Closure of the Human TKFC Active Site: Comparison of the Apoenzyme and the Complexes Formed with Either Triokinase or FMN Cyclase Substrates

 , ,
, ,
Abstract
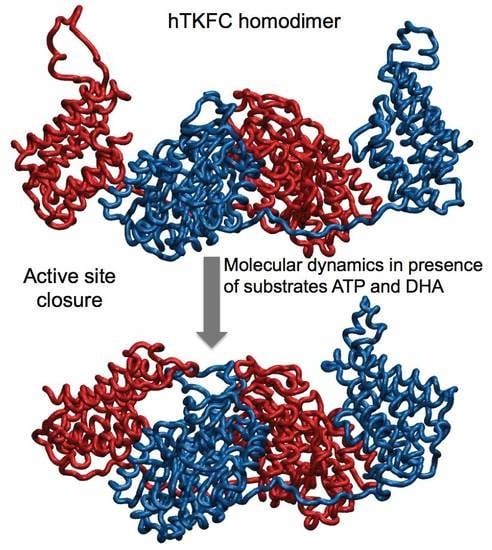
:
1. Introduction
2. Results
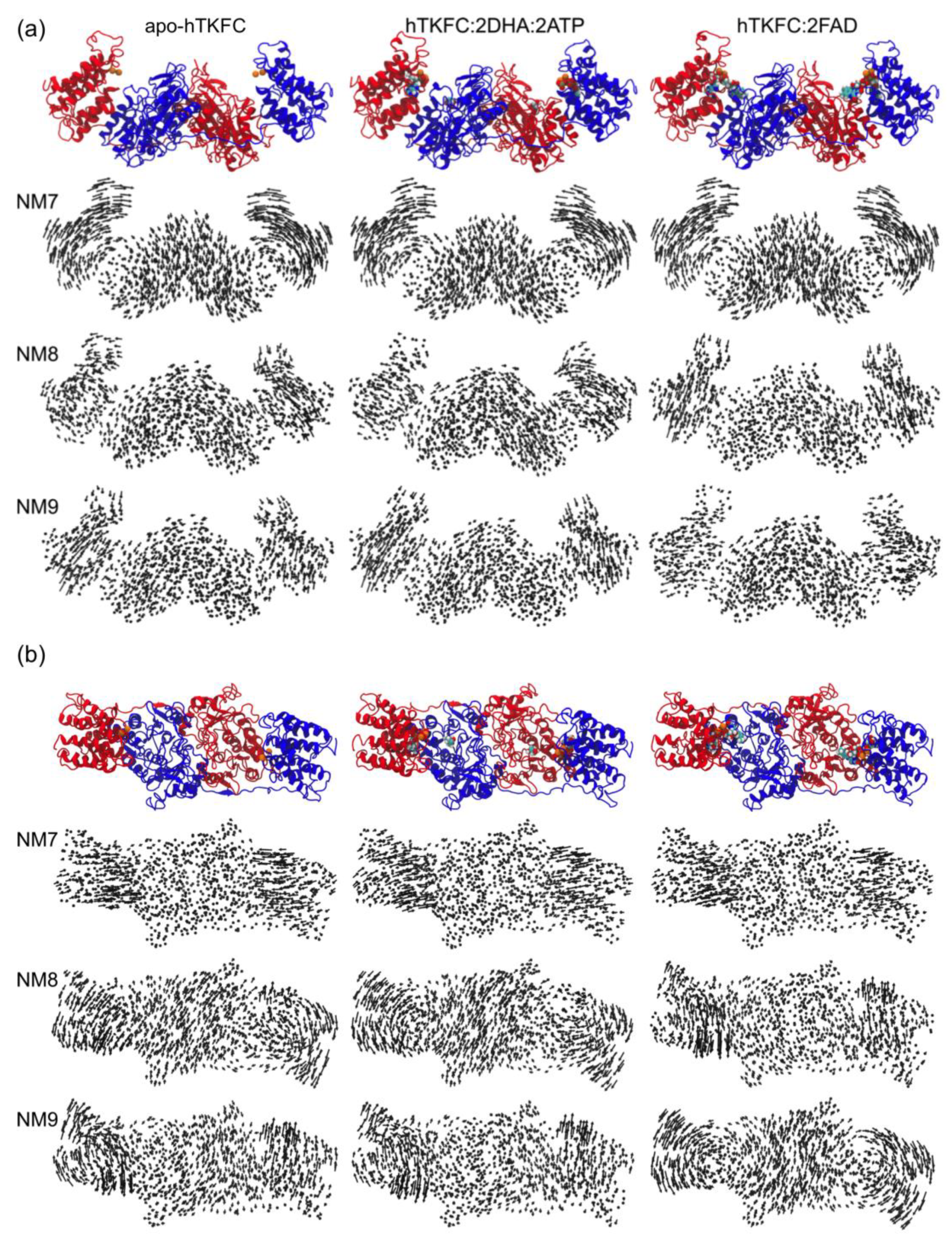
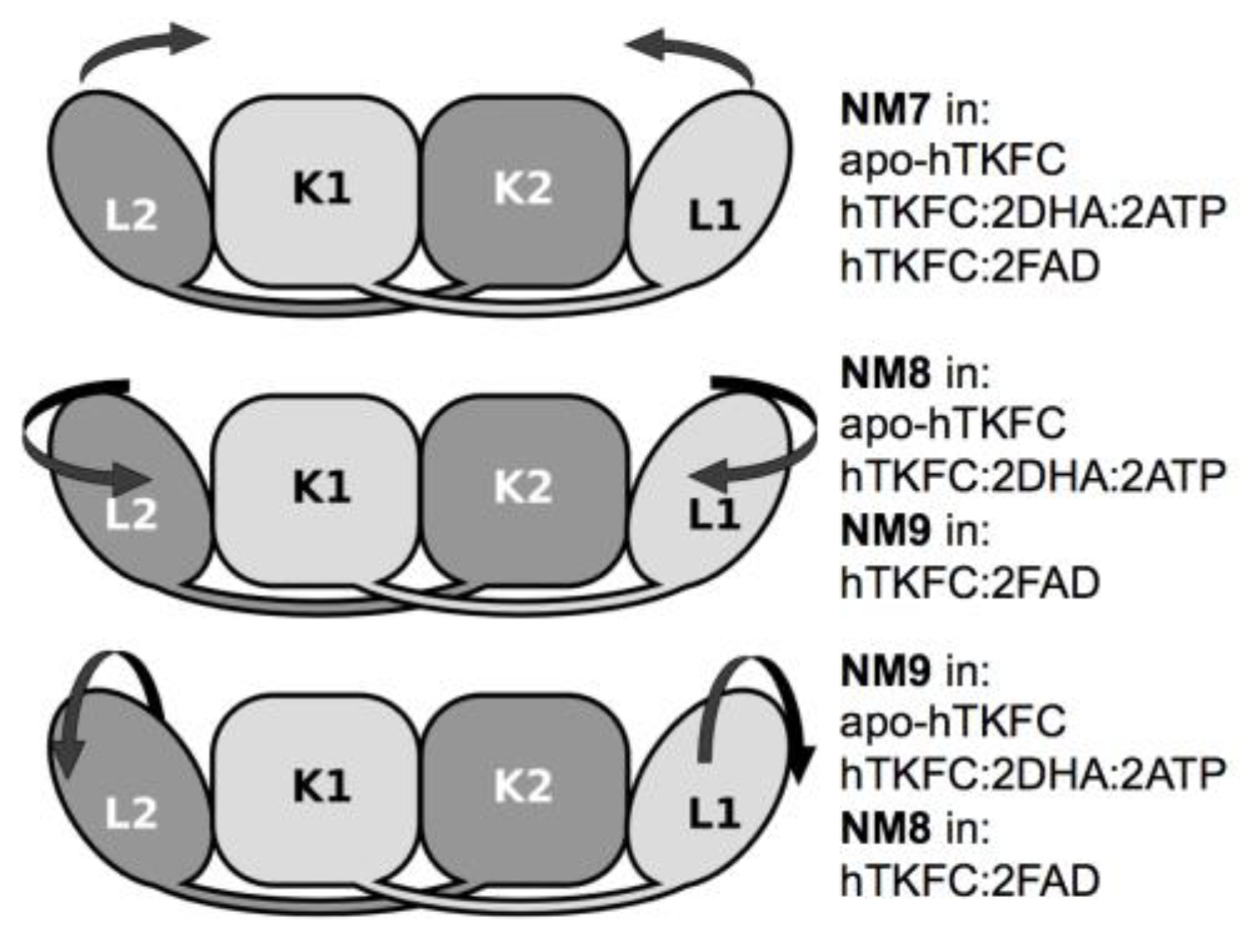
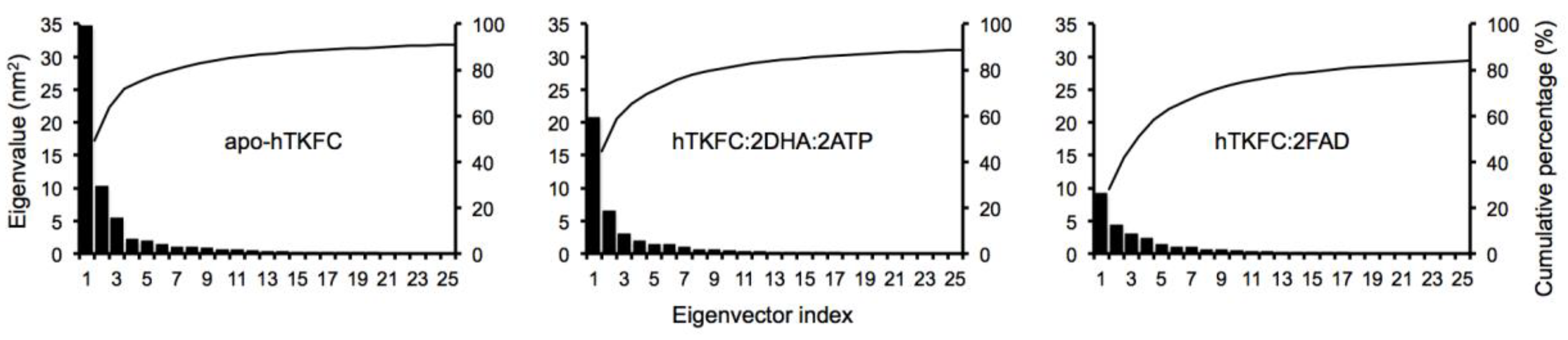
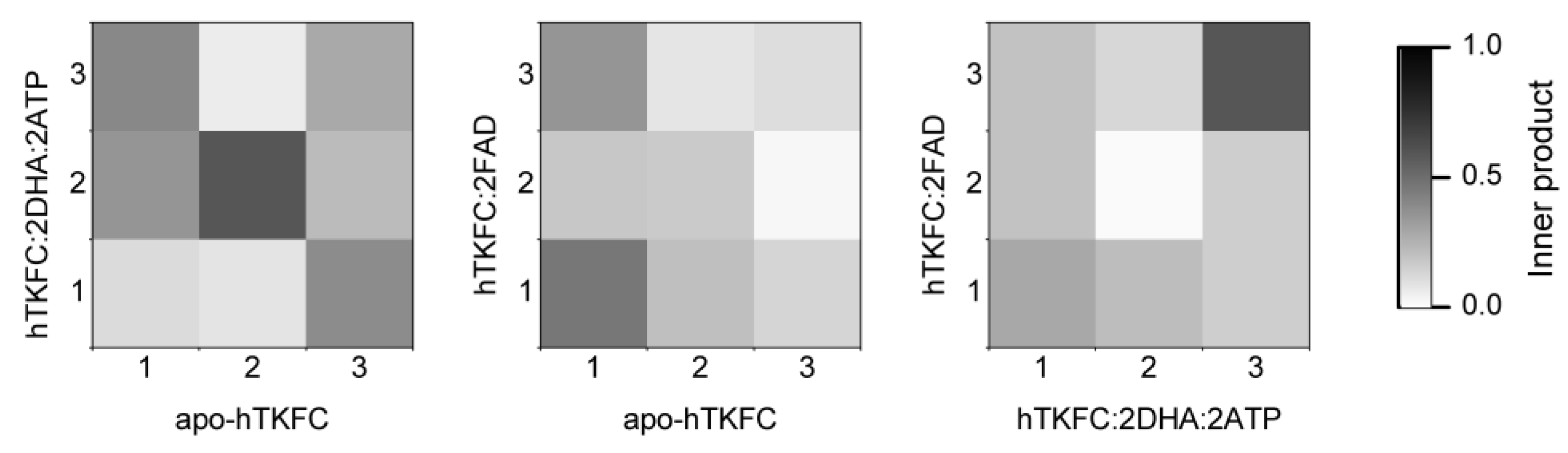
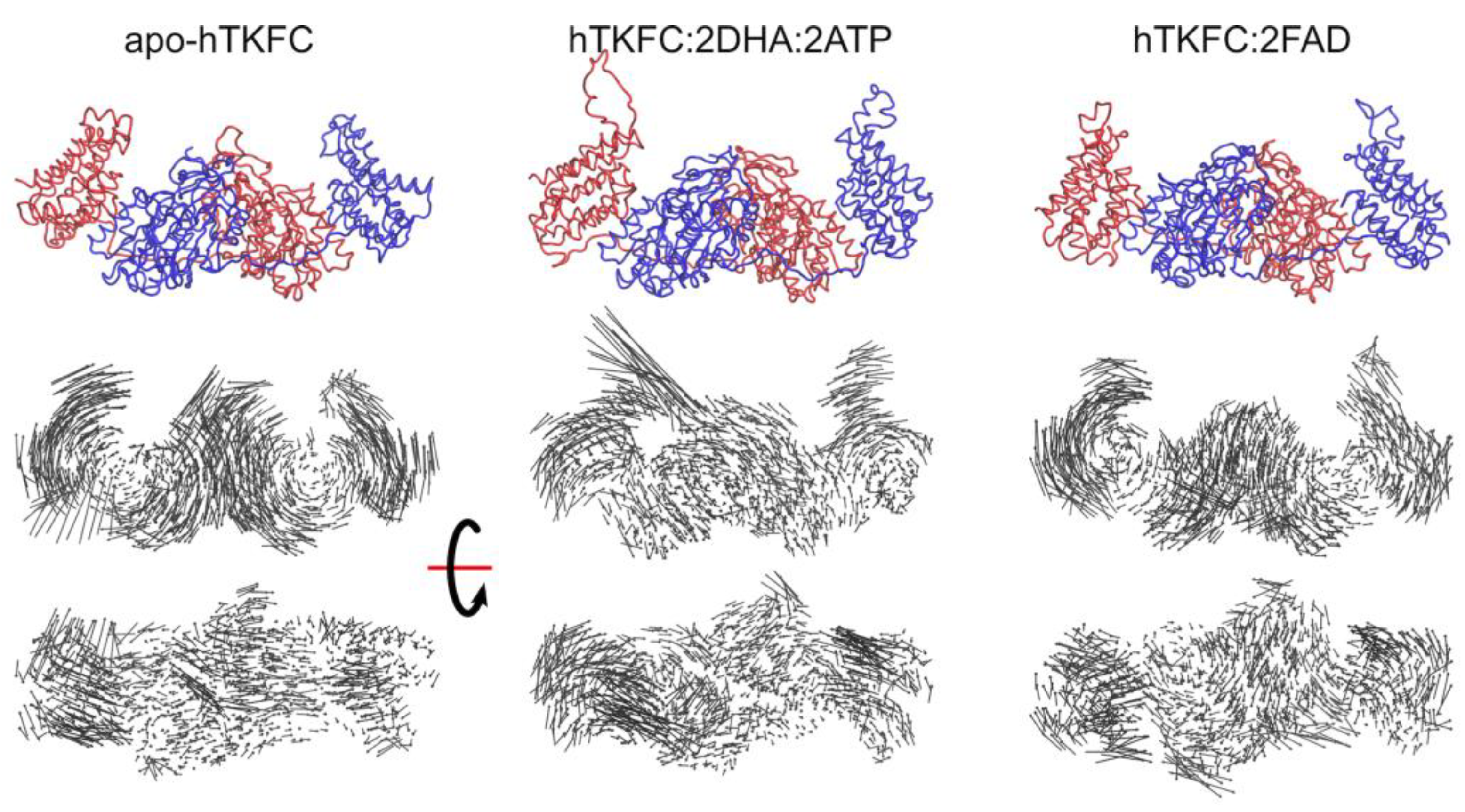
2.1. Normal-Mode Analysis of hTKFC Models Confirms the Trend of K Domains to Approach L Domains
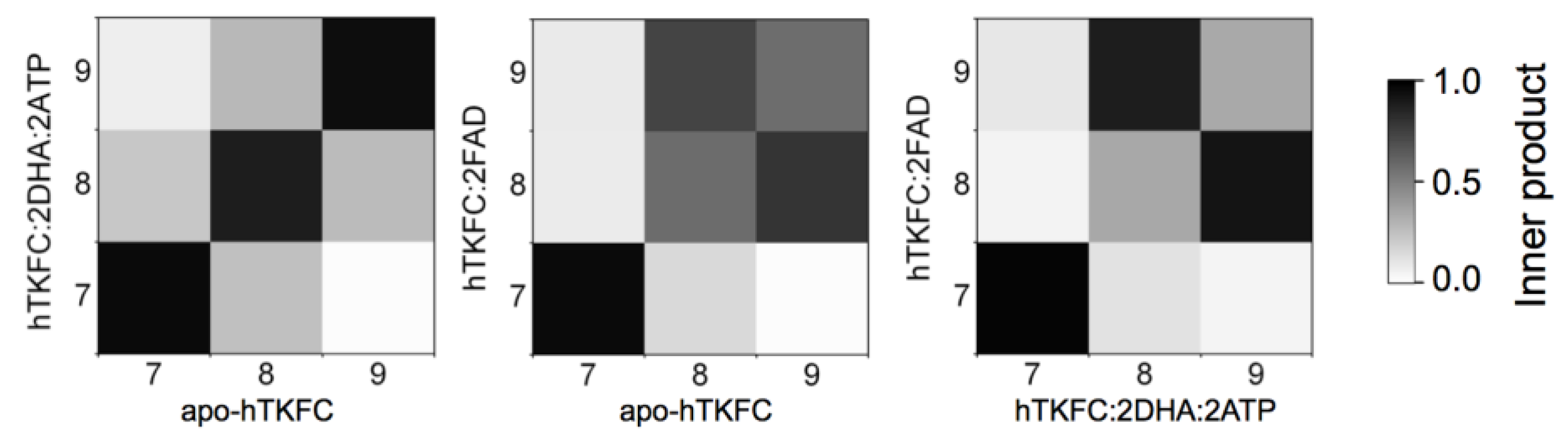
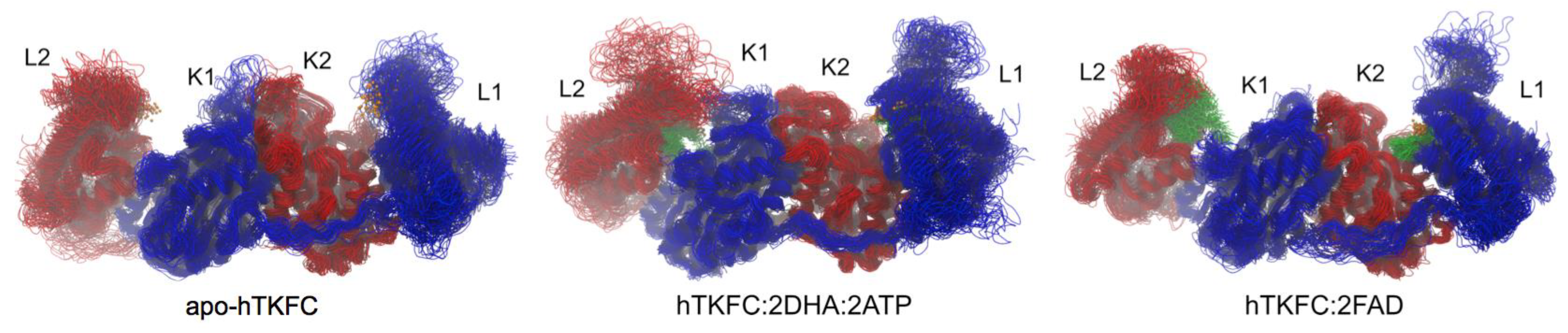
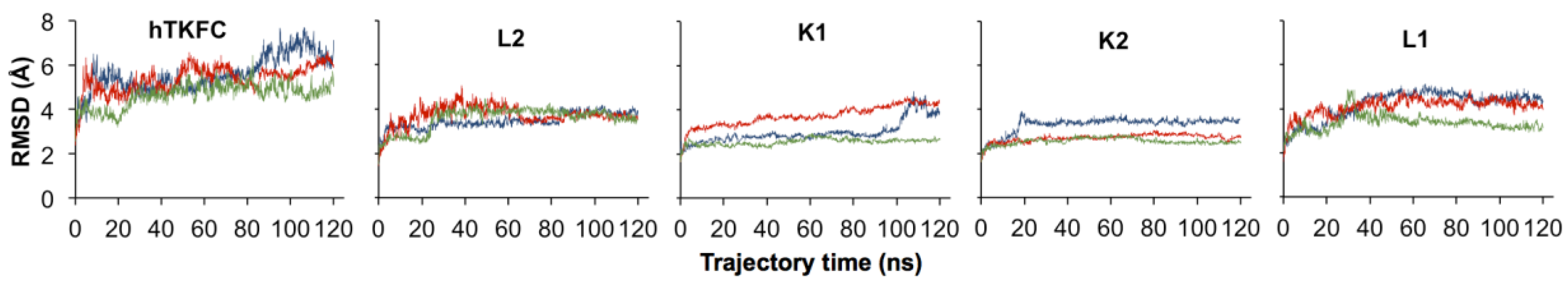
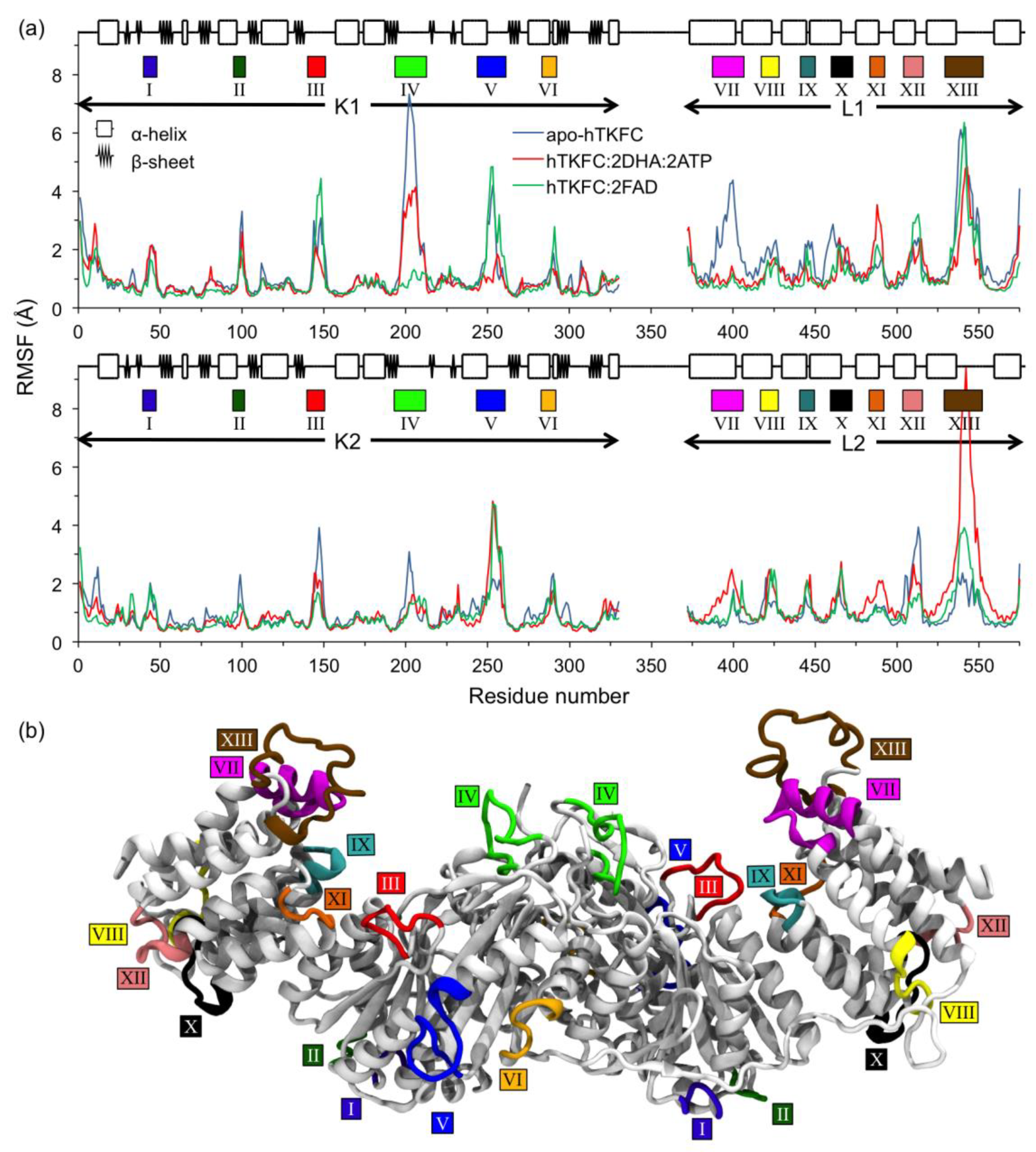
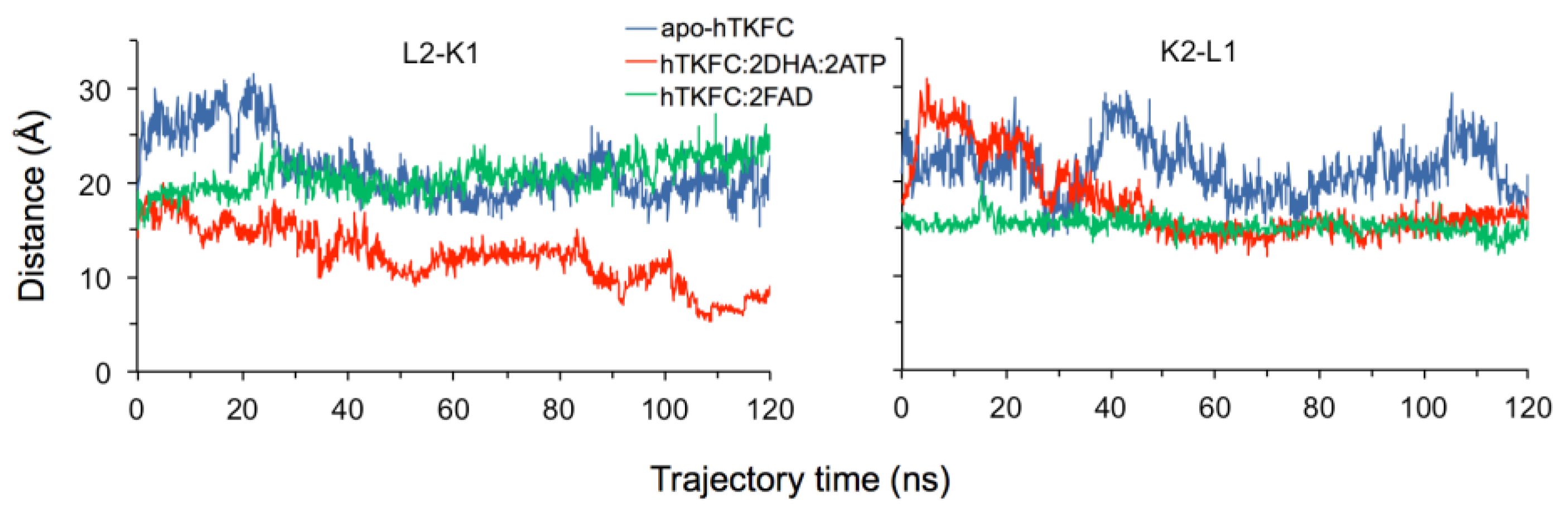
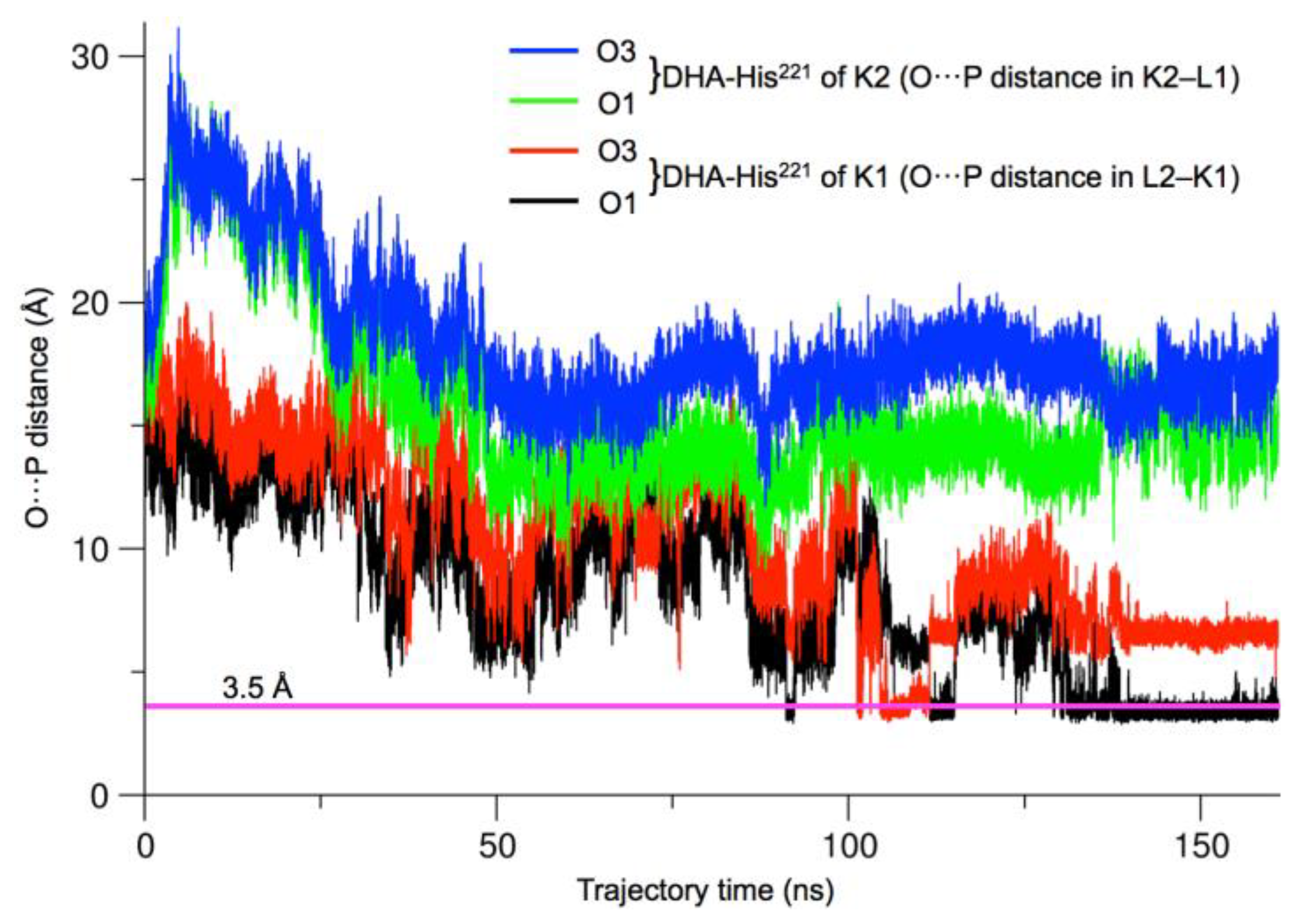
2.2. Comparison of hTKFC-2DHA-2ATP, hTKFC-2FAD and apo-hTKFC Molecular Dynamics Trajectories
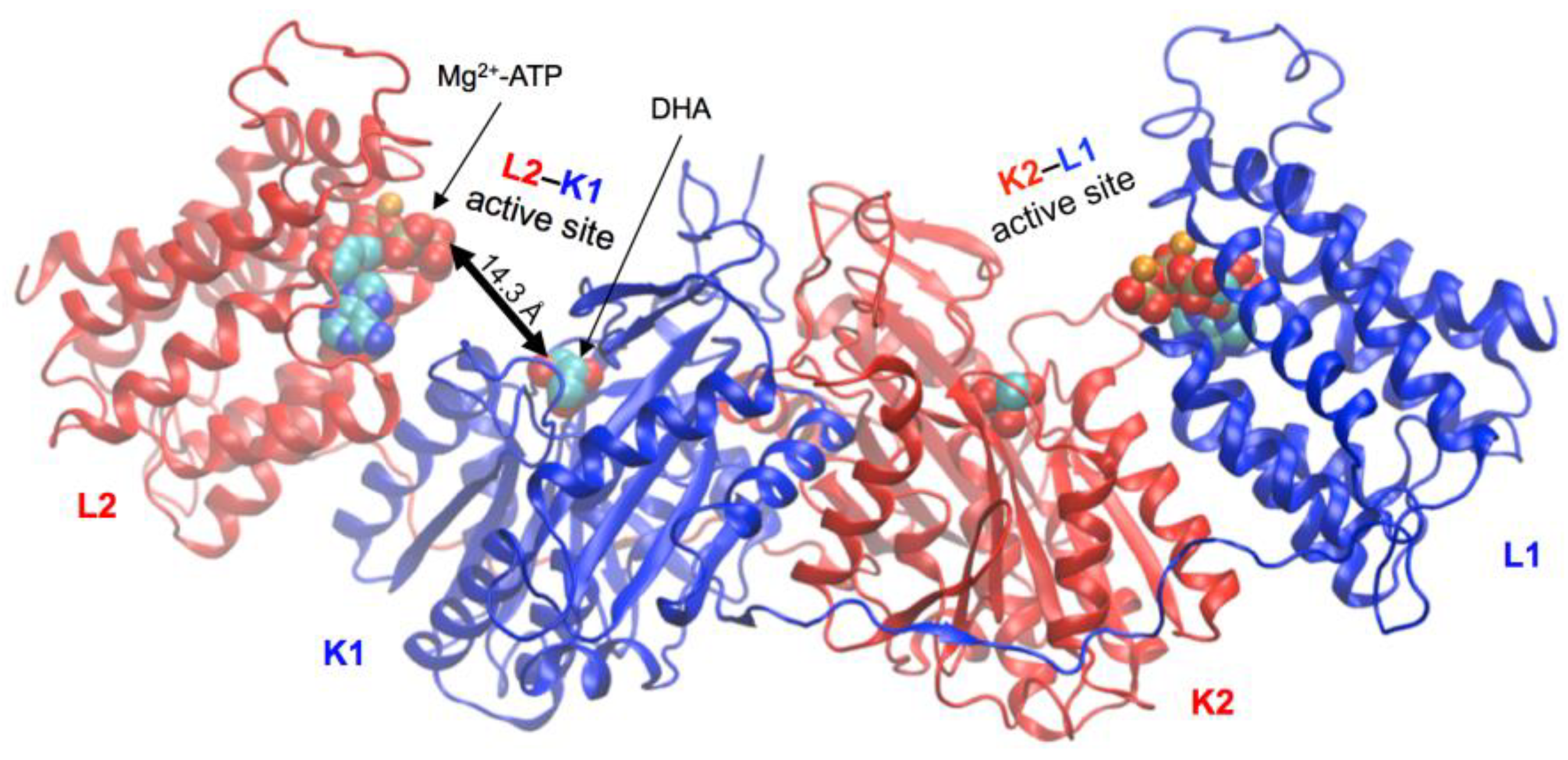
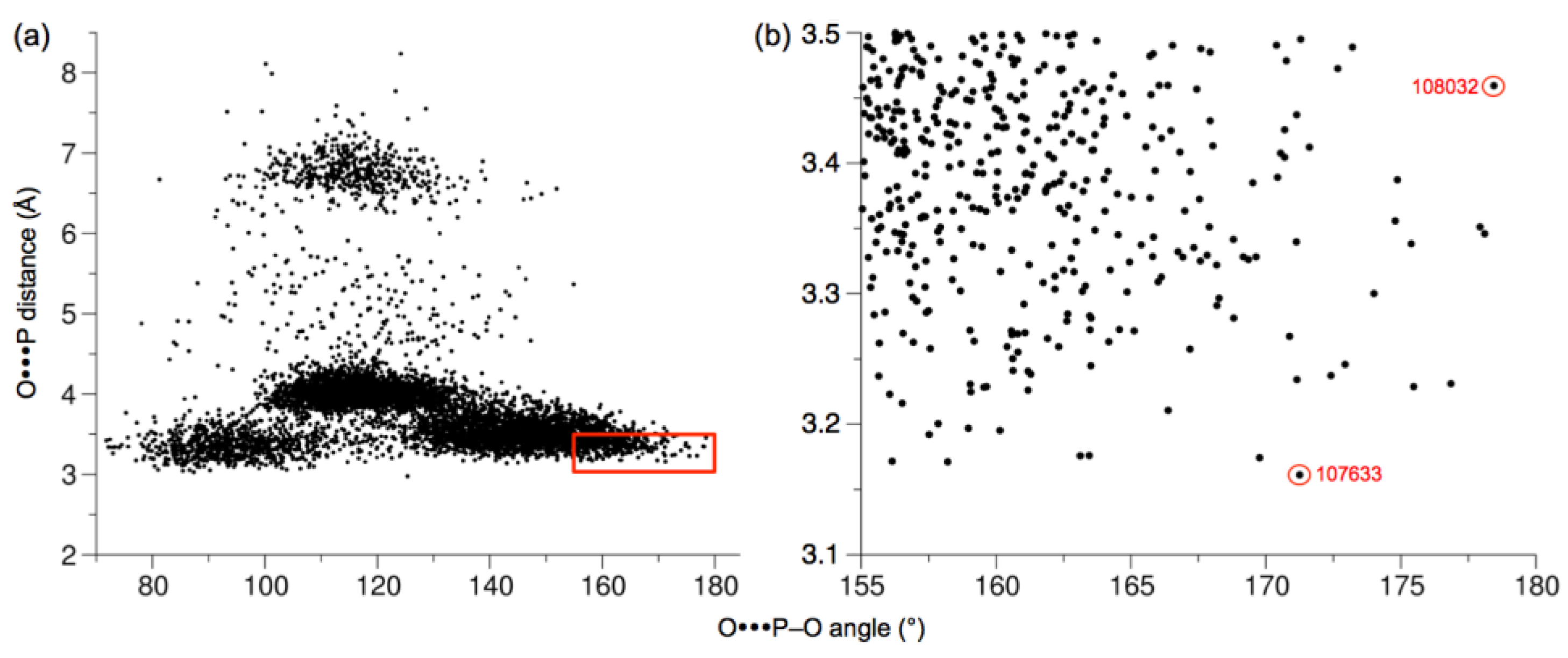
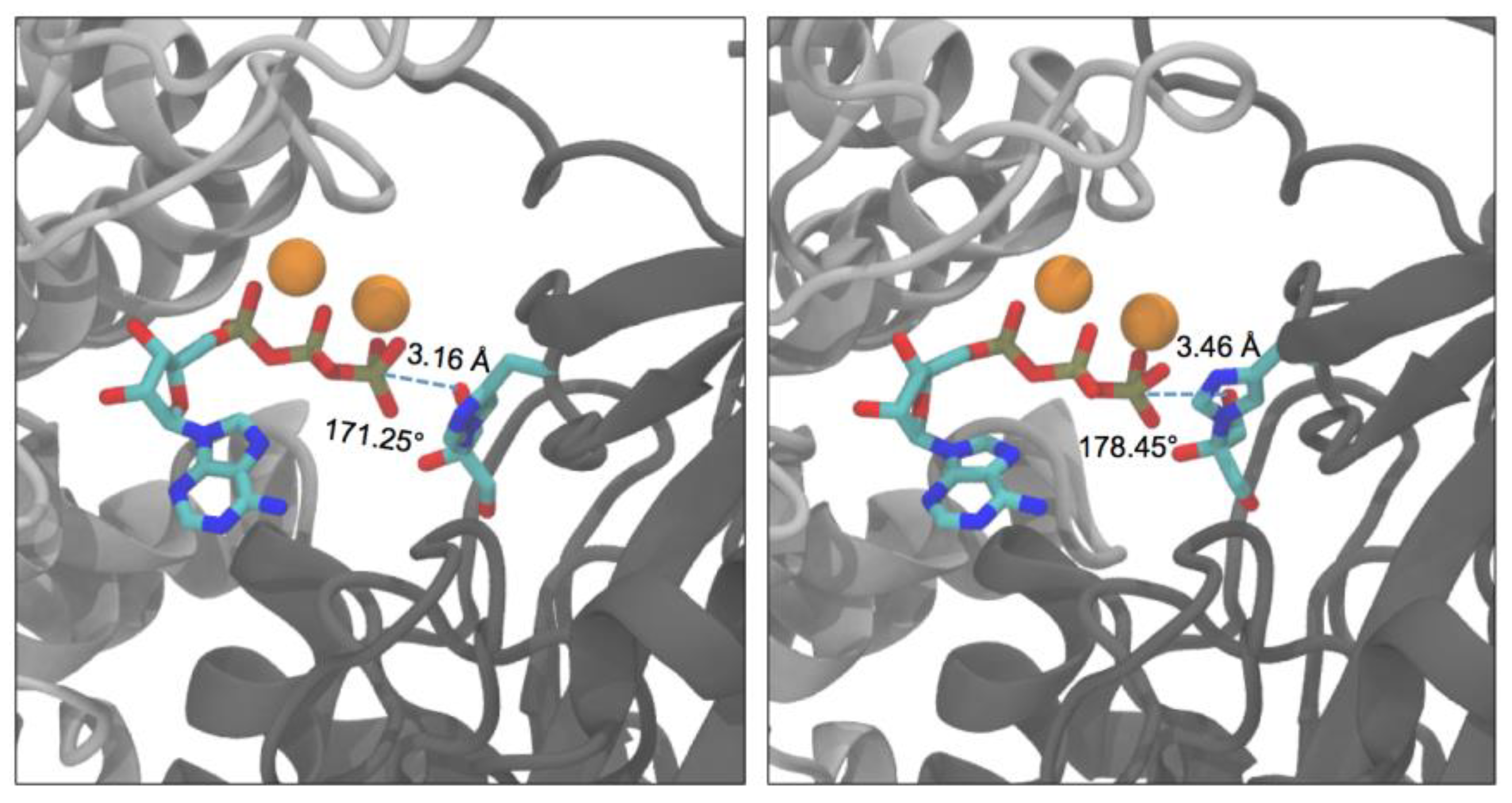
2.3. Active-Site Closure in the hTKFC-2DHA-2ATP Complex: Conformations for In-Line Nucleophilic Attack and Phosphoryl Transfer from ATP to DHA
3. Discussion
4. Materials and Methods
4.1. Structural Models of hTKFC
4.2. Normal-Mode Analyses
4.3. Molecular Dynamics
4.4. Essential Dynamics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ATP | Adenosine 5′-triphosphate |
| DHA | Dihydroxyacetone |
| DHAP | Dihydroxyacetone phosphate |
| FAD | Flavin adenine dinucleotide |
