Down-Regulation of Astrocytic Kir4.1 Channels during the Audiogenic Epileptogenesis in Leucine-Rich Glioma-Inactivated 1 (Lgi1) Mutant Rats

 ,
,
Abstract
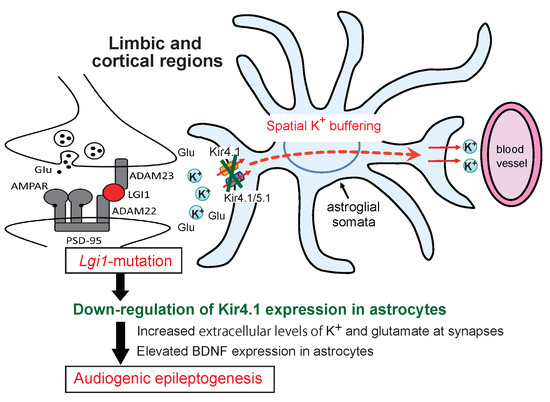
:
1. Introduction
2. Results
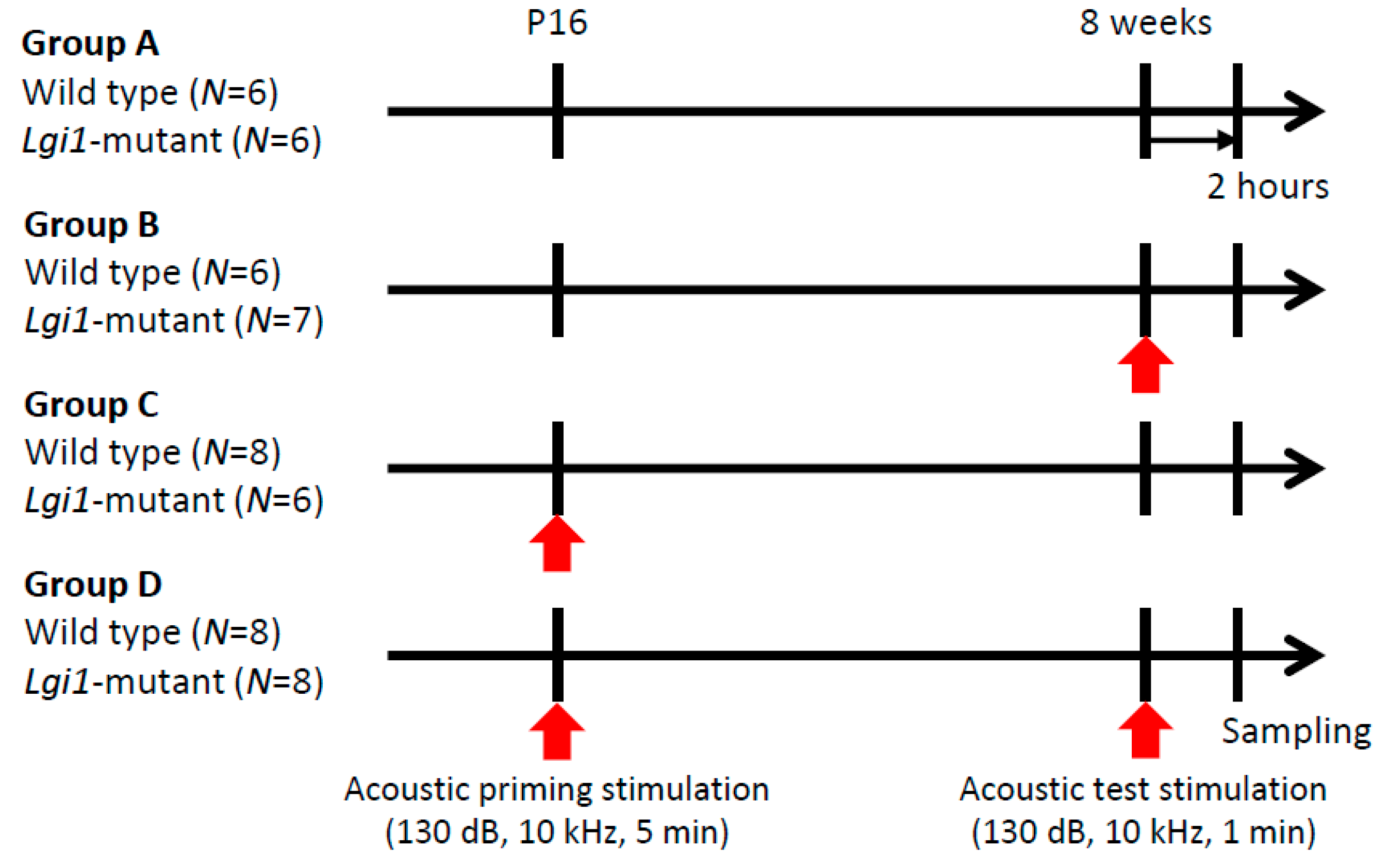
2.1. Audiogenic Seizure Induction
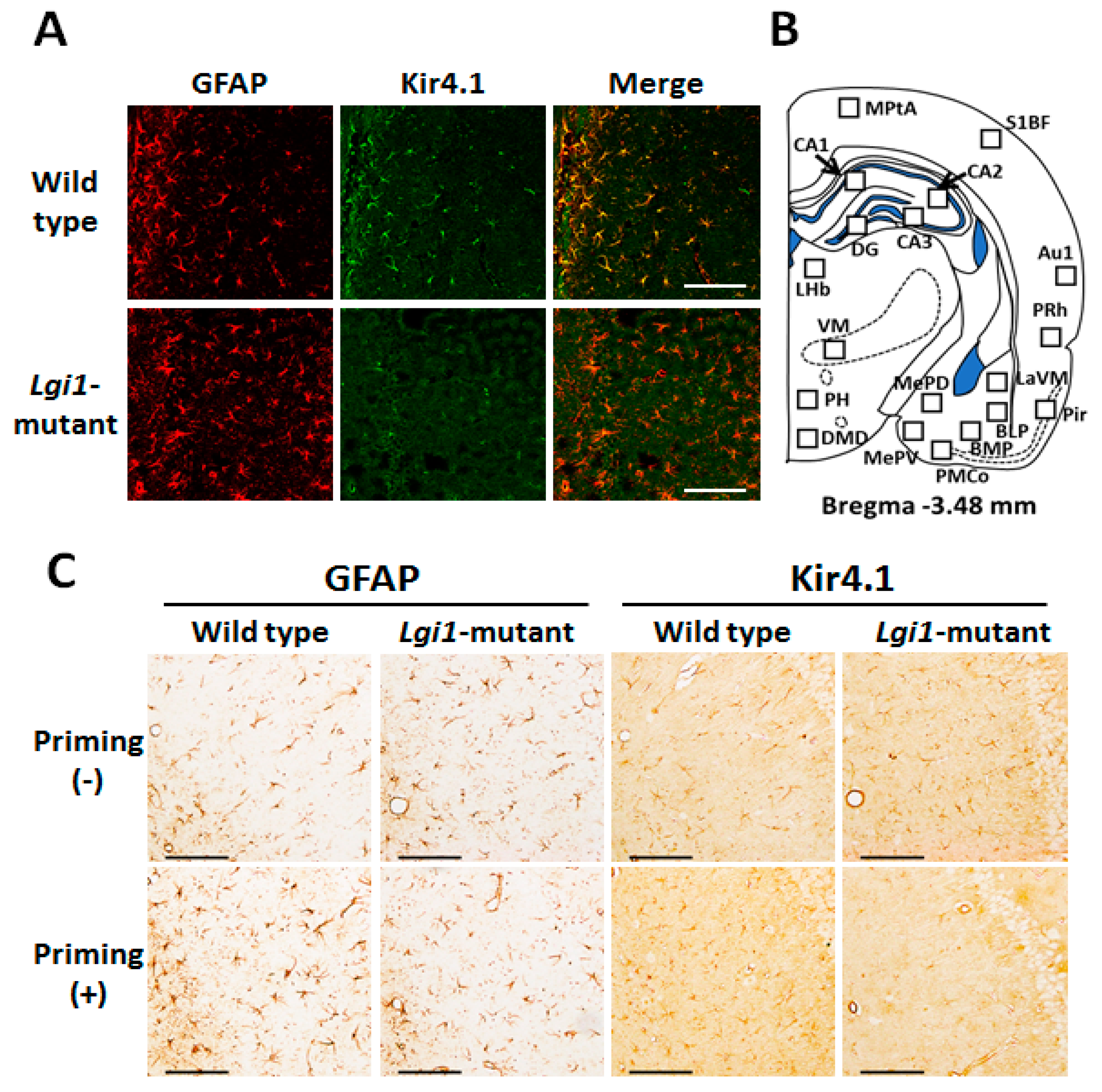
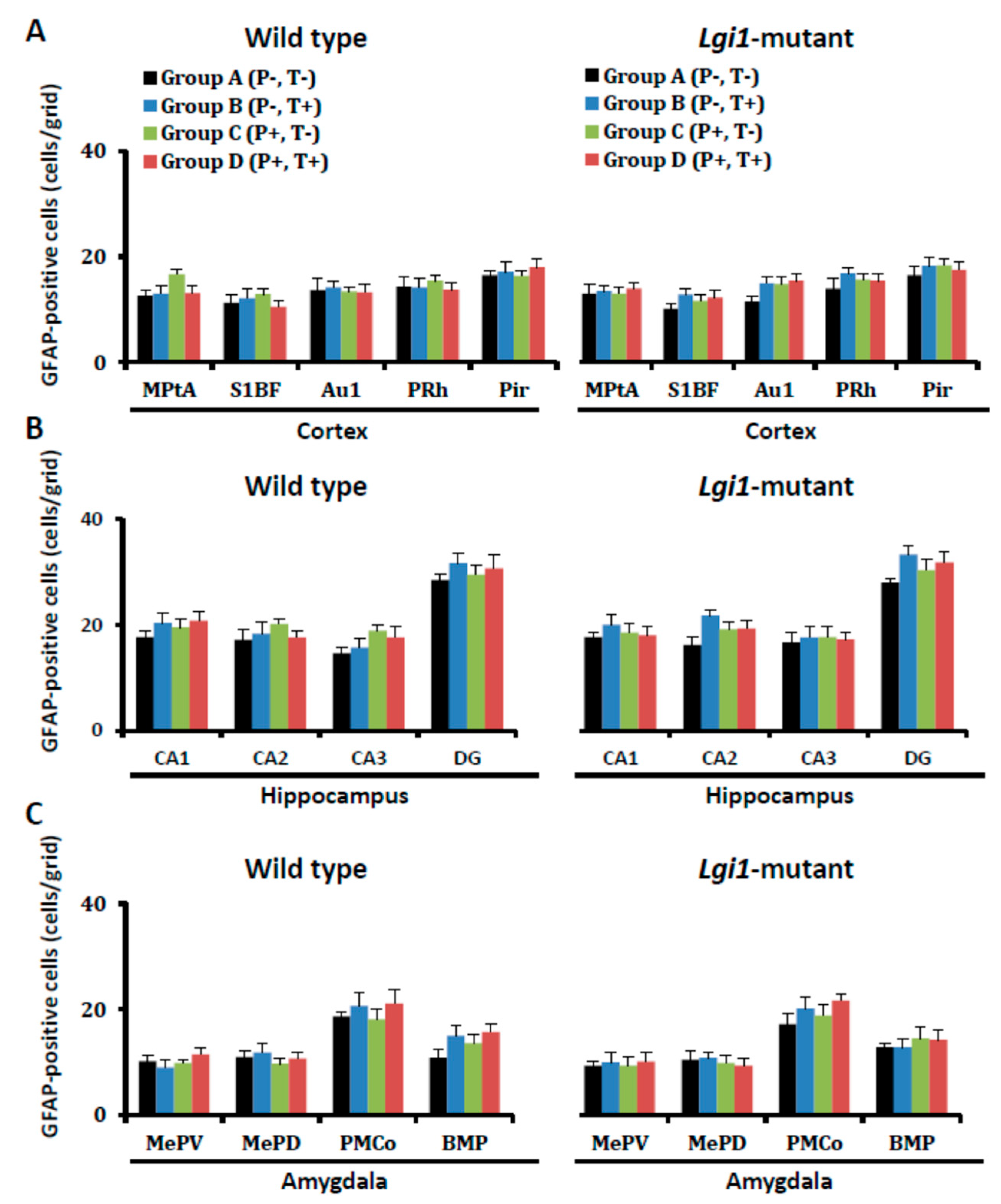
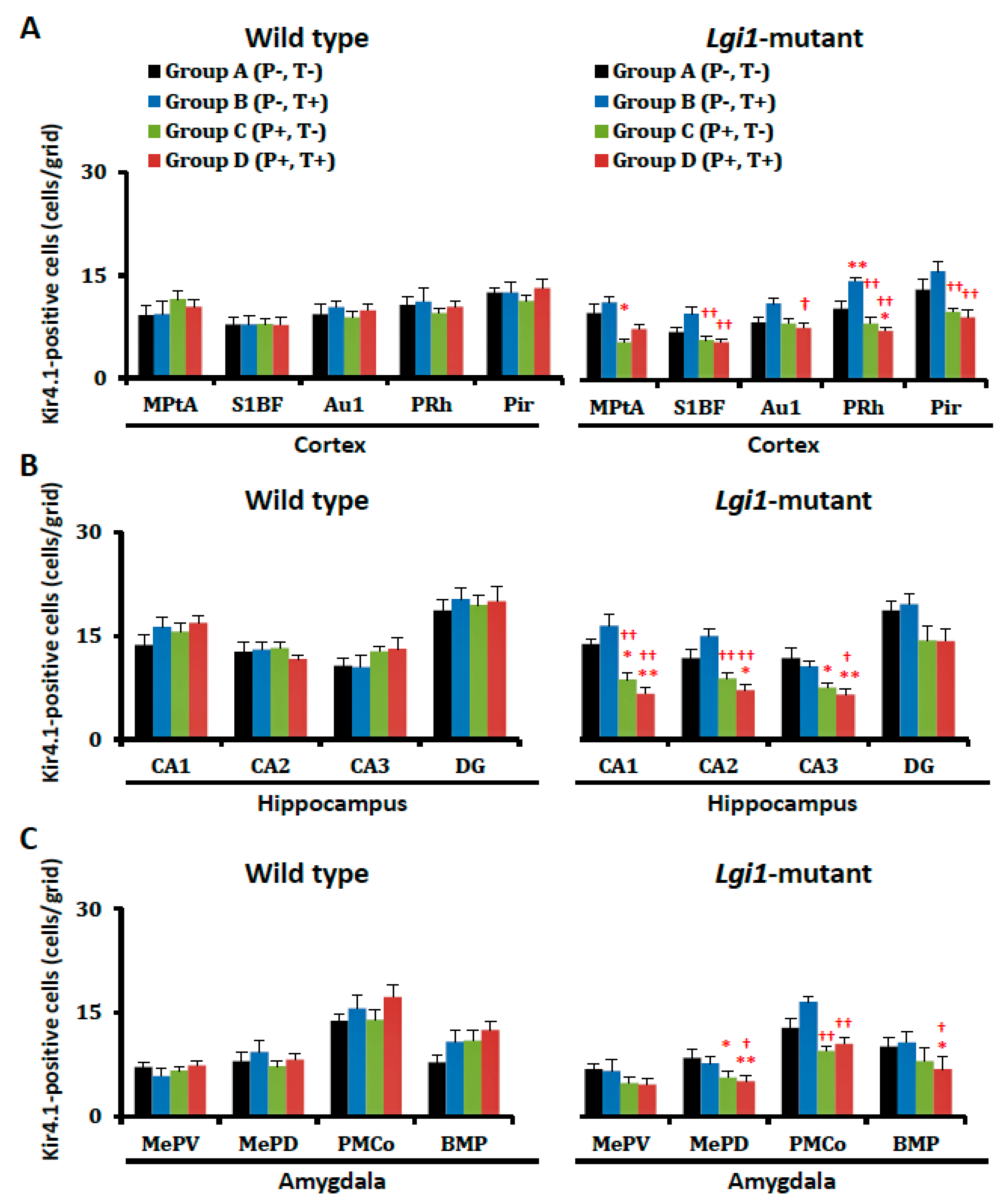
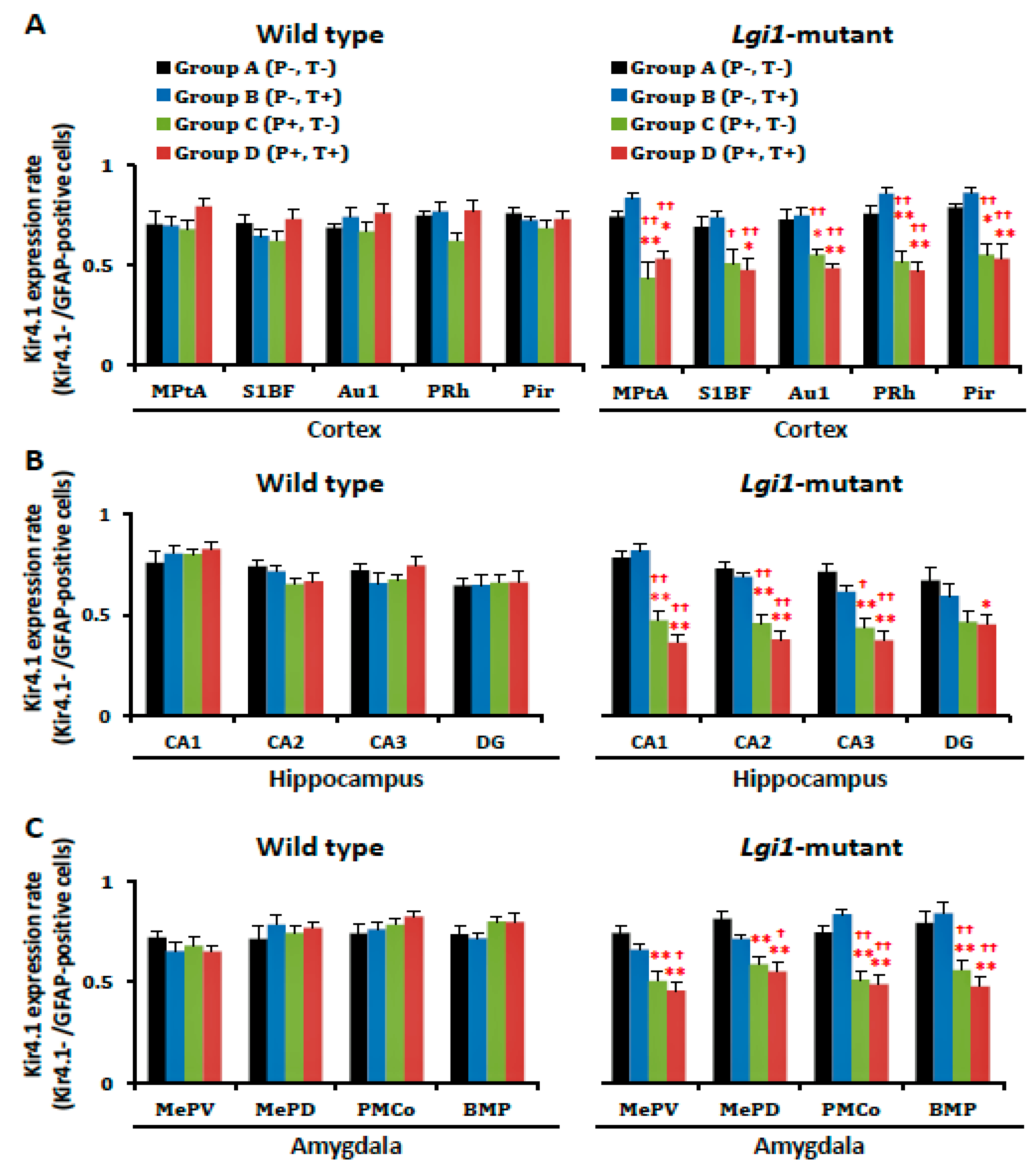
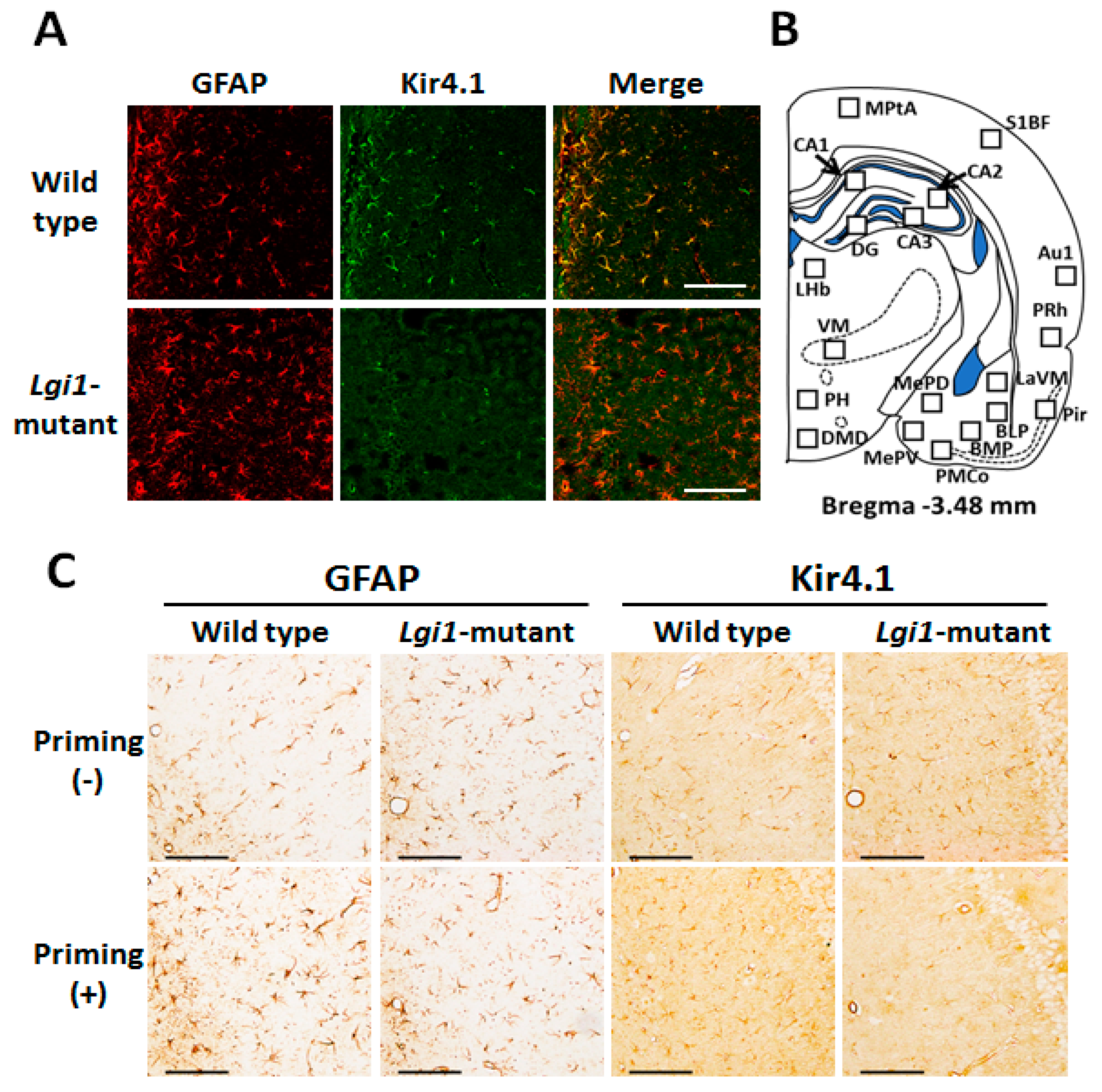
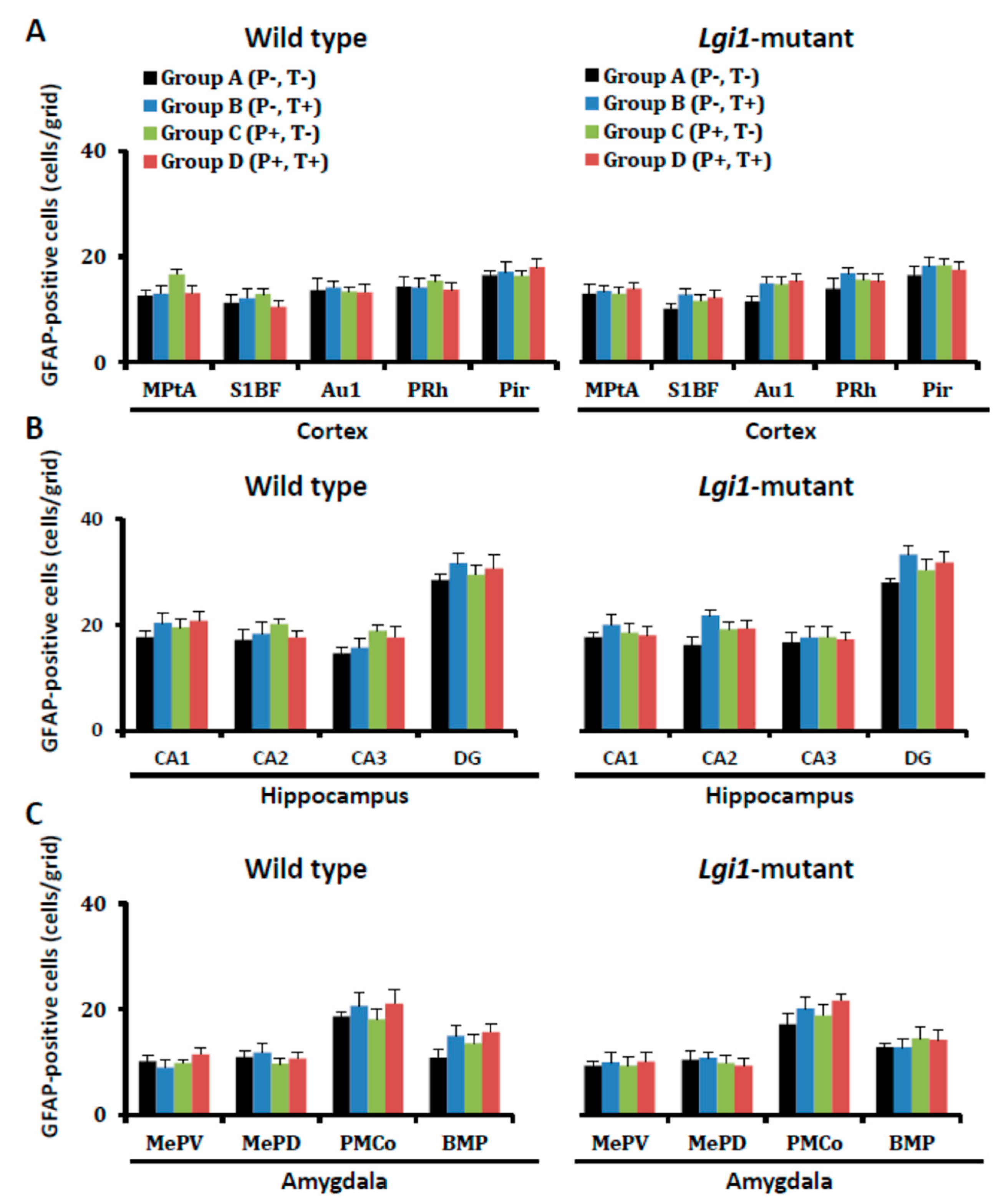
2.2. Expressional Changes in Astrocytic Kir4.1 during Audiogenic Epileptogenesis
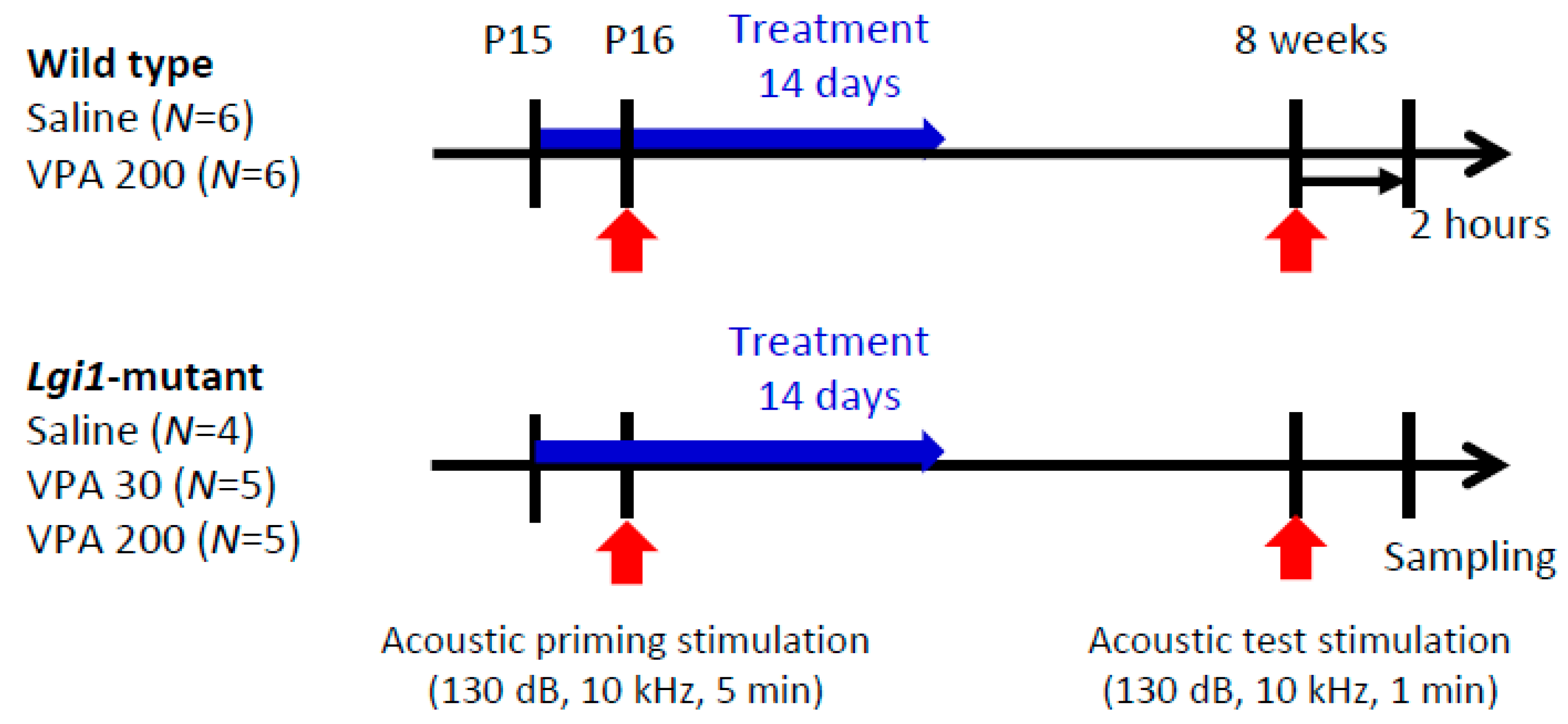
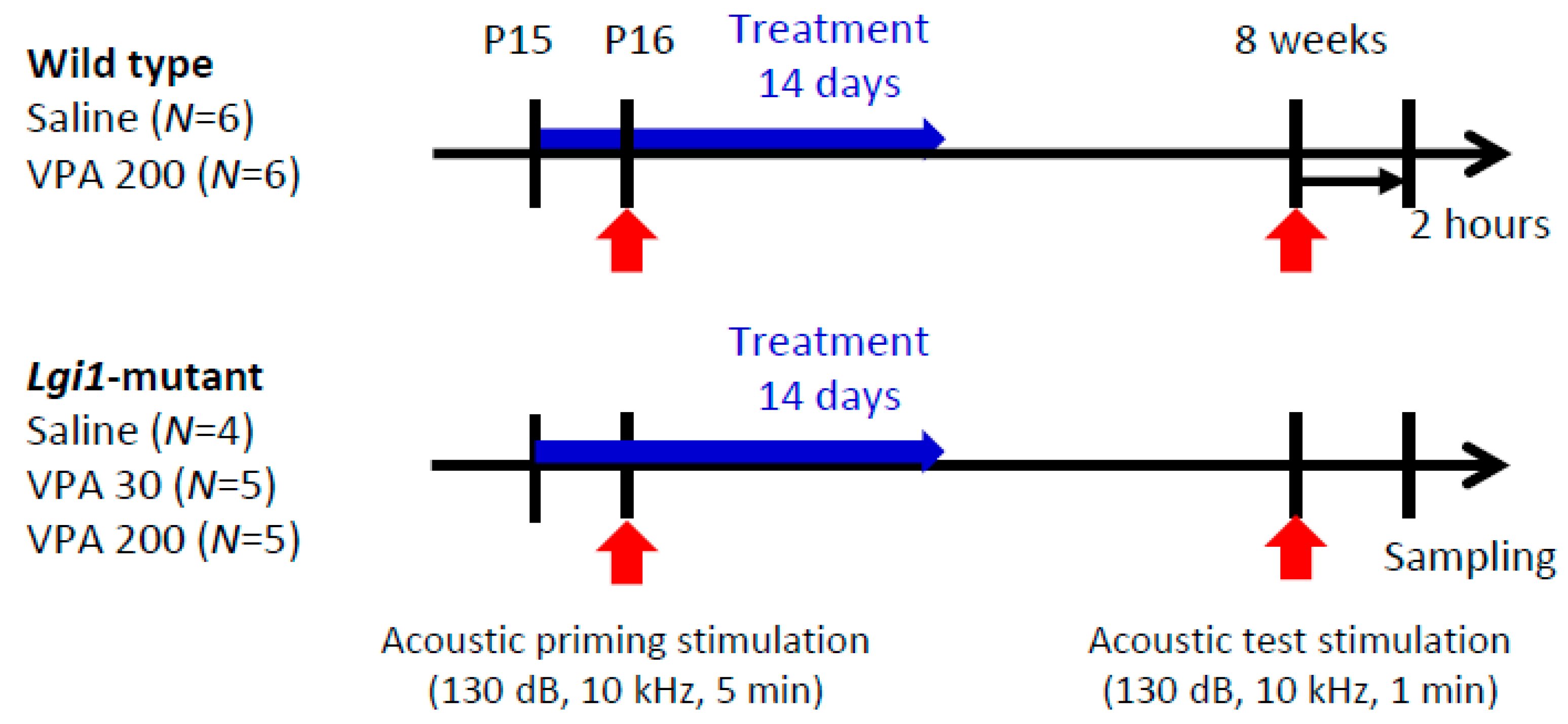
2.3. Prophylactic Actions of VPA on Seizure Susceptibility in Lgi1 Mutant Rats
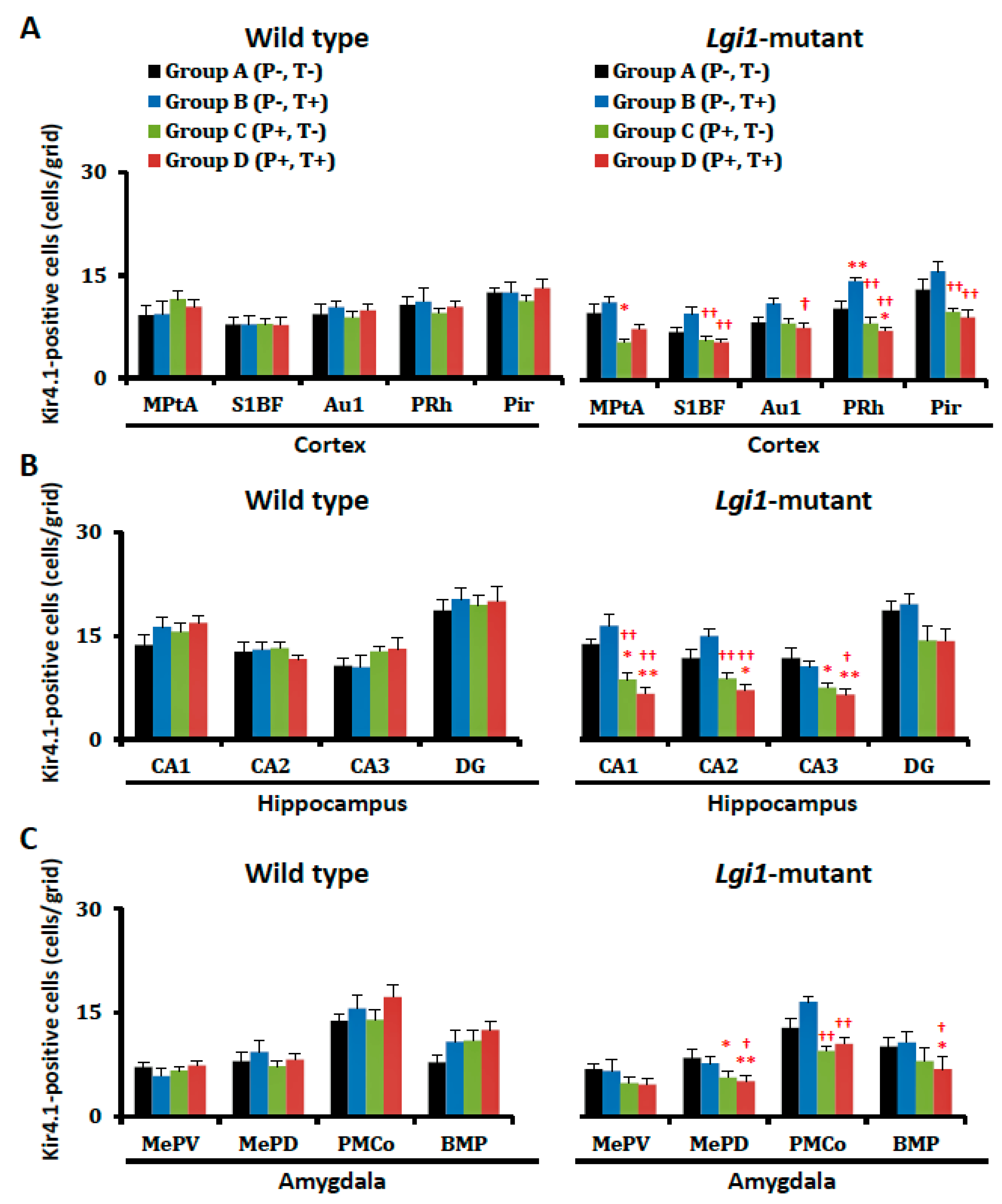
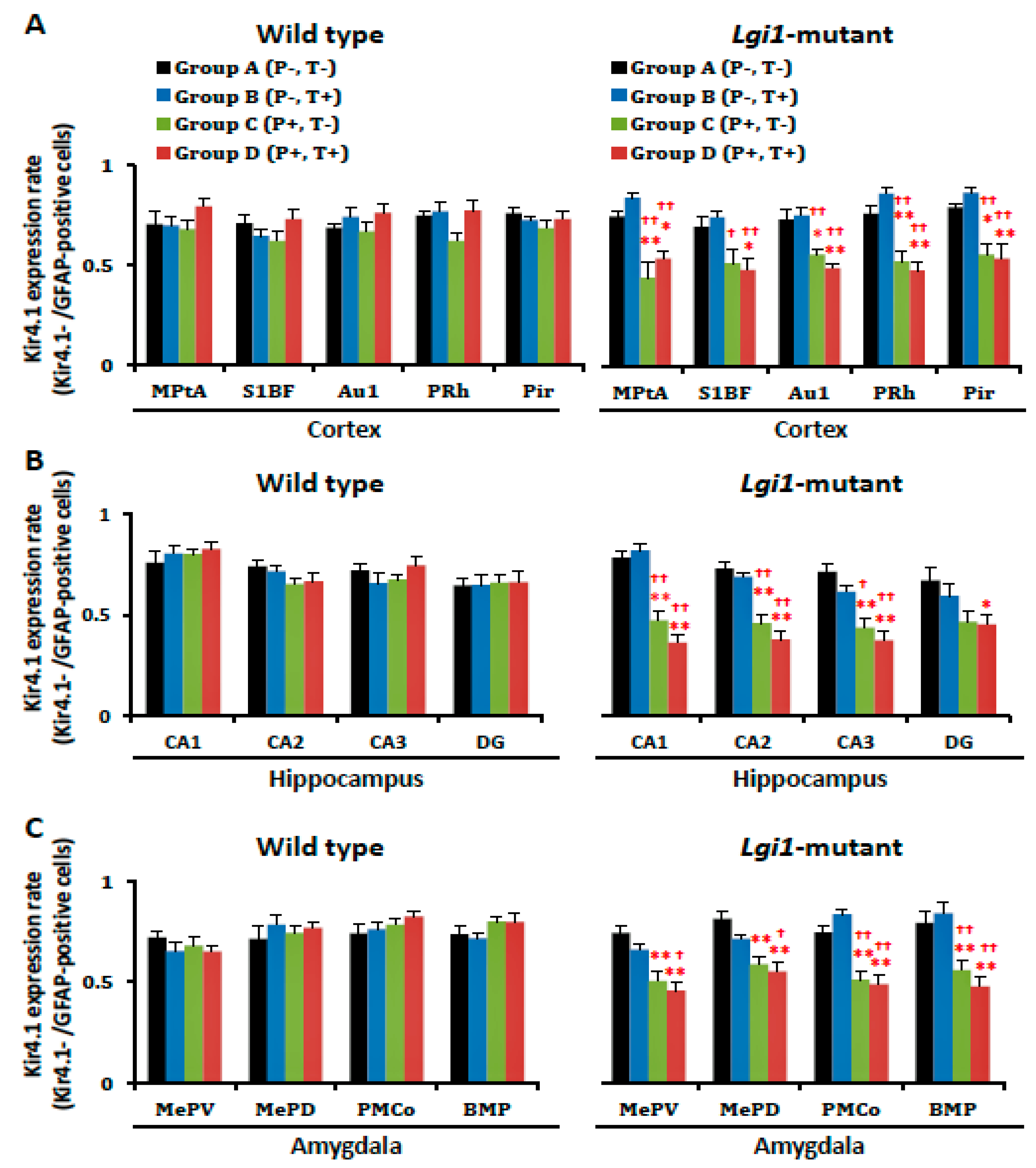
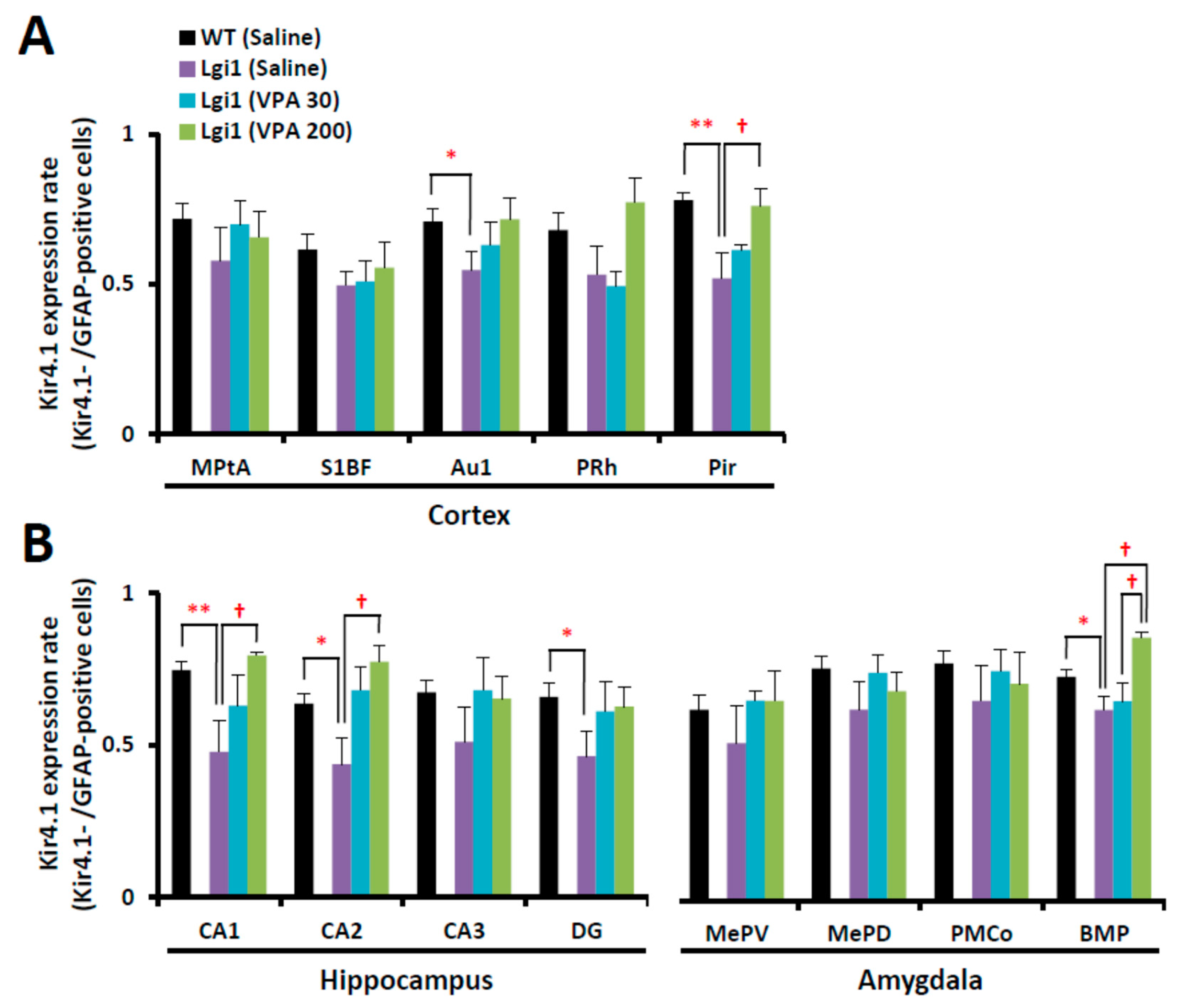
2.4. Expressional Changes in Astrocytic Kir4.1 with Treatment of VPA
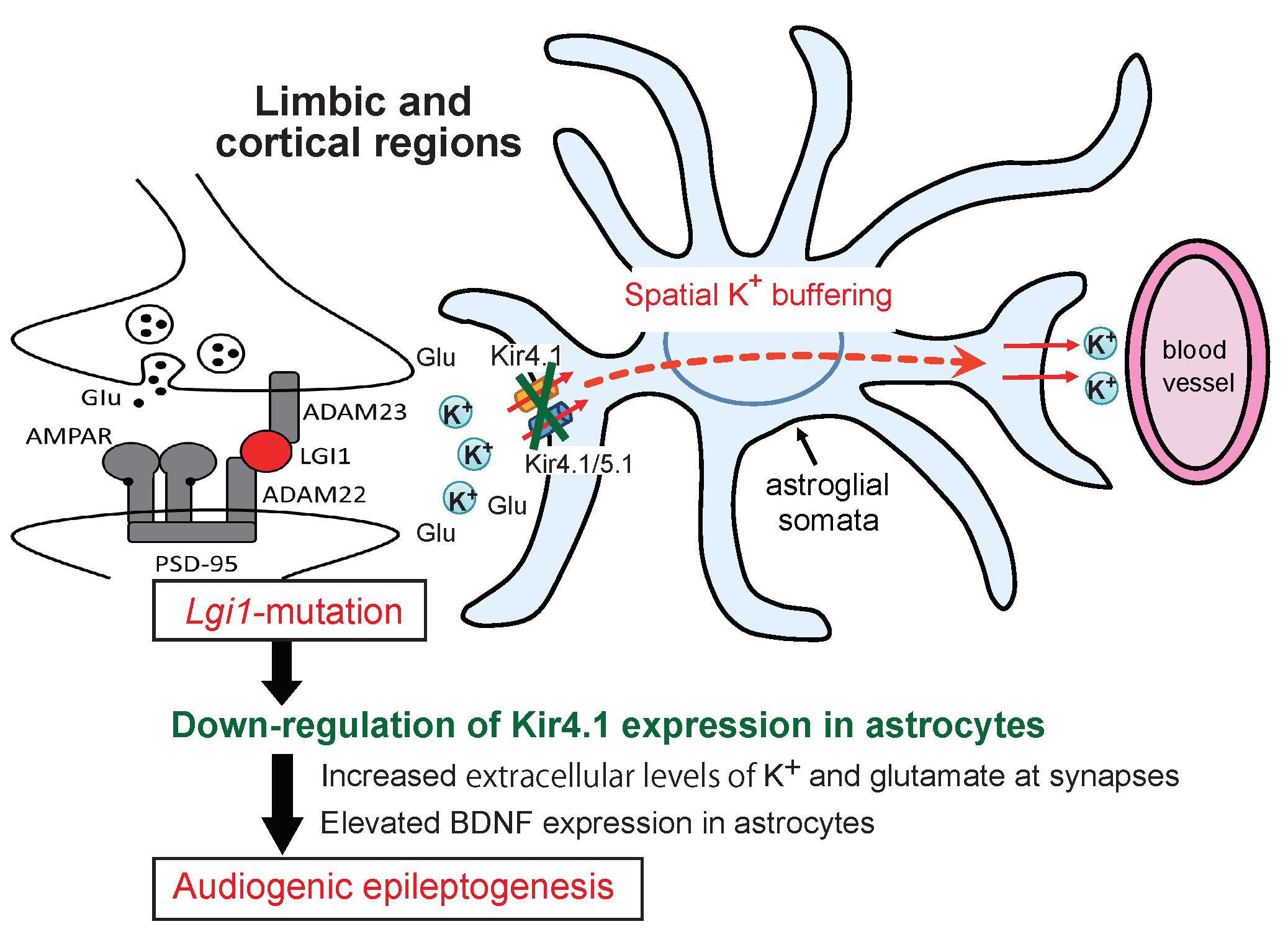
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Genotyping
4.3. Audiogenic Seizure Induction
4.4. Immunofluorescence Staining
4.5. Immunohistochemical Analysis
4.6. Drug Treatments
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Parpura, V.; Zorec, R. Gliotransmission: Exocytotic release from astrocytes. Brain Res. Rev. 2010, 63, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Devinsky, O.; Vezzani, A.; Najjar, S.; De Lanerolle, N.C.; Rogawski, M.A. Glia and epilepsy: Excitability and inflammation. Trends Neurosci. 2013, 36, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Walz, W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem. Int. 2000, 36, 291–300. [Google Scholar] [CrossRef]
