Quality Screening of Incorrectly Folded Soluble Aggregates from Functional Recombinant Proteins

Abstract
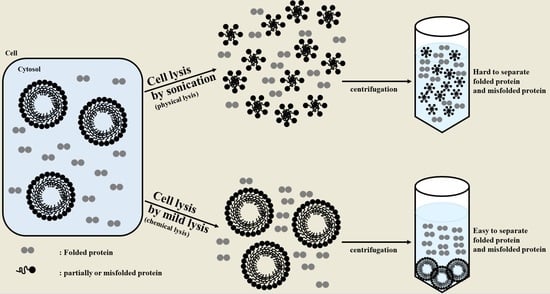
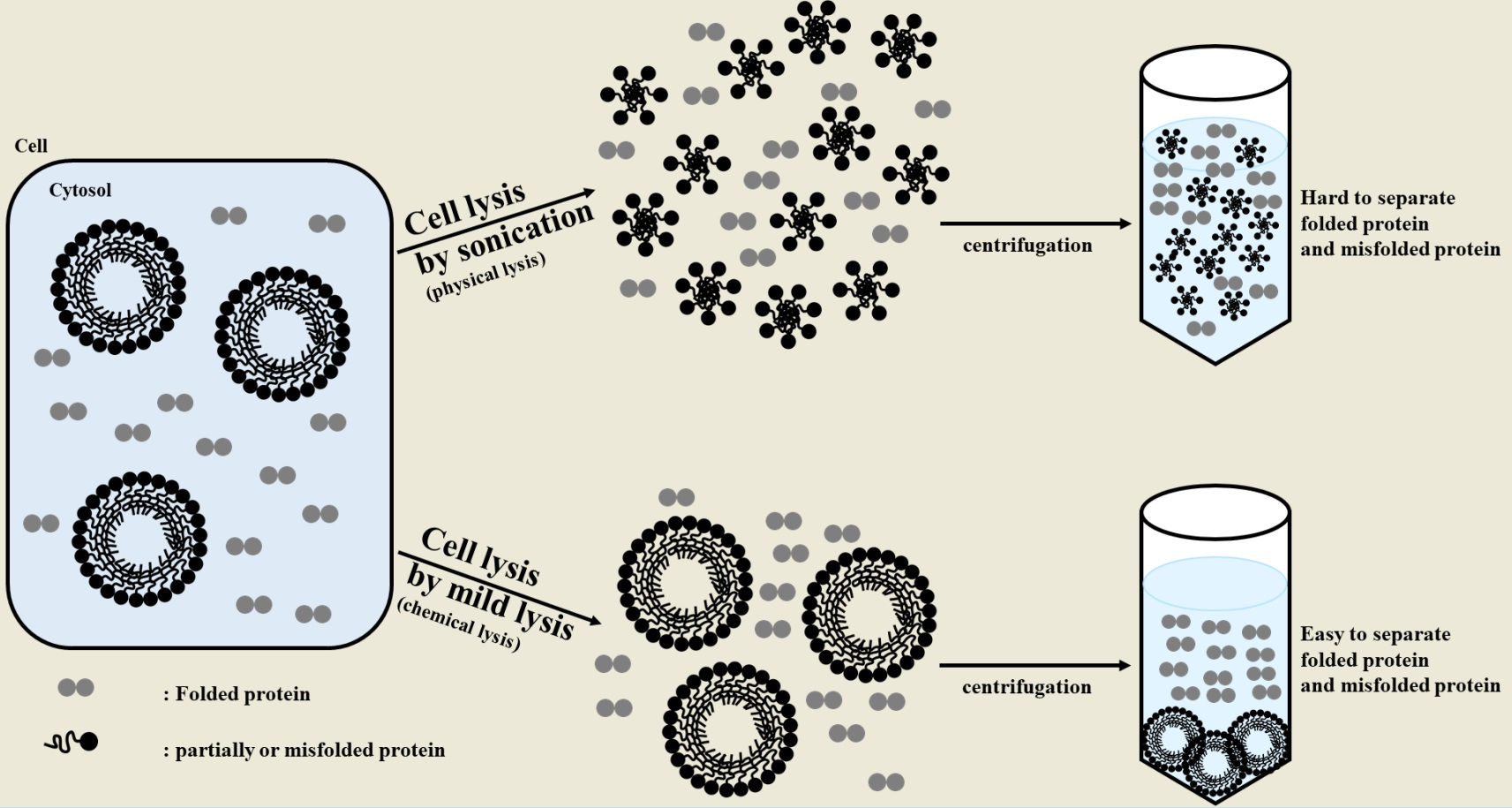
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
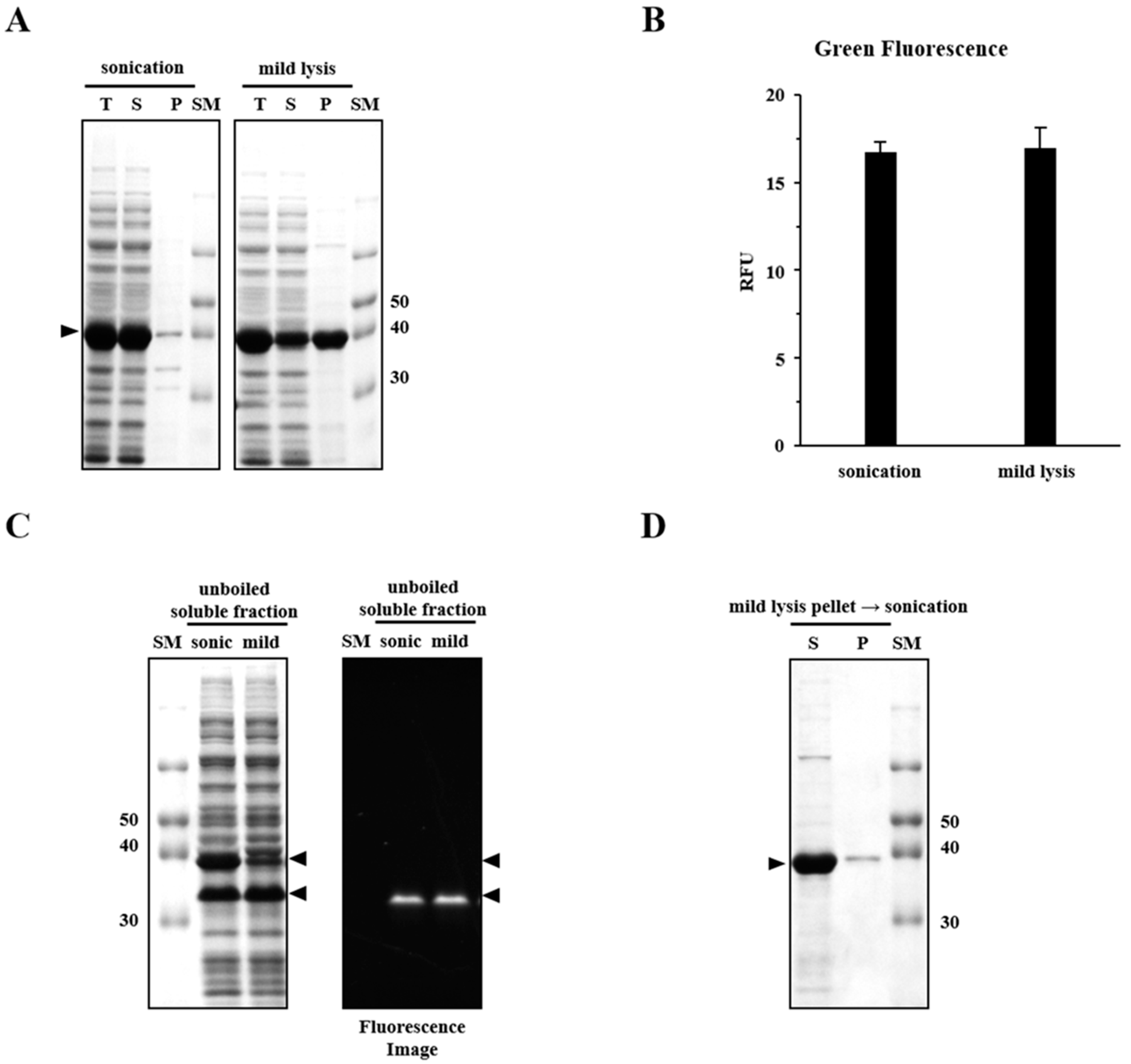
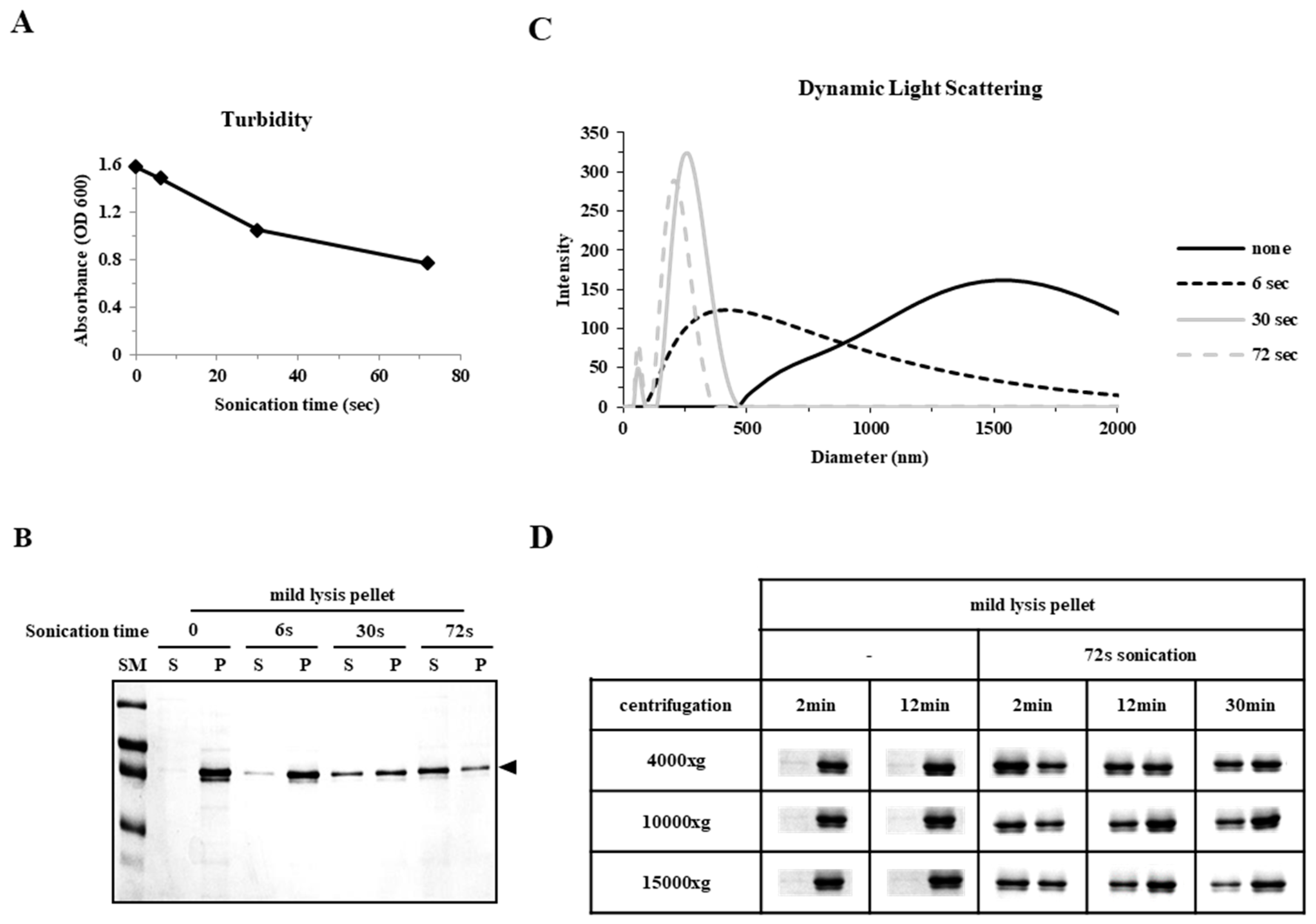
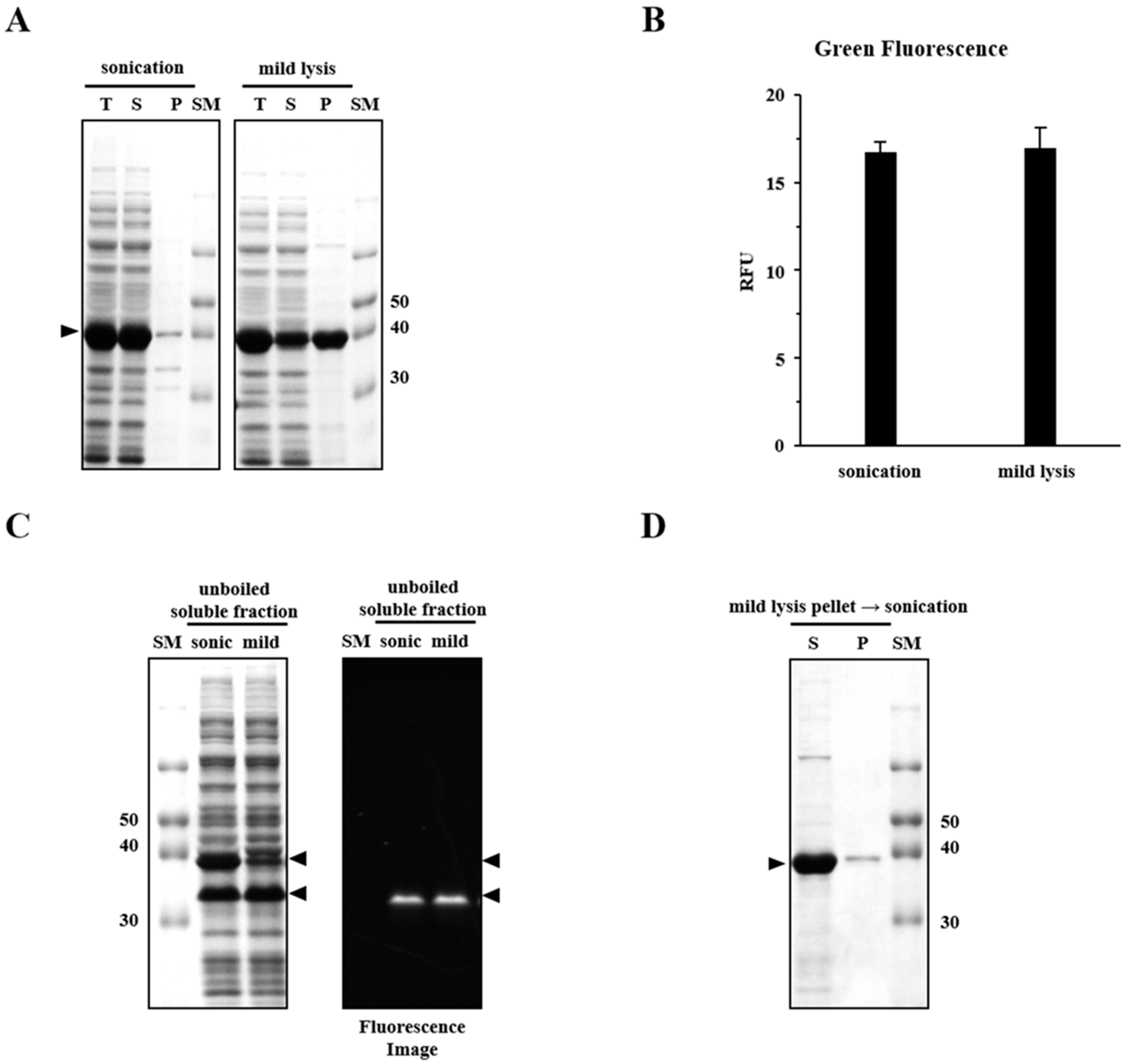
2.1. Sonication Negatively Affects Protein Quality
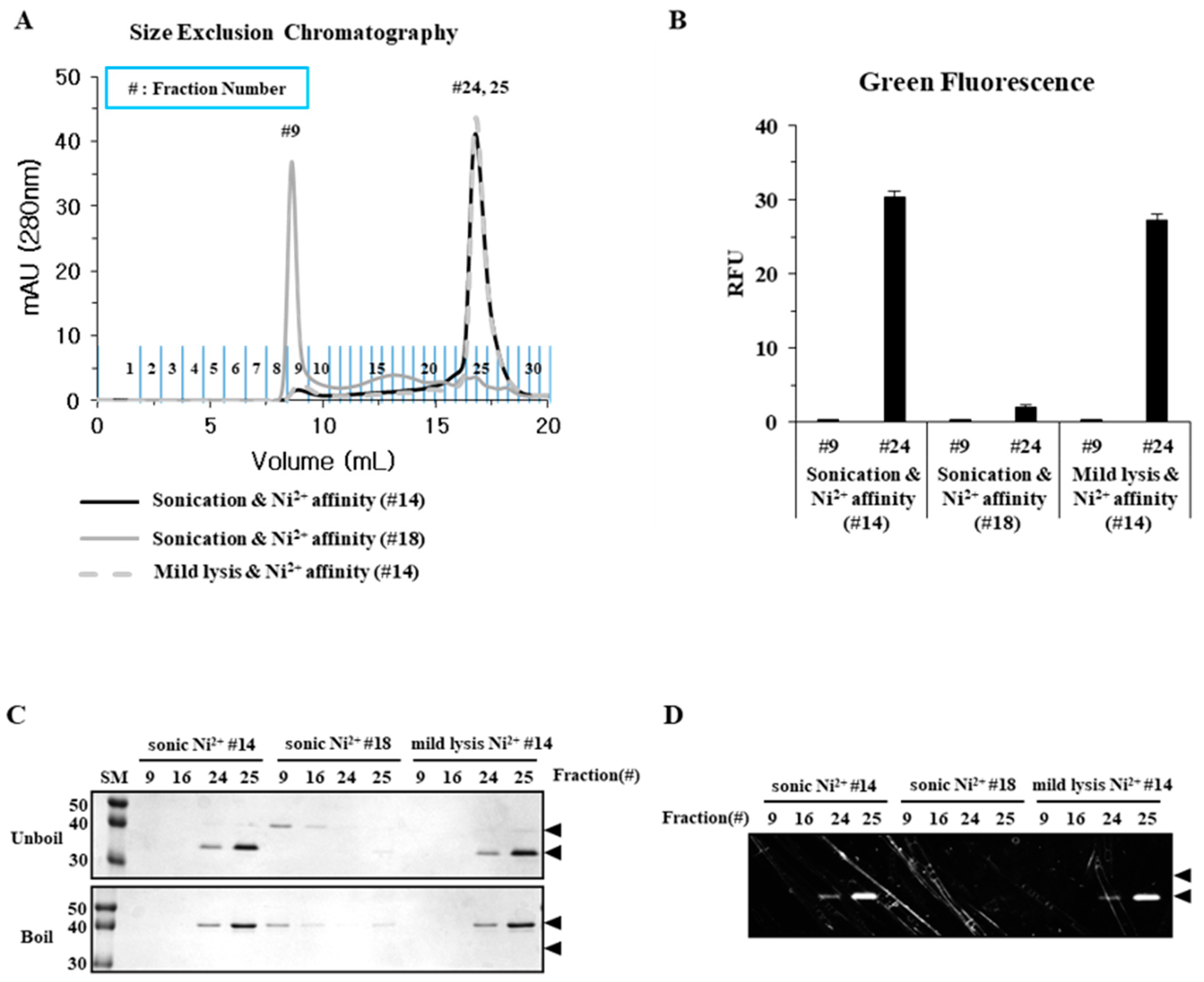
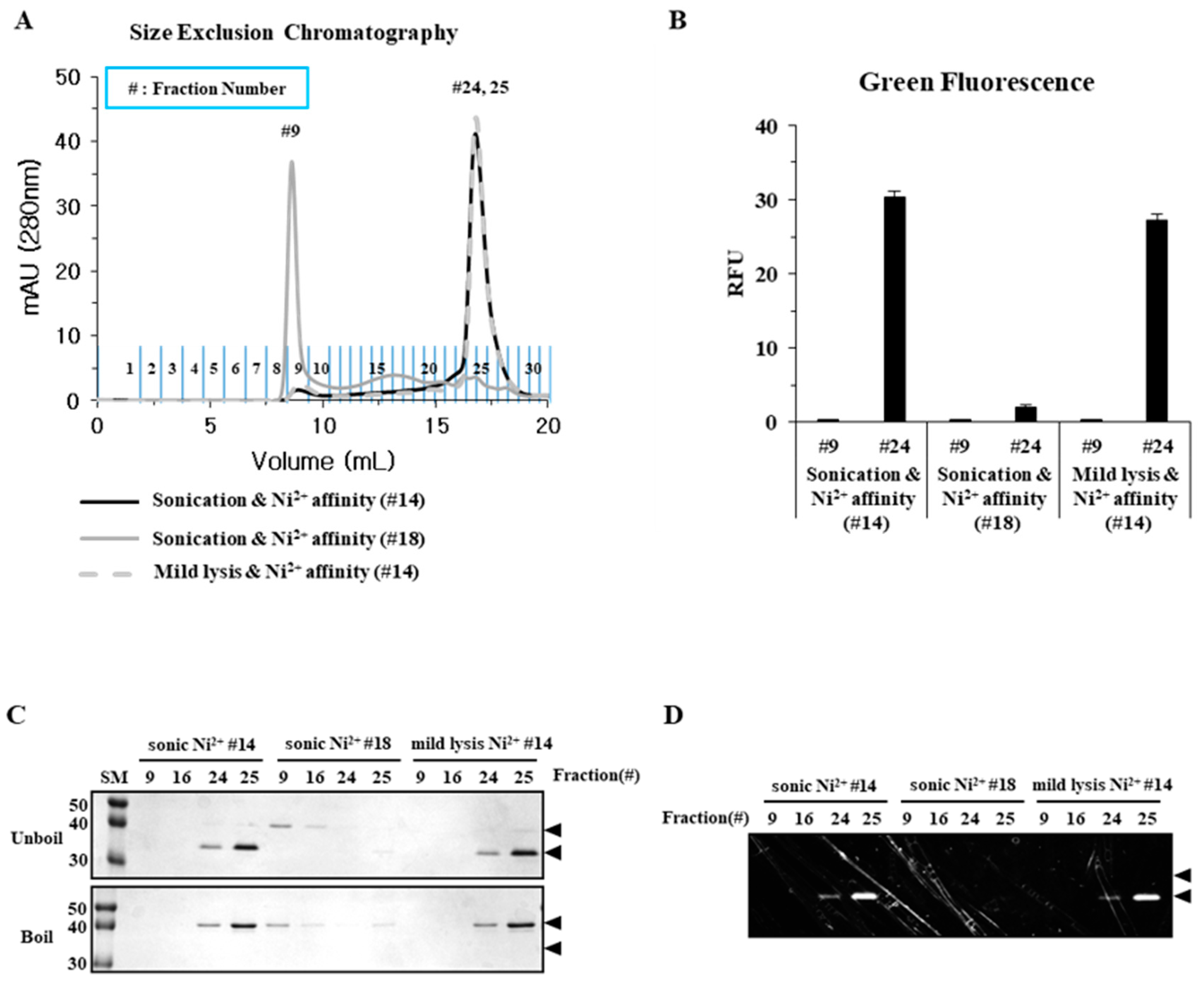
2.2. Sonication Increases the Level of Incorrectly Folded Soluble Aggregates
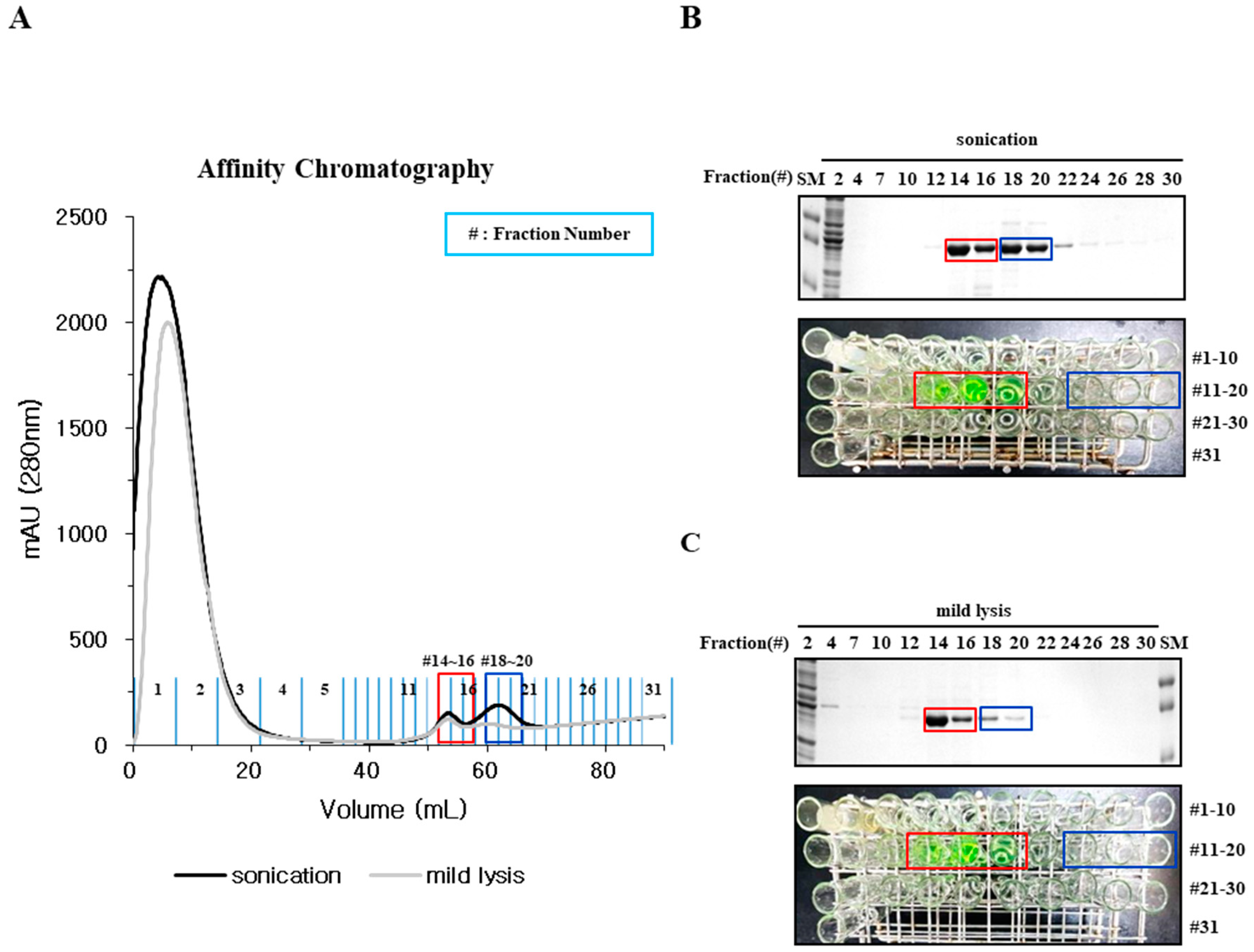
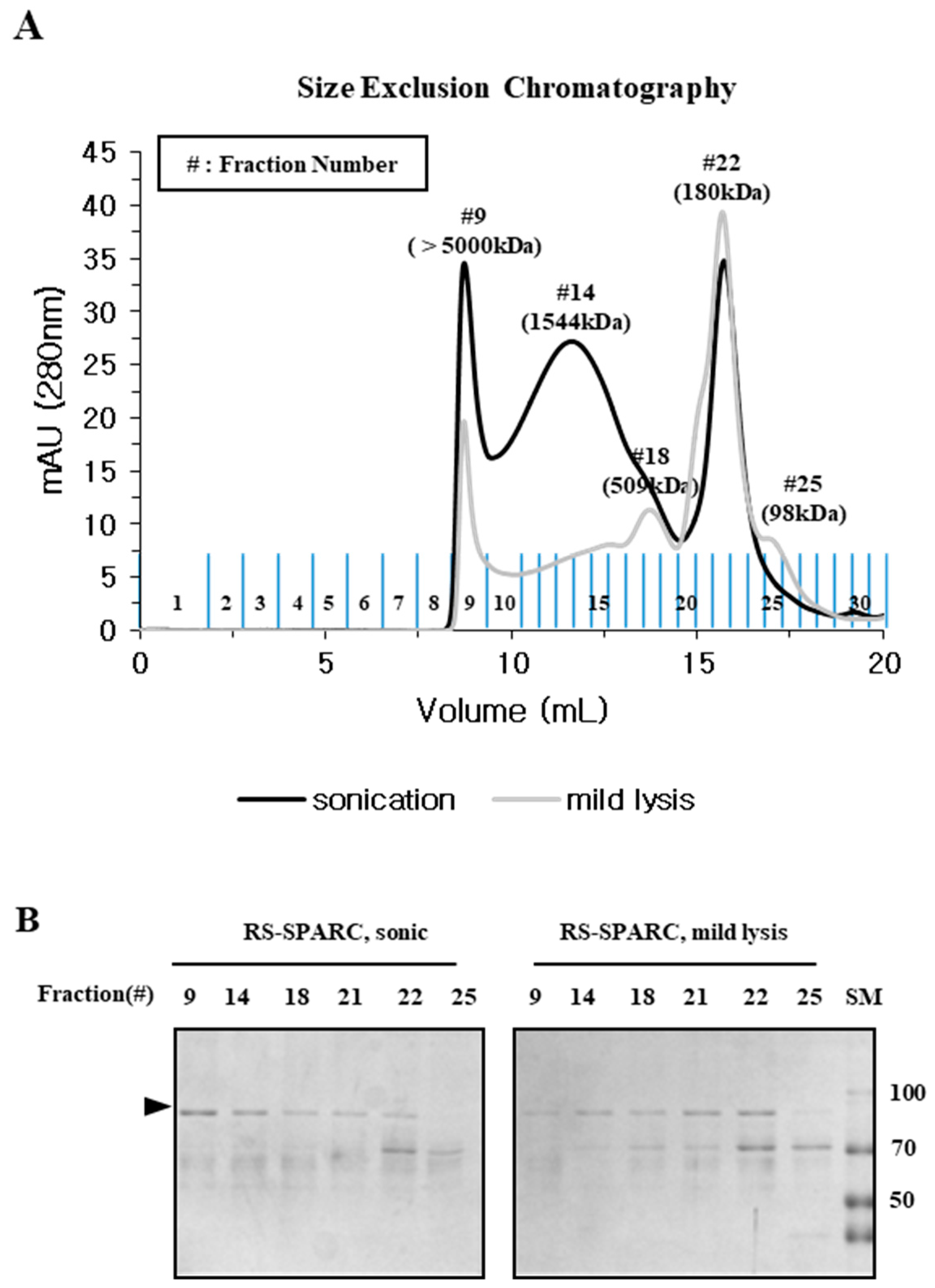
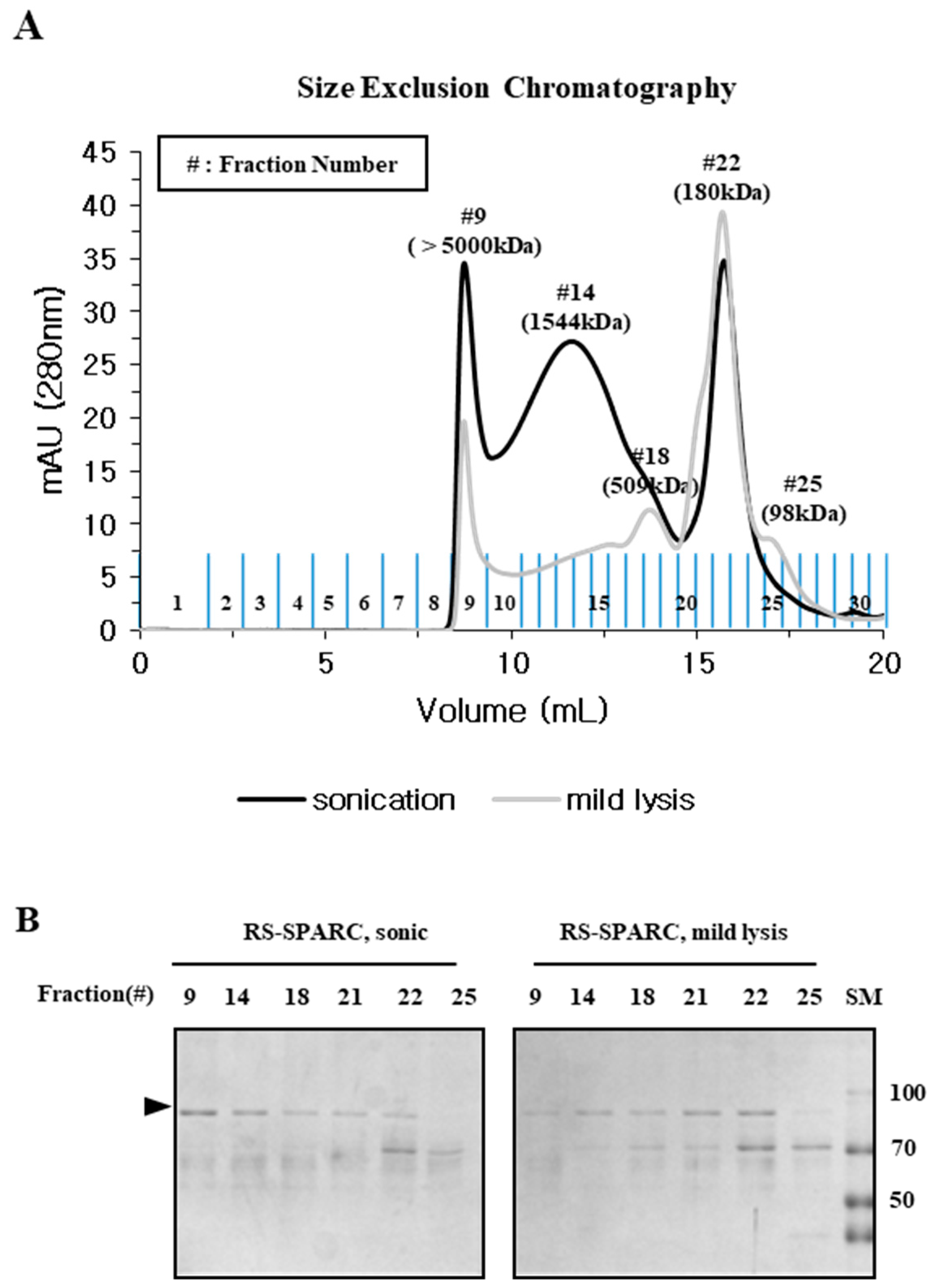
2.3. Particle Size is a Determining Factor for the Solubilization of Incorrectly Folded Aggregates
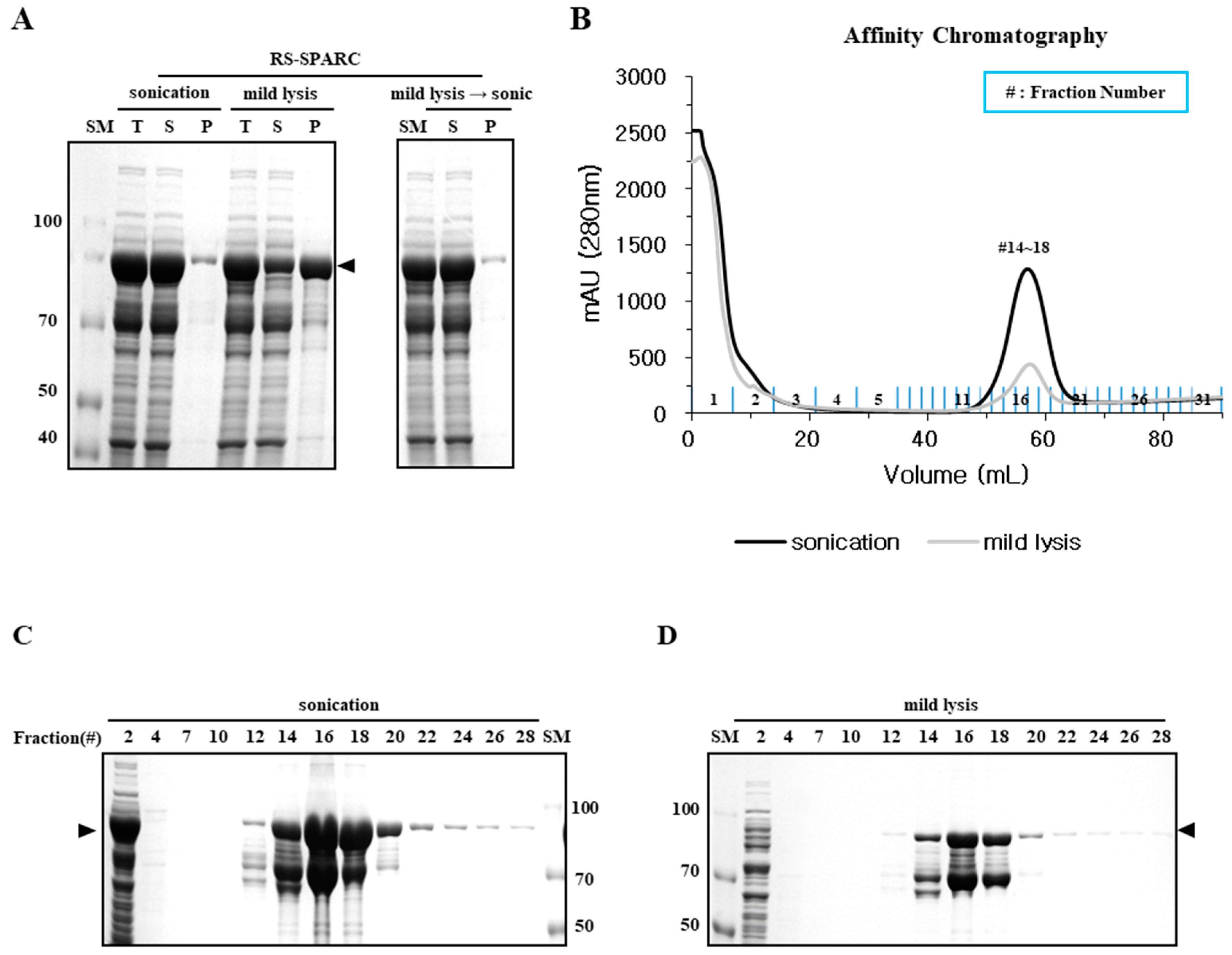
2.4. Mild Lysis Results in Better Recombinant RS-SPARC Protein Quality While Sonication Results in High Soluble Aggregate Contamination
3. Discussion
4. Materials and Methods
4.1. Construction of Protein Expression Plasmids
4.2. Protein Expression and Cell Lysis
4.3. Protein Purification
