Targeting the Iron-Response Elements of the mRNAs for the Alzheimer’s Amyloid Precursor Protein and Ferritin to Treat Acute Lead and Manganese Neurotoxicity


Abstract
:1. Amyloid Precursor Protein (APP) Translation Inhibitors for Anti-Amyloid Efficacy in Down Syndrome and Familial Alzheimer’s Disease
1.1. APP Gene Dose in Down Syndrome and in Rare Cases of Familial Alzheimer’s Disease (fAD)
1.2. APP 5’Untranslated Region Translation Blockers as Anti-Amyloid Agents In Vivo
1.3. High Throughput Screen for APP 5’UTR Inhibitors for Anti-Amyloid Efficacy
2. APP 5’UTR Activators Shield Neurons from Acute Pb/Mn Disruption of Fe-Homeostasis
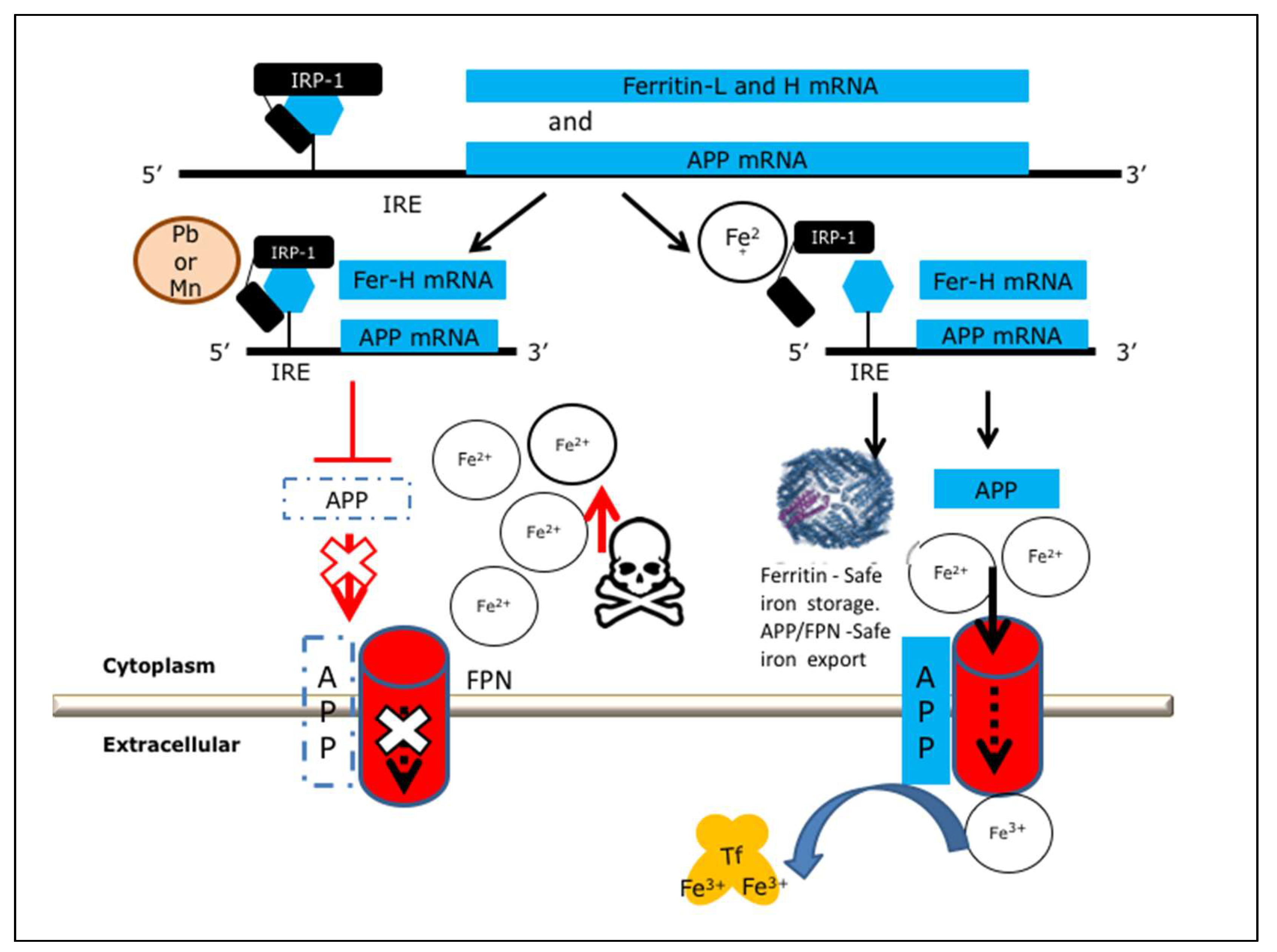
2.1. Heavy Metal Neurotoxicity by Loss of IRE/IRP Translation of APP and Ferritin-H Chain
2.2. Activation of Ferritin and APP Translation to Protect Neurons from Mn/Pb Exposure
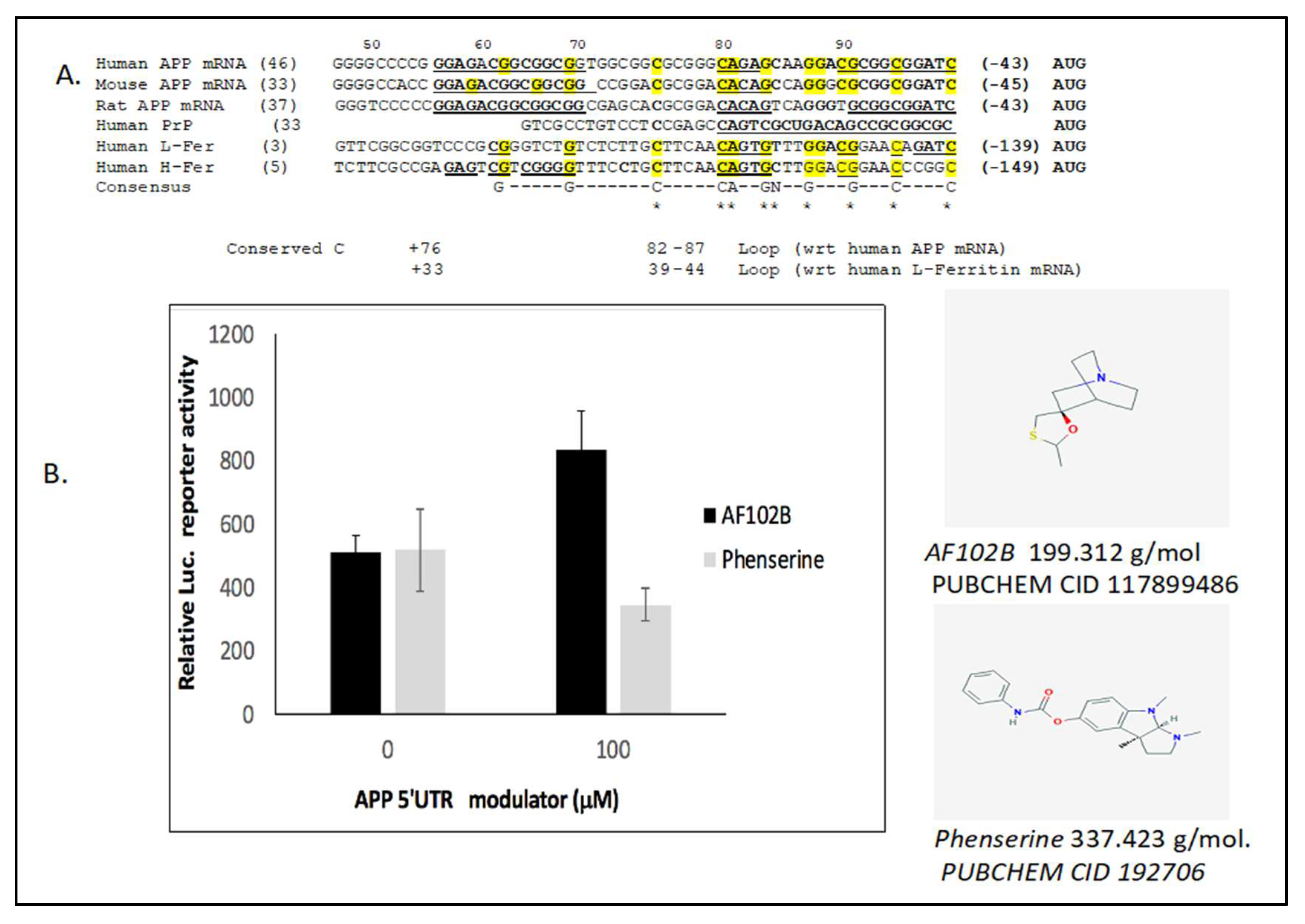
2.3. The Neuroprotective M1 Muscarinic Agonist AF102B, Which Induces Alpha-Secretase, Is an APP 5’UTR Directed Activator
2.4. Alternative to Iron Chelation: Novel High Throughput Screened APP Translation Activators in a Mode of Therapeutic Shielding of Neurons from Intracellular Fe Buildup After Pb and Mn Exposure
2.5. Model for APP Translation Activators to Shield Neurons from Pb and Mn Neurotoxicity
3. Activators of the Ferritin-H Chain 5’UTR Specific IRE to Shield Neurons from Acute Pb/Mn Disruption of Fe-Homeostasis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| APP | Amyloid precursor protein |
| Pb | Lead |
| Mn | Manganese |
| AD | Alzheimer’s disease |
| FDA | Food and Drug Administration |
| Ferritin L chain | Ferritin Light chain |
| Ferritin H Chain | Ferritin Heavy chain |
| MPTP | 1-methyl-4-phenyl-1,2,3,6 tetrahydropyridine |
| Ferroportin | FPN |
References
- Lahiri, D.K.; Chen, D.; Maloney, B.; Holloway, H.W.; Yu, Q.S.; Utsuki, T.; Giordano, T.; Sambamurti, K.; Greig, N.H. The experimental Alzheimer’s disease drug posiphen [(+)-phenserine] lowers amyloid-beta peptide levels in cell culture and mice. J. Pharmacol. Exp. Ther. 2007, 320, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Rogers, J.T. Alzheimer’s disease therapeutics targeted to the control of amyloid precursor protein translation: Maintenance of brain iron homeostasis. Biochem. Pharmacol. 2014, 88, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Salehi, A.; Delcroix, J.D.; Belichenko, P.V.; Zhan, K.; Wu, C.; Valletta, J.S.; Takimoto-Kimura, R.; Kleschevnikov, A.M.; Sambamurti, K.; Chung, P.P.; et al. Increased App Expression in a Mouse Model of Down’s Syndrome Disrupts NGF Transport and Causes Cholinergic Neuron Degeneration. Neuron 2006, 51, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Castro, P.; Zaman, S.; Holland, A. Alzheimer’s disease in people with Down’s syndrome: The prospects for and the challenges of developing preventative treatments. J. Neurol. 2017, 264, 804–813. [Google Scholar] [CrossRef] [PubMed]
- Theuns, J.; Brouwers, N.; Engelborghs, S.; Sleegers, K.; Bogaerts, V.; Corsmit, E.; De Pooter, T.; van Duijn, C.M.; De Deyn, P.P.; Van Broeckhoven, C. Promoter mutations that increase amyloid precursor-protein expression are associated with Alzheimer disease. Am. J. Hum. Genet. 2006, 78, 936–946. [Google Scholar] [CrossRef] [PubMed]
- Rovelet-Lecrux, A.; Hannequin, D.; Raux, G.; Meur, N.L.; Laquerriere, A.; Vital, A.; Dumanchin, C.; Feuillette, S.; Brice, A.; Vercelletto, M.; et al. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet. 2006, 38, 24–26. [Google Scholar] [CrossRef] [PubMed]
