Dietary Eriodictyol Alleviates Adiposity, Hepatic Steatosis, Insulin Resistance, and Inflammation in Diet-Induced Obese Mice

Abstract
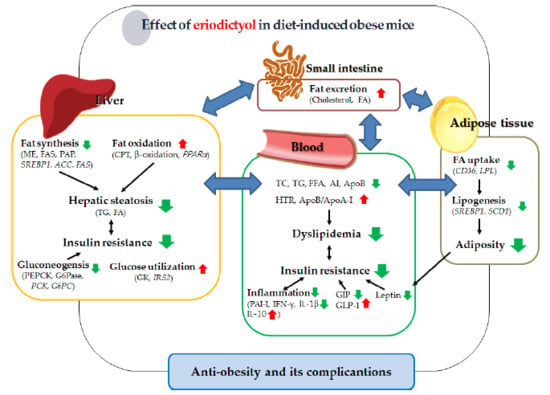
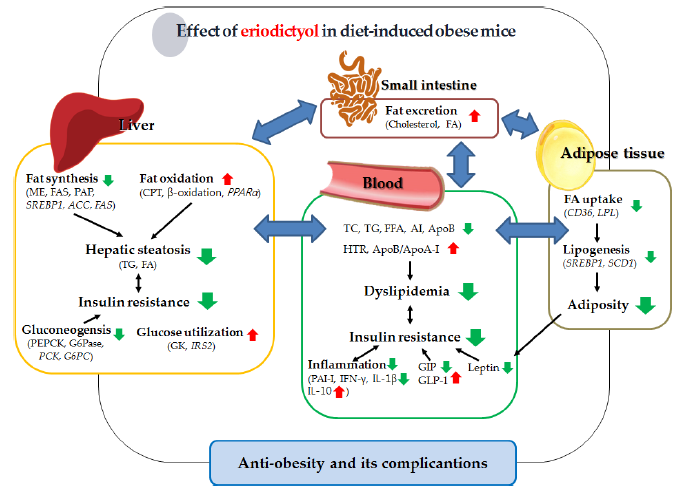
:
1. Introduction
2. Results
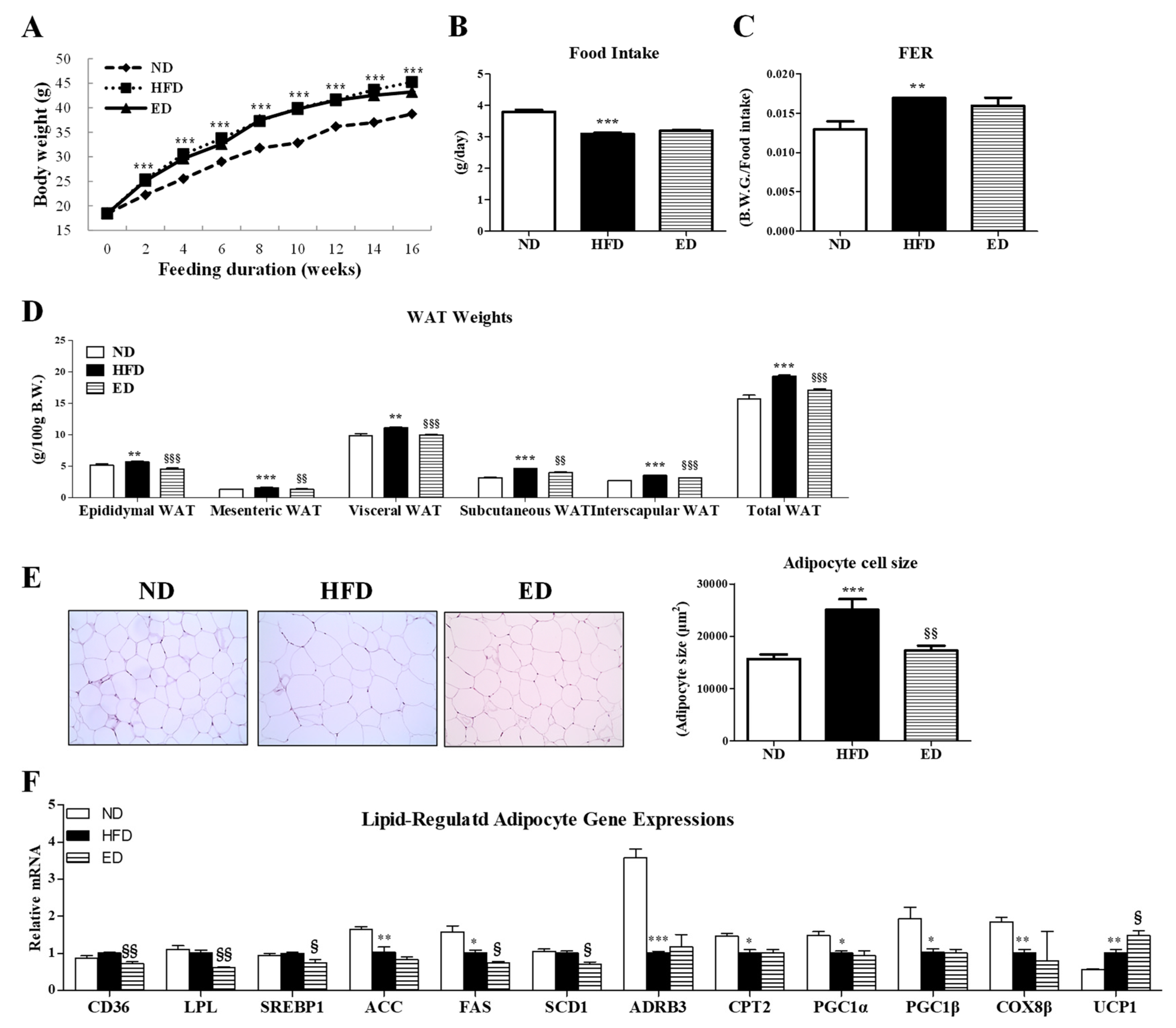
2.1. Eriodictyol Supplementation Reduced Body Fat and Deposition and Regulated Gene Expression in Adipocytes
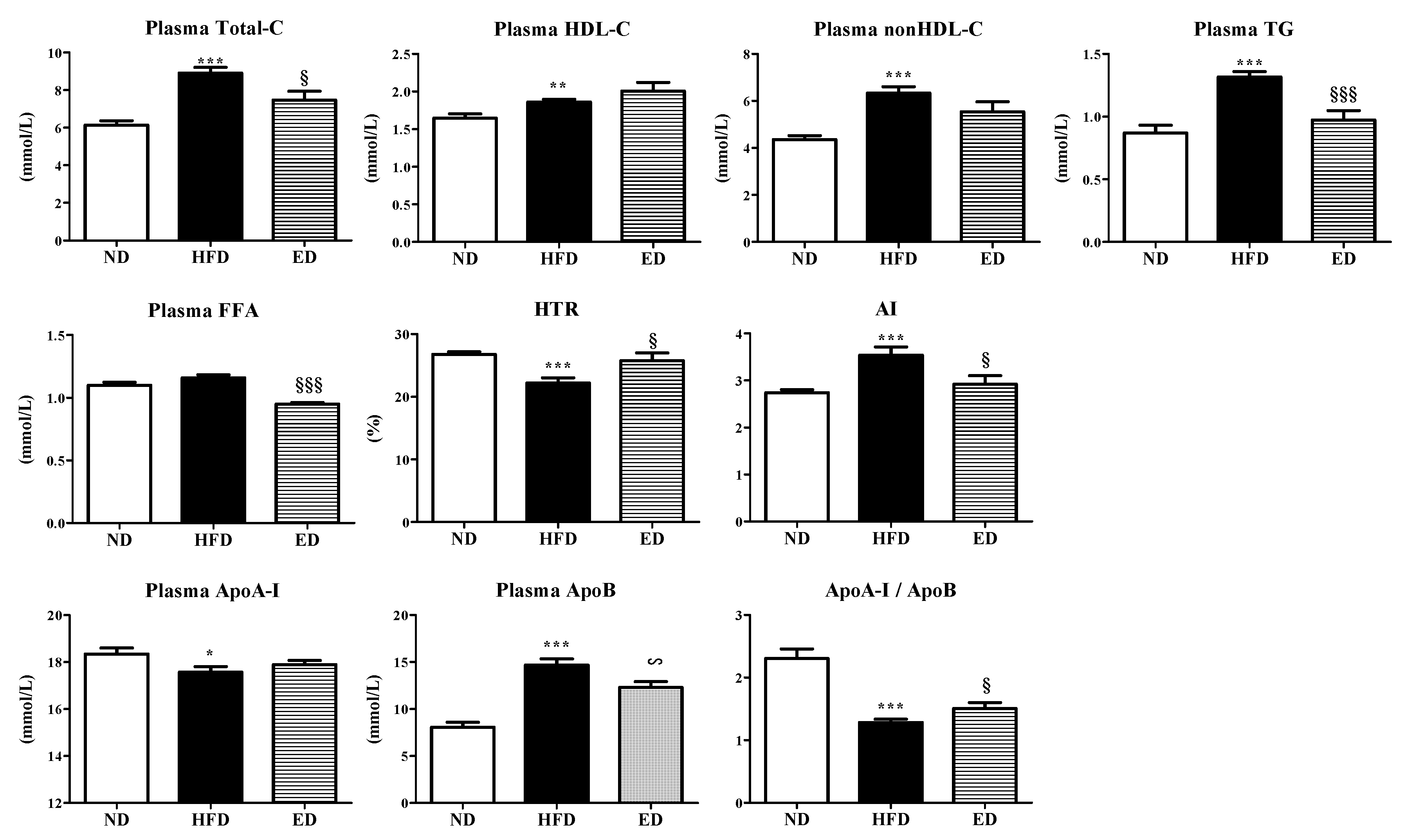
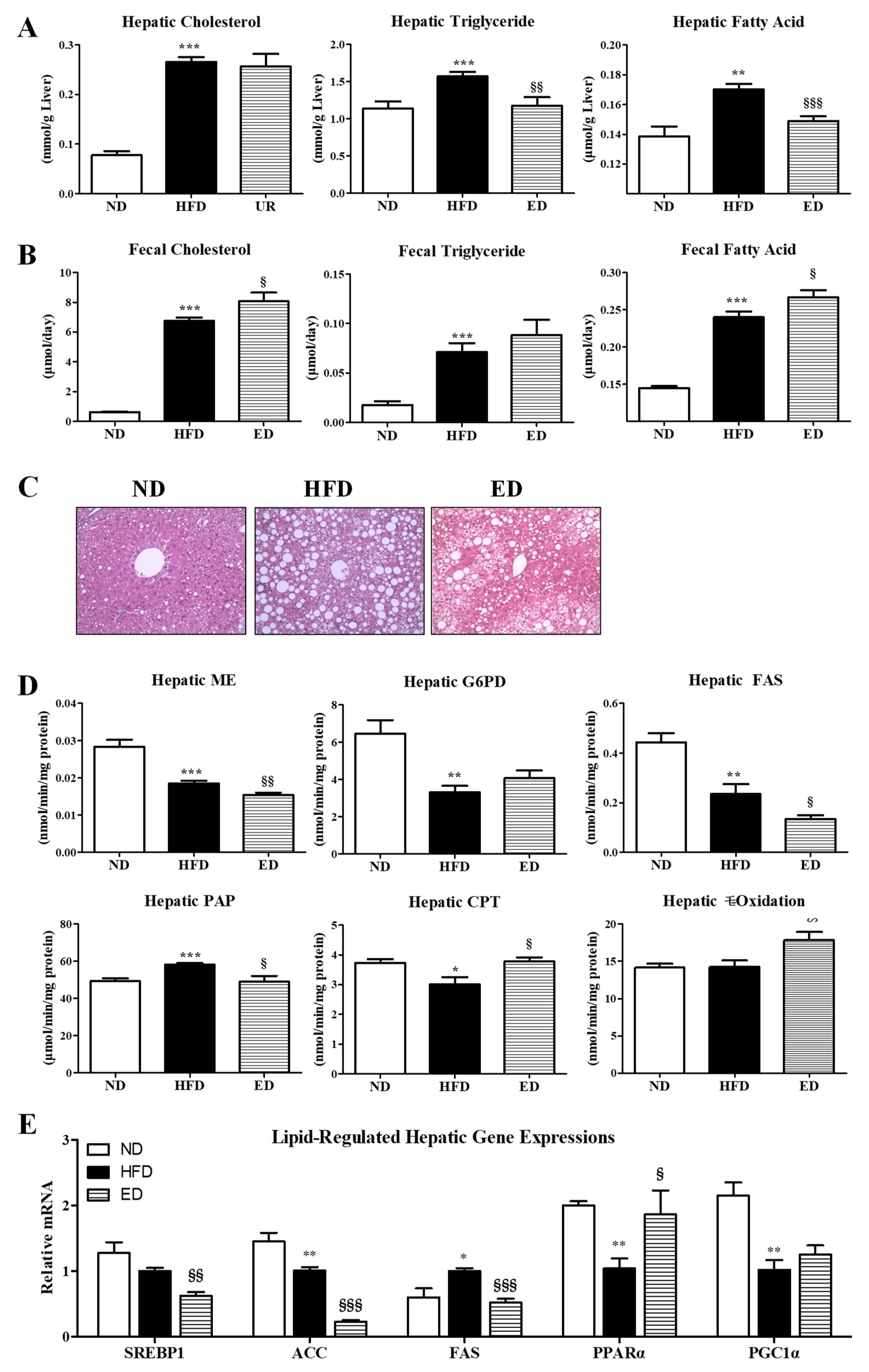
2.2. Eriodictyol Supplement Improved Plasma and Hepatic Lipid Levels, and Modulated the Expression of Hepatic Lipid-Regulating Enzymes and Genes
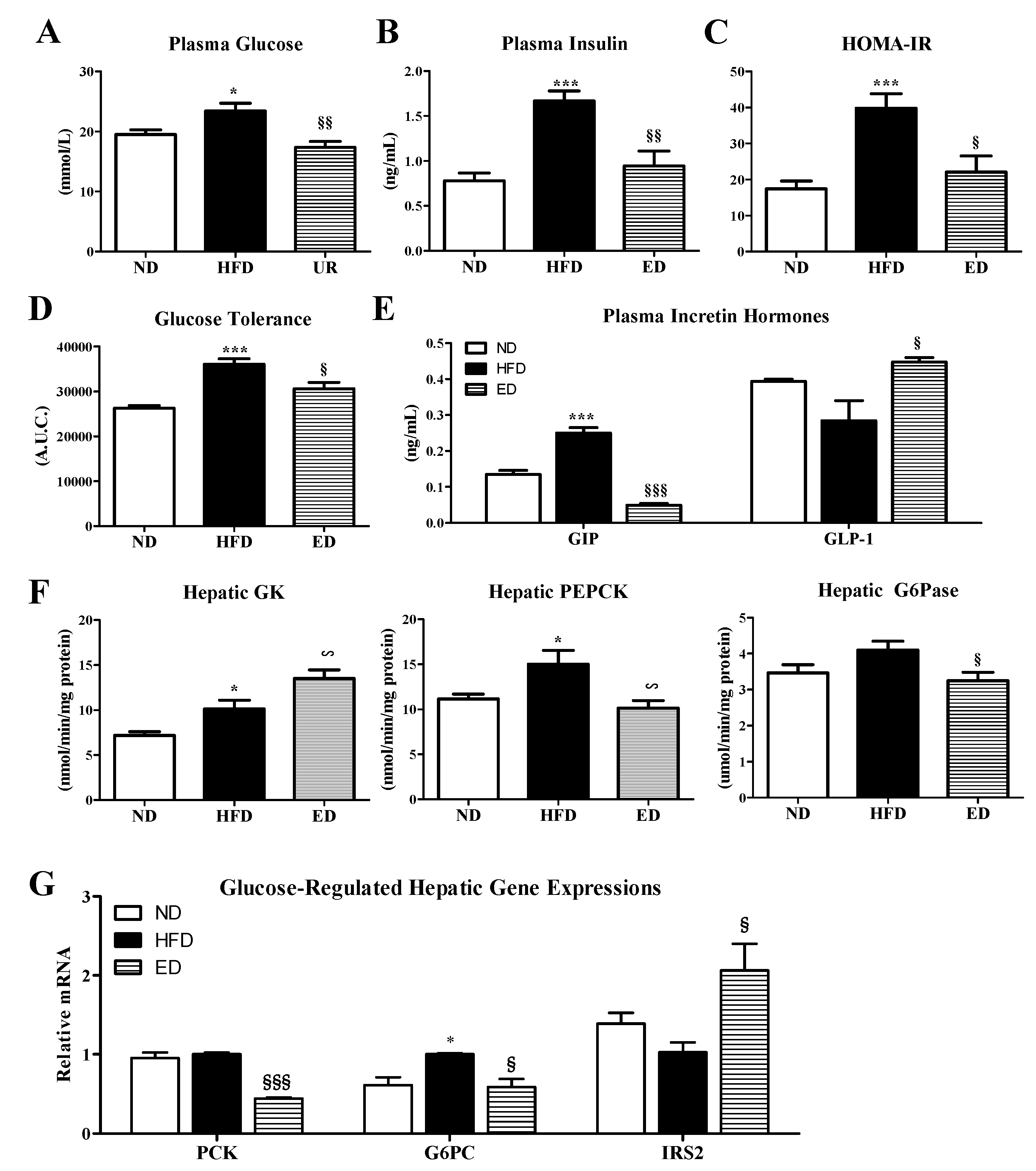
2.3. Eriodictyol Supplementation Ameliorated Insulin Resistance, Glucose Tolerance, and Adipokine/Cytokine Dysregulation
3. Discussion
4. Materials and Methods
4.1. Experimental Animals and Diet
4.2. Blood Analysis
4.3. Hepatic and Fecal Lipid Contents
4.4. Activities of Hepatic Lipid- and Glucose-Regulating Enzymes
4.5. RNA Isolation and Gene Expression Analysis
4.6. Morphological Examination of Liver and Fat Tissues
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary digital publishing institute |
| ACC | Acetyl-CoA carboxylase |
| AI | Atherogenic index |
| APO | Apolipoprotein |
| CPT | Carnitine palmitoyltransferase |
| DIO | Diet-induced obese |
| ED | Eriodictyol |
| FAS | Fatty acid synthase |
| FER | Food efficiency ratio |
| FFA | Free fatty acid |
| GIP | Gastric inhibitory polypeptide |