4.4. Dynamic Light Scattering (DLS)
4.5. Fluorescence Measurement
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Structural Genomics Consortium; China Structural Genomics Consortium; Northeast Structural Genomics Consortium; Graslund, S.; Nordlund, P.; Weigelt, J.; Hallberg, B.M.; Bray, J.; Gileadi, O.; Knapp, S.; et al. Protein production and purification. Nat. Methods 2008, 5, 135–146. [Google Scholar] [CrossRef] [Green Version]
- De Marco, A.; Deuerling, E.; Mogk, A.; Tomoyasu, T.; Bukau, B. Chaperone-based procedure to increase yields of soluble recombinant proteins produced in E. coli. BMC Biotechnol. 2007, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.H.; Kim, K.H.; Choi, S.I.; Park, E.S.; Park, S.H.; Ryu, K.; Park, Y.K.; Kwon, S.Y.; Yang, S.I.; Lee, H.C.; et al. RPS3a over-expressed in HBV-associated hepatocellular carcinoma enhances the HBx-induced NF-kappaB signaling via its novel chaperoning function. PLoS ONE 2011, 6, e22258. [Google Scholar] [CrossRef] [PubMed]
- Son, A.; Choi, S.I.; Han, G.; Seong, B.L. M1 RNA is important for the in-cell solubility of its cognate C5 protein: Implications for RNA-mediated protein folding. RNA Biol. 2015, 12, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.M.; Choi, H.S.; Seong, B.L. The folding competence of HIV-1 Tat mediated by interaction with TAR RNA. RNA Biol. 2017, 14, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Esposito, D.; Chatterjee, D.K. Enhancement of soluble protein expression through the use of fusion tags. Curr. Opin. Biotechnol. 2006, 17, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.; Kim, C.W.; Kim, B.H.; Han, K.S.; Kim, K.H.; Choi, S.I.; Seong, B.L. Assessment of substrate-stabilizing factors for DnaK on the folding of aggregation-prone proteins. Biochem. Biophys. Res. Commun. 2008, 373, 74–79. [Google Scholar] [CrossRef]
- Kyratsous, C.A.; Silverstein, S.J.; DeLong, C.R.; Panagiotidis, C.A. Chaperone-fusion expression plasmid vectors for improved solubility of recombinant proteins in Escherichia coli. Gene 2009, 440, 9–15. [Google Scholar] [CrossRef]
- Kuo, D.; Nie, M.; Courey, A.J. SUMO as a solubility tag and in vivo cleavage of SUMO fusion proteins with Ulp1. Methods Mol. Biol. 2014, 1177, 71–80. [Google Scholar]