- Buss, L.; Fisher, E.; Hardy, J.; Nizetic, D.; Groet, J.; Pulford, L.; Strydom, A. Intracerebral haemorrhage in Down syndrome: Protected or predisposed? F1000Research 2016, 5, 876. [Google Scholar] [CrossRef] [PubMed]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Lumsden, A.L.; Rogers, J.T.; Majd, S.; Newman, M.; Sutherland, G.T.; Verdile, G.; Lardelli, M. Dysregulation of Neuronal Iron Homeostasis as an Alternative Unifying Effect of Mutations Causing Familial Alzheimer’s Disease. Front. Neurosci. 2018, 12, 533. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Maloney, B.; Rogers, J.T.; Lahiri, D.K. Novel upregulation of amyloid-beta precursor protein (APP) by microRNA-346 via targeting of APP mRNA 5’-untranslated region: Implications in Alzheimer’s disease. Mol. Psychiatry 2018. [Google Scholar] [CrossRef]
- Cho, H.H.; Cahill, C.M.; Vanderburg, C.R.; Scherzer, C.R.; Wang, B.; Huang, X.; Rogers, J.T. Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J. Biol. Chem. 2010, 285, 31217–31232. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.B.; Deck, K.M.; Nizzi, C.P.; Eisenstein, R.S. A synergistic role of IRP1 and FBXL5 proteins in coordinating iron metabolism during cell proliferation. J. Biol. Chem. 2017, 292, 15976–15989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys Acta 2012, 1823, 1468–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkataramani, V.; Doeppner, T.R.; Willkommen, D.; Cahill, C.M.; Xin, Y.; Ye, G.; Liu, Y.; Southon, A.; Aron, A.; Au-Yeung, H.Y.; et al. Manganese causes neurotoxic iron accumulation via translational repression of amyloid precursor protein and H-Ferritin. J. Neurochem. 2018, 147, 831–848. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, R.C.; Park, Y.H.; Kosman, D.J. sAPP modulates iron efflux from brain microvascular endothelial cells by stabilizing the ferrous iron exporter ferroportin. EMBO Rep. 2014, 15, 809–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, C.; Steimle, B.L.; Bailey, D.K.; Kosman, D.J. The Ferroxidase Hephaestin But Not Amyloid Precursor Protein is Required for Ferroportin-Supported Iron Efflux in Primary Hippocampal Neurons. Cell Mol. Neurobiol. 2018, 38, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Carlson, E.S.; Fretham, S.J.; Unger, E.; O’Connor, M.; Petryk, A.; Schallert, T.; Rao, R.; Tkac, I.; Georgieff, M.K. Hippocampus specific iron deficiency alters competition and cooperation between developing memory systems. J. Neurodev. Disord. 2012, 2, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Carlson, E.S.; Magid, R.; Petryk, A.; Georgieff, M.K. Iron deficiency alters expression of genes implicated in Alzheimer disease pathogenesis. Brain Res. 2008, 1237, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.T.; Utsuki, T.; Rogers, J.; Yu, Q.S.; Sambamurti, K.; Brossi, A.; Ge, Y.W.; Lahiri, D.K.; Greig, N.H. Phenserine regulates translation of beta-amyloid precursor protein mRNA by a putative interleukin-1 responsive element, a target for drug development. Proc. Natl. Acad. Sci. USA 2001, 98, 7605–7610. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Cahill, C.; Balleidier, A.; Huang, C.; Lahiri, D.K.; Huang, X.; Rogers, J.T. Novel 5’ untranslated region directed blockers of iron-regulatory protein-1 dependent amyloid precursor protein translation: Implications for down syndrome and Alzheimer’s disease. PLoS ONE 2013, 8, e65978. [Google Scholar] [CrossRef] [PubMed]
- Utsuki, T.; Yu, Q.S.; Davidson, D.; Chen, D.; Holloway, H.W.; Brossi, A.; Sambamurti, K.; Lahiri, D.K.; Greig, N.H.; Giordano, T. Identification of novel small molecule inhibitors of amyloid precursor protein synthesis as a route to lower Alzheimer’s disease amyloid-beta peptide. J. Pharmacol. Exp. Ther. 2006, 318, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Maccecchini, M. Posiphen’s pharmacokinetics and mecha- 883nism of action in mild cognitive impaired patients. Alzheimer’s Dementia 2010, 6, e54. [Google Scholar] [CrossRef]
