Modulation of Obesity and Insulin Resistance by the Redox Enzyme and Adaptor Protein p66Shc


1
Veneto Institute of Molecular Medicine, 35128 Padova, Italy
2
Department of Medicine, University of Padova, 35128 Padova, Italy
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2019, 20(4), 985; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040985
Submission received: 1 February 2019
/
Revised: 15 February 2019
/
Accepted: 21 February 2019
/
Published: 24 February 2019
(This article belongs to the Special Issue Mechanisms of Insulin Resistance at the Crossroad of Obesity with Associated Metabolic Abnormalities)
Abstract
:Initially reported as a longevity-related protein, the 66 kDa isoform of the mammalian Shc1 locus has been implicated in several metabolic pathways, being able to act both as an adaptor protein and as a redox enzyme capable of generating reactive oxygen species (ROS) when it localizes to the mitochondrion. Ablation of p66Shc has been shown to be protective against obesity and the insurgence of insulin resistance, but not all the studies available in the literature agree on these points. This review will focus in particular on the role of p66Shc in the modulation of glucose homeostasis, obesity, body temperature, and respiration/energy expenditure. In view of the obesity and diabetes epidemic, p66Shc may represent a promising therapeutic target with enormous implications for human health.
Keywords:
diabetes; metabolic syndrome; adipose tissue; muscle; glucose tolerance; aging; oxidative stress1. Introduction
The p66Shc protein is encoded by the Shc1 locus, together with two shorter isoforms known as p52Shc and p46Shc [1,2]. While the last two proteins are generated by the same mRNA using different translation initiation sites [2], p66Shc is produced from a different exon arrangement at the 5′ end. The structure of p66Shc and of the Shc1 locus were extensively reviewed by different authors and will not be discussed in detail in this review [3,4,5]. The three Shc isoform proteins share a common structure, which comprises a phosphotyrosine-binding domain (PTB), a collagen homology 1 (CH1) domain rich in prolines, and a sarcoma homologous type 2 domain (SH2). Shc protein family members are present in mammals, amphibians, fishes, insects (D. Melanogaster), nematodes (C. Elegans), and yeasts, and a typical characteristic of them is to have PTB and SH2 domains in the same order from the N- to C-terminals [3,5]. From an evolutionary point of view, p66Shc appears to be the most recent isoform, as it is found in vertebrates, but not in yeasts, nematodes, and insects. Unlike p52Shc and p46Shc, p66Shc has an additional collagen homology region (CH2); moreover, p46Shc does not have a cytochrome c binding domain (CB), which is shared between p52Shc and p66Shc.
1.1. Role of p66Shc in Signal Transduction
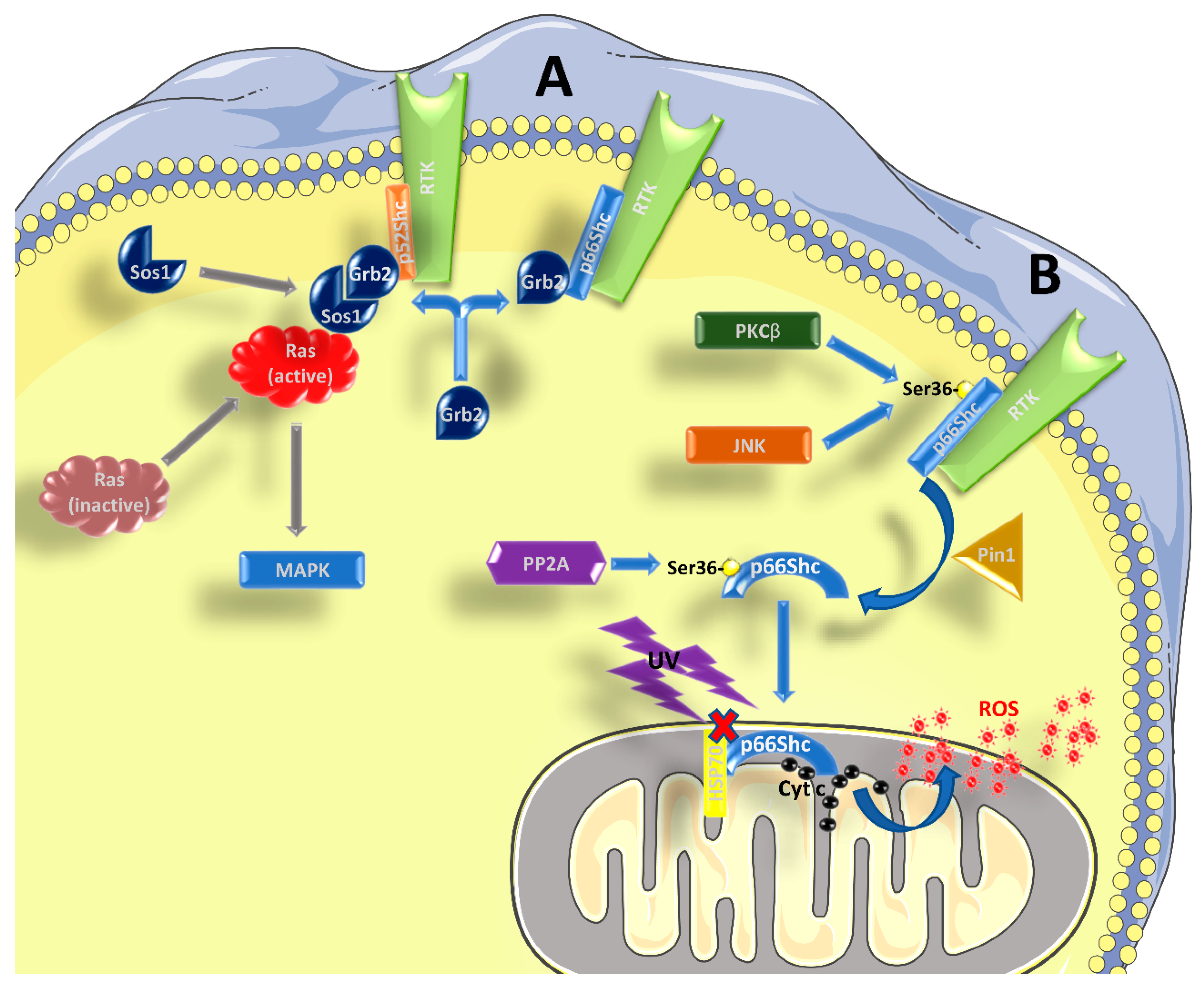
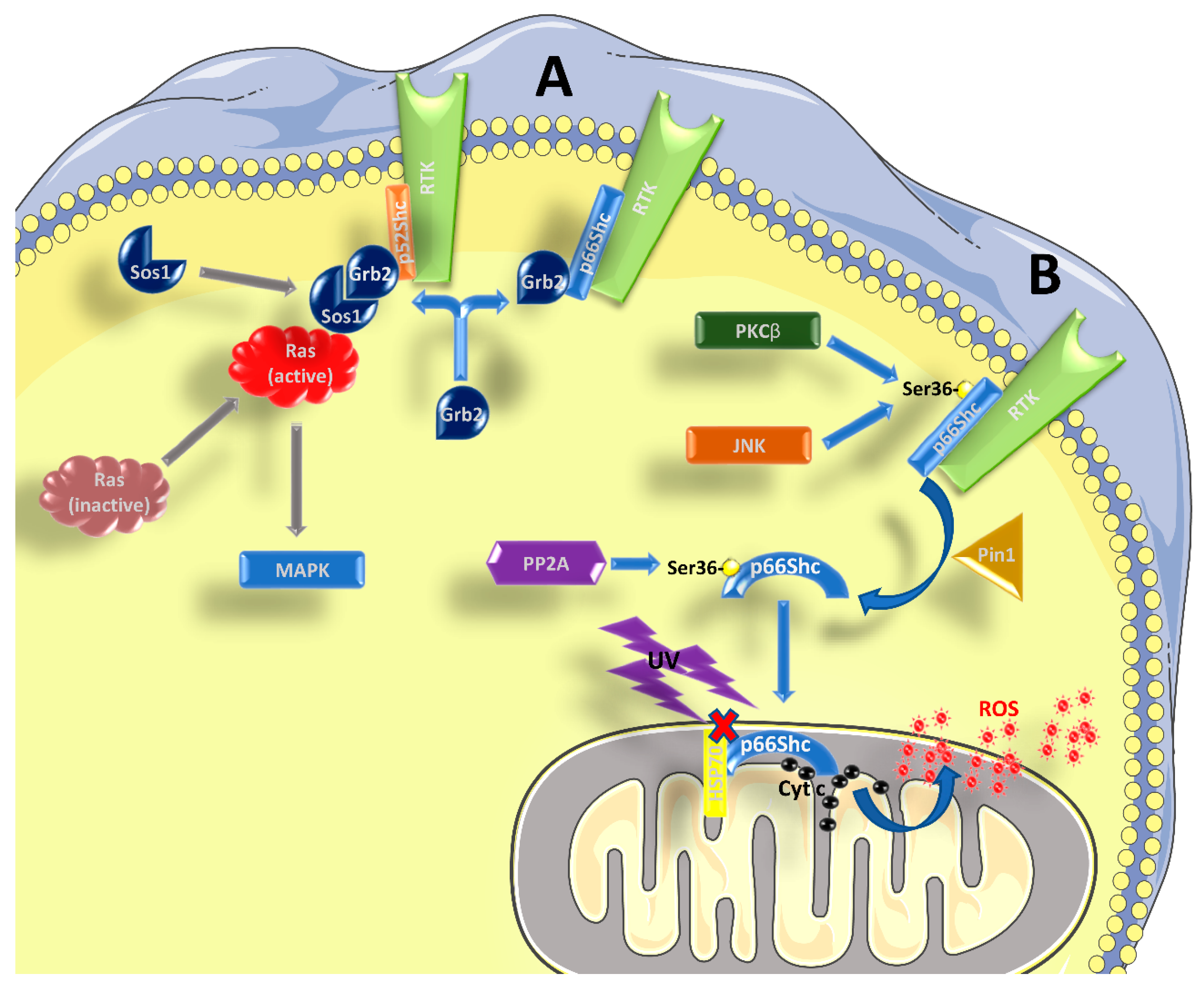
The three aforementioned Shc proteins are also different in terms of the signaling pathway wherein they are involved. It is well known that p52Shc and p46Shc are able to transduce the signal from tyrosine-kinase receptors (RTKs) to Ras and mitogen-activated protein kinase (MAPK) pathways [1,6] (Figure 1A). Shc binding to RTKs causes a phosphorylation of three tyrosine residues in their CH1 domain, which is required for the recruitment of the Grb2/Sos1 complex (growth factor receptor-bound protein 2 and son of sevenless 1) at the SH2 domain, which in turn leads to the activation of Ras [4], as Sos1 is a guanine nucleotide exchange factor (GEF). Given its structure, p66Shc should be able to form the same complexes and activate Ras. However, many