Chloride Channels in Astrocytes: Structure, Roles in Brain Homeostasis and Implications in Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
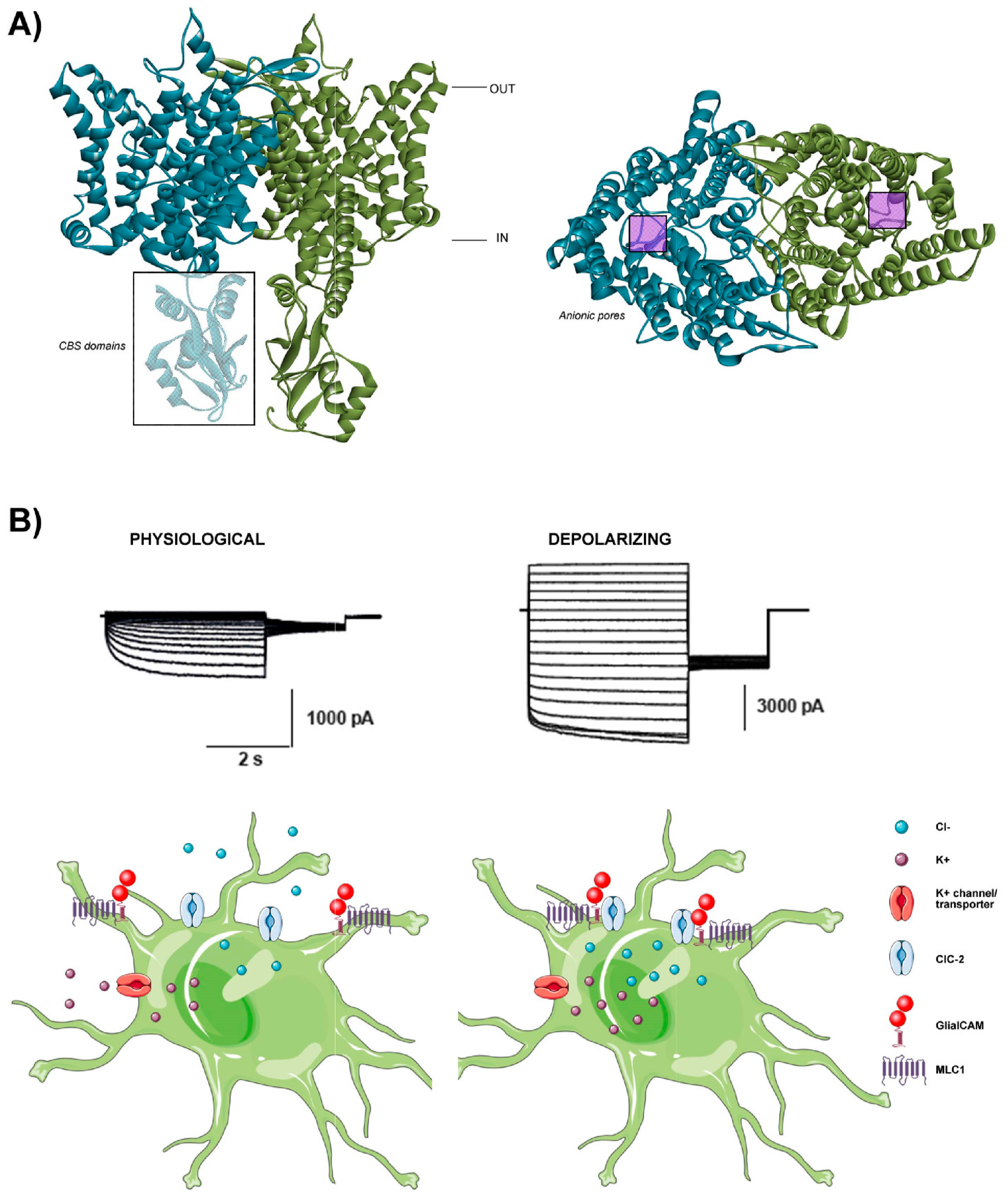
2. ClC-2
2.1. The Structure and Function of the ClC-2 Channel
2.2. The Role of ClC-2 in Glial Physiology and Implications in Disease
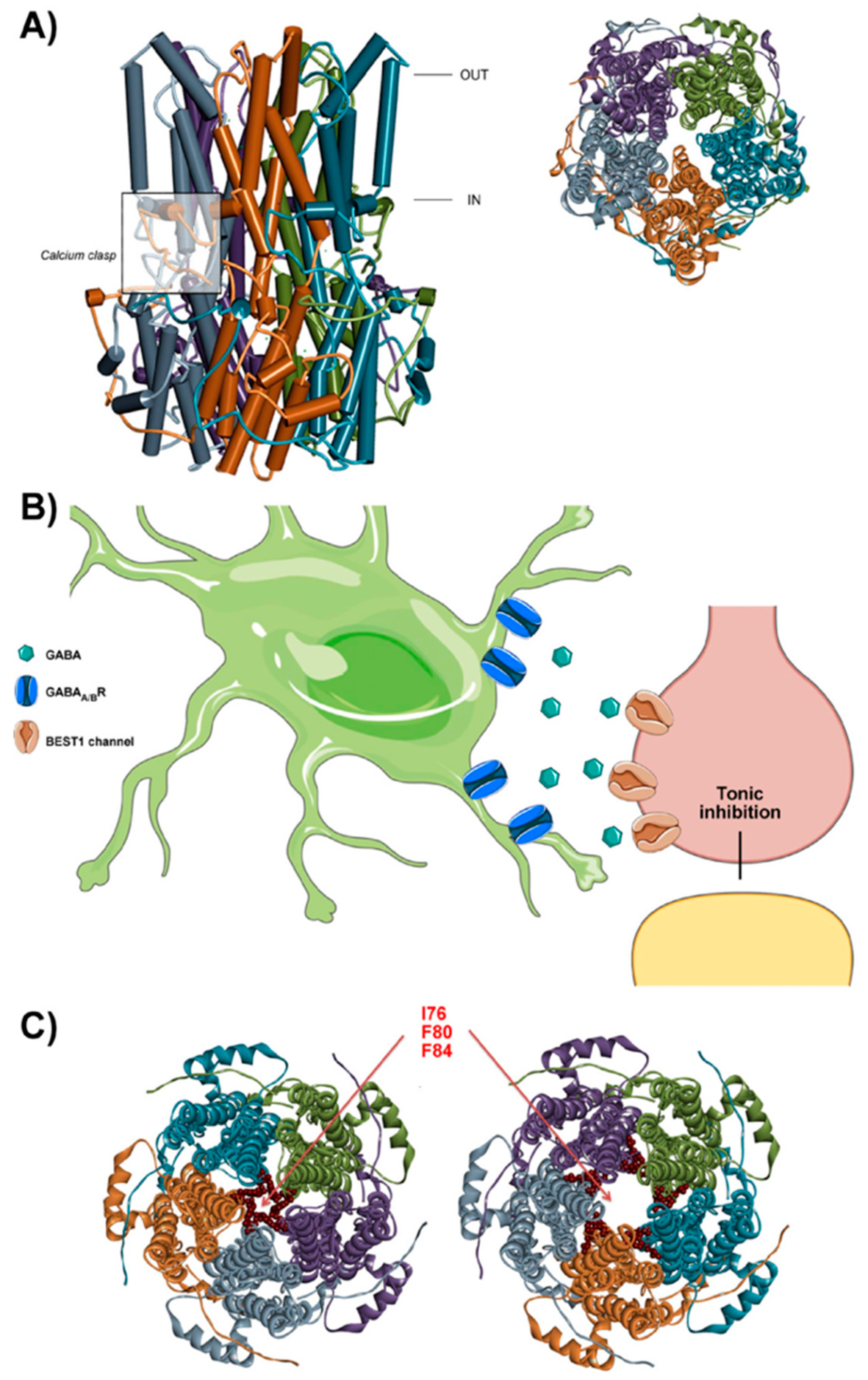
3. Bestrophin 1
3.1. Bestrophin 1 Structure and Function
3.2. Physiological Roles of Best1 in the Brain and Implications in Disease
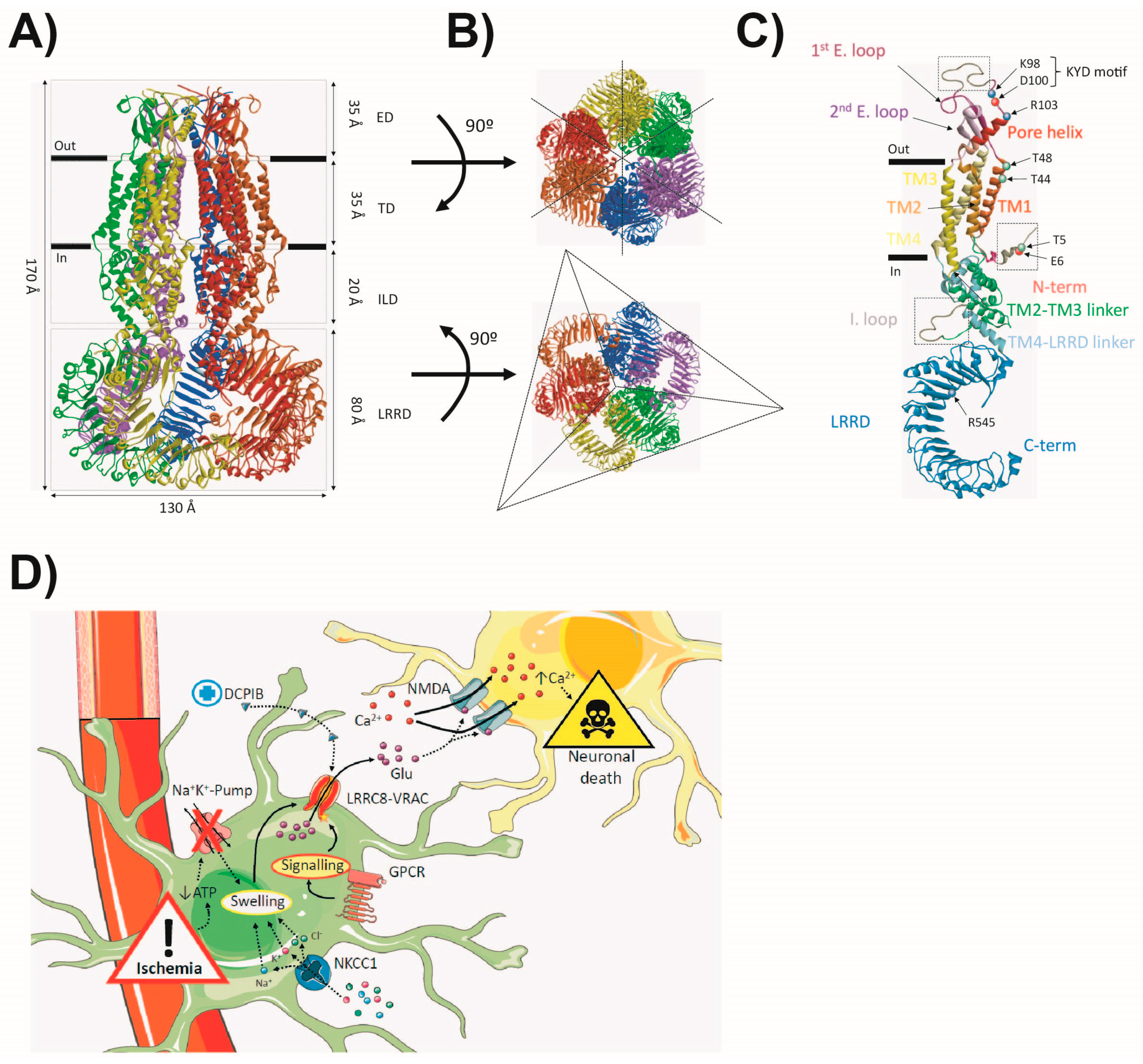
4. The Volume-Regulated Anion-Channel (VRAC)
4.1. General Features
4.2. Structure and Function
4.3. VRAC in Astrocytes
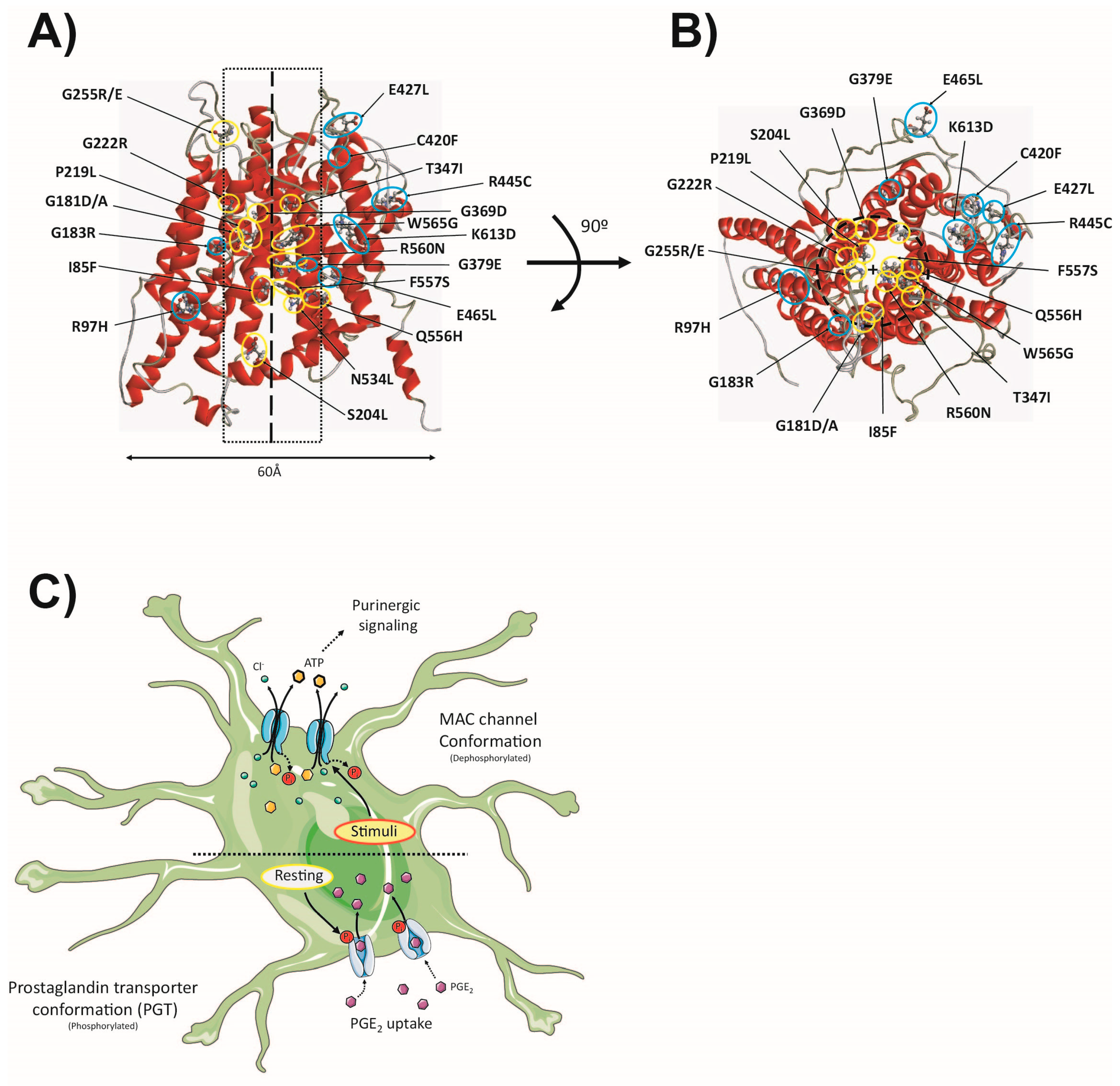
5. Maxi Cl− Channels (MAC)
5.1. General Features
5.2. Structure–Function
5.3. The MAC in Astrocytes
6. Summary and Outlook
Acknowledgments
Conflicts of Interest
References
- Jentsch, T.J.; Stein, V.; Weinreich, F.; Zdebik, A.A. Molecular structure and physiological function of chloride channels. Physiol. Rev. 2002, 82, 503–568. [Google Scholar] [CrossRef] [PubMed]
- Nilius, B.; Droogmans, G. Amazing chloride channels: an overview. Acta Physiol. Scand. 2003, 177, 119–147. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.S.; Mongin, A.A. The signaling role for chloride in the bidirectional communication between neurons and astrocytes. Neurosci. Lett. 2018, 689, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Blaesse, P.; Airaksinen, M.S.; Rivera, C.; Kaila, K. Cation-Chloride Cotransporters and Neuronal Function. Neuron 2009, 61, 820–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nedergaard, M.; Ransom, B.; Goldman, S.A. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003, 26, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Neuron-astrocyte interactions: partnership for normal function and disease in the central nervous system. Mayo Clin. Proc. 2005, 80, 1326–1338. [Google Scholar] [CrossRef] [PubMed]
- Lundgaard, I.; Osório, M.J.; Kress, B.T.; Sanggaard, S.; Nedergaard, M. White matter astrocytes in health and disease. Neuroscience 2014, 276, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howarth, C. The contribution of astrocytes to the regulation of cerebral blood flow. Front. Neurosci. 2014, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C.; Nedergaard, M. Glial regulation of the cerebral microvasculature. Nat. Neurosci. 2007, 10, 1369–1376. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Nedergaard, M. Is There a Cerebral Lymphatic System? Stroke 2013, 44, S93–S95. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Kang, H.; Xu, Q.; Chen, M.J.; Liao, Y.; Thiyagarajan, M.; O’Donnell, J.; Christensen, D.J.; Nicholson, C.; Iliff, J.J.; et al. Sleep Drives Metabolite Clearance from the Adult Brain. Science 2013, 342, 373–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, D.; Sargsyan, S.; Monk, P.N.; Shaw, P.J. Astrocyte function and role in motor neuron disease: A future therapeutic target? Glia 2009, 57, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Oberheim, N.A.; Wang, X.; Goldman, S.; Nedergaard, M. Astrocytic complexity distinguishes the human brain. Trends Neurosci. 2006, 29, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Poroca, D.R.; Pelis, R.M.; Chappe, V.M. ClC channels and transporters: Structure, physiological functions, and implications in human chloride channelopathies. Front. Pharmacol. 2017, 8, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Jentsch, T.J.; Neagoe, I.; Scheel, O. CLC chloride channels and transporters. Curr. Opin. Neurobiol. 2005, 15, 319–325. [Google Scholar] [CrossRef] [PubMed]
- Thiemann, A.; Grunder, S.; Pusch, M.; Jentsch, T.J. A chloride channel widely expressed in epithelial and non-epithelial cells. Nature 1992, 356, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Dutzler, R.; Campbell, E.B.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature 2002, 415, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P. CBS domains in CIC chloride channels implicated in myotonia and nephrolithiasis (kidney stones). J. Mol. Med. (Berl). 1997, 75, 160–163. [Google Scholar] [PubMed]
- Estévez, R.; Pusch, M.; Ferrer-Costa, C.; Orozco, M.; Jentsch, T.J. Functional and structural conservation of CBS domains from CLC chloride channels. J. Physiol. 2004, 557, 363–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.; Campbell, E.B.; MacKinnon, R. Structure of a CLC chloride ion channel by cryo-electron microscopy. Nature 2017, 541, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; MacKinnon, R. Structure of the CLC-1 chloride channel from Homo sapiens. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Stölting, G.; Fischer, M.; Fahlke, C. CLC channel function and dysfunction in health and disease. Front. Physiol. 2014, 5, 378. [Google Scholar] [PubMed]