- Teich, A.F.; Sharma, E.; Barnwell, E.; Zhang, H.; Staniszewski, A.; Utsuki, T.; Padmaraju, V.; Mazell, C.; Tzekou, A.; Sambamurti, K.; et al. Translational inhibition of APP by Posiphen: Efficacy, pharmacodynamics, and pharmacokinetics in the APP/PS1 mouse. Alzheimers Dement. (N Y) 2018, 4, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Wang, Y.; Rogers, J.T.; Wang, F. Perturbed iron distribution in Alzheimer’s disease serum, cerebrospinal fluid, and selected brain regions: A systematic review and meta-analysis. J. Alzheimers Dis. 2014, 42, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Leiter, L.M.; McPhee, J.; Cahill, C.M.; Zhan, S.S.; Potter, H.; Nilsson, L.N. Translation of the alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5’-untranslated region sequences. J. Biol. Chem. 1999, 274, 6421–6431. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Randall, J.D.; Cahill, C.M.; Eder, P.S.; Huang, X.; Gunshin, H.; Leiter, L.; McPhee, J.; Sarang, S.S.; Utsuki, T.; et al. An iron-responsive element type II in the 5’-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002, 277, 45518–45528. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Venkataramani, V.; Washburn, C.; Liu, Y.; Tummala, V.; Jiang, H.; Smith, A.; Cahill, C.M. A role for amyloid precursor protein translation to restore iron homeostasis and ameliorate lead (Pb) neurotoxicity. J. Neurochem. 2016, 138, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Rajanala, S.; Mikkilineni, S.; Cahill, C.; Brown, R.; Berry, J.; Rogers, J. The 5’-Untranslated Region of the C9orf72 mRNA Exhibits a Phylogenetic Alignment to the Cis-Aconitase Iron-Responsive Element; Novel Therapies for Amytrophic Lateral Sclerosis. Neurosci. Med. 2016, 7, 15–26. [Google Scholar] [CrossRef]
- Maccecchini, M.L.; Chang, M.Y.; Pan, C.; John, V.; Zetterberg, H.; Greig, N.H. Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid-beta peptide and tau levels: Target engagement, tolerability and pharmacokinetics in humans. J. Neurol. Neurosurg. Psychiatry 2012, 83, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.T.; Mikkilineni, S.; Cantuti-Castelvetri, I.; Smith, D.H.; Huang, X.; Bandyopadhyay, S.; Cahill, C.M.; Maccecchini, M.L.; Lahiri, D.K.; Greig, N.H. The alpha-synuclein 5’untranslated region targeted translation blockers: Anti-alpha synuclein efficacy of cardiac glycosides and Posiphen. J. Neural. Transm. (Vienna) 2011, 118, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Winblad, B.; Giacobini, E.; Frolich, L.; Friedhoff, L.T.; Bruinsma, G.; Becker, R.E.; Greig, N.H. Phenserine efficacy in Alzheimer’s disease. J. Alzheimers Dis. 2010, 22, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Jiang, H.; Lee, E.S.; Ni, M.; Erikson, K.M.; Milatovic, D.; Bowman, A.B.; Aschner, M. Ferroportin is a manganese-responsive protein that decreases manganese cytotoxicity and accumulation. J. Neurochem. 2010, 112, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.; Adlard, P.A.; Cherny, R.A.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Natl. Med. 2012, 18, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Ni, J.; Ruggiero, A.; Walshe, K.; Rogers, M.S.; Chattopadhyay, N.; Glicksman, M.A.; Rogers, J.T. A high-throughput drug screen targeted to the 5’untranslated region of Alzheimer amyloid precursor protein mRNA. J. Biomol. Screen 2006, 11, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Hartley, D.M.; Cahill, C.M.; Lahiri, D.K.; Chattopadhyay, N.; Rogers, J.T. Interleukin-1alpha stimulates non-amyloidogenic pathway by alpha-secretase (ADAM-10 and ADAM-17) cleavage of APP in human astrocytic cells involving p38 MAP kinase. J. Neurosci. Res. 2006, 84, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Thurtle, N.; Greig, J.; Cooney, L.; Amitai, Y.; Ariti, C.; Brown, M.J.; Kosnett, M.J.; Moussally, K.; Sani-Gwarzo, N.; Akpan, H.; et al. Description of 3,180 courses of chelation with dimercaptosuccinic acid in children ≤5 y with severe lead poisoning in Zamfara, Northern Nigeria: A retrospective analysis of programme data. PLoS Med. 2014, 11, e1001739. [Google Scholar] [CrossRef] [PubMed]