studies have indicated that p66Shc has an inhibitory role on the Ras-MAPK pathway, regardless its ability to bind Grb2 [2,6,7,8,9]. It has been proposed that p66Shc competes with p52Shc and p46Shc for the binding with Grb2, causing the disruption of the Grb2/Sos1 complex, and in this context it seems that the phosphorylation of Ser36 by p66Shc is required [7,10,11]. Therefore, an increased activation of p66Shc might be enough to inhibit the Ras-MAPK pathway. However, the role of p66Shc in transducing RTK signals is far from being completely understood.
There is vast literature showing that p66Shc plays a major role in the response to oxidative and environmental stress stimuli [4,12,13,14,15,16,17,18] (Figure 1B). Kinases like c-Jun N-terminal kinase (JNK) or protein kinase C β (PKCβ), which are activated in response to stress stimuli, can phosphorylate a particular serine residue (Ser36) of p66Shc within its CH2 domain [19,20]. This step is followed by a cis-trans isomerization by peptidyl-prolyl cis-trans isomerase 1 (Pin1), which allows the translocation of p66Shc into the inter-membrane mitochondrial space, after it has been dephosphorylated by protein phosphatase 2A (PP2A). A more recent paper found that Ser36 might not be the crucial phosphorylation site to mediate the PKCβ response, while Ser139, Ser213, and Thr206 might be involved [21]. At the mitochondrial level, and without pro-apoptotic stimuli (such as H2O2 or UV radiation), p66Shc is bound to high-molecular weight complexes and heat shock protein 70 (HSP70) or other proteins involved in the inter-membrane transport [22,23,24]. After stimulation, however, p66Shc can interact with cytochrome c through its CB domain, generating reactive oxygen species (ROS), by diverting electrons from the mitochondrial electron transport chain (ETC) [4,15,18,25]. In this regard, it is worth mentioning that some authors, based on the structure of p66Shc, questioned its ability to be an acceptor of electrons from the ETC (reviewed in [5,26]). However, it should be noted that, in the absence of further experimental data to corroborate this notion, this remains a mere speculation. In any case, even if the exact mechanism might be still debated, it is well known that p66Shc is involved in the production of ROS, and an excess in ROS production can interfere with many cellular processes and induce apoptosis. Apart from increasing mitochondrial ROS production, there are two other mechanisms whereby p66Shc can increase ROS levels: (i) by decreasing the production of ROS scavengers through inhibition of forkhead box O (FOXO) transcription factors and (ii) by increasing the activity of membrane NADPH oxidase via Rac1 activation (reviewed in [5,15]). The involvement of p66Shc in the induction of apoptosis is confirmed by the fact that its elimination or over-expression have opposite effects, making cells more resistant or more susceptible to apoptosis, respectively, ([12,16] and reviewed by [4,5,14,27]). However, the fact that p66Shc favors ROS formation thereby stimulating apoptosis could be a too simplistic view, since both an anti-oxidant [28] and an anti-apoptotic behavior of p66Shc [29] have been reported, albeit only in specific cell types and conditions. It was also reported that p66Shc can participate in the induction of apoptosis, acting downstream of p53 [16]. The activation of p53 in response to H2O2 confers stability to the p66Shc protein and probably an increase at the transcript level, since there is a p53-binding region within the p66Shc promoter [30]. Indeed, p53 can be activated even in the absence of p66Shc, but the cells become apoptosis-resistant in such conditions.