- Jordt, S.E.; Jentsch, T.J. Molecular dissection of gating in the ClC-2 chloride channel. EMBO J. 1997, 16, 1582–1592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemeyer, M.I.; Yusef, Y.R.; Cornejo, I.; Flores, C.A.; Sepúlveda, F.V.; Cid, L.P. Functional evaluation of human ClC-2 chloride channel mutations associated with idiopathic generalized epilepsies. Physiol. Genomics 2004, 19, 74–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jentsch, T.J.; Friedrich, T.; Schriever, A.; Yamada, H. The CLC chloride channel family. Pflugers Arch. Eur. J. Physiol. Arch. Eur. J. Physiol. 1999, 437, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, M.I.; Cid, L.P.; Yusef, Y.R.; Briones, R.; Sepulveda, F.V.; Sepúlveda, F.V. Voltage-dependent and -independent titration of specific residues accounts for complex gating of a ClC chloride channel by extracellular protons. J. Physiol. 2009, 587, 1387–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Olivares, J.; Alekov, A.; Boroumand, M.R.; Begemann, B.; Hidalgo, P.; Fahlke, C. Gating of human ClC-2 chloride channels and regulation by carboxy-terminal domains. J. Physiol. 2008, 586, 5325–5336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Santiago, J.A.; Nehrke, K.; Arreola, J. Quantitative analysis of the voltage-dependent gating of mouse parotid ClC-2 chloride channel. J. Gen. Physiol. 2005, 126, 591–603. [Google Scholar] [CrossRef] [PubMed]
- Gründer, S.; Thiemann, A.; Pusch, M.; Jentsch, T.J. Regions involved in the opening of CIC-2 chloride channel by voltage and cell volume. Nature 1992, 360, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Saviane, C.; Conti, F.; Pusch, M. The muscle chloride channel ClC-1 has a double-barreled appearance that is differentially affected in dominant and recessive myotonia. J. Gen. Physiol. 1999, 113, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Accardi, A.; Pusch, M. Fast and slow gating relaxations in the muscle chloride channel CLC-1. J. Gen. Physiol. 2000, 116, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Estévez, R.; Jentsch, T.J. CLC chloride channels: correlating structure with function. Curr. Opin. Struct. Biol. 2002, 12, 531–539. [Google Scholar] [CrossRef]
- Smith, R.L.; Clayton, G.H.; Wilcox, C.L.; Escudero, K.W.; Staley, K.J. Differential expression of an inwardly rectifying chloride conductance in rat brain neurons: a potential mechanism for cell-specific modulation of postsynaptic inhibition. J. Neurosci. 1995, 15, 4057–4067. [Google Scholar] [CrossRef] [PubMed]
- Sík, A.; Smith, R.L.; Freund, T.F. Distribution of chloride channel-2-immunoreactive neuronal and astrocytic processes in the hippocampus. Neuroscience 2000, 101, 51–65. [Google Scholar] [CrossRef]
- Jeworutzki, E.; López-Hernández, T.; Capdevila-Nortes, X.; Sirisi, S.; Bengtsson, L.; Montolio, M.; Zifarelli, G.; Arnedo, T.; Müller, C.S.; Schulte, U.; et al. GlialCAM, a Protein Defective in a Leukodystrophy, Serves as a ClC-2 Cl - Channel Auxiliary Subunit. Neuron 2012, 73, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Blanz, J.; Schweizer, M.; Auberson, M.; Maier, H.; Muenscher, A.; Hübner, C.A.; Jentsch, T.J. Leukoencephalopathy upon disruption of the chloride channel ClC-2. J. Neurosci. 2007, 27, 6581–6589. [Google Scholar] [CrossRef] [PubMed]
- Zúñiga, L.; Niemeyer, M.I.; Varela, D.; Catalán, M.; Cid, L.P.; Sepúlveda, F.V. The voltage-dependent ClC-2 chloride channel has a dual gating mechanism. J. Physiol. 2004, 555, 671–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratté, S.; Prescott, S.A. ClC-2 channels regulate neuronal excitability, not intracellular chloride levels. J. Neurosci. 2011, 31, 15838–15843. [Google Scholar] [CrossRef] [PubMed]
- Rinke, I.; Artmann, J.; Stein, V. ClC-2 voltage-gated channels constitute part of the background conductance and assist chloride extrusion. J. Neurosci. 2010, 30, 4776–4786. [Google Scholar] [CrossRef] [PubMed]
- Kleefuß-Lie, A.; Friedl, W.; Cichon, S.; Haug, K.; Warnstedt, M.; Alekov, A.; Sander, T.; Ramirez, A.; Poser, B.; Maljevic, S.; et al. CLCN2 variants in idiopathic generalized epilepsy. Nat. Genet. 2009, 41, 954–955. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, M.I.; Cid, L.P.; Sepúlveda, F.V.; Blanz, J.; Auberson, M.; Jentsch, T.J. No evidence for a role of CLCN2 variants in idiopathic generalized epilepsy. Nat. Genet. 2010, 42, 3. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, D.; Bertelli, M.; Gallo, S.; Cecchin, S.; Albiero, E.; Garofalo, P.G.; Gambardella, A.; St Hilaire, J.-M.; Kwiecinski, H.; Andermann, E.; et al. Mutations and polymorphisms of the CLCN2 gene in idiopathic epilepsy. Neurology 2004, 63, 1500–1502. [Google Scholar] [CrossRef] [PubMed]
