Cell Metabolism Control Through O-GlcNAcylation of STAT5: A Full or Empty Fuel Tank Makes a Big Difference for Cancer Cell Growth and Survival

 , and
, and {kind=link}
{kind=link}
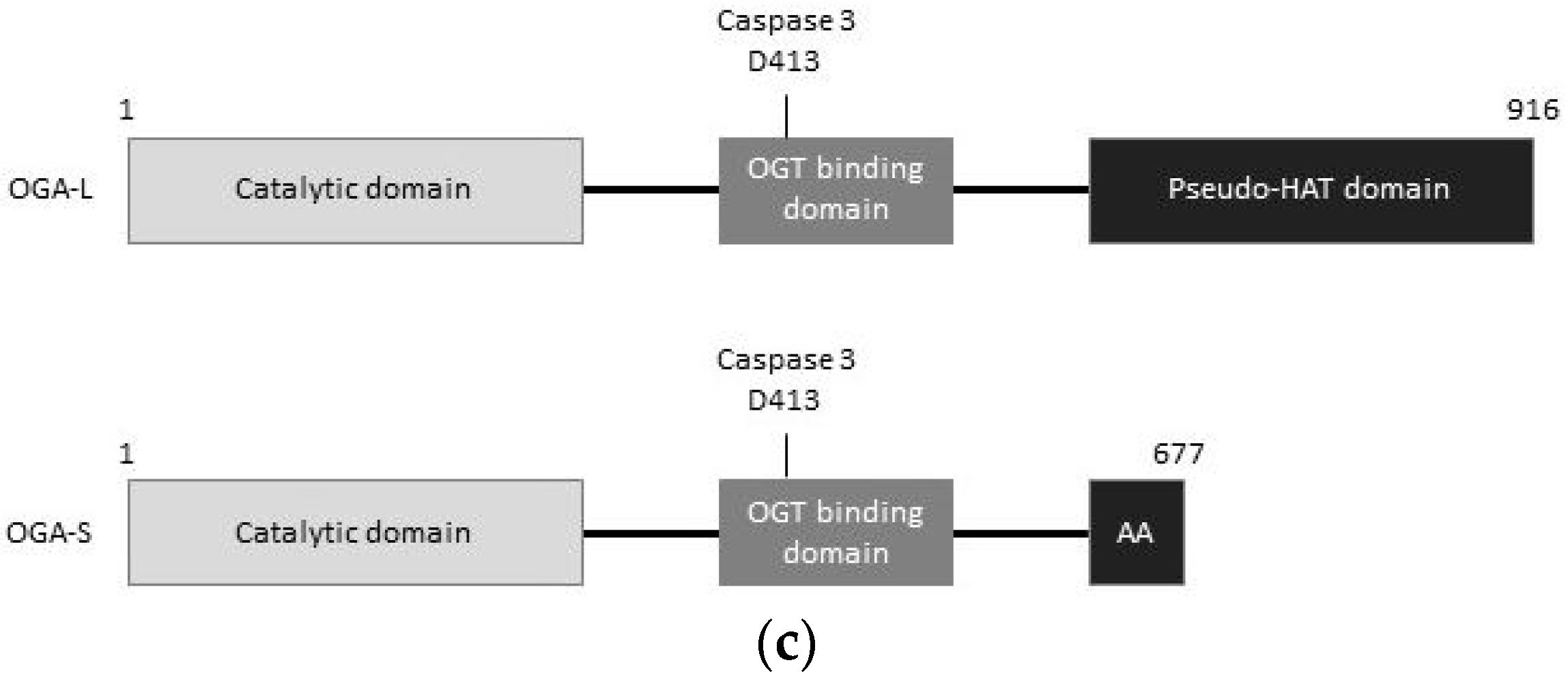{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of the Janus Kinase Signal Transducer and Activator of Transcription (JAK-STAT) Signaling Pathway
3. O-GlcNAcylation as an Essential Post-Translational Modification
4. O-GlcNAc is Crucial for JAK-STAT Pathway Functions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AcCoA | Acetyl Coenzyme A |
| AML | Acute Myeloid Leukemia |
| CALR | Calreticulin |
| CD | Catalytic domain |
| CML | Chronic Myeloid Leukemia |
| DON | 6-diazo-5-oxo-L-norleucine |
| EOGT | EGF domain-specific OGT |
| EPO | Erythropoietin |
| GFAT | Glutamine Fructose-6-phosphate Transaminase |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| GNPNAT | Glucosamine-Phosphate N-Acetyltransferase |
| GOF | Gain Of Function |
| GPI | Phosphoglucose Isomerase |
| HAT | Histone Acetyl Transferase |
| HBP | Hexosamine Biosynthetic Pathway |
| HIF | Hypoxia Inducible Factor |
| HK | Hexokinase |
| ID | Intrinsically disordered |
| IFN | Interferon |
| IL | Interleukin |
| InD | Intervening domain |
| JAK | Janus kinase |
| MCP | Monocarboxylate transporter |
| MPN | Myeloproliferative neoplasm |
| MTS | Mitochondrial targeting site |
| OGA | O-GlcNAcase |
| O-GlcNAc | O-linked β-N-acetyl glucosamine |
| OGT | O-GlcNAc transferase |
| OSM | Oncostatin M |
| PDHX | Pyruvate dehydrogenase protein X component |
| PGM3 | GlcNAc Phosphomutase |
| PIP3 | Phosphatidyl-inositol-3-phosphate |
| PPO | PIP-binding activity domain |
| PTM | Post-translational modification |
| ROS | Reactive oxygen species |
| SH | Src homology |
| SOCS | Suppressor of cytokine signaling |
| STAT | Signal transducer and activator of transcription |
| TCA | Tricarboxylic acid |
| TPO | Thrombopoietin |
| TPOR | Thrombopoietin receptor |
| TPR | Tetratricopeptide repeat |
| TYK | Tyrosine kinase |
| UDP-GlcNAc | Uridine diphosphate-GlcNAc |
| UTP | Uridine triphosphate |
| WGA | Wheat germ agglutinin |
References
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2016, 38, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature 2018, 560, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Leca, J.; Berger, T.; Mak, T.W. Parasitic Behavior of Leukemic Cells in Systemic Host Metabolism. Cell Metab. 2018, 28, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Jóźwiak, P.; Forma, E.; Bryś, M.; Krześlak, A. O-GlcNAcylation and Metabolic Reprograming in Cancer. Front. Endocrinol. 2014, 5, 145. [Google Scholar]