As discussed above, PKCβ can phosphorylate p66Shc, and a study pointed out a link between p66Shc and the autophagic pathway [31]. Autophagy is a highly regulated process through which the cells can recycle components that are either unnecessary or malfunctioning. It is well known that starvation activates autophagy, and the authors demonstrated that p66Shc can inhibit autophagy, following starvation in mouse embryonic fibroblasts (MEF) in a PKCβ-dependent manner. A recent paper investigated the induction of autophagy in vivo in the muscles of mice after downhill running, which is a type of exercise known to induce muscle damage, ROS production, and activation of the autophagic process [32]. Their data indicate that p66Shc−/− mice have higher LC3 lipidation than wild type (WT) mice, but it is not further increased after exercise and other autophagic markers are not significantly different.
1.2. p66Shc and Longevity
It was initially reported that deletion of the p66Shc gene was sufficient to cause an increase in the average and maximum longevity in mice [12]. Indeed, mice in which p66Shc was deleted had a 30% increase in their life-span compared with WT. These results were surprisingly similar to those obtained by putting mice under calorie restriction [33,34], but p66Shc−/− mice were not leaner, nor did they eat less than WT. This observation supported the idea that a decreased ROS production was protective against the accumulation of DNA damages caused by free radicals, thereby delaying ageing and promoting an increase in life-span. Despite the inhibition of apoptosis, these mice did not show an increased susceptibility toward tumorigenesis. As already mentioned, p66Shc−/− is a downstream mediator of p53 in the apoptotic pathway, but its deletion does not interfere with other p53-dependent pathways [16]. Indeed, p53−/− mice displayed increased mortality due to spontaneous tumorigenesis, which was not observed in p66Shc−/− mice. A more recent study on this matter dismantled the notion that p66Shc regulates life-span: by using a higher number of mice compared with the original study, three different mouse strains (C57BL/6J, 129Sv, and a hybrid C57BL/6J-129Sv), and animals housed in two different facilities, p66Shc−/− did not show increased life-span [35]. The authors noted how the average and maximum longevity of the WT mice were unusually low in the original study [12], and this could have been due to environmental stress. Moreover, the suspicion that p66Shc was not involved in longevity determination was already raised by a study conducted on centenarian humans, in which it was found that the expression of p66Shc in isolated fibroblasts was elevated, instead of being reduced [36].
Importantly, the role of p66Shc in the determination of lifespan was further investigated in another study, wherein a telomerase RNA component (TERC−/−) and p66Shc−/− double knockout mouse was generated. TERC−/− mice have a decreased average lifespan, and it was observed that the concomitant deletion of p66Shc was unable to restore this defect, while it was able to ameliorate other aspects, like sterility, weight loss, and multi-organ atrophy. To date, the exact phenotype of these mice has not been fully investigated [37]. In summary, it is possible that p66Shc does not truly regulate life expectancy, but it is involved in the determination of health-span, which has a very strong translational impact to human pathology.
1.3. p66Shc, Body Weight Regulation, and Obesity
According to the authors of [12], the body weight of p66Shc−/− mice was identical to that of WT mice, and so was food intake. In contrast, a more recent paper found that p66Shc−/− mice were leaner, with body weight differences being mainly due to a decreased amount of abdominal and inguinal fat, particularly evident in males, while the weight of other organs was not different [38]. Our group also showed that, under standard diet, p66Shc−/− were leaner than WT mice [39]. The observation that organ weight was not different between WT and knockout mice was confirmed by another study, in which no differences in total body weight or in fat-free mass was found, except at older ages (27-month-old animals) [40]. It was also shown that p66Shc−/− mice subjected to a 5% calorie-restriction (CR) regimen, between 4 and 18-months-of-age, are leaner than WT mice, but not with a 40% CR [35]. The effect of calorie restriction on body weight was also studied in 18-month-old animals [41]: at baseline there were no differences between WT and knockout animals, and the same results were found when a 26% CR regimen for 2 months or a 40% CR regimen for 3 days was applied. The mechanisms whereby p66Shc would regulate body weight are incompletely understood. On one side, there may be a heat dissipation from the ETC due to increased uncoupling in the adipose [38]. Furthermore, insulin is able to activate the production of H2O2 in pre-adipocytes from brown adipose tissue, but not if p66Shc is ablated [38], and this event is necessary to modulate the activity of the Akt-Foxo1 pathway. In particular, if p66Shc is missing, the phosphorylation of Akt is blunted. A proper response to insulin stimulation allows for the accumulation of triglycerides both in brown and white pre-adipocytes, by favoring their import and contemporaneously inhibiting β-oxidation processes.
Even if there is a general consensus about the fact that p66Shc deletion confers protection against obesity, some conflicting data were found. It was shown by many authors that p66Shc−/− mice are obesity-resistant, whether obesity is genetically- [39,42] or diet-induced [38,39,43]. However, in another study, a new p66Shc−/− mouse model was generated, named ShcL [44], and some contrasting data were shown. ShcL mice were susceptible to, not protected from, diet-induced obesity, becoming more obese than WT animals in response to a high-fat diet (HFD). The authors showed that ShcL mice did not have perturbations in the expression pattern of p52Shc and p46Shc, compared with the original p66Shc−/− mice (called ShcP), while p46Shc was increased in the adipose tissue of the ShcP; they reasoned that this might be the reason for the discrepancy in obesity. However, the idea that an increase in p46Shc together with the absence of p66Shc in the adipocytes, eventually coupled with a decreased expression of p52Shc, is responsible for a decreased fat accumulation was not supported by further experimental data. In addition, we did not find any increase in adipose tissue p46Shc protein expression in ShcP mice [39].
