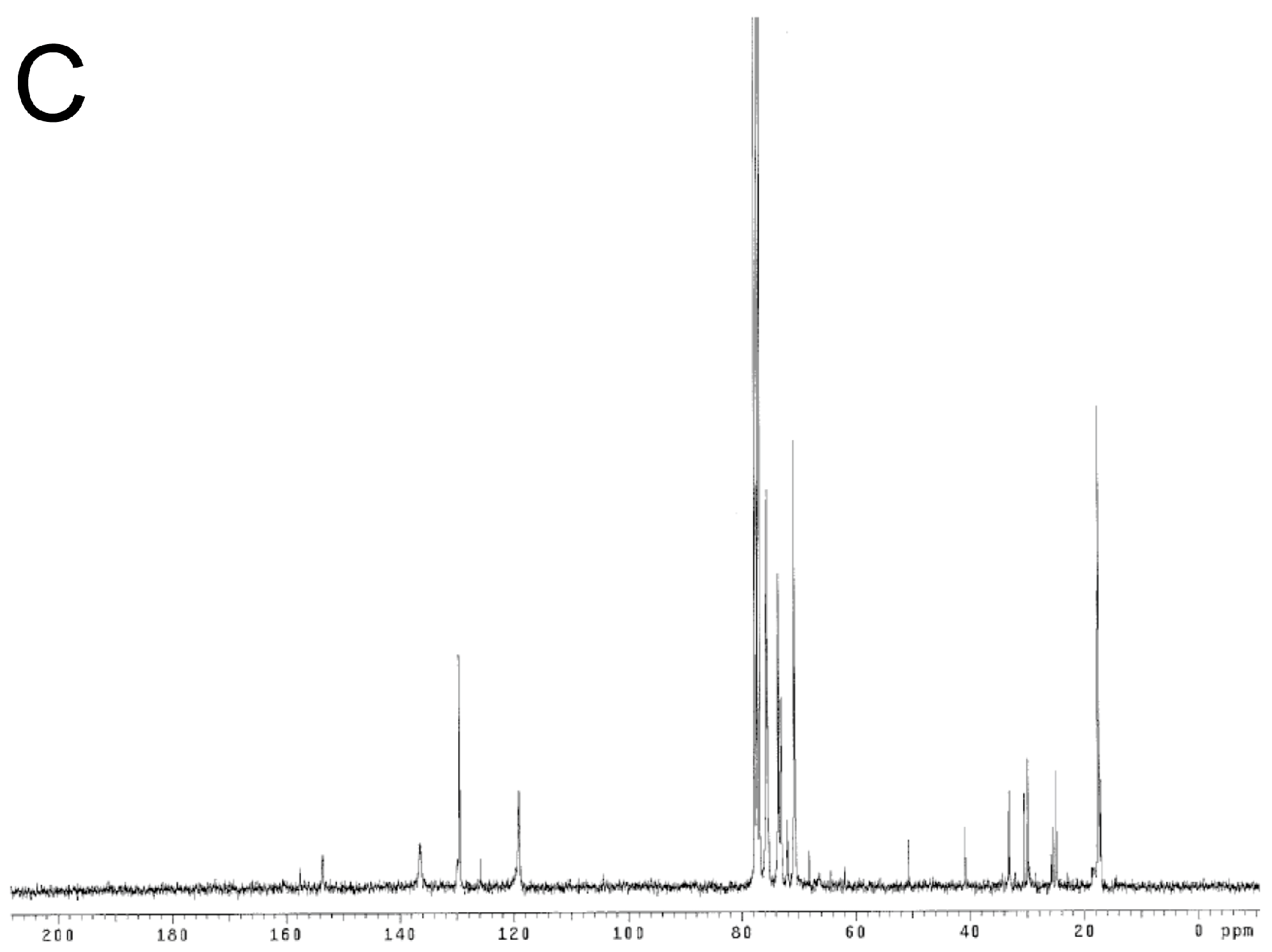
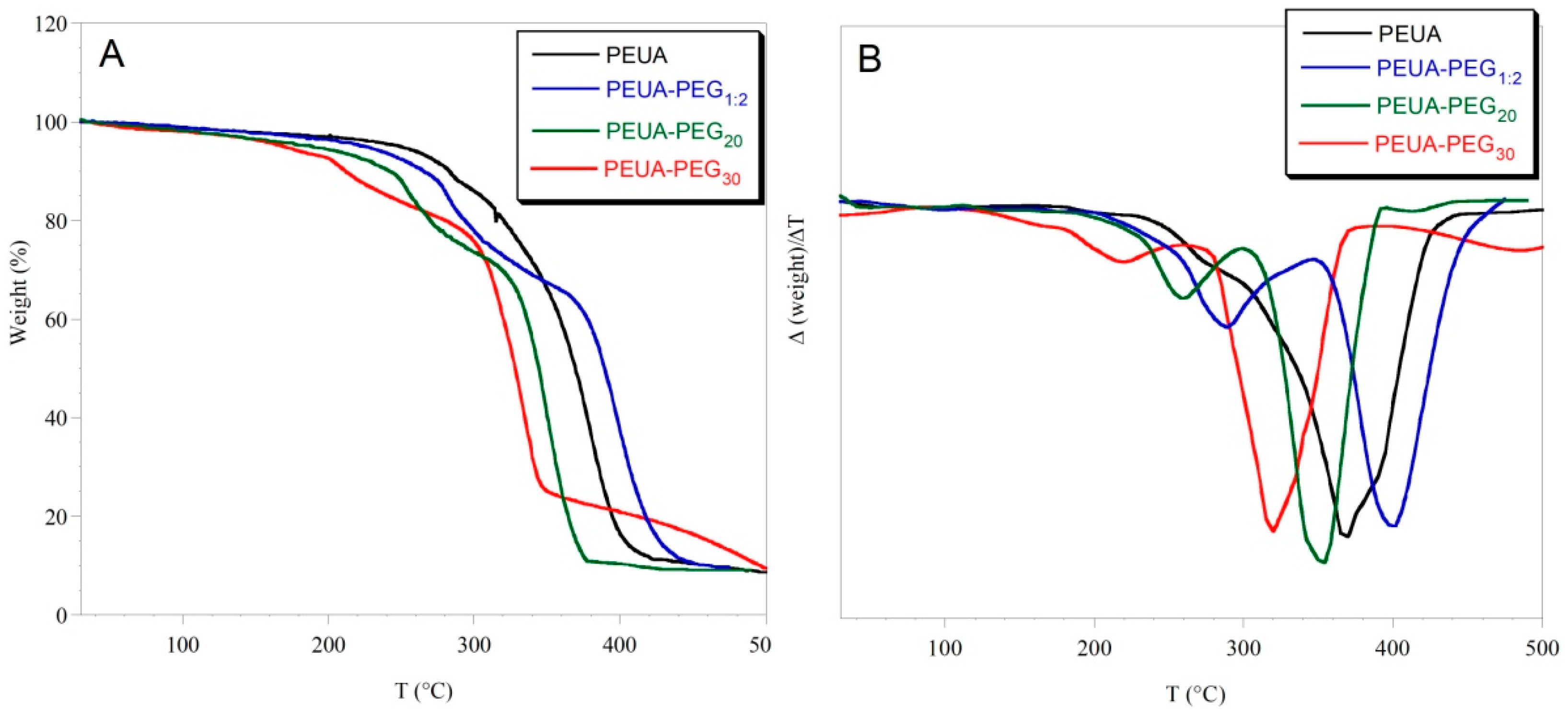
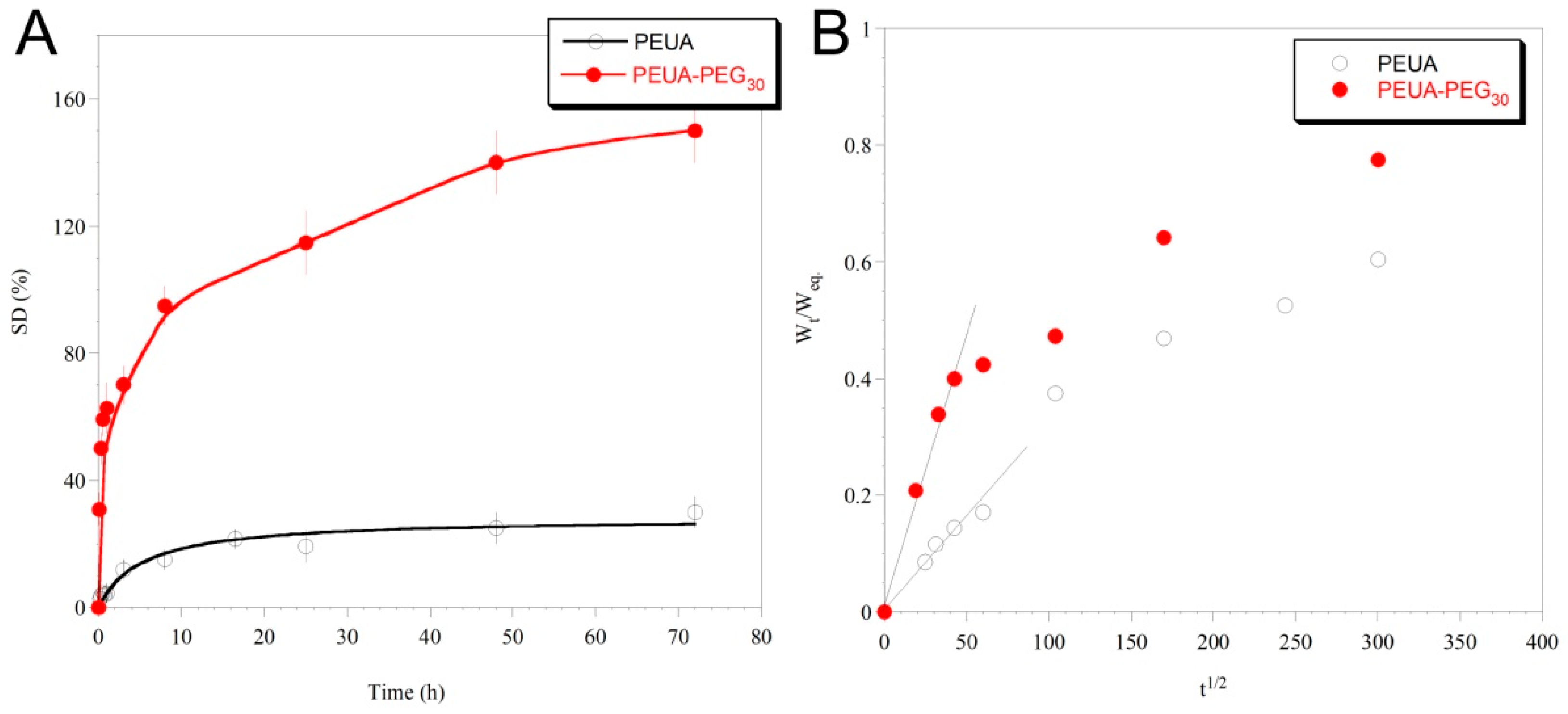
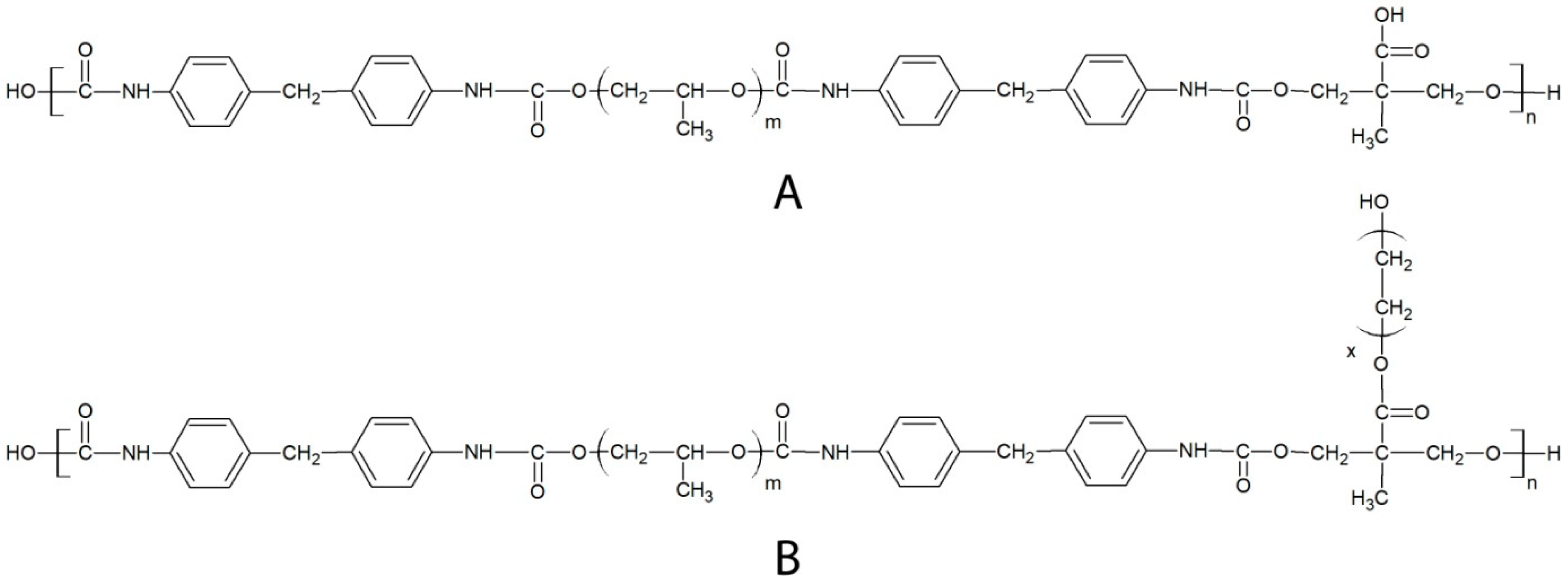
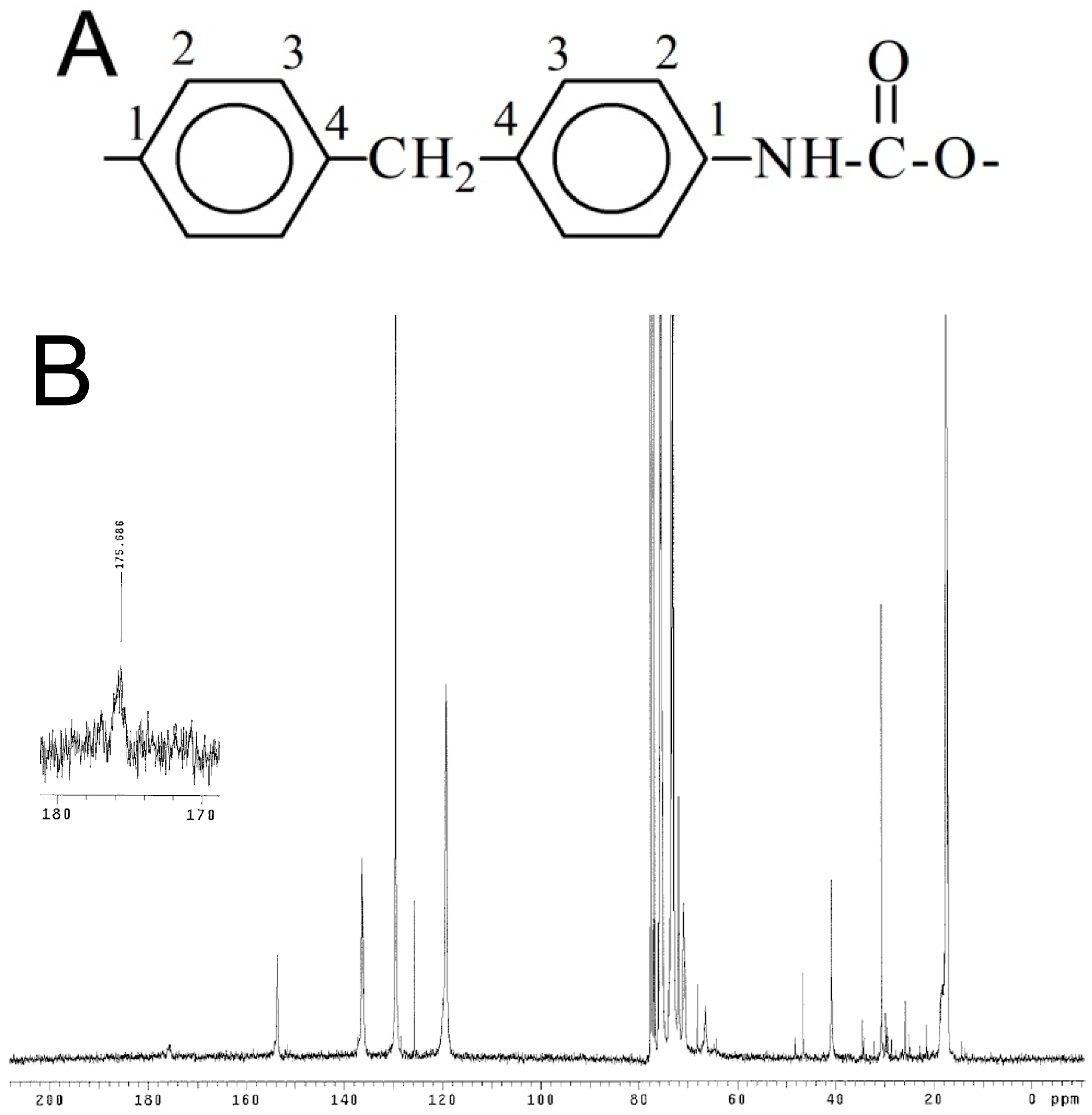
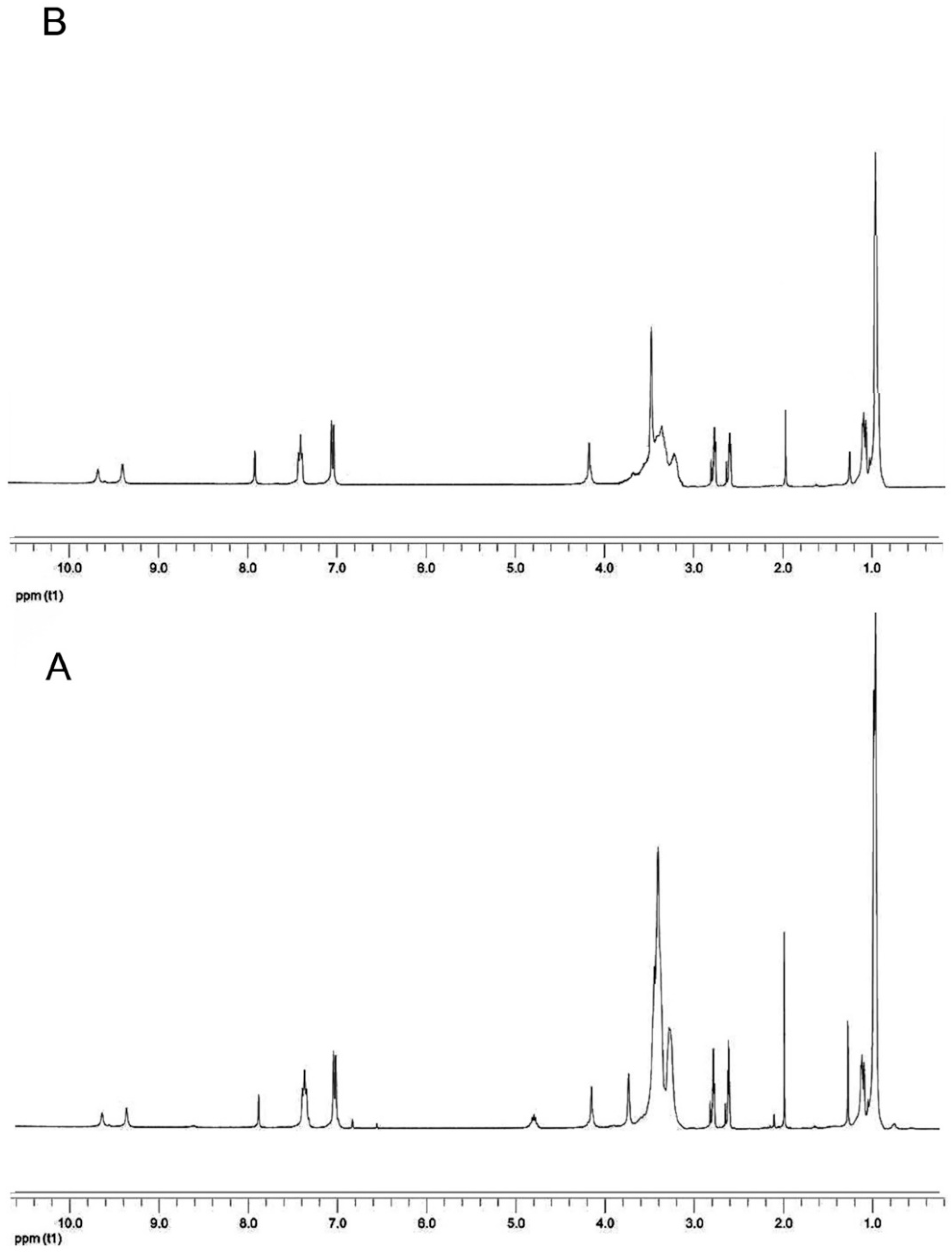
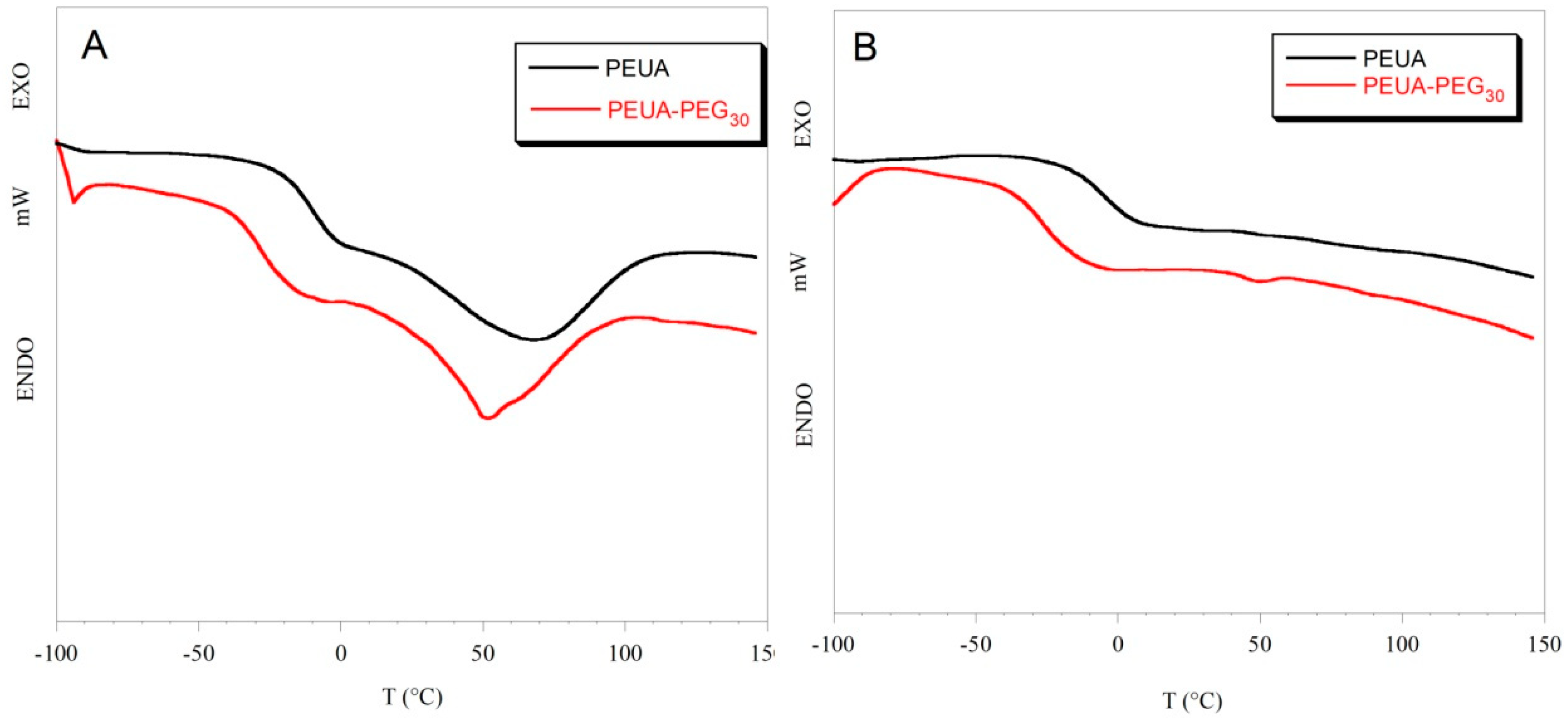
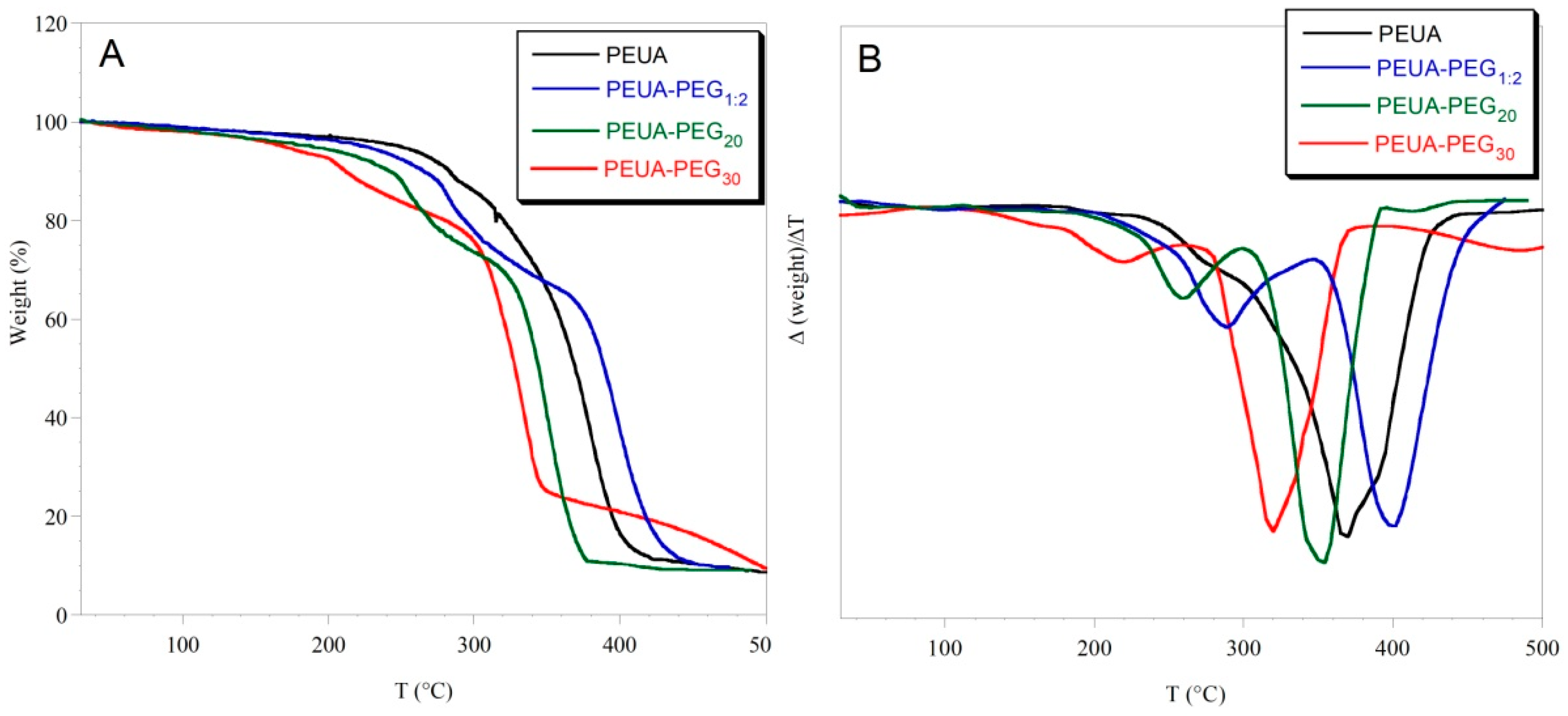
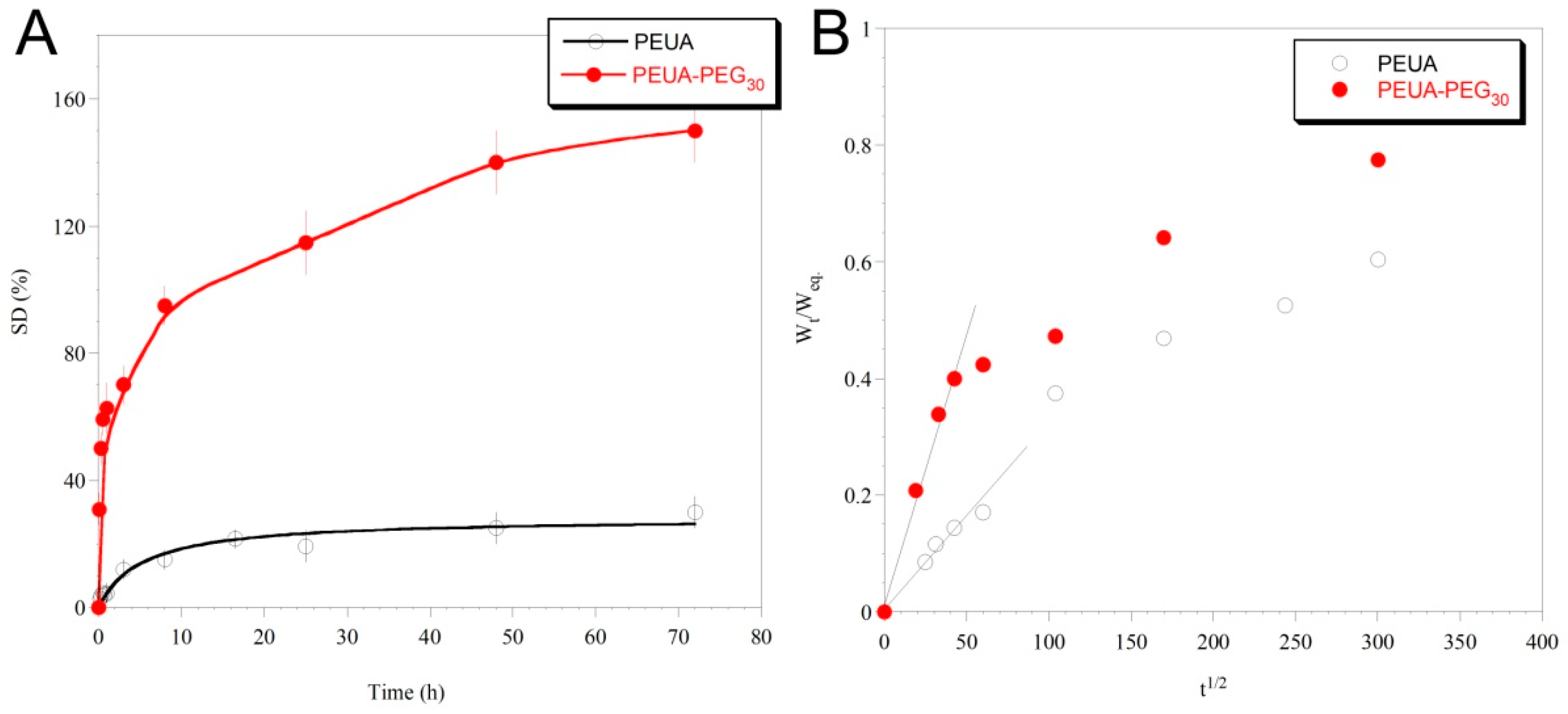
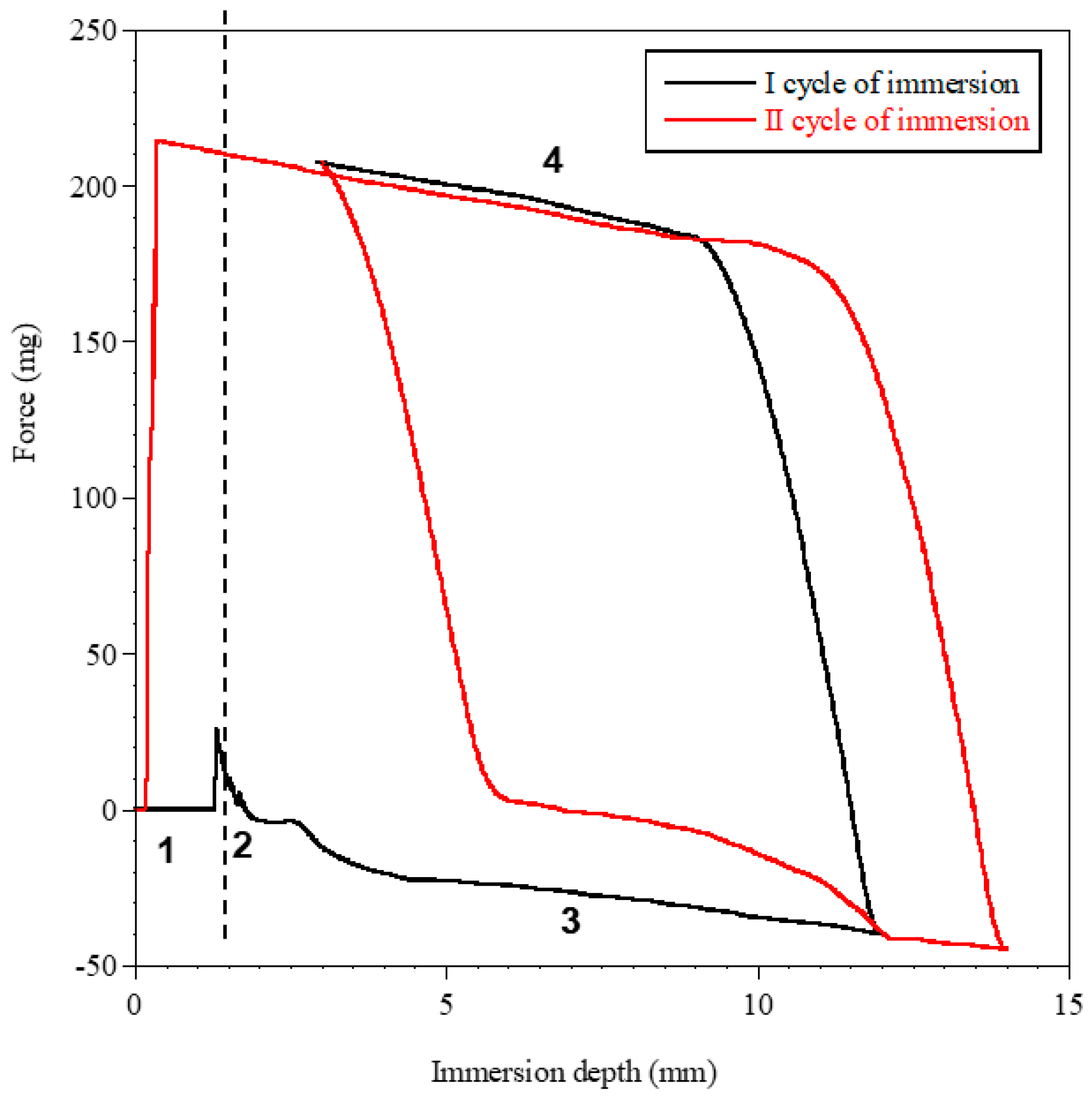
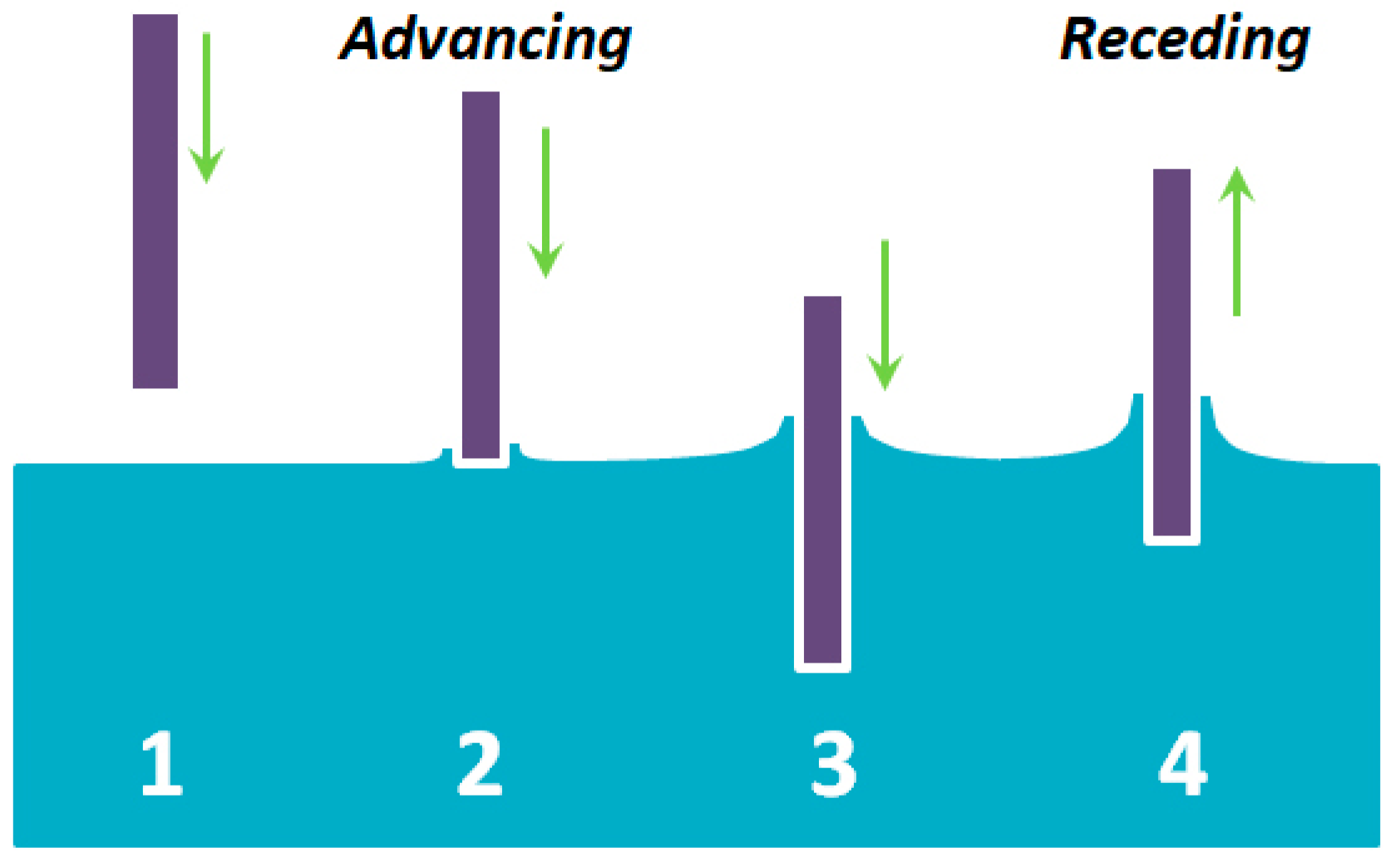
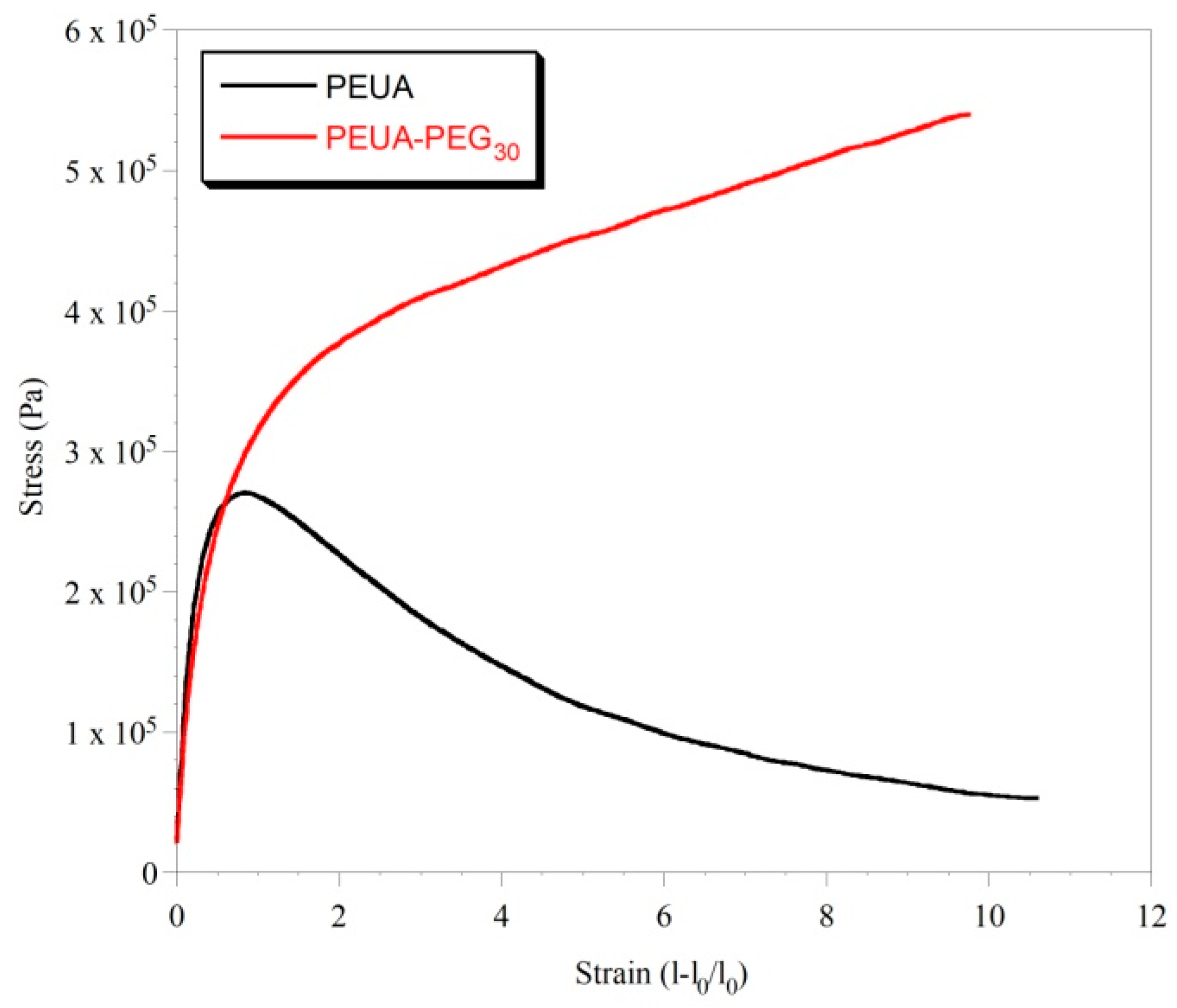
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Polymer Synthesis and Functionalization
3.3. Polymer Characterization
3.4. Bacterial Strains and Culture Medium
3.5. Assessment of the Polymer Ability to Prevent Bacterial Adhesion and Biofilm Formation
3.6. Statistics
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shin, E.J.; Choi, S.M. Advances in Waterborne Polyurethane-Based Biomaterials for Biomedical Applications. Adv. Exp. Med. Biol. 2018, 1077, 251–283. [Google Scholar] [PubMed]
- Guelcher, S.A. Biodegradable polyurethanes: Synthesis and applications in regenerative medicine. Tissue Eng. Part B Rev. 2008, 14, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.; Hasirci, N. Polyurethanes in biomedical applications. Adv. Exp. Med. Biol. 2004, 553, 83–101. [Google Scholar] [PubMed]
- Piozzi, A.; Francolini, I. Biomimetic polyurethanes. In Polymeric Materials with Antimicrobial Activity from Synthesis to Applications, 1st ed.; Muñoz-Bonilla, A., Cerrada, M., Fernández-García, M., Eds.; RSC Publishing: Cambridge, UK, 2014; pp. 224–264. [Google Scholar]
- Zdrahala, R.J.; Zdrahala, I.J. Biomedical applications of polyurethanes: A review of past promises, present realities, and a vibrant future. J. Biomater. Appl. 1999, 14, 67–90. [Google Scholar] [CrossRef]
- Bell, T.; O’Grady, N.P. Prevention of Central Line-Associated Bloodstream Infections. Infect. Dis. Clin. N. Am. 2017, 31, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Seckold, T.; Walker, S.; Dwyer, T. A comparison of silicone and polyurethane PICC lines and post-insertion complication rates: A systematic review. J. Vasc. Access 