- Choi, S.I.; Han, K.S.; Kim, C.W.; Ryu, K.S.; Kim, B.H.; Kim, K.H.; Kim, S.I.; Kang, T.H.; Shin, H.C.; Lim, K.H.; et al. Protein solubility and folding enhancement by interaction with RNA. PLoS ONE 2008, 3, e2677. [Google Scholar] [CrossRef]
- Santner, A.A.; Croy, C.H.; Vasanwala, F.H.; Uversky, V.N.; Van, Y.Y.; Dunker, A.K. Sweeping away protein aggregation with entropic bristles: Intrinsically disordered protein fusions enhance soluble expression. Biochemistry 2012, 51, 7250–7262. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, S.; Bardwell, J.C. RNAs as chaperones. RNA Biol. 2016, 13, 1228–1231. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Son, A.; Kim, J.; Kwon, S.B.; Kim, M.H.; Kim, P.; Kim, J.; Byun, Y.H.; Sung, J.; Lee, J.; et al. Chaperna-mediated assembly of ferritin-based Middle East respiratory syndrome-coronavirus nanoparticles. Front. Immunol. 2018, 9, 1093. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.W.; Jang, Y.H.; Kwon, S.B.; Lee, Y.J.; Chae, W.; Byun, Y.H.; Kim, P.; Park, C.; Lee, Y.J.; Kim, C.K.; et al. Harnessing an RNA-mediated chaperone for the assembly of influenza hemagglutinin in an immunologically relevant conformation. Faseb J. 2018, 32, 2658–2675. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.B.; Kim, P.; Woo, H.S.; Kim, T.Y.; Kim, J.Y.; Lee, H.M.; Jang, Y.S.; Kim, E.M.; Yong, T.S.; Seong, B.L. Recombinant adenylate kinase 3 from liver fluke Clonorchis sinensis for histochemical analysis and serodiagnosis of clonorchiasis. Parasitology 2018, 145, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Nomine, Y.; Ristriani, T.; Laurent, C.; Lefevre, J.F.; Weiss, E.; Trave, G. Formation of soluble inclusion bodies by hpv e6 oncoprotein fused to maltose-binding protein. Protein Expr. Purif. 2001, 23, 22–32. [Google Scholar] [CrossRef]
- Zanier, K.; Nomine, Y.; Charbonnier, S.; Ruhlmann, C.; Schultz, P.; Schweizer, J.; Trave, G. Formation of well-defined soluble aggregates upon fusion to MBP is a generic property of E6 proteins from various human papillomavirus species. Protein Expr. Purif. 2007, 51, 59–70. [Google Scholar] [CrossRef]
- Haacke, A.; Fendrich, G.; Ramage, P.; Geiser, M. Chaperone over-expression in Escherichia coli: Apparent increased yields of soluble recombinant protein kinases are due mainly to soluble aggregates. Protein Expr. Purif. 2009, 64, 185–193. [Google Scholar] [CrossRef]