- Kofuji, P.; Newman, E.A. Potassium buffering in the central nervous system. Neuroscience 2004, 129, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Simard, M.; Nedergaard, M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience 2004, 129, 877–896. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Tokudome, K.; Kunisawa, N.; Iha, H.A.; Kinboshi, M.; Mukai, T.; Serikawa, T.; Shimizu, S. Role of astroglial Kir4.1 channels in the pathogenesis and treatment of epilepsy. Ther. Targets Neurol. Dis. 2015, 2, e476. [Google Scholar] [CrossRef]
- Neusch, C.; Papadopoulos, N.; Müller, M.; Maletzki, I.; Winter, S.M.; Hirrlinger, J.; Handschuh, M.; Bähr, M.; Richter, D.W.; Kirchhoff, F.; et al. Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: Impact on extracellular K+ regulation. J. Neurophysiol. 2006, 95, 1843–1852. [Google Scholar] [CrossRef] [PubMed]
- Djukic, B.; Casper, K.B.; Philpot, B.D.; Chin, L.S.; McCarthy, K.D. Conditional knock-out of Kir4.1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J. Neurosci. 2007, 27, 11354–11365. [Google Scholar] [CrossRef] [PubMed]
- Bockenhauer, D.; Feather, S.; Stanescu, H.C.; Bandulik, S.; Zdebik, A.A.; Reichold, M.; Tobin, J.; Lieberer, E.; Sterner, C.; Landoure, G.; et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N. Engl. J. Med. 2009, 360, 1960–1970. [Google Scholar] [CrossRef] [PubMed]
- Scholl, U.I.; Choi, M.; Liu, T.; Ramaekers, V.T.; Häusler, M.G.; Grimmer, J.; Tobe, S.W.; Farhi, A.; Nelson-Williams, C.; Lifton, R.P. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc. Natl. Acad. Sci. USA 2009, 106, 5842–5847. [Google Scholar] [CrossRef] [PubMed]
- Reichold, M.; Zdebik, A.A.; Lieberer, E.; Rapedius, M.; Schmidt, K.; Bandulik, S.; Sterner, C.; Tegtmeier, I.; Penton, D.; Baukrowitz, T.; et al. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc. Natl. Acad. Sci. USA 2010, 107, 14490–14495. [Google Scholar] [CrossRef] [PubMed]