2015, 16, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Francolini, I.; Piozzi, A. Antimicrobial polyurethanes for intravascular medical devices. In Advances in Polyurethane Biomaterials; Cooper, S.L., Guan, J., Eds.; Woodhead Publishing (Elsevier Ltd.): Sawston, UK, 2016; Chapter 12; pp. 349–385. [Google Scholar]
- Sheng, W.H.; Ko, W.J.; Wang, J.T.; Chang, S.C.; Hsueh, P.R.; Luh, K.T. Evaluation of antiseptic-impregnated central venous catheters for prevention of catheter-related infection in intensive care unit patients. Diagn. Microbiol. Infect. Dis. 2000, 38, 1–5. [Google Scholar] [CrossRef]
- Yorganci, K.; Krepel, C.; Weigelt, J.A.; Edmiston, C.E. In vitro evaluation of the antibacterial activity of three different central venous catheters against gram-positive bacteria. Eur. J. Clin. Microbiol. Infect. Dis. 2002, 21, 379–384. [Google Scholar] [PubMed]
- Donelli, G.; Francolini, I.; Ruggeri, V.; Guaglianone, E.; D’Ilario, L.; Piozzi, A. Pore formers promoted release of an antifungal drug from functionalized polyurethanes to inhibit Candida colonization. J. Appl. Microbiol. 2006, 100, 615–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowatzki, P.J.; Koepsel, R.R.; Stoodley, P.; Min, K.; Harper, A.; Murata, H.; Donfack, J.; Hortelano, E.R.; Ehrlich, G.D.; Russell, A.J. Salicylic acid-releasing polyurethane acrylate polymers as anti-biofilm urological catheter coatings. Acta Biomater. 2012, 8, 1869–1880. [Google Scholar] [CrossRef] [PubMed]
- Francolini, I.; Donelli, G.; Crisante, F.; Taresco, V.; Piozzi, A. Antimicrobial polymers for anti-biofilm medical devices: State-of-art and perspectives. Adv. Exp. Med. Biol. 2015, 831, 93–117. [Google Scholar] [PubMed]
- Huang, K.S.; Yang, C.H.; Huang, S.L.; Chen, C.Y.; Lu, Y.Y.; Lin, Y.S. Recent Advances in Antimicrobial Polymers: A Mini-Review. Int. J. Mol. Sci. 2016, 17, 1578. [Google Scholar] [CrossRef] [PubMed]
- Cattò, C.; Villa, F.; Cappitelli, F. Recent progress in bio-inspired biofilm-resistant polymeric surfaces. Crit. Rev. Microbiol. 2018, 44, 633–652. [Google Scholar] [CrossRef] [PubMed]