| GK | Glucokinase |
| GLP-1 | Glucagon-like peptide-1 |
| G6Pase | Glucose-6-phosphatase |
| G6PD | Glucose-6-phosphate dehydrogenase |
| HFD | High-fat diet |
| HOMA-IR | Homeostasis model assessment of insulin resistance |
| HTR | HDL-cholesterol-to-TC ratio |
| H&E | Hematoxylin and eosin |
| IFN-γ | Interferon-gamma |
| IL | Interleukin |
| IPGTT | Intraperitoneal glucose tolerance test |
| IR | Insulin resistance |
| IRS2 | Insulin receptor substrate 2 |
| ME | Malic enzyme |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PAP | Phosphatidate phosphohydrolase |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| PGC1α | PPAR-gamma coactivator 1 alpha |
| PPARα | Peroxisome proliferator-activated receptor alpha |
| SREBP | Sterol regulatory element-binding transcription factor |
| TC | Total-cholesterol |
| TG | Triglyceride |
| WAT | White adipose tissue |
References
- Guh, D.P.; Zhang, W.; Bansback, N.; Amarsi, Z.; Birmingham, C.L.; Anis, A.H. The incidence of co-morbidities related to obesity and overweight: A systematic review and meta-analysis. BMC. Public Health. 2009, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.; Catalano, K.J.; Ananthnarayan, S.; Kim, S.P.; van Citters, G.W.; Dea, M.K.; Bergman, R.N. Molecular evidence supporting the portal theory: A causative link between visceral adiposity and hepatic insulin resistance. Am. J. Physiol. Endocrinol. MeTable. 2005, 288, E454–E461. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, L.M.; de Carvalho, J.L.; da Silva, H.C.; Lemos, T.L.; Arriaga, A.M.; Braz-Filho, R.; Militão, G.C.; Silva, T.D.; Ribeiro, P.R.; Santiago, G.M. New cytotoxic bibenzyl and other constituents from Bauhinia Ungulata L. (Fabaceae). Chem. Biodivers. 2016, 13, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Milella, L.; Milazzo, S.; De Leo, M.; Vera Saltos, M.B.; Faraone, I.; Tuccinardi, T.; Lapillo, M.; De Tommasi, N.; Braca, A. α-Glucosidase and α-amylase inhibitors from arcytophyllum thymifolium. J. Nat. Prod. 2016, 79, 2104–2112. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.D.; Feng, Y.; Li, H.M.; Xia, X.S.; Li, R.T. A new flavonoid glycoside from Elsholtzia bodinieri. Nat. Prod. Res. 2016, 30, 2278–2284. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Chen, K.; Du, P.; Wang, S.S.; Ren, B.; Ren, Y.L.; Yan, H.S.; Liang, Y.; Wu, F.H. Phenolic compounds from Clinopodium chinense (Benth.) O. Kuntze and their inhibitory effects on α-Glucosidase and vascular endothelial cells injury. Chem. Biodivers. 2016, 13, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Ji, R.; Han, M. Eriodictyol protects H9c2 cardiomyocytes against the injury induced by hypoxia/reoxygenation by improving the dysfunction of mitochondria. Exp. Ther. Med. 2019, 17, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y. Obesity and inflammation: The linking mechanism and the complications. Arch. Med. Sci. 2017, 13, 851–863. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Kumazoe, M.; Nakamura, Y.; Won, Y.S.; Bae, J.; Yamashita, S.; Tachibana, H. The combination of green tea extract and eriodictyol inhibited high-fat/high-sucrose diet-induced cholesterol upregulation is accompanied by suppression of cholesterol synthesis enzymes. J. Nutr. Sci. Vitaminol (Tokyo). 2016, 62, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Terauchi, Y. Lessons from mouse models of high-fat diet-induced NAFLD. Int. J. Mol. Sci. 2013, 14, 21240–21257. [Google Scholar] [CrossRef] [PubMed]
- Jensen, V.S.; Hvid, H.; Damgaard, J.; Nygaard, H.; Ingvorsen, C.; Wulff, E.M.; Lykkesfeldt, J.; Fledelius, C. Dietary fat stimulates development of NAFLD more potently than dietary fructose in Sprague-Dawley rats. Diabetol. Metab. Syndr. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Souza, M.R.; Diniz, M.F.; Medeiros, J.E.; Araujo, M.S. Metabolic syndrome and risk factors for non-alcoholic fatty liver disease. Arq. Gastroenterol. 2012, 49, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuschwander-Tetri, B.A. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology. 2010, 52, 774–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perry, R.J.; Samuel, V.T.; Petersen, K.F.; Shulman, G.I. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014, 510, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016, 65, 1038–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, N.; Flatt, P.R. Evidence for beneficial effects of compromised gastric inhibitory polypeptide action in obesity-related diabetes and possible therapeutic implications. Diabetologia. 2009, 52, 1724–1731. [Google Scholar] [CrossRef] [PubMed]