- Greig, J.; Thurtle, N.; Cooney, L.; Ariti, C.; Ahmed, A.O.; Ashagre, T.; Ayela, A.; Chukwumalu, K.; Criado-Perez, A.; Gomez-Restrepo, C.; et al. Association of blood lead level with neurological features in 972 children affected by an acute severe lead poisoning outbreak in Zamfara State, northern Nigeria. PLoS ONE 2014, 9, e93716. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhao, Q.; Slavkovich, V.; Aschner, M.; Graziano, J.H. Alteration of iron homeostasis following chronic exposure to manganese in rats. Brain Res. 1999, 833, 125–132. [Google Scholar] [CrossRef]
- Peres, T.V.; Schettinger, M.R.; Chen, P.; Carvalho, F.; Avila, D.S.; Bowman, A.B.; Aschner, M. Manganese-induced neurotoxicity: A review of its behavioral consequences and neuroprotective strategies. BMC Pharmacol. Toxicol. 2016, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Andruska, K.M.; Racette, A.B. Neuromythology of Manganism. Curr. Epidemiol. Rep. 2015, 2, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdalkader, M.; Lampinen, R.; Kanninen, K.M.; Malm, T.M.; Liddell, J.R. Targeting Nrf2 to Suppress Ferroptosis and Mitochondrial Dysfunction in Neurodegeneration. Front. Neurosci. 2018, 12, 466. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Zhang, N.; Wang, Y.Q.; Wu, Q.; Yu, P.; Shi, Z.H.; Duan, X.L.; Zhao, B.L.; Wu, W.S.; Chang, Y.Z. Mitochondrial Ferritin Protects Hydrogen Peroxide-Induced Neuronal Cell Damage. Aging Dis. 2017, 8, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Sundsmo, M.; Roch, J.M.; Kimura, N.; Cole, G.; Schubert, D.; Oltersdorf, T.; Schenk, D.B. Secreted form of amyloid beta protein precursor is involved in the growth regulation of fibroblasts. Cell 1989, 58, 615–622. [Google Scholar] [CrossRef]
- Jin, L.W.; Ninomiya, H.; Roch, J.M.; Schubert, D.; Masliah, E.; Otero, D.A.; Saitoh, T. Peptides containing the RERMS sequence of amyloid beta/A4 protein precursor bind cell surface and promote neurite extension. J. Neurosci. 1994, 14, 5461–5470. [Google Scholar] [CrossRef] [PubMed]
- Bowes, M.; Masliah, E.; Otero, D.A.C.; Zivin, J.A.; Saitoh, T. Reduction of Neurological Damage by a Peptide Segement of the Amyloid-b.A4 Protein Precursor in a Rabbit Spinal cord Ischemia Model. Experiment. Neurol. 1994, 129, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Goldstein, L.E.; Lahiri, D.K.; Rogers, J.T. Role of the APP Non-Amyloidogenic Signaling Pathway and Targeting alpha-Secretase as an Alternative Drug Target for Treatment of Alzheimer’s Disease. Curr. Med. Chem. 2007, 14, 2848–2864. [Google Scholar] [CrossRef] [PubMed]
- Basha, M.R.; Wei, W.; Bakheet, S.A.; Benitez, N.; Siddiqi, H.K.; Ge, Y.W.; Lahiri, D.K.; Zawia, N.H. The fetal basis of amyloidogenesis: Exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J. Neurosci. 2005, 25, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Basha, M.R.; Brock, B.; Cox, D.P.; Cardozo-Pelaez, F.; McPherson, C.A.; Harry, J.; Rice, D.C.; Maloney, B.; Chen, D.; et al. Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): Evidence for a developmental origin and environmental link for AD. J. Neurosci. 2008, 28, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.; Brandeis, R.; Pittel, Z.; Karton, I.; Sapir, M.; Dachir, S.; Levy, A.; Heldman, E. (±)-cis-2-methyl-spiro(1,3-oxathiolane-5,3’) quinuclidine (AF102B): A new M1 agonist attenuates cognitive dysfunctions in AF64A-treated rats. Neurosci. Lett 1989, 102, 325–331. [Google Scholar] [CrossRef]