- Adlhart, C.; Verran, J.; Azevedo, N.F.; Olmez, H.; Keinänen-Toivola, M.M.; Gouveia, I.; Melo, L.F.; Crijns, F. Surface modifications for antimicrobial effects in the healthcare setting: A critical overview. J. Hosp. Infect. 2018, 99, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Bonilla, A.; Fernández-García, M. Polymeric materials with antimicrobial activity. Prog. Polym. Sci. 2012, 37, 281–339. [Google Scholar] [CrossRef]
- Campoccia, D.; Montanaro, L.; Arciola, C.R. A review of the biomaterials technologies for infection-resistant surfaces. Biomaterials 2013, 34, 8533–8554. [Google Scholar] [CrossRef] [PubMed]
- Francolini, I.; Vuotto, C.; Piozzi, A.; Donelli, G. Antifouling and antimicrobial biomaterials: An overview. APMIS 2017, 125, 392–417. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Ribeiro, A.M.; de Melo Carrasco, L.D. Cationic antimicrobial polymers and their assemblies. Int. J. Mol. Sci. 2013, 14, 9906–9946. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Alfredo, N.; Rodríguez Hernández, J. Microstructured polymer blend surfaces produced by spraying functional copolymers and their blends. Materials 2016, 9, 431. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.C.; Siedlecki, C.A. Protein adsorption, platelet adhesion, and bacterial adhesion to polyethylene-glycol-textured polyurethane biomaterial surfaces. J. Biomed. Mater. Res. B Appl. Biomater. 2017, 105, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Mendez, A.R.; Tan, T.Y.; Low, H.Y.; Otto, K.H.; Tan, H.; Khoo, X. Micro-textured films for reducing microbial colonization in a clinical setting. J. Hosp. Infect. 2018, 98, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Alfredo, N.; Martínez-Campos, E.; Santos-Coquillat, A.; Dorronsoro, A.; Cortajarena, A.L.; Del Campo, A.; Rodríguez-Hernández, J. Fabrication of biocompatible and efficient antimicrobial porous polymer surfaces by the Breath Figures approach. J. Colloid Interface Sci. 2018, 513, 820–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostuni, E.; Chapman, R.G.; Holmlin, E.; Takayama, S.; Whitesides, G.M. A survey of structure-property relationships of surfaces that resist the adsorption of protein. Langmuir 2001, 17, 5605–5620. [Google Scholar] [CrossRef]