- Tan, E.P.; Villar, M.T.; Lezi, E.; Lu, J.; Selfridge, J.E.; Artigues, A.; Swerdlow, R.H.; Slawson, C. Altering O-Linked β-N-Acetylglucosamine Cycling Disrupts Mitochondrial Function. J. Biol. Chem. 2014, 289, 14719–14730. [Google Scholar] [CrossRef] [PubMed]
- Freund, P.; Kerenyi, M.A.; Hager, M.; Wagner, T.; Wingelhofer, B.; Pham, H.T.; Elabd, M.; Han, X.; Valent, P.; Gouilleux, F.; et al. O-GlcNAcylation of STAT5 controls tyrosine phosphorylation and oncogenic transcription in STAT5-dependent malignancies. Leukemia 2017, 31, 2132–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’sullivan, L.A.; Liongue, C.; Lewis, R.S.; Stephenson, S.E.; Ward, A.C. Cytokine receptor signaling through the Jak–Stat–Socs pathway in disease. Mol. Immunol. 2007, 44, 2497–2506. [Google Scholar] [CrossRef] [PubMed]
- Dodington, D.W.; Desai, H.R.; Woo, M. JAK/STAT—Emerging Players in Metabolism. Trends Endocrinol. Metab. 2018, 29, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Farrell, B.; Breeze, A.L. Structure, activation and dysregulation of fibroblast growth factor receptor kinases: Perspectives for clinical targeting. Biochem. Soc. Trans. 2018, 46, 1753–1770. [Google Scholar] [CrossRef] [PubMed]
- Laurence, A. JAK Kinases in Health and Disease: An Update. Open Rheumatol. J. 2012, 6, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Silvennoinen, O.; Ungureanu, D.; Niranjan, Y.; Hammaren, H.; Bandaranayake, R.; Hubbard, S.R. New insights into the structure and function of the pseudokinase domain in JAK2. Biochem. Soc. Trans. 2013, 41, 1002–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babon, J.J.; Lucet, I.S.; Murphy, J.M.; Nicola, N.A.; Varghese, L.N. The molecular regulation of Janus kinase (JAK) activation. Biochem. J. 2014, 462, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.; Sexl, V.; Valent, P.; Moriggl, R. Inhibition of STAT5: A therapeutic option in BCR-ABL1-driven leukemia. Oncotarget 2014, 5, 9564–9576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa-Pereira, A.P.; Bonito, N.A.; Seckl, M.J. Dysregulation of janus kinases and signal transducers and activators of transcription in cancer. Am. J. Cancer Res. 2011, 1, 806–816. [Google Scholar] [PubMed]