- Fisher, A.; Michaelson, D.M.; Brandeis, R.; Haring, R.; Chapman, S.; Pittel, Z. M1 muscarinic agonists as potential disease-modifying agents in Alzheimer’s disease. Rationale and perspectives. Ann. N. Y. Acad. Sci. 2000, 920, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A. Cholinergic modulation of amyloid precursor protein processing with emphasis on M1 muscarinic receptor: Perspectives and challenges in treatment of Alzheimer’s disease. J. Neurochem. 2012, 120, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Georgievska, B.; Mattsson, A.; Isacson, O. Cognitive changes and modified processing of amyloid precursor protein in the cortical and hippocampal system after cholinergic synapse loss and muscarinic receptor activation. Proc. Natl. Acad. Sci. USA 1999, 96, 12108–12113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitsch, R.M.; Slack, B.E.; Wurtman, R.J.; Growdon, J.H. Release of Alzheimer amyloid precursor derivatives stimulated by activation of muscarinic acetylcholine receptors. Science 1992, 258, 304–307. [Google Scholar] [CrossRef] [PubMed]
- Nitsch, R.M.; Deng, M.; Tennis, M.; Schoenfeld, D.; Growdon, J.H. The Selective Muscarinic M1 AgonistAF102B Decreases levels of totalAbeta in the Cerebrospinal Fluid of patients with Alzheimer’s Disease. Ann. Neurol. 2000, 48, 913–918. [Google Scholar] [CrossRef]
- Nitsch, R.M.; Kim, C.; Growdon, J.H. Vasopressin and bradykinin regulate secretory processing of the amyloid protein precursor of Alzheimer’s disease. Neurochem. Res. 1998, 23, 807–814. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Greig, N.H.; Lahiri, D.; Fisher, A. Translation and Processing of the Amyloid Precursor Protein in response to an M1 muscaric agonist and an acetyl cholinesterase inhibitor. In Recent Progress in Alzheimer’s and Parkinson’s Diseases; Taylor & Francis Group: London, England, 2006; pp. 267–278. [Google Scholar]
- Fisher, A. Therapeutic strategies in Alzheimer’s Disease: M1 Muscaric Agonists. Jpn. J. Pharmacol. 2000, 84, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Walden, W.E.; Selezneva, A.I.; Dupuy, J.; Volbeda, A.; Fontecilla-Camps, J.C.; Theil, E.C.; Volz, K. Structure of dual function iron regulatory protein 1 complexed with ferritin IRE-RNA. Science 2006, 314, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Ma, J.; Walden, W.E.; Merrick, W.C.; Theil, E.C.; Goss, D.J. Rapid kinetics of iron responsive element (IRE) RNA/iron regulatory protein 1 and IRE-RNA/eIF4F complexes respond differently to metal ions. Nucleic Acids Res. 2014, 42, 6567–6577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maloney, B.; Ge, Y.W.; Greig, N.; Lahiri, D.K. Presence of a “CAGA box” in the APP gene unique to amyloid plaque-forming species and absent in all APLP-1/2 genes: Implications in Alzheimer’s disease. FASEB J. 2004, 18, 1288–1290. [Google Scholar] [CrossRef] [PubMed]
- Goforth, J.B.; Anderson, S.A.; Nizzi, C.P.; Eisenstein, R.S. Multiple determinants within iron-responsive elements dictate iron regulatory protein binding and regulatory hierarchy. RNA 2010, 16, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.A.; Nizzi, C.P.; Chang, Y.I.; Deck, K.M.; Schmidt, P.J.; Galy, B.; Damnernsawad, A.; Broman, A.T.; Kendziorski, C.; Hentze, M.W.; et al. The IRP1-HIF-2alpha axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab. 2013, 17, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Hahl, P.; Davis, T.; Washburn, C.; Rogers, J.T.; Smith, A. Mechanisms of neuroprotection by hemopexin: Modeling the control of heme and iron homeostasis in brain neurons in inflammatory states. J. Neurochem. 2013, 125, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Wu, Q.; Monga, J.; Xie, E.; Wang, H.; Wang, S.; Zhang, H.; Wang, Z.Y.; Zhou, T.; Shi, Y.; et al. HDAC1 Governs Iron Homeostasis Independent of Histone Deacetylation in Iron-Overload Murine Models. Antioxid Redox Signal. 2018, 28, 1224–1237. [Google Scholar] [CrossRef] [PubMed]
- Crapper McLachlan, D.R.; Dalton, A.J.; Kruck, T.P.; Bell, M.Y.; Smith, W.L.; Kalow, W.; Andrews, D.F. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 1991, 337, 1304–1308. [Google Scholar] [CrossRef]