- Cortez, M.A.; Li, C.; Whitehead, S.N.; Dhani, S.U.; D’Antonio, C.; Huan, L.J.; Bennett, S.A.; Snead, O.C., 3rd; Bear, C.E. Disruption of ClC-2 expression is associated with progressive neurodegeneration in aging mice. Neuroscience 2010, 167, 154–162. [Google Scholar] [CrossRef] [PubMed]
- van der Knaap, M.S.S.; Boor, I.; Estevez, R.; Estévez, R. Megalencephalic leukoencephalopathy with subcortical cysts: chronic white matter oedema due to a defect in brain ion and water homoeostasis. Lancet Neurol. 2012, 11, 973–985. [Google Scholar] [CrossRef]
- Scheper, G.C.; van Berkel, C.G.M.; Leisle, L.; de Groot, K.E.; Errami, A.; Jentsch, T.J.; Van der Knaap, M.S. Analysis of CLCN2 as Candidate Gene for Megalencephalic Leukoencephalopathy with Subcortical Cysts. Genet. Test. Mol. Biomarkers 2010, 14, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Leegwater, P.A.; Yuan, B.Q.; van der Steen, J.; Mulders, J.; Konst, A.A.; Boor, P.K.; Mejaski-Bosnjak, V.; van der Maarel, S.M.; Frants, R.R.; Oudejans, C.B.; et al. Mutations of MLC1 (KIAA0027), encoding a putative membrane protein, cause megalencephalic leukoencephalopathy with subcortical cysts. Am. J. Hum. Genet. 2001, 68, 831–838. [Google Scholar] [CrossRef] [PubMed]
- López-Hernández, T.; Ridder, M.C.; Montolio, M.; Capdevila-Nortes, X.; Polder, E.; Sirisi, S.S.; Duarri, A.; Schulte, U.; Fakler, B.; Nunes, V.; et al. Mutant GlialCAM causes megalencephalic leukoencephalopathy with subcortical cysts, benign familial macrocephaly, and macrocephaly with retardation and autism. Am. J. Hum. Genet. 2011, 88, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Jeworutzki, E.; Lagostena, L.; Elorza-Vidal, X.; López-Hernández, T.; Estévez, R.; Pusch, M. GlialCAM, a CLC-2 Cl(-) channel subunit, activates the slow gate of CLC chloride channels. Biophys. J. 2014, 107, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Bugiani, M.; Dubey, M.; Breur, M.; Postma, N.L.; Dekker, M.P.; ter Braak, T.; Boschert, U.; Abbink, T.E.M.; Mansvelder, H.D.; Min, R.; et al. Megalencephalic leukoencephalopathy with cysts: the Glialcam -null mouse model. Ann. Clin. Transl. Neurol. 2017, 4, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Hoegg-Beiler, M.B.; Sirisi, S.; Orozco, I.J.; Ferrer, I.; Hohensee, S.; Auberson, M.; Gödde, K.; Vilches, C.; De Heredia, M.L.; Nunes, V.; et al. Disrupting MLC1 and GlialCAM and ClC-2 interactions in leukodystrophy entails glial chloride channel dysfunction. Nat. Commun. 2014, 5, 3475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirisi, S.; Elorza-Vidal, X.; Arnedo, T.; Armand-Ugón, M.; Callejo, G.; Capdevila-Nortes, X.; López-Hernández, T.; Schulte, U.; Barrallo-Gimeno, A.; Nunes, V.; et al. Depolarization causes the formation of a ternary complex between GlialCAM, MLC1 and ClC-2 in astrocytes: implications in megalencephalic leukoencephalopathy. Hum. Mol. Genet. 2017, 26, 2436–2450. [Google Scholar] [CrossRef] [PubMed]
- Estévez, R.; Elorza-Vidal, X.; Gaitán-Peñas, H.; Pérez-Rius, C.; Armand-Ugón, M.; Alonso-Gardón, M.; Xicoy-Espaulella, E.; Sirisi, S.; Arnedo, T.; Capdevila-Nortes, X.; et al. Megalencephalic leukoencephalopathy with subcortical cysts: A personal biochemical retrospective. Eur. J. Med. Genet. 2018, 61, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Zeydan, B.; Uygunoglu, U.; Altintas, A.; Saip, S.; Siva, A.; Abbink, T.E.M.; van der Knaap, M.S.; Yalcinkaya, C. Identification of 3 Novel Patients with CLCN2-Related Leukoencephalopathy due to CLCN2 Mutations. Eur. Neurol. 2017, 78, 125–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depienne, C.; Bugiani, M.; Dupuits, C.; Galanaud, D.; Touitou, V.; Postma, N.; van Berkel, C.; Polder, E.; Tollard, E.; Darios, F.; et al. Brain white matter oedema due to ClC-2 chloride channel deficiency: An observational analytical study. Lancet. Neurol. 2013, 12, 659–668. [Google Scholar] [CrossRef]
- Giorgio, E.; Vaula, G.; Benna, P.; Lo Buono, N.; Eandi, C.M.; Dino, D.; Mancini, C.; Cavalieri, S.; Di Gregorio, E.; Pozzi, E.; et al. A novel homozygous change of CLCN2 (p.His590Pro) is associated with a subclinical form of leukoencephalopathy with ataxia (LKPAT). J. Neurol. Neurosurg. Psychiatry 2017, 88, 894–896. [Google Scholar] [CrossRef] [PubMed]
- Di Bella, D.; Pareyson, D.; Savoiardo, M.; Farina, L.; Ciano, C.; Caldarazzo, S.; Sagnelli, A.; Bonato, S.; Nava, S.; Bresolin, N.; et al. Subclinical leukodystrophy and infertility in a man with a novel homozygous CLCN2 mutation. Neurology 2014, 83, 1217–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanagasi, H.A.; Bilgiç, B.; Abbink, T.E.M.; Hanagasi, F.; Tüfekçioğlu, Z.; Gürvit, H.; Başak, N.; van der Knaap, M.S.; Emre, M. Secondary paroxysmal kinesigenic dyskinesia associated with CLCN2 gene mutation. Parkinsonism Relat. Disord. 2015, 21, 544–546. [Google Scholar] [CrossRef] [PubMed]