Very few studies have focused on adipokines. In particular, it was demonstrated that in lean p66Shc−/− mice there were decreased plasma levels of leptin and adiponectin [39,45]. The same was also demonstrated in obese knockout animals for adiponectin [39] and leptin [45]. One study, in contrast, found no differences in plasma leptin concentration between WT and p66Shc−/− mice, but only increased plasma leptin levels in females compared with males, regardless of genotype, when animals were prenatally exposed to HFD [46]. Regarding adiponectin, it was higher in knockout females compared with WT, and also in females versus males, but only in p66Shc−/−. Adiponectin was also measured in primary brown adipocytes [45] or in the adipose tissue [46], finding a decrease in p66Shc−/− adipocytes or an increase only in p66Shc−/− females, respectively. The concentration of circulating plasminogen activator inhibitor 1 (PAI-1) was similar between WT and knockout animals, which was increased in obese Lepob/ob animals, regardless of genotype [39], while its expression in white adipose tissue, measured by quantitative PCR, was decreased [45]. Finally, TNFα production was decreased also in p66Shc−/− mice, both in the plasma of obese animals and in primary brown adipocytes [45].
1.4. p66Shc, Diabetes, and the IGF-1 Axis
The possibility that p66Shc regulates body weight and that its deletion improves obesity, made p66Shc a possible candidate gene against obesity-related diseases. [47]
Furthermore, based on the role of p66Shc in ROS production, there is a consensus about the ability of p66Shc−/− mice to counteract many side-effects of pathologies commonly attributed to oxidative stress, including chronic diabetic complications. In fact, it has been demonstrated that the absence of p66Shc confers protection toward diabetes-induced endothelial damage [48,49], diabetic nephropathy [46], and diabetic cardiomyopathy [50] and improves the healing of diabetic ulcers [51]. The exact mechanisms at work have never been clearly dissected, and there may be several pathways affected by p66Shc deletion in addition to the regulation of cellular oxidant status.
It was reported that the insulin-like growth factor 1 receptor (IGF1-receptor) can phosphorylate p66Shc, and in MEFs derived from mice where the expression of IGF1-receptor was reduced (IGF-1R+/− mice) there was also a reduced tyrosine phosphorylation of p66Shc and p52Shc [52]. IGF1 stimulation is also able to induce the phosphorylation of p66Shc in tyrosine residues in L6 myoblasts [53], and the silencing of p66Shc leads to abnormal phosphorylation of extracellular signal-regulated kinase 1/2 (ERK1/2), which is elevated in basal conditions and blunted after IGF1 stimulation. Moreover, the reduced expression of p66Shc caused an increased glucose uptake in basal conditions, preeminently due to an ERK-mediated remodeling of the actin cytoskeleton, but also to an increase of GLUT1 and GLUT3, both at the protein and mRNA levels [54].
1.5. Role of p66Shc in the Regulation of Glucose Homeostasis
Many studies tried to shed light on the role of p66Shc in insulin and glucose metabolism. This is reasonable, as Shc adapter proteins can interact and transduce the signaling evoked by insulin receptors (reviewed in [55]). Moreover, it was demonstrated that IRS-1, p66Shc, and S6K can associate to form a complex (reviewed in [56]). However, as already noted for body weight and obesity, contrasting data can be found in the literature in regard to the role of p66Shc in glucose and insulin tolerance at the whole-body level. As reported above, GLUT1 and GLUT3 were upregulated in L6 myoblasts with reduced p66Shc expression. Moreover, basal glucose transport was increased, while the adenoviral-mediated overexpression of p66Shc produced opposite effects [54]. Using different cell types (HeLa and MEFs), it was confirmed that p66Shc deficiency enhances glucose uptake [57]. Also, cells have an increased proportion of metabolites for fatty acids biosynthesis. Another partial confirmation came from a study conducted in the skeletal muscles of p66Shc−/− mice, wherein glucose uptake was not studied in detail, but it was found that glucose content was similar to that of fed WT animals, and glycogen was more abundant in knockout muscles, either in fed or starved conditions [58], indicating that glucose uptake in knockout animals was probably not impaired. In primary adipocytes, basal glucose uptake was found to be identical between WT and p66Shc−/− but increased after insulin stimulation only in knockout cells [42]. Using the aforementioned ShcL p66Shc−/− knockout mice, it was confirmed that insulin-stimulated glucose uptake was increased in cultured adipocytes, compared with that of WT-derived adipocytes [44]. However, this matter may be more complicated than it seems. In the same paper [44], using the initial p66Shc−/− mice (ShcP) as in [42], opposing results were found regarding insulin-stimulated glucose uptake in adipocytes, which was decreased in the first and increased in the latter, respectively. An attempt to reconcile these contrasting data was made in a more recent study [56], where the authors discussed the possibility that p66Shc plays both a positive and a negative role on the insulin pathway, by acting upstream and downstream of mTOR/S6K. Basically, it was proposed that in WT obese mice there is insulin desensitization in the adipose tissue due to constitutive activation of S6K (and its substrate S6), which leads to IRS-1 degradation. In parallel, decreased PI3K recruitment to IRS-1 impairs downstream Akt signaling. Finally, we also reported that glucose uptake in isolated skeletal muscles was lower in p66Shc−/− mice, compared that with WT, after insulin stimulation [39].
Some studies reported an increased lactate production in response to p66Shc deletion. This was demonstrated in immortalized p66Shc−/− MEFs as a result of increased anaerobic glycolysis and decreased mitochondrial respiration [59], and confirmed in the same cell type and also in HeLa cells [57]. In the latter paper, the authors showed that p66Shc deficiency in HeLa cells enhances the glycolytic metabolism, favored by the concomitant activation of the pentose phosphate and hexosamine pathways, which contributes to the maintenance of a proper redox balance within the cell (by provision of NAPH) and provides a positive feedback on the signaling [57]. On the other hand, when p66Shc expression was restored in p66Shc−/− MEF cells, glycolytic metabolism was impaired. Interestingly, this paper identified a link between p66Shc and mTOR signaling and it was shown that activation of mTOR is associated with an increased anabolic metabolism and protein synthesis. In particular, both S6K and Akt, targets of mTORC1 and mTORC2, respectively, were phosphorylated in p66Shc−/− compared with control HeLa cells after serum stimulation, but not if cells were pre-incubated with the mTOR inhibitor Torin. Consistently, an increased production of lactate and citrate was also observed in the skeletal muscles of p66Shc−/− mice [58], but in this case the authors showed a decreased glycolytic capacity, both in a fed and fasted state, as suggested by a decreased activity of key glycolytic enzymes, such as hexokinase, phosphofructokinase, and pyruvate kinase.