| FMN | Flavin mononucleotide |
| GA | d-Glyceraldehyde |
| hTKFC | Human triokinase/FMN cyclase |
| RMSD | Root-mean-square deviation |
| PTS | Phosphoenolpyruvate:sugar phosphotransferase system |
References
- Cabezas, A.; Costas, M.J.; Pinto, R.M.; Couto, A.; Cameselle, J.C. Identification of human and rat FAD-AMP lyase (cyclic FMN forming) as ATP-dependent dihydroxyacetone kinases. Biochem. Biophys. Res. Commun. 2005, 338, 1682–1689. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, J.R.; Couto, A.; Cabezas, A.; Pinto, R.M.; Ribeiro, J.M.; Canales, J.; Costas, M.J.; Cameselle, J.C. Bifunctional homodimeric triokinase/FMN cyclase: Contribution of protein domains to the activities of the human enzyme and molecular dynamics simulation of domain movements. J. Biol. Chem. 2014, 289, 10620–10636. [Google Scholar] [CrossRef] [PubMed]
- Hers, H.G.; Kusaka, T. Le metabolisme du fructose-1-phosphate dans le foie. Biochim. Biophys. Acta 1953, 11, 427–437. [Google Scholar] [CrossRef]
- Timson, D.J. Mammalian triokinase and dihydroxyacetone kinase are the same enzyme. In Fructose. Synthesis, Functions and Health Implications; Johnston, B.M., Collins, A.B., Eds.; Nova Science Publishers, Inc.: New York, NY, USA, 2012; pp. 153–160. ISBN 978-1-62081-126-9. [Google Scholar]