- van der Knaap, M.S.; Depienne, C.; Sedel, F.; Abbink, T.E.M. CLCN2-Related Leukoencephalopathy. In GeneReviews(R); University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Gaitán-Peñas, H.; Apaja, P.M.; Arnedo, T.; Castellanos, A.; Elorza-Vidal, X.; Soto, D.; Gasull, X.; Lukacs, G.L.; Estévez, R. Leukoencephalopathy-causing CLCN2 mutations are associated with impaired Cl− channel function and trafficking. J. Physiol. 2017, 595, 6993–7008. [Google Scholar] [CrossRef] [PubMed]
- Benfenati, V.; Ferroni, S. Water transport between CNS compartments: functional and molecular interactions between aquaporins and ion channels. Neuroscience 2010, 168, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, P.; Newman, E.A. Potassium buffering in the central nervous system. Neuroscience 2004, 129, 1045–1056. [Google Scholar] [CrossRef] [PubMed]
- Rash, J.E. Molecular disruptions of the panglial syncytium block potassium siphoning and axonal saltatory conduction: pertinence to neuromyelitis optica and other demyelinating diseases of the central nervous system. Neuroscience 2010, 168, 982–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butt, A.; Kalsi, A. Inwardly rectifying potassium channels (KIR) in central nervous system glia: A special role for KIR 4.1 in glial functions. J. Cell. Mol. Med. 2006, 10, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Olsen, M.L.; Higashimori, H.; Campbell, S.L.; Hablitz, J.J.; Sontheimer, H. Functional expression of Kir4.1 channels in spinal cord astrocytes. Glia 2006, 53, 516–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ambrosio, R.; Gordon, D.S.; Winn, H.R. Differential role of KIR channel and Na(+)/K(+)-pump in the regulation of extracellular K(+) in rat hippocampus. J. Neurophysiol. 2002, 87, 87–102. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Xu, Q.; Wang, W.; Takano, T.; Nedergaard, M. Bergmann glia modulate cerebellar Purkinje cell bistability via Ca2+-dependent K+ uptake. Proc. Natl. Acad. Sci. USA 2012, 109, 7911–7916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, B.R.; Assentoft, M.; Cotrina, M.L.; Hua, S.Z.; Nedergaard, M.; Kaila, K.; Voipio, J.; Macaulay, N. Contributions of the Na+/K+-ATPase, NKCC1, and Kir4.1 to hippocampal K+ clearance and volume responses. Glia 2014, 62, 608–622. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.I.; Rash, J.E. Astrocyte and oligodendrocyte connexins of the glial syncytium in relation to astrocyte anatomical domains and spatial buffering. Cell Commun. Adhes. 2003, 10, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Orellana, J.A.; Stehberg, J. Hemichannels: new roles in astroglial function. Front. Physiol. 2014, 5, 193. [Google Scholar] [CrossRef] [PubMed]
- Nagelhus, E.A.; Mathiisen, T.M.; Ottersen, O.P. Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience 2004, 129, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Haj-Yasein, N.N.; Jensen, V.; Østby, I.; Omholt, S.W.; Voipio, J.; Kaila, K.; Ottersen, O.P.; Hvalby, Ø.; Nagelhus, E.A. Aquaporin-4 regulates extracellular space volume dynamics during high-frequency synaptic stimulation: A gene deletion study in mouse hippocampus. Glia 2012, 60, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Dietzel, I.; Heinemann, U.; Lux, H.D. Relations between slow extracellular potential changes, glial potassium buffering, and electrolyte and cellular volume changes during neuronal hyperactivity in cat brain. Glia 1989, 2, 25–44. [Google Scholar] [CrossRef] [PubMed]
- Krämer, F.; Stöhr, H.; Weber, B.H.F. Cloning and characterization of the murine Vmd2 RFP-TM gene family. Cytogenet. Genome Res. 2004, 105, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Dickson, V.K.; Pedi, L.; Long, S.B. Structure and insights into the function of a Ca2+-activated Cl-channel. Nature 2014, 516, 13–218. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Liu, Q.; Kloss, B.; Bruni, R.; Kalathur, R.C.; Guo, Y.; Kloppmann, E.; Rost, B.; Colecraft, H.M.; Hendrickson, W.A. Structure and selectivity in bestrophin ion channels. Science 2014, 346, 355–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.-J.; Lee, C.J. Distribution and Function of the Bestrophin-1 (Best1) Channel in the Brain. Exp. Neurobiol. 2017, 26, 113–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milenkovic, V.M.; Röhrl, E.; Weber, B.H.F.; Strauss, O. Disease-associated missense mutations in bestrophin-1 affect cellular trafficking and anion conductance. J. Cell Sci. 2011, 124, 2988–2996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaisey, G.; Miller, A.N.; Long, S.B. Distinct regions that control ion selectivity and calcium-dependent activation in the bestrophin ion channel. Proc. Natl. Acad. Sci. USA 2016, 113, E7399–E7408. [Google Scholar] [CrossRef] [PubMed]
- Hartzell, H.C.; Qu, Z.; Yu, K.; Xiao, Q.; Chien, L.-T. Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies. Physiol. Rev. 2008, 88, 639–672. [Google Scholar] [CrossRef] [PubMed]