- Desai, N.P.; Hossainya, S.F.A.; Hubbell, J.A. Surface-immobilized polyethylene oxide for bacterial repellence. Biomaterials 1992, 13, 417–420. [Google Scholar] [CrossRef]
- Chen, S.F.; Li, L.Y.; Zhao, C.; Zheng, J. Surface hydration: Principles and applications toward low-fouling/nonfouling biomaterials. Polymer 2010, 51, 5283–5293. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.I.; Lee, J.H.; Andrade, J.D.; De Gennes, P.G. Protein-surface interactions in the presence of polyethylene oxide ii. Effect of protein size. J. Colloid Interface Sci. 1991, 142, 159–166. [Google Scholar] [CrossRef]
- Razatos, A.; Ong, Y.; Boulay, F.; Elbert, D.L.; Hubbell, J.A.; Sharma, M.M.; Georgiou, G. Force measurements between bacteria and poly(ethylene glycol)-coated surfaces. Langmuir 2000, 16, 9155–9158. [Google Scholar] [CrossRef]
- Corneillie, S.; Lan, P.N.; Schacht, E.; Davies, M.; Shard, A.; Green, R.; Denyer, S.; Wassall, M.; Whitfield, H.; Choong, S. Polyethylene glycol-containing polyurethanes for biomedical applications. Polym. Int. 1998, 46, 251–259. [Google Scholar] [CrossRef]
- Chen, X.; Liu, W.; Zhao, Y.; Jiang, L.; Xu, H.; Yang, X. Preparation and characterization of PEG-modified polyurethane pressure-sensitive adhesives for transdermal drug delivery. Drug Dev. Ind. Pharm. 2009, 35, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Lee, S.Y.; Cho, J.W. Synthesis and characterization of biocompatible poly(ethylene glycol)-functionalized polyurethane using click chemistry. Polym. Bull. 2010, 64, 401–411. [Google Scholar] [CrossRef]
- Orban, J.M.; Chapman, T.M.; Wagner, W.R.; Jankowski, R. Easily grafted polyurethanes with reactive main chain functional groups. Synthesis, characterization, and antithrombogenicity of poly(ethylene glycol)-grafted poly(urethanes). J. Polym. Sci. A Polym. Chem. 1999, 37, 3441–3448. [Google Scholar] [CrossRef]
- Park, K.D.; Suzuki, K.; Lee, W.K.; Lee, J.E.; Kim, Y.H.; Sakurai, Y.; Okano, T. Platelet adhesion and activation on polyethylene glycol modified polyurethane surfaces. Measurement of cytoplasmic calcium. ASAIO J. 1996, 42, M876–M880. [Google Scholar] [CrossRef] [PubMed]