- Harrison, S.T. Bacterial cell disruption: A key unit operation in the recovery of intracellular products. Biotechnol. Adv. 1991, 9, 217–240. [Google Scholar] [CrossRef]
- Stathopulos, P.B.; Scholz, G.A.; Hwang, Y.M.; Rumfeldt, J.A.; Lepock, J.R.; Meiering, E.M. Sonication of proteins causes formation of aggregates that resemble amyloid. Protein Sci. 2004, 13, 3017–3027. [Google Scholar] [CrossRef]
- Craggs, T.D. Green fluorescent protein: Structure, folding and chromophore maturation. Chem. Soc. Rev. 2009, 38, 2865–2875. [Google Scholar] [CrossRef]
- Aoki, T.; Tahara, T.; Satoh, K.; Fujino, H.; Watabe, H. General properties of GFP-display, an electrophoretic analysis for single amino acid changes in target polypeptides. Anal. Biochem. 2003, 317, 107–115. [Google Scholar] [CrossRef]
- Aoki, T.; Takahashi, Y.; Koch, K.S.; Leffert, H.L.; Watabe, H. Construction of a fusion protein between protein A and green fluorescent protein and its application to western blotting. FEBS Lett. 1996, 384, 193–197. [Google Scholar] [CrossRef]
- Jang, Y.H.; Cho, S.H.; Son, A.; Lee, Y.H.; Lee, J.; Lee, K.H.; Seong, B.L. High-yield soluble expression of recombinant influenza virus antigens from Escherichia coli and their potential uses in diagnosis. J. Virol. Methods 2014, 196, 56–64. [Google Scholar] [CrossRef]
- Onesti, S.; Miller, A.D.; Brick, P. The crystal structure of the lysyl-tRNA synthetase (LysU) from Escherichia coli. Structure 1995, 3, 163–176. [Google Scholar] [CrossRef]
- Kwon, S.B.; Yu, J.E.; Park, C.; Lee, J.; Seong, B.L. Nucleic acid-dependent structural transition of the intrinsically disordered N-terminal appended domain of human Lysyl-tRNA synthetase. Int. J. Mol. Sci. 2018, 19, 3016. [Google Scholar] [CrossRef] [PubMed]
- Thommen, M.; Holtkamp, W.; Rodnina, M.V. Co-translational protein folding: Progress and methods. Curr. Opin. Struct. Biol. 2017, 42, 83–89. [Google Scholar] [CrossRef]
- Kim, C.W.; Han, K.S.; Ryu, K.S.; Kim, B.H.; Kim, K.H.; Choi, S.I.; Seong, B.L. N-terminal domains of native multidomain proteins have the potential to assist de novo folding of their downstream domains in vivo by acting as solubility enhancers. Protein Sci. 2007, 16, 635–643. [Google Scholar] [CrossRef]
- Chen, A.; Zhang, L.; Gu, S.; Tang, R.; Xie, Y.; Ji, C. Investigation of TtrD, an expressing recombinant fusion tag, in Escherichia coli. Protein Expr. Purif. 2016, 120, 65–71. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, S.B.; Yu, J.E.; Kim, J.; Oh, H.; Park, C.; Lee, J.; Seong, B.L. Quality Screening of Incorrectly Folded Soluble Aggregates from Functional Recombinant Proteins. Int. J. Mol. Sci. 2019, 20, 907. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040907
Kwon SB, Yu JE, Kim J, Oh H, Park C, Lee J, Seong BL. Quality Screening of Incorrectly Folded Soluble Aggregates from Functional Recombinant Proteins. International Journal of Molecular Sciences. 2019; 20(4):907. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040907
Chicago/Turabian StyleKwon, Soon Bin, Ji Eun Yu, Jihoon Kim, Hana Oh, Chan Park, Jinhee Lee, and Baik L. Seong. 2019. "Quality Screening of Incorrectly Folded Soluble Aggregates from Functional Recombinant Proteins" International Journal of Molecular Sciences 20, no. 4: 907. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040907