- Ortmann, R.A.; Cheng, T.; Visconti, R.; Frucht, D.M.; O’Shea, J.J. Janus kinases and signal transducers and activators of transcription: Their roles in cytokine signaling, development and immunoregulation. Arthritis Res. 2000, 2, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.; London, L.; Bandach, I.; Segev, Y. The hypoxia inducible factor/erythropoietin (EPO)/EPO receptor pathway is disturbed in a rat model of chronic kidney disease related anemia. PLoS ONE 2018, 13, e0196684. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.C.; Touw, I.; Yoshimura, A. The Jak-Stat pathway in normal and perturbed hematopoiesis. Blood 2000, 95, 19–29. [Google Scholar] [PubMed]
- Pellegrini, S.; Dusanter-Fourt, I. The structure, regulation and function of the Janus kinases (JAKs) and the signal transducers and activators of transcription (STATs). Eur. J. Biochem. 1997, 248, 615–633. [Google Scholar] [CrossRef] [PubMed]
- Liongue, C.; O’Sullivan, L.A.; Trengove, M.C.; Ward, A.C. Evolution of JAK-STAT Pathway Components: Mechanisms and Role in Immune System Development. PLoS ONE 2012, 7, e32777. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Wang, B.; Liu, X.; Liu, X.; He, Y.; Zhang, Q.; Wang, X. Comparative transcriptome analysis of ovary and testis reveals potential sex-related genes and pathways in spotted knifejaw Oplegnathus punctatus. Gene 2017, 637, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ma, P.; Li, Q.; Xiao, Q.; Sun, H.; Olasege, B.S.; Wang, Q.; Pan, Y. Exploring the Genetic Correlation between Growth and Immunity Based on Summary Statistics of Genome-Wide Association Studies. Front. Genet. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Auernhammer, C.J.; Chesnokova, V.; Bousquet, C.; Melmed, S. Pituitary Corticotroph SOCS-3: Novel Intracellular Regulation of Leukemia-Inhibitory Factor-Mediated Proopiomelanocortin Gene Expression and Adrenocorticotropin Secretion. Mol. Endocrinol. 1998, 12, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Herrera, S.C.; Bach, E.A. JAK/STAT signaling in stem cells and regeneration: From Drosophila to vertebrates. Development 2019, 146, dev167643. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, J.W.; Grebien, F.; Kerenyi, M.A.; Friedbichler, K.; Kovacic, B.; Zankl, B.; Hoelbl, A.; Nivarti, H.; Beug, H.; Sexl, V.; et al. The different functions of Stat5 and chromatin alteration through Stat5 proteins. Front. Biosci. J. Virtual Libr. 2008, 13, 6237–6254. [Google Scholar] [CrossRef]
- Briscoe, J.; Guschin, D.; Müller, M. Signal transduction. Just another signalling pathway. Curr. Biol. CB 1994, 4, 1033–1035. [Google Scholar] [CrossRef]
- SMART Servier Medical Art—3000 Free Medical Images. Available online: https://smart.servier.com/ (accessed on 16 January 2019).