- Han, D.K.; Park, K.; Park, K.D.; Ahn, K.D.; Kim, Y.H. In vivo biocompatibility of sulfonated PEO-grafted polyurethanes for polymer heart valve and vascular graft. Artif. Organs 2006, 30, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hu, X.; Zhang, Y.; Li, D.; Wu, Z.; Zhang, T. Effect of chain density and conformation on protein adsorption at PEG-grafted polyurethane surfaces. Colloids Surf. B Biointerfaces 2008, 61, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Suh, H.; Lee, J.H.; Park, S.N.; Shin, S.H.; Kim, Y.H.; Chung, S.M.; Kim, H.K.; Lim, J.Y.; Kim, H.S. A polyethylene glycol grafted bi-layered polyurethane scaffold: Preliminary study of a new candidate prosthesis for repair of a partial tracheal defect. Eur. Arch. Oto-Rhino-Laryngol. 2008, 265, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Rao, L.; Zhou, H.; Li, T.; Li, C.; Duan, Y.Y. Polyethylene glycol-containing polyurethane hydrogel coatings for improving the biocompatibility of neural electrodes. Acta Biomater. 2012, 8, 2233–2242. [Google Scholar] [CrossRef] [PubMed]
- Mei, T.; Zhu, Y.; Ma, T.; He, T.; Li, L.; Wei, C.; Xu, K. Synthesis, characterization, and biocompatibility of alternating block polyurethanes based on PLA and PEG. J. Biomed. Mater. Res. A 2014, 102, 3243–3254. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Cho, Y.W.; Kwon, I.C.; Jeong, S.Y.; Bae, Y.H. Assessment of PEO/PTMO multiblock copolymer/segmented polyurethane blends as coating materials for urinary catheters: In vitro bacterial adhesion and encrustation behavior. Biomaterials 2002, 23, 3991–4000. [Google Scholar] [CrossRef]
- Patel, J.D.; Ebert, M.; Ward, R.; Anderson, J.M. S. epidermidis biofilm formation: Effects of biomaterial surface chemistry and serum proteins. J. Biomed. Mater. Res. A 2007, 80, 742–751. [Google Scholar] [CrossRef] [PubMed]