- Ritchie, C.W.; Bush, A.I.; Mackinnon, A.; Macfarlane, S.; Mastwyk, M.; MacGregor, L.; Kiers, L.; Cherny, R.; Li, Q.X.; Tammer, A.; et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Arch. Neurol. 2003, 60, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Tanzi, R.E. Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics 2008, 5, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.M.; Forsberg, A.C.; Stroebel, B.M.; Faltesek, K.A.; Verden, D.R.; Hamel, K.A.; Raney, E.B.; Crow, J.M.; Haase, L.R.; Knutzen, K.E.; et al. Intranasal deferoxamine affects memory loss, oxidation, and the insulin pathway in the streptozotocin rat model of Alzheimer’s disease. J. Neurol. Sci. 2017, 380, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Masaldan, S.; Clatworthy, S.A.S.; Gamell, C.; Meggyesy, P.M.; Rigopoulos, A.T.; Haupt, S.; Haupt, Y.; Denoyer, D.; Adlard, P.A.; Bush, A.I.; et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol. 2017, 14, 100–115. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, D.I.; Billings, J.L.; Adlard, P.A.; Ayton, S.; Sedjahtera, A.; Masters, C.L.; Wilkins, S.; Shackleford, D.M.; Charman, S.A.; Bal, W.; et al. The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol. Commun. 2017, 5, 53. [Google Scholar] [CrossRef] [PubMed]
- Prasanthi, J.R.; Schrag, M.; Dasari, B.; Marwarha, G.; Dickson, A.; Kirsch, W.M.; Ghribi, O. Deferiprone reduces amyloid-beta and tau phosphorylation levels but not reactive oxygen species generation in hippocampus of rabbits fed a cholesterol-enriched diet. J. Alzheimers Dis. 2012, 30, 167–182. [Google Scholar] [CrossRef] [PubMed]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- Doraiswamy, P.M.; Xiong, G.L. Pharmacological strategies for the prevention of Alzheimer’s disease. Expert Opin. Pharmacother. 2006, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Cherny, R.A.; Finkelstein, D.I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J.P.; Perez, K.; et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 2008, 59, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.; Ramos, E.; de Los Rios, C.; Egea, J.; Del Pino, J.; Reiter, R.J. A review of metal-catalyzed molecular damage: Protection by melatonin. J. Pineal Res. 2014, 56, 343–370. [Google Scholar] [CrossRef] [PubMed]
- Llorens, F.; Thune, K.; Schmitz, M.; Ansoleaga, B.; Frau-Mendez, M.A.; Cramm, M.; Tahir, W.; Gotzmann, N.; Berjaoui, S.; Carmona, M.; et al. Identification of new molecular alterations in fatal familial insomnia. Hum. Mol. Genet. 2016, 25, 2417–2436. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.; Bezprozvanny, I.; Wu, L.; Ryskamp, D.A.; Bar-Ner, N.; Natan, N.; Brandeis, R.; Elkon, H.; Nahum, V.; Gershonov, E.; et al. AF710B, a Novel M1/sigma1 Agonist with Therapeutic Efficacy in Animal Models of Alzheimer’s Disease. Neurodegener Dis. 2016, 16, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Welt, T.; Kulic, L.; Hoey, S.E.; McAfoose, J.; Spani, C.; Chadha, A.S.; Fisher, A.; Nitsch, R.M. Acute Effects of Muscarinic M1 Receptor Modulation on AbetaPP Metabolism and Amyloid-beta Levels in vivo: A Microdialysis Study. J. Alzheimers Dis. 2015, 46, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Fisher, A.; Pittel, Z.; Haring, R.; Bar-Ner, N.; Kliger-Spatz, M.; Natan, N.; Egozi, I.; Sonego, H.; Marcovitch, I.; Brandeis, R. M1 muscarinic agonists can modulate some of the hallmarks in Alzheimer’s disease: Implications in future therapy. J. Mol. Neurosci. 2003, 20, 349–356. [Google Scholar] [CrossRef]