The relationship between p66Shc and glucose homeostasis was also studied in obese mice, and conflicting results have been obtained even in this case. p66Shc−/−LepOb/Ob double knockout mice on a mixed genetic background (C57Bl/6J and 129Sv) had improved insulin sensitivity (similar to that of WT lean mice) and glucose tolerance compared with p66ShcWT LepOb/Ob animals, but were still glucose intolerant [42]. No differences in fasting glucose between lean WT and p66Shc−/− mice were reported, nor in glucose tolerance tests (GTT) and insulin tolerance tests (ITT). Another study, however, found that lean p66Shc−/− were more insulin sensitive and glucose tolerant than WT animals [44] at 3 months of age. At 24 months, only insulin sensitivity was improved, in comparison with WT animals. Moreover, insulin sensitivity was reported to be higher in muscle samples from both ShcL and ShcP mice than from control WT. The same paper reported an improved insulin sensitivity in ShcP mice even after a high fat diet (HFD), which was lost in ShcL mice, at least in the liver. However, our data depicted a different scenario in mice on a pure C57Bl/6J background: worsened glucose tolerance in 18-week-old and more insulin resistance in 30-week-old p66Shc−/− lean compared with WT lean animals, while obese p66Shc−/− were more insulin resistant and equally glucose intolerant [39]. We further confirmed that obese p66Shc−/− are not protected from insulin resistance and glucose intolerance [43]. These data on mice were supported by data on human samples, in which a decreased expression of p66Shc in the visceral adipose tissue was associated with a lower BMI, but without any improvement in diabetes, dyslipidemia, or hypertension [39]. We and others found that the muscles of obese p66Shc−/− mice have increased ectopic fat accumulation, which in part can explain these results [32,39,44]. Moreover, we also demonstrated that the microbiota hosted by p66Shc−/− mice is different from that of WT controls, and that this might explain the differences between studies [43]. Finally, a recent paper studied the metabolic effect on the offspring of WT and p66Shc−/− where dams were given a HFD before and during pregnancy. In this experiment, p66Shc−/− progeny was protected from the deleterious effect of the diet, and that was especially evident for females [46]. However, p66Shc−/− mice were more insulin resistant than WT animals (9 weeks after birth), and this was worsened in animals exposed to pre-natal HFD. WT and knockout males showed a similar response to glucose in GTT, while p66Shc−/− females had an improved glucose tolerance, especially evident in the HFD group.
1.6. Body Temperature Regulation, Respiration, and Energy Expenditure
Interestingly, some studies indicated a possible link between p66Shc and body temperature regulation. In the paper of Berniakovich and colleagues [38], it was shown that p66Shc−/− mice had a higher basal body temperature (at 22 °C external temperature), compared with WT animals. This might be due to an increased metabolic activity in the brown adipose tissue (BAT), since p66Shc−/− express higher amounts of uncoupling protein 1 (UCP1) in this tissue [38]. In the same work, it was demonstrated that the deletion of p66Shc had a dramatic impact on cold adaptation. When housed at 5 °C, the body temperature of these mice dropped by about 6 °C, compared with a halved change in WT animals. The trough was reached more rapidly in knockout than in WT animals: after 3 or 4 h, respectively [38]. However, body temperature returned to normal values within 6 h, both in WT and p66Shc−/− mice, so even if cold adaptation was affected, thermogenesis was not impaired. A possible explanation to this phenomenon comes from a lower thermal insulation in the knockout mice, due to a decreased white fat mass (as previously discussed). A more recent study confirmed this notion by demonstrating a negative selection toward p66Shc−/− mice when left in large outdoor enclosures for one year [45]. This study is particularly relevant, as it sheds light on the reason why p66Shc was phylogenetically conserved, despite its role in the induction of the oxidative stress. p66Shc might be important to promote survival in conditions of environmental stress, whilst its metabolic role might be competitively disadvantageous or detrimental in modern life-style conditions, favoring the development of obesity and metabolic syndrome. In this regard, p66Shc may be considered a typical thrifty gene [60].
Concerning respiration and energy expenditure, p66Shc−/− male mice have a higher oxygen consumption at basal level, and a slightly higher energy expenditure than wild-type animals [38]. A more recent study found rather different results, as the respiratory quotient (calculated as the ratio between the volume of CO2 produced and the volume of the O2 consumed) was increased in p66Shc−/− mice compared with WT animals, which is consistent with an increased glucose utilization in knockout mice [40]. In this study, energy expenditure was also measured, and p66Shc−/− mice displayed lower values than WT mice. However, when fat-free mass was taken into account instead of total body weight, this difference was no more statistically significant. The response of p66Shc−/− mice to calorie restriction was also investigated in 18-month-old animals [41]. When a 26% CR was applied for 2 months there were no differences between WT and knockout mice. However, after 24 h during the 3 days of 40% CR there was a significantly lower energy expenditure during the light phase, compared with the WT animals, but in this case, fat-free mass was not measured.
2. Conclusions and Perspectives
The great majority of the in vivo studies on p66Shc were conducted on one single p66Shc−/− mouse strain [12]. This mouse model was originally generated in the 129Sv strain, and it was afterwards backcrossed to the C57BL/6J strain, and then crossed with other mouse models, such as p53−/−, TERC−/−, and LepOb/Ob [16,35,37,39,42]. The availability of this knockout was surely fundamental in the exploration of the metabolic role of p66Shc and in confirming results that were produced in cell lines, wherein p66Shc was silenced or deleted. However, relying on a single mouse model can be a limitation, and independent confirmations are required. Another p66Shc−/−, named ShcL, was made available recently, and some results were surprisingly in contrast with previous data [44]. To date, this mouse was used only in the referred paper. More importantly, it will be extremely helpful to develop tissue-specific and inducible p66Shc-knockout mice to better dissect the role of p66Shc in different tissues and at different developmental stages or before/after environmental stresses (e.g., HFD). As shown in the present review, looking at different ages with different duration of HFD regimens, starting the diet at different ages can lead to contrasting results, which sometimes appear difficult to reconcile (Table 1). As a final complication, we showed that p66Shc depletion can also influence gut microbiota, which in turns affects the metabolism of the mouse. Altogether, these notions encourage exploring further the role of p66Shc in the regulation of body weight and metabolism. In view of the obesity and diabetes epidemic, p66Shc may represent a promising therapeutic target with enormous implications for human health.