- Inyushin, M.; Kucheryavykh, L.Y.; Kucheryavykh, Y.V.; Nichols, C.G.; Buono, R.J.; Ferraro, T.N.; Skatchkov, S.N.; Eaton, M.J. Potassium channel activity and glutamate uptake are impaired in astrocytes of seizure-susceptible DBA/2 mice. Epilepsia 2010, 51, 1707–1713. [Google Scholar] [CrossRef] [PubMed]
- Harada, Y.; Nagao, Y.; Shimizu, S.; Serikawa, T.; Terada, R.; Fujimoto, M.; Okuda, A.; Mukai, T.; Sasa, M.; Kurachi, Y.; et al. Expressional analysis of inwardly rectifying Kir4.1 channels in Noda epileptic rat (NER). Brain Res. 2013, 1517, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Wallace, G.C.; Holmes, C.; McDowell, M.L.; Smith, J.A.; Marshall, J.D.; Bonilha, L.; Edwards, J.C.; Glazier, S.S.; Ray, S.K.; et al. Hippocampal tissue of patients with refractory temporal lobe epilepsy is associated with astrocyte activation, inflammation, and altered expression of channels and receptors. Neuroscience 2012, 220, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Heuser, K.; Eid, T.; Lauritzen, F.; Thoren, A.E.; Vindedal, G.F.; Taubøll, E.; Gjerstad, L.; Spencer, D.D.; Ottersen, O.P.; Nagelhus, E.A.; et al. Loss of perivascular Kir4.1 potassium channels in the sclerotic hippocampus of patients with mesial temporal lobe epilepsy. J. Neuropathol. Exp. Neurol. 2012, 71, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Steinhäuser, C.; Seifert, G.; Bedner, P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia 2012, 60, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Kinboshi, M.; Mukai, T.; Nagao, Y.; Matsuba, Y.; Tsuji, Y.; Tanaka, S.; Tokudome, K.; Shimizu, S.; Ito, H.; Ikeda, A.; et al. Inhibition of inwardly rectifying potassium (Kir) 4.1 channels facilitates brain-derived neurotrophic factor (BDNF) expression in astrocytes. Front. Mol. Neurosci. 