- Hannou, S.A.; Haslam, D.E.; McKeown, N.M.; Herman, M.A. Fructose metabolism and metabolic disease. J. Clin. Investig. 2018, 128, 545–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molin, M.; Norbeck, J.; Blomberg, A. Dihydroxyacetone kinases in Saccharomyces cerevisiae are involved in detoxification of dihydroxyacetone. J. Biol. Chem. 2003, 278, 1415–1423. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Chi, S.W.; Lee, P.Y.; Kang, S.; Cho, S.; Lee, C.K.; Bae, K.H.; Park, B.C.; Park, S.G. Reduced formation of advanced glycation endproducts via interactions between glutathione peroxidase 3 and dihydroxyacetone kinase 1. Biochem. Biophys. Res. Commun. 2009, 389, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Hers, H.G.; Hue, L. Gluconeogenesis and related aspects of glycolysis. Annu. Rev. Biochem. 1983, 52, 617–653. [Google Scholar] [CrossRef] [PubMed]
- Moreno, K.X.; Satapati, S.; DeBerardinis, R.J.; Burgess, S.C.; Malloy, C.R.; Merritt, M.E. Real-time detection of hepatic gluconeogenic and glycogenolytic states using hyperpolarized [2-13C]dihydroxyacetone. J. Biol. Chem. 2014, 289, 35859–35867. [Google Scholar] [CrossRef] [PubMed]
- Erni, B.; Siebold, C.; Christen, S.; Srinivas, A.; Oberholzer, A.; Baumann, U. Small substrate, big surprise: Fold, function and phylogeny of dihydroxyacetone kinases. Cell. Mol. Life. Sci. 2006, 63, 890–900. [Google Scholar] [CrossRef] [PubMed]