- Johnson, A.A.; Guziewicz, K.E.; Lee, C.J.; Kalathur, R.C.; Pulido, J.S.; Marmorstein, L.Y.; Marmorstein, A.D. Bestrophin 1 and retinal disease. Prog. Retin. Eye Res. 2017, 58, 45–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Oh, S.-J.; Han, K.-S.; Woo, D.H.; Park, H.; Mannaioni, G.; Traynelis, S.F.; Lee, C.J. Bestrophin-1 Encodes for the Ca2+-Activated Anion Channel in Hippocampal Astrocytes. J. Neurosci. 2009, 29, 13063–13073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Yoon, B.-E.; Berglund, K.; Oh, S.-J.; Park, H.; Shin, H.-S.; Augustine, G.J.; Lee, C.J. Channel-Mediated Tonic GABA Release from Glia. Science 2010, 330, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Han, K.S.; Oh, S.J.; Jo, S.; Woo, J.; Yoon, B.E.; Lee, C.J. High glutamate permeability and distal localization of Best1 channel in CA1 hippocampal astrocyte. Mol. Brain 2013, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Min, J.O.; Kang, D.-S.; Kim, Y.S.; Jung, G.H.; Park, H.J.; Kim, S.; An, H.; Kwon, J.; Kim, J.; et al. Control of motor coordination by astrocytic tonic GABA release through modulation of excitation/inhibition balance in cerebellum. Proc. Natl. Acad. Sci. USA 2018, 115, 5004–5009. [Google Scholar] [CrossRef] [PubMed]
- Woo, D.H.; Han, K.-S.; Shim, J.W.; Yoon, B.-E.; Kim, E.; Bae, J.Y.; Oh, S.-J.; Hwang, E.M.; Marmorstein, A.D.; Bae, Y.C.; et al. TREK-1 and Best1 Channels Mediate Fast and Slow Glutamate Release in Astrocytes upon GPCR Activation. Cell 2012, 151, 25–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, S.; Yarishkin, O.; Hwang, Y.J.; Chun, Y.E.; Park, M.; Woo, D.H.; Bae, J.Y.; Kim, T.; Lee, J.; Chun, H.; et al. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat. Med. 2014, 20, 886–896. [Google Scholar] [CrossRef] [PubMed]
- Olsen, M.L.; Khakh, B.S.; Skatchkov, S.N.; Zhou, M.; Lee, C.J.; Rouach, N. New Insights on Astrocyte Ion Channels: Critical for Homeostasis and Neuron-Glia Signaling. J. Neurosci. 2015, 35, 13827–13835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, S.J.; Woo, J.; Lee, Y.S.; Cho, M.; Kim, E.; Cho, N.C.; Park, J.Y.; Pae, A.N.; Justin Lee, C.; Hwang, E.M. Direct interaction with 14-3-3$γ$ promotes surface expression of Best1 channel in astrocyte. Mol. Brain 2017, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mackintosh, C. Dynamic interactions between 14-3-3 proteins and phosphoproteins regulate diverse cellular processes. Biochem. J. 2004, 381, 329–342. [Google Scholar] [CrossRef] [PubMed]
- Berg, D.; Holzmann, C.; Riess, O. 14-3-3 proteins in the nervous system. Nat. Rev. Neurosci. 2003, 4, 752–762. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, K.E.; Leblanc, N.; Hatton, W.J.; Britton, F.C. Functional properties of murine bestrophin 1 channel. Biochem. Biophys. Res. Commun. 2009, 384, 476–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.N.; Vaisey, G.; Long, S.B. Molecular mechanisms of gating in the calcium-activated chloride channel bestrophin. Elife 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, E.K.; Lambert, I.H.; Pedersen, S.F. Physiology of Cell Volume Regulation in Vertebrates. Physiol. Rev. 2009, 89, 193–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jentsch, T.J. VRACs and other ion channels and transporters in the regulation of cell volume and beyond. Nat. Rev. Mol. Cell Biol. 2016, 17, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Hisadome, K.; Koyama, T.; Kimura, C.; Droogmans, G.; Ito, Y.; Oike, M. Volume-regulated anion channels serve as an auto/paracrine nucleotide release pathway in aortic endothelial cells. J. Gen. Physiol. 2002, 119, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Burow, P.; Klapperstück, M.; Markwardt, F. Activation of ATP secretion via volume-regulated anion channels by sphingosine-1-phosphate in RAW macrophages. Pflugers Arch. Eur. J. Physiol. 2015, 467, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Voss, F.K.; Ullrich, F.; Münch, J.; Lazarow, K.; Lutter, D.; Mah, N.; Andrade-Navarro, M.A.; von Kries, J.P.; Stauber, T.; Jentsch, T.J.; et al. Identification of LRRC8 Heteromers as an Essential Component of the Volume-Regulated Anion Channel VRAC. Science 2014, 344, 634–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Z.; Dubin, A.E.; Mathur, J.; Tu, B.; Reddy, K.; Miraglia, L.J.; Reinhardt, J.; Orth, A.P.; Patapoutian, A. SWELL1, a Plasma Membrane Protein, Is an Essential Component of Volume-Regulated Anion Channel. Cell 2014, 157, 447–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syeda, R.; Qiu, Z.; Dubin, A.E.; Murthy, S.E.; Florendo, M.N.; Mason, D.E.; Mathur, J.; Cahalan, S.M.; Peters, E.C.; Montal, M.; et al. LRRC8 Proteins Form Volume-Regulated Anion Channels that Sense Ionic Strength. Cell 2016, 164, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Gaitán-Peñas, H.; Gradogna, A.; Laparra-Cuervo, L.; Solsona, C.; Fernández-Dueñas, V.; Barrallo-Gimeno, A.; Ciruela, F.; Lakadamyali, M.; Pusch, M.; Estévez, R. Investigation of LRRC8-Mediated Volume-Regulated Anion Currents in Xenopus Oocytes. Biophys. J. 2016, 111, 1429–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutter, D.; Ullrich, F.; Lueck, J.C.; Kempa, S.; Jentsch, T.J. Selective transport of neurotransmitters and –modulators by distinct volume-regulated LRRC8 anion channels. J. Cell Sci. 2017, 130, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Hyzinski-García, M.C.; Rudkouskaya, A.; Mongin, A.A. LRRC8A protein is indispensable for swelling-activated and ATP-induced release of excitatory amino acids in rat astrocytes. J. Physiol. 2014, 592, 4855–4862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schober, A.L.; Wilson, C.S.; Mongin, A.A. Molecular composition and heterogeneity of the LRRC8-containing swelling-activated osmolyte channels in primary rat astrocytes. J. Physiol. 2017, 595, 6939–6951. [Google Scholar] [CrossRef] [PubMed]
- Hazama, A.; Shimizu, T.; Ando-Akatsuka, Y.; Hayashi, S.; Tanaka, S.; Maeno, E.; Okada, Y. Swelling-induced, CFTR-independent ATP release from a human epithelial cell line: lack of correlation with volume-sensitive cl(-) channels. J. Gen. Physiol. 1999, 114, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R. LRRC8 proteins share a common ancestor with pannexins, and may form hexameric channels involved in cell-cell communication. Bioessays 2012, 34, 551–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasuya, G.; Nakane, T.; Yokoyama, T.; Jia, Y.; Inoue, M.; Watanabe, K.; Nakamura, R.; Nishizawa, T.; Kusakizako, T.; Tsutsumi, A.; et al. Cryo-EM structures of the human volume-regulated anion channel LRRC8. Nat. Struct. Mol. Biol. 2018, 25, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Kern, D.M.; Oh, S.; Hite, R.K.; Brohawn, S.G.; Biology, C.; Berkeley, C.; Berkeley, C.; Program, S.B.; Sloan, M.; Cancer, K.; et al. Cryo-EM structures of the DCPIB-inhibited volume-regulated anion channel LRRC8A in lipid nanodiscs. bioRxiv 2018, Jan, 442–459. [Google Scholar] [CrossRef] [PubMed]
- Deneka, D.; Sawicka, M.; Lam, A.K.M.; Paulino, C.; Dutzler, R. Structure of a volume-regulated anion channel of the LRRC8 family. Nature 2018, 558, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Kefauver, J.M.; Saotome, K.; Dubin, A.E.; Pallesen, J.; Cottrell, C.A.; Cahalan, S.M.; Qiu, Z.; Hong, G.; Crowley, C.S.; Whitwam, T.; et al. Structure of the human volume regulated anion channel. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Nakagawa, S.; Suga, M.; Yamashita, E.; Oshima, A.; Fujiyoshi, Y.; Tsukihara, T. Structure of the connexin 26 gap junction channel at 3.5 Å resolution. Nature 2009, 458, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Oshima, A.; Tani, K.; Fujiyoshi, Y. Atomic structure of the innexin-6 gap junction channel determined by cryo-EM. Nat. Commun. 2016, 7, 13681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowens, N.H.; Dohare, P.; Kuo, Y.-H.; Mongin, A.A. DCPIB, the proposed selective blocker of volume-regulated anion channels, inhibits several glutamate transport pathways in glial cells. Mol. Pharmacol. 2013, 83, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.C.; Oberheim, N.; Kettenmann, H.; Ransom, B.R. Pharmacological “cross-inhibition” of connexin hemichannels and swelling activated anion channels. Glia 2009, 57, 258–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, L.; Samuels, S.; Locovei, S.; Macagno, E.R.; Muller, K.J.; Dahl, G. Innexins form two types of channels. FEBS Lett. 2007, 581, 5703–5708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akita, T.; Okada, Y. Characteristics and roles of the volume-sensitive outwardly rectifying (VSOR) anion channel in the central nervous system. Neuroscience 2014, 275, 211–231. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, F.; Reincke, S.M.; Voss, F.K.; Stauber, T.; Jentsch, T.J. Inactivation and Anion Selectivity of Volume-regulated Anion Channels (VRACs) Depend on C-terminal Residues of the First Extracellular Loop. J. Biol. Chem. 2016, 291, 17040–17048. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Polovitskaya, M.M.; Jentsch, T.J. LRRC8 N termini influence pore properties and gating of volume-regulated anion channels (VRACs). J. Biol. Chem. 2018, 293, 13440–13451. [Google Scholar] [CrossRef] [PubMed]