- Francolini, I.; Donelli, G.; Vuotto, C.; Baroncini, F.A.; Stoodley, P.; Taresco, V.; Martinelli, A.; D’Ilario, L.; Piozzi, A. Antifouling polyurethanes to fight device-related staphylococcal infections: Synthesis, characterization, and antibiofilm efficacy. Pathog. Dis. 2014, 70, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Marconi, W.; Martinelli, A.; Piozzi, A.; Zane, D. Direct synthesis of carboxylated polyurethanes. Europ. Polymer. J. 1991, 27, 135–139. [Google Scholar] [CrossRef]
- Piozzi, A.; Francolini, I.; Occhiaperti, L.; Di Rosa, R.; Ruggeri, V.; Donelli, G. Polyurethanes loaded with antibiotics: Influence of polymer-antibiotic interactions on in vitro activity against Staphylococcus epidermidis. J. Chemother. 2004, 16, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Francolini, I.; Ruggeri, V.; Martinelli, A.; D’Ilario, L.; Piozzi, A. Novel metal-polyurethane complexes with enhanced antimicrobial activity. Macromol. Rapid. Commun. 2006, 27, 233–237. [Google Scholar] [CrossRef]
- Schneider, N.S.; Sung, C.S.P. Transition behavior and phase segregation in TDI polyurethanes. Polym. Eng. Sci. 1977, 17, 73–80. [Google Scholar] [CrossRef]
- Faucheris, J.A. The dependence of glass transition temperature on molecular weight for poly(propylene oxide) and poly(butylene oxide). Polym. Lett. 1965, 3, 143–145. [Google Scholar] [CrossRef]
- Okkema, A.Z.; Cooper, S.L. The effect of carboxylate and/or sulfonate ions on phase segregation in polyurethanes. Biomaterials 1991, 12, 668. [Google Scholar] [CrossRef]
- Song, F.; Koo, H.; Ren, D. Effects of material properties on bacterial adhesion and biofilm formation. J. Dent. Res. 2015, 94, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, L.; Levanen, E. Superhydrophobic surfaces for the reduction of bacterial adhesion. RSC Adv. 2013, 3, 12003–12020. [Google Scholar] [CrossRef]
- Ko, J.; Cho, K.; Han, S.W.; Sung, H.K.; Baek, S.W.; Koh, W.G.; Yoon, J.S. Hydrophilic surface modification of poly(methyl methacrylate)-based ocular prostheses using poly(ethylene glycol) grafting. Colloids Surf. B Biointerfaces 2017, 158, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Pissis, P.; Apekis, L.; Christodoulides, C.; Niaounakis, M.; Kyritsis, A.; Nebdal, J. Water effects in polyurethane block