- Canales, J.; Cabezas, A.; Pinto, R.M.; Cameselle, J.C. Fluorimetric HPLC detection of endogenous riboflavin 4′,5′-cyclic phosphate in rat liver at nanomolar concentrations. Anal. Biochem. 2005, 341, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Moreno, I.; Iturrate, L.; Martín-Hoyos, R.; Jimeno, M.L.; Mena, M.; Bastida, A.; García-Junceda, E. From kinase to cyclase: An unusual example of catalytic promiscuity modulated by metal switching. Chembiochem 2009, 10, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Gauss, D.; Sánchez-Moreno, I.; Oroz-Guinea, I.; García-Junceda, E.; Wohlgemuth, R. Phosphorylation catalyzed by dihydroxyacetone kinase. Eur. J. Org. Chem. 2018, 2018, 2892–2895. [Google Scholar] [CrossRef]
- Sánchez-Moreno, I.; García-García, J.F.; Bastida, A.; García-Junceda, E. Multienzyme system for dihydroxyacetone phosphate-dependent aldolase catalyzed C-C bond formation from dihydroxyacetone. Chem. Commun. 2004, 1634–1635. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Moreno, I.; Bordes, I.; Castillo, R.; Ruiz-Pernia, J.J.; Moliner, V.; García-Junceda, E. Tuning the phosphoryl donor specificity of dihydroxyacetone kinase from ATP to inorganic polyphosphate. An insight from computational studies. Int. J. Mol. Sci. 2015, 16, 27835–27849. [Google Scholar] [CrossRef] [PubMed]
- Siebold, C.; Arnold, I.; Garcia-Alles, L.F.; Baumann, U.; Erni, B. Crystal structure of the Citrobacter freundii dihydroxyacetone kinase reveals an eight-stranded alpha-helical barrel ATP-binding domain. J. Biol. Chem. 2003, 278, 48236–48244. [Google Scholar] [CrossRef] [PubMed]