Funding
This research was funded by the University of Padova, grant No. 2019-UNPD0Z9-0058438, and by the Italian Ministry of Education and University, grant PRIN No. 2015ZTT5KB.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pelicci, G.; Lanfrancone, L.; Grignani, F.; McGlade, J.; Cavallo, F.; Forni, G.; Nicoletti, I.; Grignani, F.; Pawson, T.; Pelicci, P.G. A novel transforming protein (SHC) with an SH2 domain is implicated in mitogenic signal transduction. Cell 1992, 70, 93–104. [Google Scholar] [CrossRef]
- Migliaccio, E.; Mele, S.; Salcini, A.E.; Pelicci, G.; Lai, K.M.; Superti-Furga, G.; Pawson, T.; Di Fiore, P.P.; Lanfrancone, L.; Pelicci, P.G. Opposite effects of the p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signalling pathway. EMBO J. 1997, 16, 706–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luzi, L.; Confalonieri, S.; Di Fiore, P.P.; Pelicci, P.G. Evolution of Shc functions from nematode to human. Curr. Opin. Genet. Dev. 2000, 10, 668–674. [Google Scholar] [CrossRef]
- Bhat, S.S.; Anand, D.; Khanday, F.A. p66Shc as a switch in bringing about contrasting responses in cell growth: Implications on cell proliferation and apoptosis. Mol. Cancer 2015, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Galimov, E.R. The Role of p66shc in Oxidative Stress and Apoptosis. Acta Nat. 2010, 2, 44–51. [Google Scholar]
- Bonfini, L.; Migliaccio, E.; Pelicci, G.; Lanfrancone, L.; Pelicci, P.G. Not all Shc’s roads lead to Ras. Trends Biochem. Sci. 1996, 21, 257–261. [Google Scholar] [CrossRef]
- Okada, S.; Kao, A.W.; Ceresa, B.P.; Blaikie, P.; Margolis, B.; Pessin, J.E. The 66-kDa Shc isoform is a negative regulator of the epidermal growth factor-stimulated mitogen-activated protein kinase pathway. J. Biol. Chem. 1997, 272, 28042–28049. [Google Scholar] [CrossRef] [PubMed]
- Pacini, S.; Pellegrini, M.; Migliaccio, E.; Patrussi, L.; Ulivieri, C.; Ventura, A.; Carraro, F.; Naldini, A.; Lanfrancone, L.; Pelicci, P.; et al. p66SHC promotes apoptosis and antagonizes mitogenic signaling in T cells. Mol. Cell. Biol. 2004, 24, 1747–1757. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Shen, X.; Clemmons, D.R. p66shc negatively regulates insulin-like growth factor I signal transduction via inhibition of p52shc binding to Src homology 2 domain-containing protein tyrosine phosphatase substrate-1 leading to impaired growth factor receptor-bound protein-2 membrane recruitment. Mol. Endocrinol. 2008, 22, 2162–2175. [Google Scholar] [PubMed]
- Arany, I.; Faisal, A.; Nagamine, Y.; Safirstein, R.L. p66shc inhibits pro-survival epidermal growth factor receptor/ERK signaling during severe oxidative stress in mouse renal proximal tubule cells. J. Biol. Chem. 2008, 283, 6110–6117. [Google Scholar] [CrossRef] [PubMed]
- Khanday, F.A.; Santhanam, L.; Kasuno, K.; Yamamori, T.; Naqvi, A.; Dericco, J.; Bugayenko, A.; Mattagajasingh, I.; Disanza, A.; Scita, G.; et al. Sos-mediated activation of rac1 by p66shc. J. Cell Biol. 2006, 172, 817–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliaccio, E.; Giorgio, M.; Mele, S.; Pelicci, G.; Reboldi, P.; Pandolfi, P.P.; Lanfrancone, L.; Pelicci, P.G. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature 1999, 402, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Rizzuto, R. p66Shc, oxidative stress and aging: Importing a lifespan determinant into mitochondria. Cell Cycle 2008, 7, 304–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natalicchio, A.; Tortosa, F.; Perrini, S.; Laviola, L.; Giorgino, F. p66Shc, a multifaceted protein linking Erk signalling, glucose metabolism, and oxidative stress. Arch. Physiol. Biochem. 2011, 117, 116–124. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, E.; Baldassari, F.; Bononi, A.; Wieckowski, M.R.; Pinton, P. Oxidative stress in cardiovascular diseases and obesity: Role of p66Shc and protein kinase C. Oxid. Med. Cell. Longev. 2013, 2013, 564961. [Google Scholar] [CrossRef] [PubMed]
- Trinei, M.; Giorgio, M.; Cicalese, A.; Barozzi, S.; Ventura, A.; Migliaccio, E.; Milia, E.; Padura, I.M.; Raker, V.A.; Maccarana, M.; et al. A p53-p66Shc signalling pathway controls intracellular redox status, levels of oxidation-damaged DNA and oxidative stress-induced apoptosis. Oncogene 2002, 21, 3872–3878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinei, M.; Migliaccio, E.; Bernardi, P.; Paolucci, F.; Pelicci, P.; Giorgio, M. p66Shc, mitochondria, and the generation of reactive oxygen species. Methods Enzymol. 2013, 528, 99–110. [Google Scholar] [PubMed]
- Di Lisa, F.; Giorgio, M.; Ferdinandy, P.; Schulz, R. New aspects of p66Shc in ischemia reperfusion injury and cardiovascular diseases. Br. J. Pharmacol. 2017, 174, 1690–1703. [Google Scholar] [CrossRef] [PubMed]
- Le, S.; Connors, T.J.; Maroney, A.C. c-Jun N-terminal kinase specifically phosphorylates p66ShcA at serine 36 in response to ultraviolet irradiation. J. Biol. Chem. 2001, 276, 48332–48336. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Rimessi, A.; Marchi, S.; Orsini, F.; Migliaccio, E.; Giorgio, M.; Contursi, C.; Minucci, S.; Mantovani, F.; Wieckowski, M.R.; et al. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life-span determinant p66Shc. Science 2007, 315, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Haller, M.; Khalid, S.; Kremser, L.; Fresser, F.; Furlan, T.; Hermann, M.; Guenther, J.; Drasche, A.; Leitges, M.; Giorgio, M.; et al. Novel Insights into the PKCbeta-dependent Regulation of the Oxidoreductase p66Shc. J. Biol. Chem. 2016, 291, 23557–23568. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Migliaccio, E.; Moroni, M.; Contursi, C.; Raker, V.A.; Piccini, D.; Martin-Padura, I.; Pelliccia, G.; Trinei, M.; Bono, M.; et al. The life span determinant p66Shc localizes to mitochondria where it associates with mitochondrial heat shock protein 70 and regulates trans-membrane potential. J. Biol. Chem. 2004, 279, 25689–25695. [Google Scholar] [CrossRef] [PubMed]