copolymers. J. Polym. Sci. Part B Polym. Phys. 1996, 34, 1529–1539. [Google Scholar] [CrossRef]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release. II Fickian and anomalous release from swellable devices. J. Control. Release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Wang, C.; Nair, S.; Wynne, K.J. Wilhelmy balance characterization beyond contact angles: Differentiating leaching from nanosurface reorganization and optimizing surface modification. Polymer 2017, 116, 565–571. [Google Scholar] [CrossRef]
- Blodgett, K.B.; Langmuir, I. Built-up films of barium stearate and their optical properties. Phys. Rev. 1937, 51, 964–982. [Google Scholar] [CrossRef]
- Kaganer, V.M.; Mohwald, H.; Dutta, P. Structure and phase transitions in Langmuir monolayers. Rev. Mod. Phys. 1999, 71, 779–819. [Google Scholar] [Green Version]
- Grassi, L.; Maisetta, G.; Esin, S.; Batoni, G. Combination strategies to enhance the efficacy antimicrobial peptides against bacterial biofilms. Front. Microbiol. 2017, 8, 2409. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, K.A.; Wenke, J.C. Current therapies in treatment and prevention of fracture wound biofilms: Why a multifaceted approach is essential for resolving persistent infections. J. Bone Jt. Infect. 2018, 3, 50–67. [Google Scholar] [CrossRef] [PubMed]
- Neises, B.; Steglich, W. Simple method for the esterification of carboxylic acids. Angew. Chem. 1978, 7, 522–524. [Google Scholar] [CrossRef]
- Long, J.; Hyder, M.N.; Huang, R.Y.M.; Chen, P. Thermodynamic modeling of contact angles on rough, heterogeneous surfaces. Adv. Colloid Interface Sci. 2005, 118, 173–190. [Google Scholar] [CrossRef] [PubMed]
- De Gennes, P.G. Wetting: Statics and dynamics. Rev. Mod. Phys. 1985, 57, 827–863. [Google Scholar] [CrossRef]
- Donelli, G.; Francolini, I.; Romoli, D.; Guaglianone, E.; Piozzi, A.; Ragunath, C.; Kaplan, J.B. Synergistic activity of dispersin B and cefamandole nafate in inhibition of staphylococcal biofilm growth on polyurethanes. Antimicrob. Agents Chemother. 2007, 51, 2733–2740. [Google Scholar] [CrossRef] [PubMed]