- Bond, M.R.; Hanover, J.A. A little sugar goes a long way: The cell biology of O-GlcNAc. J. Cell Biol. 2015, 208, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Love, D.C.; Hanover, J.A. The Hexosamine Signaling Pathway: Deciphering the “O-GlcNAc Code”. Sci. Signal. 2005, 2005, re13. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, N.; Zachara, N.E.; Hart, G.W.; Marth, J.D. Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Mol. Cell. Biol. 2004, 24, 1680–1690. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.R.; Song, M.; Lee, H.; Jeon, Y.; Choi, E.J.; Jang, H.J.; Moon, H.Y.; Byun, H.Y.; Kim, E.K.; Kim, D.H. O-GlcNAcase is essential for embryonic development and maintenance of genomic stability: Genomic instability by dysregulation O-GlcNAcylation. Aging Cell 2012, 11, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.B.; Singh, J.P.; Li, M.D.; Wu, J.; Yang, X. Cracking the O-GlcNAc code in metabolism. Trends Endocrinol. Metab. 2013, 24, 301–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Hart, G.W. O-GlcNAc profiling: From proteins to proteomes. Clin. Proteom. 2014, 11, 8. [Google Scholar] [CrossRef] [PubMed]
- Levine, Z.G.; Fan, C.; Melicher, M.S.; Orman, M.; Benjamin, T.; Walker, S. O-GlcNAc Transferase Recognizes Protein Substrates Using an Asparagine Ladder in the Tetratricopeptide Repeat (TPR) Superhelix. J. Am. Chem. Soc. 2018, 140, 3510–3513. [Google Scholar] [CrossRef] [PubMed]
- Chiaradonna, F.; Ricciardiello, F.; Palorini, R. The Nutrient-Sensing Hexosamine Biosynthetic Pathway as the Hub of Cancer Metabolic Rewiring. Cells 2018, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Hardivillé, S.; Hart, G.W. Nutrient Regulation of Signaling, Transcription, and Cell Physiology by O-GlcNAcylation. Cell Metab. 2014, 20, 208–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aquino-Gil, M.; Pierce, A.; Perez-Cervera, Y.; Zenteno, E.; Lefebvre, T. OGT: A short overview of an enzyme standing out from usual glycosyltransferases. Biochem. Soc. Trans. 2017, 45, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Nagel, A.K.; Ball, L.E. O-GlcNAc transferase and O-GlcNAcase: Achieving target substrate specificity. Amino Acids 2014, 46, 2305–2316. [Google Scholar] [CrossRef] [PubMed]
- Nagel, A.K.; Ball, L.E. Intracellular protein O-GlcNAc modification integrates nutrient status with transcriptional and metabolic regulation. Adv. Cancer Res. 2015, 126, 137–166. [Google Scholar] [PubMed]
- Nishikawa, I.; Nakajima, Y.; Ito, M.; Fukuchi, S.; Homma, K.; Nishikawa, K. Computational Prediction of O-linked Glycosylation Sites that Preferentially Map on Intrinsically Disordered Regions of Extracellular Proteins. Int. J. Mol. Sci. 2010, 11, 4991–5008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathak, S.; Alonso, J.; Schimpl, M.; Rafie, K.; Blair, D.E.; Borodkin, V.S.; Schüttelkopf, A.W.; Albarbarawi, O.; Van Aalten, D.M. The active site of O-GlcNAc transferase imposes constraints on substrate sequence. Nat. Struct. Mol. Biol. 2015, 22, 744–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapannone, R.; Mariappa, D.; Ferenbach, A.T.; van Aalten, D.M. Nucleocytoplasmic human O-GlcNAc transferase is sufficient for O-GlcNAcylation of mitochondrial proteins. Biochem. J. 2016, 473, 1693–1702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, M.; Furukawa, K.; Okajima, T. Extracellular O-linked β-N-acetylglucosamine: Its biology and relationship to human disease. World J. Biol. Chem. 2014, 5, 224–230. [Google Scholar] [PubMed]