- Fisher, A.; Brandeis, R.; Bar-Ner, R.H.; Kliger-Spatz, M.; Natan, N.; Sonego, H.; Marcovitch, I.; Pittel, Z. AF150(S) and AF267B: M1 muscarinic agonists as innovative therapies for Alzheimer’s disease. J. Mol. Neurosci. 2002, 19, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Neha; Ansari, M.M.; Khan, H.A. Yohimbine hydrochloride ameliorates collagen type-II-induced arthritis targeting oxidative stress and inflammatory cytokines in Wistar rats. Environ. Toxicol. 2017, 32, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Tibodeau, J.D.; Fox, P.M.; Ropp, P.A.; Theil, E.C.; Thorp, H.H. The up-regulation of ferritin expression using a small-molecule ligand to the native mRNA. Proc. Natl. Acad. Sci. USA 2006, 103, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Balla, G.; Jacob, H.S.; Balla, J.; Rosenberg, M.; Nath, K.; Apple, F.; Eaton, J.W.; Vercellotti, G.M. Ferritin: A cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 1992, 267, 18148–18153. [Google Scholar] [PubMed]
- Singh, A.; Haldar, S.; Horback, K.; Tom, C.; Zhou, L.; Meyerson, H.; Singh, N. Prion protein regulates iron transport by functioning as a ferrireductase. J. Alzheimers Dis. 2013, 35, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Wu, Q.; Wu, W.; Li, H.; Guo, Y.; Yu, P.; Gao, G.; Shi, Z.; Zhao, B.; Chang, Y.Z. Mitochondrial Ferritin Deletion Exacerbates beta-Amyloid-Induced Neurotoxicity in Mice. Oxid. Med. Cell. Longev. 2017, 2017, 1020357. [Google Scholar] [CrossRef] [PubMed]
- Kaur, D.; Yantiri, F.; Rajagopalan, S.; Kumar, J.; Mo, J.Q.; Boonplueang, R.; Viswanath, V.; Jacobs, R.; Yang, L.; Beal, M.F.; et al. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: A novel therapy for Parkinson’s disease. Neuron 2003, 37, 899–909. [Google Scholar] [CrossRef]
- Kosmidis, S.; Missirlis, F.; Botella, J.A.; Schneuwly, S.; Rouault, T.A.; Skoulakis, E.M. Behavioral decline and premature lethality upon pan-neuronal ferritin overexpression in Drosophila infected with a virulent form of Wolbachia. Front. Pharmacol. 2014, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Chang, S.Y.; Wu, Q.; Gou, Y.J.; Jia, L.; Cui, Y.M.; Yu, P.; Shi, Z.H.; Wu, W.S.; Gao, G.; et al. The Protective Role of Mitochondrial Ferritin on Erastin-Induced Ferroptosis. Front. Aging Neurosci. 2016, 8, 308. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.A.; Botella, J.A.; Metzendorf, C.; Lind, M.I.; Schneuwly, S. Mitoferrin modulates iron toxicity in a Drosophila model of Friedreich’s ataxia. Free Radic. Biol. Med. 2015, 85, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Buss, J.L.; Torti, F.M.; Torti, S.V. The role of iron chelation in cancer therapy. Curr. Med. Chem. 2003, 10, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shachar, D.; Youdim, M.B. Iron, melanin and dopamine interaction: Relevance to Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 1993, 17, 139–150. [Google Scholar] [CrossRef]
- Planalp, R.P.; Przyborowska, A.M.; Park, G.; Ye, N.; Lu, F.H.; Rogers, R.D.; Broker, G.A.; Torti, S.V.; Brechbiel, M.W. Novel cytotoxic chelators that bind iron(II) selectively over zinc(II) under aqueous aerobic conditions. Biochem. Soc. Trans. 2002, 30, 758–762. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, S.; Huang, X.; Cho, H.; Greig, N.H.; Youdim, M.B.; Rogers, J.T. Metal specificity of an iron-responsive element in Alzheimer’s APP mRNA 5’untranslated region, tolerance of SH-SY5Y and H4 neural cells to desferrioxamine, clioquinol, VK-28, and a piperazine chelator. J. Neural. Transm. Suppl. 2006, 71, 237–247. [Google Scholar]
- Singh, A.; Kong, Q.; Luo, X.; Petersen, R.B.; Meyerson, H.; Singh, N. Prion protein (PrP) knock-out mice show altered iron metabolism: A functional role for PrP in iron uptake and transport. PLoS ONE 2009, 4, e6115. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Haldar, S.; Tripathi, A.K.; McElwee, M.K.; Horback, K.; Beserra, A. Iron in neurodegenerative disorders of protein misfolding: A case of prion disorders and Parkinson’s disease. Antioxid Redox Signal. 2014, 21, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Geller, L.N.; Potter, H. Chromosome missegregation and trisomy 21 mosaicism in Alzheimer’s disease. Neurobiol. Dis. 1999, 6, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.; Myers, A.; Hardy, J. The law of mass action applied to neurodegenerative disease: A hypothesis concerning the etiology and pathogenesis of complex diseases. Hum. Mol. Genet. 2004, 13, R123–R126. [Google Scholar] [CrossRef] [PubMed]