- Mongin, A.A. Volume-regulated anion channel--a frenemy within the brain. Pflugers Arch. 2016, 468, 421–441. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Perez, C.J.; Kim, J.; Zhang, H.; Murphy, C.J.; Hamidi, T.; Jaubert, J.; Platt, C.D.; Chou, J.; Deng, M.; et al. Deficient LRRC8A-dependent volume-regulated anion channel activity is associated with male infertility in mice. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Bellot-Saez, A.; Kékesi, O.; Morley, J.W.; Buskila, Y. Astrocytic modulation of neuronal excitability through K+ spatial buffering. Neurosci. Biobehav. Rev. 2017, 77, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Formaggio, F.; Saracino, E.; Mola, M.G.; Rao, S.B.; Amiry-Moghaddam, M.; Muccini, M.; Zamboni, R.; Nicchia, G.P.; Caprini, M.; Benfenati, V. LRRC8A is essential for swelling-activated chloride current and for regulatory volume decrease in astrocytes. FASEB J. 2019, 33, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Elorza-Vidal, X.; Sirisi, S.; Gaitán-Peñas, H.; Pérez-Rius, C.; Alonso-Gardón, M.; Armand-Ugón, M.; Lanciotti, A.; Brignone, M.S.; Prat, E.; Nunes, V.; et al. GlialCAM/MLC1 modulates LRRC8/VRAC currents in an indirect manner: Implications for megalencephalic leukoencephalopathy. Neurobiol. Dis. 2018, 119, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Kimelberg, H.K.; Feustel, P.J.; Jin, Y.; Paquette, J.; Boulos, A.; Keller, J.; Tranmer, B.I. Acute treatment with tamoxifen reduces ischemic damage following middle cerebral artery occlusion. Neuroreport 2000, 11, 2675–2679. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H.; Feustel, P.J.; Kimelberg, H.K. DCPIB, a specific inhibitor of volume regulated anion channels (VRACs), reduces infarct size in MCAo and the release of glutamate in the ischemic cortical penumbra. Exp. Neurol. 2008, 210, 514–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ridder, M.C.; Boor, I.; Lodder, J.C.; Postma, N.L.; Capdevila-Nortes, X.; Duarri, A.; Brussaard, A.B.; Estévez, R.; Scheper, G.C.; Mansvelder, H.D.; et al. Megalencephalic leucoencephalopathy with cysts: Defect in chloride currents and cell volume regulation. Brain 2011, 134, 3342–3354. [Google Scholar] [CrossRef] [PubMed]
- Capdevila-Nortes, X.; López-Hernández, T.; Apaja, P.M.; de Heredia, M.L.; Sirisi, S.; Callejo, G.; Arnedo, T.; Nunes, V.; Lukacs, G.L.; Gasull, X.; et al. Insights into MLC pathogenesis: GlialCAM is an MLC1 chaperone required for proper activation of volume-regulated anion currents. Hum. Mol. Genet. 2013, 22, 4405–4416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabirov, R.Z.; Merzlyak, P.G.; Islam, M.R.; Okada, T.; Okada, Y. The properties, functions, and pathophysiology of maxi-anion channels. Pflügers Arch. - Eur. J. Physiol. 2016, 468, 405–420. [Google Scholar] [CrossRef] [PubMed]
- Sabirov, R.Z.; Dutta, A.K.; Okada, Y. Volume-Dependent Atp-Conductive Large-Conductance Anion Channel as a Pathway for Swelling-Induced Atp Release. J. Gen. Physiol. 2001, 118, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Sabirov, R.Z.; Merzlyak, P.G.; Okada, T.; Islam, M.R.; Uramoto, H.; Mori, T.; Makino, Y.; Matsuura, H.; Xie, Y.; Okada, Y. The organic anion transporter SLCO2A1 constitutes the core component of the Maxi-Cl channel. EMBO J. 2017, 36, 3309–3324. [Google Scholar] [CrossRef] [PubMed]
- Kanai, N.; Lu, R.; Satriano, J.A.; Bao, Y.; Wolkoff, A.W.; Schuster, V.L. Identification and characterization of a prostaglandin transporter. Science 1995, 268, 866–869. [Google Scholar] [CrossRef]
- Sabirov, R.Z.; Okada, Y. ATP-Conducting Maxi-Anion Channel: A New Player in Stress-Sensory Transduction. Jpn. J. Physiol. 2004, 54, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.S.; Satriano, J.A.; Schuster, V.L. Mapping the Substrate Binding Site of the Prostaglandin Transporter PGT by Cysteine Scanning Mutagenesis. J. Biol. Chem. 1999, 274, 25564–25570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.S.; Bao, Y.; Schuster, V.L. Role of Conserved Transmembrane Cationic Amino Acids in the Prostaglandin Transporter PGT. Biochemistry 2002, 41, 9215–9221. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xia, W.; He, J.; Zhang, Z.; Ke, Y.; Yue, H.; Wang, C.; Zhang, H.; Gu, J.; Hu, W.; et al. Exome Sequencing Identifies SLCO2A1 Mutations as a Cause of Primary Hypertrophic Osteoarthropathy. Am. J. Hum. Genet. 2012, 90, 125–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; He, J.; Fu, W.; Zhang, C.; Zhang, Z. Two novel mutations in the SLCO2A1 gene in a Chinese patient with primary hypertrophic osteoarthropathy. Gene 2014, 534, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Jalonen, T. Single-channel characteristics of the large-conductance anion channel in rat cortical astrocytes in primary culture. Glia 1993, 9, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-T.; Tashmukhamedov, B.A.; Inoue, H.; Okada, Y.; Sabirov, R.Z. Roles of two types of anion channels in glutamate release from mouse astrocytes under ischemic or osmotic stress. Glia 2006, 54, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Sabirov, R.Z.; Okada, Y. Oxygen-glucose deprivation induces ATP release via maxi-anion channels in astrocytes. Purinergic Signal. 2008, 4, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Diggle, C.P.; Parry, D.A.; Logan, C.V.; Laissue, P.; Rivera, C.; Mart, C.; Fonseca, D.J.; Morgan, J.E.; Allanore, Y.; Fontenay, M.; et al. Prostaglandin Transporter Mutations Cause Pachydermoperiostosis with Myelofibrosis. Hum. Mutat. 2012, 33, 1175–1181. [Google Scholar] [CrossRef] [PubMed]