2017, 10, 408. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Kinboshi, M.; Shimizu, S. Inwardly rectifying potassium channel Kir4.1 as a novel modulator of BDNF expression in astrocytes. Int. J. Mol. Sci. 2018, 19, 3313. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Kinboshi, M.; Nagao, Y.; Shimizu, S.; Ono, A.; Sakagami, Y.; Okuda, A.; Fujimoto, M.; Ito, H.; Ikeda, A.; et al. Antiepileptic drugs elevate astrocytic Kir4.1 expression in the rat limbic region. Front. Pharmacol. 2018, 9, 845. [Google Scholar] [CrossRef] [PubMed]
- Kalachikov, S.; Evgrafov, O.; Ross, B.; Winawer, M.; Barker-Cummings, C.; Martinelli Boneschi, F.; Choi, C.; Morozov, P.; Das, K.; Teplitskaya, E.; et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat. Genet. 2002, 30, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Morante-Redolat, J.M.; Gorostidi-Pagola, A.; Piquer-Sirerol, S.; Sáenz, A.; Poza, J.J.; Galán, J.; Gesk, S.; Sarafidou, T.; Mautner, V.F.; Binelli, S.; et al. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum. Mol. Genet. 2002, 11, 1119–1128. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Oses-Prieto, J.; Kim, M.Y.; Horresh, I.; Peles, E.; Burlingame, A.L.; Trimmer, J.S.; Meijer, D.; Rasband, M.N. ADAM22, a Kv1 channel-interacting protein, recruits membrane-associated guanylate kinases to juxtaparanodes of myelinated axons. J. Neurosci. 2010, 30, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Lovero, K.L.; Fukata, Y.; Granger, A.J.; Fukata, M.; Nicoll, R.A. The LGI1-ADAM22 protein complex directs synapse maturation through regulation of PSD-95 function. Proc. Natl. Acad. Sci. USA 2015, 112, E4129–E4137. [Google Scholar] [CrossRef] [PubMed]