- Varshney, S.; Stanley, P. EOGT and O-GlcNAc on secreted and membrane proteins. Biochem. Soc. Trans. 2017, 45, 401–408. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Roth, C.; Turkenburg, J.P.; Davies, G.J. Three-dimensional structure of a Streptomyces sviceus GNAT acetyltransferase with similarity to the C-terminal domain of the human GH84 O-GlcNAcase. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Kia-Ki, H.; Martinage, A. Post-translational chemical modification(S) of proteins. Int. J. Biochem. 1992, 24, 19–28. [Google Scholar] [CrossRef]
- Han, K.-K.; Martinage, A. Post-translational chemical modifications of proteins—III. Current developments in analytical procedures of identification and quantitation of post-translational chemically modified amino acid(s) and its derivatives. Int. J. Biochem. 1993, 25, 957–970. [Google Scholar] [CrossRef]
- Krämer, O.H.; Moriggl, R. Acetylation and sumoylation control STAT5 activation antagonistically. JAK-STAT 2012, 1, 203–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Qian, C.; Cao, X. Post-Translational Modification Control of Innate Immunity. Immunity 2016, 45, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Enchev, R.I.; Schulman, B.A.; Peter, M. Protein neddylation: Beyond cullin–RING ligases. Nat. Rev. Mol. Cell Biol. 2015, 16, 30–44. [Google Scholar] [CrossRef] [PubMed]
- Alleyn, M.; Breitzig, M.; Lockey, R.; Kolliputi, N. The dawn of succinylation: A posttranslational modification. Am. J. Physiol. Cell Physiol. 2018, 314, C228–C232. [Google Scholar] [CrossRef] [PubMed]
- Van Nguyen, T.; Angkasekwinai, P.; Dou, H.; Lin, F.M.; Lu, L.S.; Cheng, J.; Chin, Y.E.; Dong, C.; Yeh, E.T. SUMO-specific protease 1 is critical for early lymphoid development through regulation of STAT5 activation. Mol. Cell 2012, 45, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Rani, A.; Murphy, J.J. STAT5 in Cancer and Immunity. J. Interferon Cytokine Res. 2016, 36, 226–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlova, A.; Wingelhofer, B.; Neubauer, H.A.; Maurer, B.; Berger-Becvar, A.; Keserű, G.M.; Gunning, P.T.; Valent, P.; Moriggl, R. Emerging therapeutic targets in myeloproliferative neoplasms and peripheral T-cell leukemia and lymphomas. Expert Opin. Ther. Targets 2018, 22, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.-H.; Chi, J.-T.; Boyce, M. Functional crosstalk among oxidative stress and O-GlcNAc signaling pathways. Glycobiology 2018, 28, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.G.; Park, S.Y.; Ji, S.; Jang, I.; Park, S.; Kim, H.S.; Kim, S.M.; Yook, J.I.; Park, Y.I.; Roth, J.; et al. O-GlcNAc Protein Modification in Cancer Cells Increases in Response to Glucose Deprivation through Glycogen Degradation. J. Biol. Chem. 2009, 284, 34777–34784. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.P.; Geisler, T.S.; Chambers, J.H.; McClain, D.A. Up-regulation of O -GlcNAc Transferase with Glucose Deprivation in HepG2 Cells Is Mediated by Decreased Hexosamine Pathway Flux. J. Biol. Chem. 2009, 284, 3425–3432. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, Y.; Pan, X.; Shi, M.; Wu, Q.; Huang, T.; Jiang, H.; Li, W.; Zhang, J. O-GlcNAc elevation through activation of the hexosamine biosynthetic pathway enhances cancer cell chemoresistance. Cell Death Dis. 2018, 9, 485. [Google Scholar] [CrossRef] [PubMed]