- Cheek, S.; Ginalski, K.; Zhang, H.; Grishin, N.V. A comprehensive update of the sequence and structure classification of kinases. BMC Struct. Biol. 2005, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.P.; Roth, R.L.; van der Westhuyzen, C.W.; Parkinson, C.J. Conserved phosphoryl transfer mechanisms within kinase families and the role of the C8 proton of ATP in the activation of phosphoryl transfer. BMC Res. Notes 2012, 5, 131. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Bahar, I.E. Normal Mode Analysis: Theory and Applications to Biological and Chemical Systems; Chapman & Hall/CRC: Boca Raton, FL, USA, 2006; ISBN 978-1-58488-472-9. [Google Scholar]
- Skjaerven, L.; Hollup, S.M.; Reuter, N. Normal mode analysis for proteins. J. Mol. Struct. (Theochem) 2009, 898, 42–48. [Google Scholar] [CrossRef]
- Mahajan, S.; Sanejouand, Y.H. On the relationship between low-frequency normal modes and the large-scale conformational changes of proteins. Arch. Biochem. Biophys. 2015, 567, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Ma, J. Usefulness and limitations of normal mode analysis in modeling dynamics of biomolecular complexes. Structure 2005, 13, 373–380. [Google Scholar] [CrossRef] [PubMed]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [CrossRef] [PubMed]
- Mildvan, A.S. Mechanisms of signaling and related enzymes. Proteins 1997, 29, 401–416. [Google Scholar] [CrossRef]