- Francolini, I.; Piozzi, A.; Donelli, G. Efficacy evaluation of antimicrobial drug-releasing polymer matrices. In Microbial Biofilms; Humana Press: New York, NY, USA, 2014; Volume 1147, pp. 215–225. [Google Scholar]
- Merritt, J.H.; Kadouri, D.E.; O’Toole, G.A. Growing and analyzing static biofilms. Curr. Protoc. Microbiol. 2005, 22. [Google Scholar] [CrossRef]
- Azeredo, J.; Azevedo, N.F.; Briandet, R.; Cerca, N.; Coenye, T.; Costa, A.R.; Desvaux, M.; Di Bonaventura, G.; Hébraud, M.; Jaglic, Z.; et al. Critical review on biofilm methods. Crit. Rev. Microbiol. 2017, 43, 313–351. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | Tg (°C) | ΔCp (J/g*K) |
|---|---|---|
| PEUA | −11 ± 2 | 0.50 ± 0.02 |
| PEUA-PEG1:2 (*) | −33 ± 2 | 0.39 ± 0.02 |
| PEUA-PEG20 | −29 ± 2 | 0.40 ± 0.03 |
| PEUA-PEG30 | −27 ± 2 | 0.43 ± 0.03 |
| Soft phase (PPO) | −67 | - |
| Polymer | I cycle | II cycle | H (I cycle) | H (II cycle) | ||
|---|---|---|---|---|---|---|
| Θadv | Θrec | Θadv | Θrec | |||
| PEUA | 93 ± 3 | 47 ± 3 | 92 ± 1 | 50 ± 3 | 46 | 42 |
| PEUA-PEG30 | 94 ± 2 | 35 ± 3 | 83 ± 2 | 37 ± 1 | 59 | 46 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francolini, I.; Silvestro, I.; Di Lisio, V.; Martinelli, A.; Piozzi, A. Synthesis, Characterization, and Bacterial Fouling-Resistance Properties of Polyethylene Glycol-Grafted Polyurethane Elastomers. Int. J. Mol. Sci. 2019, 20, 1001. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20041001
Francolini I, Silvestro I, Di Lisio V, Martinelli A, Piozzi A. Synthesis, Characterization, and Bacterial Fouling-Resistance Properties of Polyethylene Glycol-Grafted Polyurethane Elastomers. International Journal of Molecular Sciences. 2019; 20(4):1001. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20041001
Chicago/Turabian StyleFrancolini, Iolanda, Ilaria Silvestro, Valerio Di Lisio, Andrea Martinelli, and Antonella Piozzi. 2019. "Synthesis, Characterization, and Bacterial Fouling-Resistance Properties of Polyethylene Glycol-Grafted Polyurethane Elastomers" International Journal of Molecular Sciences 20, no. 4: 1001. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20041001