- Orsini, F.; Moroni, M.; Contursi, C.; Yano, M.; Pelicci, P.; Giorgio, M.; Migliaccio, E. Regulatory effects of the mitochondrial energetic status on mitochondrial p66Shc. Biol. Chem. 2006, 387, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, F.; Francia, P.; Camici, G.G.; Pelicci, P.G.; Luscher, T.F.; Volpe, M. Final common molecular pathways of aging and cardiovascular disease: Role of the p66Shc protein. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Migliaccio, E.; Orsini, F.; Paolucci, D.; Moroni, M.; Contursi, C.; Pelliccia, G.; Luzi, L.; Minucci, S.; Marcaccio, M.; et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 2005, 122, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.Y.; Kushnareva, Y.E.; Murphy, A.N.; Starkov, A.A. Mitochondrial ROS Metabolism: 10 Years Later. Biochemistry (Mosc) 2015, 80, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Gertz, M.; Steegborn, C. The mitochondrial apoptosis pathway and p66Shc--a regulatory redox enzyme or an adapter protein snuggling around? Cell Cycle 2010, 9, 4425–4426. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, M.; Tsuji, Y. Evidence for a novel antioxidant function and isoform-specific regulation of the human p66Shc gene. Mol. Biol. Cell 2014, 25, 2116–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansone, P.; Storci, G.; Giovannini, C.; Pandolfi, S.; Pianetti, S.; Taffurelli, M.; Santini, D.; Ceccarelli, C.; Chieco, P.; Bonafe, M. p66Shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem Cells 2007, 25, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Jung, S.B.; Naqvi, A.; Hoffman, T.A.; DeRicco, J.; Yamamori, T.; Cole, M.P.; Jeon, B.H.; Irani, K. p53 impairs endothelium-dependent vasomotor function through transcriptional upregulation of p66shc. Circ. Res. 2008, 103, 1441–1450. [Google Scholar] [CrossRef] [PubMed]
- Patergnani, S.; Marchi, S.; Rimessi, A.; Bonora, M.; Giorgi, C.; Mehta, K.D.; Pinton, P. PRKCB/protein kinase C, beta and the mitochondrial axis as key regulators of autophagy. Autophagy 2013, 9, 1367–1385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granatiero, V.; Gherardi, G.; Vianello, M.; Salerno, E.; Zecchini, E.; Toniolo, L.; Pallafacchina, G.; Murgia, M.; Blaauw, B.; Rizzuto, R.; et al. Role of p66shc in skeletal muscle function. Sci. Rep. 2017, 7, 6283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weindruch, R.; Walford, R.L. Dietary restriction in mice beginning at 1 year of age: Effect on life-span and spontaneous cancer incidence. Science 1982, 215, 1415–1418. [Google Scholar] [CrossRef] [PubMed]
- Weindruch, R.; Walford, R.L.; Fligiel, S.; Guthrie, D. The retardation of aging in mice by dietary restriction: Longevity, cancer, immunity and lifetime energy intake. J. Nutr. 1986, 116, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, J.J.; Tran, D.; Giorgio, M.; Griffey, S.M.; Koehne, A.; Laing, S.T.; Taylor, S.L.; Kim, K.; Cortopassi, G.A.; Lloyd, K.C.; et al. The influence of Shc proteins on life span in mice. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, S.; Bonafe, M.; Di Tella, L.; Tiberi, L.; Salvioli, S.; Monti, D.; Sorbi, S.; Franceschi, C. p66(shc) is highly expressed in fibroblasts from centenarians. Mech. Ageing Dev. 2005, 126, 839–844. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Stendardo, M.; Migliaccio, E.; Pelicci, P.G. P66SHC deletion improves fertility and progeric phenotype of late-generation TERC-deficient mice but not their short lifespan. Aging Cell 2016, 15, 446–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berniakovich, I.; Trinei, M.; Stendardo, M.; Migliaccio, E.; Minucci, S.; Bernardi, P.; Pelicci, P.G.; Giorgio, M. p66Shc-generated oxidative signal promotes fat accumulation. J Biol Chem 2008, 283, 34283–34293. [Google Scholar] [CrossRef] [PubMed]
- Ciciliot, S.; Albiero, M.; Menegazzo, L.; Poncina, N.; Scattolini, V.; Danesi, A.; Pagnin, E.; Marabita, M.; Blaauw, B.; Giorgio, M.; et al. p66Shc deletion or deficiency protects from obesity but not metabolic dysfunction in mice and humans. Diabetologia 2015, 58, 2352–2360. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.H.; Kim, K.; Ramsey, J.J. The influence of shc proteins and aging on whole body energy expenditure and substrate utilization in mice. PLoS ONE 2012, 7, e48790. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.H.; Kim, K.; Ramsey, J.J. The influence of acute, late-life calorie restriction on whole body energy metabolism in p66Shc(−/−) mice. Mech. Ageing Dev. 2012, 133, 414–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranieri, S.C.; Fusco, S.; Panieri, E.; Labate, V.; Mele, M.; Tesori, V.; Ferrara, A.M.; Maulucci, G.; De Spirito, M.; Martorana, G.E.; et al. Mammalian life-span determinant p66shcA mediates obesity-induced insulin resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 13420–13425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciciliot, S.; Albiero, M.; Campanaro, S.; Poncina, N.; Tedesco, S.; Scattolini, V.; Dalla Costa, F.; Cignarella, A.; Vettore, M.; Di Gangi, I.M.; et al. Interplay between gut microbiota and p66Shc affects obesity-associated insulin resistance. FASEB J. 2018, 32, 4004–4015. [Google Scholar] [CrossRef] [PubMed]
- Tomilov, A.A.; Ramsey, J.J.; Hagopian, K.; Giorgio, M.; Kim, K.M.; Lam, A.; Migliaccio, E.; Lloyd, K.C.; Berniakovich, I.; Prolla, T.A.; et al. The Shc locus regulates insulin signaling and adiposity in mammals. Aging Cell 2011, 10, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, M.; Berry, A.; Berniakovich, I.; Poletaeva, I.; Trinei, M.; Stendardo, M.; Hagopian, K.; Ramsey, J.J.; Cortopassi, G.; Migliaccio, E.; et al. The p66Shc knocked out mice are short lived under natural condition. Aging Cell 2012, 11, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Bellisario, V.; Berry, A.; Capoccia, S.; Raggi, C.; Panetta, P.; Branchi, I.; Piccaro, G.; Giorgio, M.; Pelicci, P.G.; Cirulli, F. Gender-dependent resiliency to stressful and metabolic challenges following prenatal exposure to high-fat diet in the p66(Shc−/−) mouse. Front. Behav. Neurosci. 2014, 8, 285. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Ceolotto, G.; Pagnin, E.; de Kreutzenberg, S.; Avogaro, A. At the crossroads of longevity and metabolism: The metabolic syndrome and lifespan determinant pathways. Aging Cell 2011, 10, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Chen, H.Z.; Wan, Y.Z.; Zhang, Q.J.; Wei, Y.S.; Huang, S.; Liu, J.J.; Lu, Y.B.; Zhang, Z.Q.; Yang, R.F.; et al. Repression of P66Shc expression by SIRT1 contributes to the prevention of hyperglycemia-induced endothelial dysfunction. Circ. Res. 2011, 109, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Kim, Y.R.; Vikram, A.; Kumar, S.; Kassan, M.; Gabani, M.; Lee, S.K.; Jacobs, J.S.; Irani, K. P66Shc-Induced MicroRNA-34a Causes Diabetic Endothelial Dysfunction by Downregulating Sirtuin1. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2394–2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rota, M.; LeCapitaine, N.; Hosoda, T.; Boni, A.; De Angelis, A.; Padin-Iruegas, M.E.; Esposito, G.; Vitale, S.; Urbanek, K.; Casarsa, C.; et al. Diabetes promotes cardiac stem cell aging and heart failure, which are prevented by deletion of the p66shc gene. Circ. Res. 2006, 99, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Fadini, G.P.; Albiero, M.; Menegazzo, L.; Boscaro, E.; Pagnin, E.; Iori, E.; Cosma, C.; Lapolla, A.; Pengo, V.; Stendardo, M.; et al. The redox enzyme p66Shc contributes to diabetes and ischemia-induced delay in cutaneous wound healing. Diabetes 2010, 59, 2306–2314. [Google Scholar] [CrossRef] [PubMed]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Natalicchio, A.; Laviola, L.; De Tullio, C.; Renna, L.A.; Montrone, C.; Perrini, S.; Valenti, G.; Procino, G.; Svelto, M.; Giorgino, F. Role of the p66Shc isoform in insulin-like growth factor I receptor signaling through MEK/Erk and regulation of actin cytoskeleton in rat myoblasts. J. Biol. Chem. 2004, 279, 43900–43909. [Google Scholar] [CrossRef] [PubMed]