- Fukata, Y.; Yokoi, N.; Miyazaki, Y.; Fukata, M. The LGI1-ADAM22 protein complex in synaptic transmission and synaptic disorders. Neurosci. Res. 2017, 116, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Petit-Pedrol, M.; Sell, J.; Planagumà, J.; Mannara, F.; Radosevic, M.; Haselmann, H.; Ceanga, M.; Sabater, L.; Spatola, M.; Soto, D.; et al. LGI1 antibodies alter Kv1.1 and AMPA receptors changing synaptic excitability, plasticity and memory. Brain 2018, 141, 3144–3159. [Google Scholar] [CrossRef] [PubMed]
- Baulac, S.; Ishida, S.; Mashimo, T.; Boillot, M.; Fumoto, N.; Kuwamura, M.; Ohno, Y.; Takizawa, A.; Aoto, T.; Ueda, M.; et al. A rat model for LGI1-related epilepsies. Hum. Mol. Genet. 2012, 21, 3546–3557. [Google Scholar] [CrossRef] [PubMed]
- Fumoto, N.; Mashimo, T.; Masui, A.; Ishida, S.; Mizuguchi, Y.; Minamimoto, S.; Ikeda, A.; Takahashi, R.; Serikawa, T.; Ohno, Y. Evaluation of seizure foci and genes in the Lgi1(L385R/+) mutant rat. Neurosci. Res. 2014, 80, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Nagao, Y.; Harada, Y.; Mukai, T.; Shimizu, S.; Okuda, A.; Fujimoto, M.; Ono, A.; Sakagami, Y.; Ohno, Y. Expressional analysis of the astrocytic Kir4.1 channel in a pilocarpine-induced temporal lobe epilepsy model. Front. Cell. Neurosci. 2013, 7, 104. [Google Scholar] [CrossRef] [PubMed]
- Herranz-Pérez, V.; Olucha-Bordonau, F.E.; Morante-Redolat, J.M.; Pérez-Tur, J. Regional distribution of the leucine-rich glioma inactivated (LGI) gene family transcripts in the adult mouse brain. Brain Res. 2010, 1307, 177–194. [Google Scholar] [CrossRef] [PubMed]
- Curia, G.; Gualtieri, F.; Bartolomeo, R.; Vezzali, R.; Biagini, G. Resilience to audiogenic seizures is associated with p-ERK1/2 dephosphorylation in the subiculum of Fmr1 knockout mice. Front. Cell. Neurosci. 2013, 7, 46. [Google Scholar] [CrossRef] [PubMed]