- Castori, M.; Sinibaldi, L.; Mingarelli, R.; Rs, L.; Dl, R. Pachydermoperiostosis: an update. Clin. Genet. 2005, 68, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Locker, J.; Lu, R.; Schuster, V.L. Failure of Postnatal Ductus Arteriosus Closure in Prostaglandin Transporter-Deficient Mice. Circulation 2010, 121, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Kanai, N.; Bao, Y.; Schuster, V.L. Cloning, in vitro expression, and tissue distribution of a human prostaglandin transporter cDNA(hPGT). J. Clin. Invest. 1996, 98, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Abdullaev, I.F.; Rudkouskaya, A.; Schools, G.P.; Kimelberg, H.K.; Mongin, A.A.; Bal-Price, A.; Moneer, Z.; Brown, G.C.; et al. Pharmacological comparison of swelling-activated excitatory amino acid release and Cl− currents in cultured rat astrocytes. J. Physiol. 2006, 572, 677–689. [Google Scholar]
- Wang, Z.; Haydon, P.G.; Yeung, E.S. Direct Observation of Calcium-Independent Intercellular ATP Signaling in Astrocytes. Anal. Chem. 2000, 72, 2001–2007. [Google Scholar] [CrossRef] [PubMed]
- Stout, C.E.; Costantin, J.L.; Naus, C.C.G.; Charles, A.C. Intercellular Calcium Signaling in Astrocytes via ATP Release through Connexin Hemichannels. J. Biol. Chem. 2002, 277, 10482–10488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coco, S.; Calegari, F.; Pravettoni, E.; Pozzi, D.; Taverna, E.; Rosa, P.; Matteoli, M.; Verderio, C. Storage and Release of ATP from Astrocytes in Culture. J. Biol. Chem. 2003, 278, 1354–1362. [Google Scholar] [CrossRef] [PubMed]
- Suadicani, S.O.; Brosnan, C.F.; Scemes, E. P2X7 receptors mediate ATP release and amplification of astrocytic intercellular Ca2+ signaling. J. Neurosci. 2006, 26, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Cotrina, M.L.; Lin, J.H.-C.; Alves-Rodrigues, A.; Liu, S.; Li, J.; Azmi-Ghadimi, H.; Kang, J.; Naus, C.C.G.; Nedergaard, M. Connexins regulate calcium signaling by controlling ATP release. Proc. Natl. Acad. Sci. USA 1998, 95, 15735–15740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, G.R.J.; Baimoukhametova, D.V.; Hewitt, S.A.; Rajapaksha, W.R.; Fisher, T.E.; Bains, J.S. Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat. Neurosci. 2005, 8, 1078–1086. [Google Scholar] [CrossRef] [PubMed]
- Bal-Price, A.; Moneer, Z.; Brown, G.C. Nitric oxide induces rapid, calcium-dependent release of vesicular glutamate and ATP from cultured rat astrocytes. Glia 2002, 40, 312–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chever, O.; Lee, C.-Y.; Rouach, N. Astroglial Connexin43 Hemichannels Tune Basal Excitatory Synaptic Transmission. J. Neurosci. 2014, 34, 11228–11232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, L.; Madar, A.; Lacroix, M.M.; Yi, C.; Benchenane, K.; Giaume, C. Astroglial Connexin 43 Hemichannels Modulate Olfactory Bulb Slow Oscillations. J. Neurosci. 2015, 35, 15339–15352. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, R.; Dahl, G.; Qiu, F.; Spray, D.C.; Scemes, E. Pannexin 1: the molecular substrate of astrocyte “hemichannels”. J. Neurosci. 2009, 29, 7092–7097. [Google Scholar] [CrossRef] [PubMed]
- Suadicani, S.O.; Iglesias, R.; Wang, J.; Dahl, G.; Spray, D.C.; Scemes, E. ATP signaling is deficient in cultured pannexin1-null mouse astrocytes. Glia 2012, 60, 1106–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Toychiev, A.H.; Takahashi, N.; Sabirov, R.Z.; Okada, Y. Maxi-anion channel as a candidate pathway for osmosensitive ATP release from mouse astrocytes in primary culture. Cell Res. 2008, 18, 558–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elorza-Vidal, X.; Gaitán-Peñas, H.; Estévez, R. Chloride Channels in Astrocytes: Structure, Roles in Brain Homeostasis and Implications in Disease. Int. J. Mol. Sci. 2019, 20, 1034. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051034
Elorza-Vidal X, Gaitán-Peñas H, Estévez R. Chloride Channels in Astrocytes: Structure, Roles in Brain Homeostasis and Implications in Disease. International Journal of Molecular Sciences. 2019; 20(5):1034. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051034
Chicago/Turabian StyleElorza-Vidal, Xabier, Héctor Gaitán-Peñas, and Raúl Estévez. 2019. "Chloride Channels in Astrocytes: Structure, Roles in Brain Homeostasis and Implications in Disease" International Journal of Molecular Sciences 20, no. 5: 1034. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051034