- Gewinner, C.; Hart, G.; Zachara, N.; Cole, R.; Beisenherz-Huss, C.; Groner, B. The Coactivator of Transcription CREB-binding Protein Interacts Preferentially with the Glycosylated Form of Stat5. J. Biol. Chem. 2004, 279, 3563–3572. [Google Scholar] [CrossRef] [PubMed]
- Pfitzner, E.; Jahne, R.; Wissler, M.; Stoecklin, E.; Groner, B. p300/CREB-Binding Protein Enhances the Prolactin-Mediated Transcriptional Induction through Direct Interaction with the Transactivation Domain of Stat5, but Does Not Participate in the Stat5-Mediated Suppression of the Glucocorticoid Response. Mol. Endocrinol. 1998, 12, 1582–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollmann, S.; Grundschober, E.; Maurer, B.; Warsch, W.; Grausenburger, R.; Edlinger, L.; Huuhtanen, J.; Lagger, S.; Hennighausen, L.; Valent, P.; et al. Twins with different personalities: STAT5B—But not STAT5A—Has a key role in BCR/ABL-induced leukemia. Leukemia 2019. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 Supports Ras-Dependent Oncogenic Transformation. Science 2009, 324, 1713–1716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laarse SAM van der Leney, A.C.; Heck, A.J.R. Crosstalk between phosphorylation and O-GlcNAcylation: Friend or foe. FEBS J. 2018, 285, 3152–3167. [Google Scholar] [CrossRef] [PubMed]
- Kaasik, K.; Kivimäe, S.; Allen, J.J.; Chalkley, R.J.; Huang, Y.; Baer, K.; Kissel, H.; Burlingame, A.L.; Shokat, K.M.; Ptáček, L.J.; et al. Glucose Sensor O-GlcNAcylation Coordinates with Phosphorylation to Regulate Circadian Clock. Cell Metab. 2013, 17, 291–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Li, X.; Nai, S.; Geng, Q.; Liao, J.; Xu, X.; Li, J. Checkpoint kinase 1–induced phosphorylation of O-linked β-N-acetylglucosamine transferase regulates the intermediate filament network during cytokinesis. J. Biol. Chem. 2017, 292, 19548–19555. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Salgado, O.C.; Singh, S.; Hippen, K.L.; Maynard, J.C.; Burlingame, A.L.; Ball, L.E.; Blazar, B.R.; Farrar, M.A.; Hogquist, K.A.; et al. The lineage stability and suppressive program of regulatory T cells require protein O-GlcNAcylation. Nat. Commun. 2019, 10, 354. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rauth, M.; Freund, P.; Orlova, A.; Grünert, S.; Tasic, N.; Han, X.; Ruan, H.-B.; Neubauer, H.A.; Moriggl, R. Cell Metabolism Control Through O-GlcNAcylation of STAT5: A Full or Empty Fuel Tank Makes a Big Difference for Cancer Cell Growth and Survival. Int. J. Mol. Sci. 2019, 20, 1028. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051028
Rauth M, Freund P, Orlova A, Grünert S, Tasic N, Han X, Ruan H-B, Neubauer HA, Moriggl R. Cell Metabolism Control Through O-GlcNAcylation of STAT5: A Full or Empty Fuel Tank Makes a Big Difference for Cancer Cell Growth and Survival. International Journal of Molecular Sciences. 2019; 20(5):1028. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051028
Chicago/Turabian StyleRauth, Manuel, Patricia Freund, Anna Orlova, Stefan Grünert, Nikola Tasic, Xiaonan Han, Hai-Bin Ruan, Heidi A. Neubauer, and Richard Moriggl. 2019. "Cell Metabolism Control Through O-GlcNAcylation of STAT5: A Full or Empty Fuel Tank Makes a Big Difference for Cancer Cell Growth and Survival" International Journal of Molecular Sciences 20, no. 5: 1028. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051028