- Natalicchio, A.; De Stefano, F.; Perrini, S.; Laviola, L.; Cignarelli, A.; Caccioppoli, C.; Quagliara, A.; Melchiorre, M.; Leonardini, A.; Conserva, A.; et al. Involvement of the p66Shc protein in glucose transport regulation in skeletal muscle myoblasts. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E228–E237. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z. Adapter proteins regulate insulin resistance and lipid metabolism in obesity. Sci. Bull. 2016, 61, 8. [Google Scholar] [CrossRef]
- Ranieri, S.C.; Fusco, S.; Pani, G. p66(ShcA): Linking mammalian longevity with obesity-induced insulin resistance. Vitam. Horm. 2013, 91, 219–241. [Google Scholar] [PubMed]
- Soliman, M.A.; Abdel Rahman, A.M.; Lamming, D.W.; Birsoy, K.; Pawling, J.; Frigolet, M.E.; Lu, H.; Fantus, I.G.; Pasculescu, A.; Zheng, Y.; et al. The adaptor protein p66Shc inhibits mTOR-dependent anabolic metabolism. Sci. Signal. 2014, 7, ra17. [Google Scholar] [CrossRef] [PubMed]
- Hagopian, K.; Tomilov, A.A.; Kim, K.; Cortopassi, G.A.; Ramsey, J.J. Key glycolytic enzyme activities of skeletal muscle are decreased under fed and fasted states in mice with knocked down levels of Shc proteins. PLoS ONE 2015, 10, e0124204. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, S.; Combs, C.A.; French, S.; Ahn, B.H.; Fergusson, M.M.; Balaban, R.S.; Finkel, T. The mammalian longevity-associated gene product p66shc regulates mitochondrial metabolism. J. Biol. Chem. 2006, 281, 10555–10560. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.N.; Barker, D.J. The thrifty phenotype hypothesis. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Role of p66Shc in signal transduction. (A) p52Shc and p46Shc are activated by phosphorylation in tyrosine residues within their CH1 domain, when bound to RTKs and possibly other receptors. Subsequently, the recruitment of the Grb2/Sos1 complex allows for the activation of Ras and the MAPK pathway. p66Shc can compete with the other two isoforms for the binding of Grb2, interfering with Ras activation. (B) After being activated by RTKs, and the concomitant phosphorylation in Ser36 by kinases such as PKCβ or JNK, p66Shc is subjected to cis-trans isomerization by Pin1. It then translocates to the inter-membrane space of the mitochondrion, after being dephosphorylated by PP2A. Without stimulation, p66Shc is bound to other proteins, like HSP70, and therefore is inactive. After stimulation with UV-light or H2O2, p66Shc can bind to cytochrome c and contribute to the formation of ROS. See the main text for further details.
Figure 1.
Role of p66Shc in signal transduction. (A) p52Shc and p46Shc are activated by phosphorylation in tyrosine residues within their CH1 domain, when bound to RTKs and possibly other receptors. Subsequently, the recruitment of the Grb2/Sos1 complex allows for the activation of Ras and the MAPK pathway. p66Shc can compete with the other two isoforms for the binding of Grb2, interfering with Ras activation. (B) After being activated by RTKs, and the concomitant phosphorylation in Ser36 by kinases such as PKCβ or JNK, p66Shc is subjected to cis-trans isomerization by Pin1. It then translocates to the inter-membrane space of the mitochondrion, after being dephosphorylated by PP2A. Without stimulation, p66Shc is bound to other proteins, like HSP70, and therefore is inactive. After stimulation with UV-light or H2O2, p66Shc can bind to cytochrome c and contribute to the formation of ROS. See the main text for further details.

{kind=link}
Table 1.
Summary of the contrasting results obtained on the role of p66Shc in physiological conditions and metabolic diseases. WT = wild type.
Table 1.
Summary of the contrasting results obtained on the role of p66Shc in physiological conditions and metabolic diseases. WT = wild type.
| Condition | In Favor | Against |
|---|---|---|
| Increased longevity | Deletion of p66Shc increases average and maximum longevity [12] | No increase in lifespan of p66Shc−/− mice [35] High expression of p66Shc in centenarians [36] Deletion of p66Shc does not rescue TREC deficiency [37] |
| Bodyweight regulation | p66Shc−/− mice are leaner than age-matched WT [35,38,39,40,43] | p66Shc deletion has no influence on bodyweight [12,40,41] |
| Protection from obesity | Knockout mice gain less weight than WT controls [38,39,42,43] | Knockout mice gain more weight than WT controls [44] |
| Improved glucose homeostasis | Glucose uptake is increased in the absence of p66Shc [42,44,54,57,58] Increased glycolysis [57,59] Improved glucose tolerance and/or insulin sensitivity in lean [44] and obese knockout animals [42] | Glucose uptake is not increased [42] or even decreased [39,44] in the absence of p66Shc Decreased glycolysis [58] Similar or worsened glucose tolerance and/or insulin sensitivity in lean [39,43,44] and obese knockout animals [39,43,44] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ciciliot, S.; Fadini, G.P. Modulation of Obesity and Insulin Resistance by the Redox Enzyme and Adaptor Protein p66Shc. Int. J. Mol. Sci. 2019, 20, 985. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040985
AMA Style
Ciciliot S, Fadini GP. Modulation of Obesity and Insulin Resistance by the Redox Enzyme and Adaptor Protein p66Shc. International Journal of Molecular Sciences. 2019; 20(4):985. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040985
Chicago/Turabian StyleCiciliot, Stefano, and Gian Paolo Fadini. 2019. "Modulation of Obesity and Insulin Resistance by the Redox Enzyme and Adaptor Protein p66Shc" International Journal of Molecular Sciences 20, no. 4: 985. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040985
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.