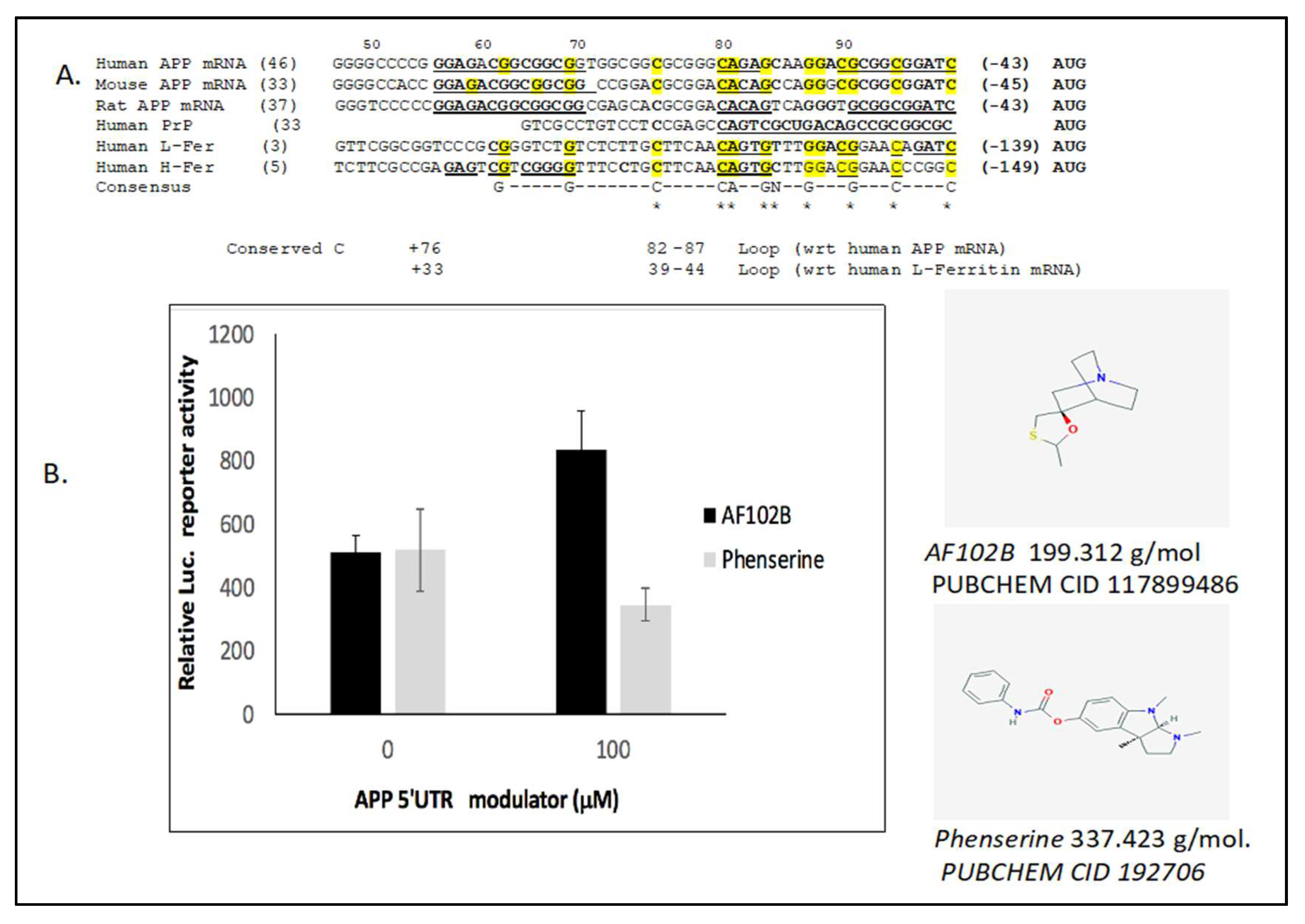

{kind=link}
{kind=link}
| APP 5’UTR Inhibitors (In Two Clinical Trials [24,29,31]) | HTS APP 5’UTR Inhibitors |
|---|---|
| Phenserine—Anticholinesterase [31] Enantiomer = Posiphen [1] APP 5’UTR translation blockers [20]. Anti-amyloid. LD-50; 50 µM in SH-SY5Y neuroblastoma cells. Phase-III clinical trials for AD [24,31]. Typical of DUP–APP specific orphan disease drugs to restore APP dose/Fe balance in fAD. | JTR-009 (PUBCHEM AID 1285) was high throughput screened (HTS) in a transfection-based assay to inhibit APP 5’UTR translation conferred to a luciferase reported gene [21,34,35] PubChem AID: 1285 (Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/1285) JTR-009 can be medicinally optimized to reduce Aβ specific anti-amyloid especially when generated in fAD cases of duplicated over-expression of the APP gene in DS/Dup–APP subjects [21]. |
| APP 5’UTR Activators (FDA-Approved) | HTS APP 5’UTR Activators |
|---|---|
| AF102B M1-muscaric agonist [78]. APP(s) and APP 5’UTR translation activator [79]. Neurotrophin in the clinic for Sjogren’s disease [80,81]. Rogers et al. [57]. | PUBCHEM-AID-1276 (Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/1276). Agents predicted to promote iron export to rescue neurons from acute Pb- and Mn-induced iron overload and neurotoxicity [14]. |
| A. Ferritin IRE Activator | H-ferritin IRE Inhibitor |
| Activates ferritin translation. Yohimbine—Natural product is a neurotrophin that reduces Fe catalyzed oxidative stress. Ferritin-H chain 5’UTR activator, Tibodeau et al. [83]. | Iron chelators inhibit ferritin translation. Desferrioxamine [91,92]/Deferriprone treat conditions of iron overload and redox-associated stress in neurodegeneration [27,93,94] and cases of iron overload, β-thalassemia transfusion [64]. Melatonin chelates Fe from oxidative/nitrosative damage to cells [76]. |
| B. HTS Prion Protein 5’UTR Activators | HTS Prion Protein Inhibitors |
| PUBCHEM AID 1999 (Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/1999) Predicted to influence Iron REDOX [85,95,96] | PUBCHEM AID 488862 (Available online: https://pubchem.ncbi.nlm.nih.gov/bioassay/488894#section=Top Top PrP 5’UTR blocker is “BL-1” which counter-induced L- and H-ferritin translation to enhance intracellular Fe-storage & cell-based viability. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogers, J.T.; Xia, N.; Wong, A.; Bakshi, R.; Cahill, C.M. Targeting the Iron-Response Elements of the mRNAs for the Alzheimer’s Amyloid Precursor Protein and Ferritin to Treat Acute Lead and Manganese Neurotoxicity. Int. J. Mol. Sci. 2019, 20, 994. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040994
Rogers JT, Xia N, Wong A, Bakshi R, Cahill CM. Targeting the Iron-Response Elements of the mRNAs for the Alzheimer’s Amyloid Precursor Protein and Ferritin to Treat Acute Lead and Manganese Neurotoxicity. International Journal of Molecular Sciences. 2019; 20(4):994. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040994
Chicago/Turabian StyleRogers, Jack T., Ning Xia, Angela Wong, Rachit Bakshi, and Catherine M. Cahill. 2019. "Targeting the Iron-Response Elements of the mRNAs for the Alzheimer’s Amyloid Precursor Protein and Ferritin to Treat Acute Lead and Manganese Neurotoxicity" International Journal of Molecular Sciences 20, no. 4: 994. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040994