- Kokaia, M.; Ernfors, P.; Kokaia, Z.; Elmér, E.; Jaenisch, R.; Lindvall, O. Suppressed epileptogenesis in BDNF mutant mice. Exp. Neurol. 1995, 133, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.E.; Shannon, H.E. The seizure-related phenotype of brain-derived neurotrophic factor knockdown mice. Neuroscience 2005, 136, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, C.; Lähteinen, S.; Suzuki, F.; Anne-Marie, L.; Huber, S.; Häussler, U.; Haas, C.; Larmet, Y.; Castren, E.; Depaulis, A. Increase in BDNF-mediated TrkB signaling promotes epileptogenesis in a mouse model of mesial temporal lobe epilepsy. Neurobiol. Dis. 2011, 42, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Grabenstatter, H.L.; Russek, S.J.; Brooks-Kayal, A.R. Molecular pathways controlling inhibitory receptor expression. Epilepsia 2012, 53, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.K.; Routbort, M.J.; Ryan, T.E.; Yancopoulos, G.D.; McNamara, J.O. Selective inhibition of kindling development by intraventricular administration of TrkB receptor body. J. Neurosci. 1999, 19, 1424–1436. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Gu, B.; He, X.P.; Joshi, R.B.; Wackerle, H.D.; Rodriguiz, R.M.; Wetsel, W.C.; McNamara, J.O. Transient inhibition of TrkB kinase following status epilepticus prevents development of temporal lobe epilepsy. Neuron 2013, 79, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Lukasiuk, K.; Dudek, F.E.; Staley, K.J. Epileptogenesis. Cold Spring Harb. Perspect. Med. 2015, 5, a022822. [Google Scholar] [CrossRef] [PubMed]
- Skowrońska, K.; Obara-Michlewska, M.; Czarnecka, A.; Dąbrowska, K.; Zielińska, M.; Albrecht, J. Persistent overexposure to N-methyl-d-aspartate (NMDA) calcium-dependently downregulates glutamine synthetase, aquaporin 4, and Kir4.1 channel in mouse cortical astrocytes. Neurotox. Res. 2018, 35, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.E.; Wen, L.; Silva, J.; Li, Z.; Head, K.; Sossey-Alaoui, K.; Pao, A.; Mei, L.; Cowell, J.K. Lgi1 null mutant mice exhibit myoclonic seizures and CA1 neuronal hyperexcitability. Hum. Mol. Genet. 2010, 19, 1702–1711. [Google Scholar] [CrossRef] [PubMed]