- Bächler, C.; Flukiger-Bruhwiler, K.; Schneider, P.; Bähler, P.; Erni, B. From ATP as substrate to ADP as coenzyme: Functional evolution of the nucleotide binding subunit of dihydroxyacetone kinases. J. Biol. Chem. 2005, 280, 18321–18325. [Google Scholar] [CrossRef] [PubMed]
- Oberholzer, A.E.; Schneider, P.; Baumann, U.; Erni, B. Crystal structure of the nucleotide-binding subunit DhaL of the Escherichia coli dihydroxyacetone kinase. J. Mol. Biol. 2006, 359, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; McDonald, L.; Cui, Q.; Matte, A.; Cygler, M.; Ekiel, I. Structural and mechanistic insight into covalent substrate binding by Escherichia coli dihydroxyacetone kinase. Proc. Natl. Acad. Sci. USA 2011, 108, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Zurbriggen, A.; Jeckelmann, J.M.; Christen, S.; Bieniossek, C.; Baumann, U.; Erni, B. X-ray structures of the three Lactococcus lactis dihydroxyacetone kinase subunits and of a transient intersubunit complex. J. Biol. Chem. 2008, 283, 35789–35796. [Google Scholar] [CrossRef] [PubMed]
- Gutknecht, R.; Beutler, R.; Garcia-Alles, L.F.; Baumann, U.; Erni, B. The dihydroxyacetone kinase of Escherichia coli utilizes a phosphoprotein instead of ATP as phosphoryl donor. EMBO J. 2001, 20, 2480–2486. [Google Scholar] [CrossRef] [PubMed]
- Bordes, I.; Castillo, R.; Moliner, V. A theoretical study of the phosphoryl transfer reaction from ATP to Dha catalyzed by DhaK from Escherichia coli. J. Phys. Chem. B 2017, 121, 8878–8892. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.N.; Dunaway-Mariano, D. Phosphoryl group transfer: Evolution of a catalytic scaffold. Trends Biochem. Sci. 2004, 29, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Lassila, J.K.; Zalatan, J.G.; Herschlag, D. Biological phosphoryl-transfer reactions: Understanding mechanism and catalysis. Annu. Rev. Biochem. 2011, 80, 669–702. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.H. Models for biological phosphoryl transfer. Biochim. Biophys. Acta 2004, 1697, 279–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleland, W.W.; Hengge, A.C. Enzymatic mechanisms of phosphate and sulfate transfer. Chem. Rev. 2006, 106, 3252–3278. [Google Scholar] [CrossRef] [PubMed]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 1999, 12, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krieger, E.; Nabuurs, A.B.; Vriend, G. Homology modeling. In Structural Bioinformatics; Bourne, P.E., Weissig, H., Eds.; Wiley-Liss, Inc.: Hoboken, NJ, USA, 2003; pp. 509–523. [Google Scholar]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Dupradeau, F.Y.; Cézard, C.; Lelong, R.; Stanislawiak, É.; Pêcher, J.; Delepine, J.C.; Cieplak, P. R.E.DD.B.: A database for RESP and ESP atomic charges, and force field libraries. Nucleic Acids Res. 2008, 36, D360–D367. [Google Scholar] [CrossRef] [PubMed]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| apo-hTKFC | hTKFC:2DHA:2ATP | hTKFC:2FAD | |
|---|---|---|---|