- Boillot, M.; Lee, C.Y.; Allene, C.; Leguern, E.; Baulac, S.; Rouach, N. LGI1 acts presynaptically to regulate excitatory synaptic transmission during early postnatal development. Sci. Rep. 2016, 6, 21769. [Google Scholar] [CrossRef] [PubMed]
- Vajda, F.J.; Eadie, M.J. The clinical pharmacology of traditional antiepileptic drugs. Epileptic Disord. 2014, 16, 395–408. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.M.; Shin, C.; McNamara, J.O. Antiepileptogenic effects of conventional anticonvulsants in the kindling model of epilepsy. Ann. Neurol. 1991, 29, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Bolanos, A.R.; Sarkisian, M.; Yang, Y.; Hori, A.; Helmers, S.L.; Mikati, M.; Tandon, P.; Stafstrom, C.E.; Holmes, G.L. Comparison of valproate and phenobarbital treatment after status epilepticus in rats. Neurology 1998, 51, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, Y.; Araki, H.; Futagami, K.; Kawasaki, H.; Gomita, Y. Effects of valproate, phenytoin, and zonisamide on clonic and tonic seizures induced by acute and repeated exposure of mice to flurothyl. Physiol. Behav. 2003, 78, 465–469. [Google Scholar] [CrossRef]
- Vinet, J.; Costa, A.M.; Salinas-Navarro, M.; Leo, G.; Moons, L.; Arckens, L.; Biagini, G. A Hydroxypyrone-based inhibitor of metalloproteinase-12 displays neuroprotective properties in both status epilepticus and optic nerve crush animal models. Int. J. Mol. Sci. 2018, 19, 2178. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotype | Acoustic Stimulation | Responses to Test Stimulation (No. of Animals) | ||||
|---|---|---|---|---|---|---|
| Priming | Test | Total | None | WR | WR+ GTCS | |
| Group B Wild type | - | + | 6 | 6 | 0 | 0 |
| Group B Lgi1-mutant | - | + | 7 | 7 | 0 | 0 |
| Group D Wild type | + | + | 8 | 0 | 8 | 0 |
| Group D Lgi1-mutant | + | + | 8 | 0 | 1 | 7 (88%) |
| Genotype | Drug (mg/kg) | Responses to Test Stimulation (No. of Animals) | |||
|---|---|---|---|---|---|
| Total | None | WR | WR + GTCS | ||
| Wild type | Saline | 6 | 0 | 6 | 0 |
| Wild type | VPA 200 | 6 | 0 | 4 | 0 |
| Lgi1-mutant | Saline | 4 | 0 | 1 | 3 (75%) |
| Lgi1-mutant | VPA 30 | 5 | 0 | 4 | 1 (20%) |
| Lgi1-mutant | VPA 200 | 5 | 0 | 4 | 1 (20%) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinboshi, M.; Shimizu, S.; Mashimo, T.; Serikawa, T.; Ito, H.; Ikeda, A.; Takahashi, R.; Ohno, Y. Down-Regulation of Astrocytic Kir4.1 Channels during the Audiogenic Epileptogenesis in Leucine-Rich Glioma-Inactivated 1 (Lgi1) Mutant Rats. Int. J. Mol. Sci. 2019, 20, 1013. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051013
Kinboshi M, Shimizu S, Mashimo T, Serikawa T, Ito H, Ikeda A, Takahashi R, Ohno Y. Down-Regulation of Astrocytic Kir4.1 Channels during the Audiogenic Epileptogenesis in Leucine-Rich Glioma-Inactivated 1 (Lgi1) Mutant Rats. International Journal of Molecular Sciences. 2019; 20(5):1013. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051013
Chicago/Turabian StyleKinboshi, Masato, Saki Shimizu, Tomoji Mashimo, Tadao Serikawa, Hidefumi Ito, Akio Ikeda, Ryosuke Takahashi, and Yukihiro Ohno. 2019. "Down-Regulation of Astrocytic Kir4.1 Channels during the Audiogenic Epileptogenesis in Leucine-Rich Glioma-Inactivated 1 (Lgi1) Mutant Rats" International Journal of Molecular Sciences 20, no. 5: 1013. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051013