| Components used for both procedures | |||
| hTKFC chains | 2 | 2 | 2 |
| Mg2+ | 4 | 4 | 4 |
| DHA | 0 | 2 | 0 |
| ATP | 0 | 2 | 0 |
| FAD | 0 | 0 | 2 |
| Components used only for molecular dynamics | |||
| Na+ | 84 | 84 | 84 |
| Cl− | 94 | 86 | 90 |
| H2O | 87,364 | 87,570 | 87,304 |
| Total number of atoms | |||
| In normal mode analysis | 16,814 | 16,924 | 16,982 |
| In molecular dynamics | 279,084 | 279,804 | 279,068 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodrigues, J.R.; Cameselle, J.C.; Cabezas, A.; Ribeiro, J.M. Closure of the Human TKFC Active Site: Comparison of the Apoenzyme and the Complexes Formed with Either Triokinase or FMN Cyclase Substrates. Int. J. Mol. Sci. 2019, 20, 1099. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051099
Rodrigues JR, Cameselle JC, Cabezas A, Ribeiro JM. Closure of the Human TKFC Active Site: Comparison of the Apoenzyme and the Complexes Formed with Either Triokinase or FMN Cyclase Substrates. International Journal of Molecular Sciences. 2019; 20(5):1099. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051099
Chicago/Turabian StyleRodrigues, Joaquim Rui, José Carlos Cameselle, Alicia Cabezas, and João Meireles Ribeiro. 2019. "Closure of the Human TKFC Active Site: Comparison of the Apoenzyme and the Complexes Formed with Either Triokinase or FMN Cyclase Substrates" International Journal of Molecular Sciences 20, no. 5: 1099. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051099