- Creutzfeldt, W.; Ebert, R.; Willms, B.; Frerichs, H.; Brown, J.C. Gastric inhibitory polypeptide (GIP) and insulin in obesity: Increased response to stimulation and defective feedback control of serum levels. Diabetologia. 1978, 14, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Nordlie, R.C.; Foster, J.D. Regulation of glucose production by the liver. Annu. Rev. Nutr. 1999, 19, 379–406. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Singh, N.; Akanksha; Jayendra; Maurya, R.; Srivastava, A.K. Coagulanolide modulates hepatic glucose metabolism in C57BL/KsJ-db/db mice. Hum. Exp. Toxicol. 2012, 31, 1056–1065. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, C.M.; Ueki, K.; Kahn, R. Complementary roles of IRS-1 and IRS-2 in the hepatic regulation of metabolism. J. Clin. Invest. 2005, 115, 718–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourlier, V.; Zakaroff-Girard, A.; Miranville, A.; De Barros, S.; Maumus, M.; Sengenes, C.; Galitzky, J.; Lafontan, M.; Karpe, F.; Frayn, K.N.; et al. Remodeling phenotype of human subcutaneous adipose tissue macrophages. Circulation. 2008, 117, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Henegar, C.; Tordjman, J.; Achard, V.; Lacasa, D.; Cremer, I.; Guerre-Millo, M.; Poitou, C.; Basdevant, A.; Stich, V.; Viguerie, N.; et al. Adipose tissue transcriptomic signature highlights the pathological relevance of extracellular matrix in human obesity. Genome. Biol. 2008, 9, R14. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Kataria, M.A.; Saini, V.; Yadav, A. Role of leptin and adiponectin in insulin resistance. Clin. Chim. Acta. 2013, 417, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Considine, R.V.; Sinha, M.K.; Heiman, M.L.; Kriauciunas, A.; Stephens, T.W.; Nyce, M.R.; Ohannesian, J.P.; Marco, C.C.; McKee, L.J.; Bauer, T.L.; et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N. Engl. J. Med. 1996, 334, 292–295. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Aronis, K.N.; Kountouras, J.; Raptis, D.D.; Vasiloglou, M.F.; Mantzoros, C.S. Circulating leptin in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Diabetologia. 2016, 59, 30–43. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Zhao, C.Y.; Wang, W.; Wang, Y.D.; Sun, H.; Cao, W.; Yu, W.Y.; Zhang, L.; Ji, R.; Li, M.; et al. The relationship between hepatic resistin overexpression and inflammation in patients with nonalcoholic steatohepatitis. BMC Gastroenterol. 2014, 14, 39. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane-Stanley, G.H. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Walton, P.A.; Possmayer, F. Mg2-dependent phosphatidate phosphohydrolase of rat lung: Development of an assay employing a defined chemical substrate which reflects the phosphohydrolase activity measured using membrane-bound substrate. Anal. Biochem. 1985, 151, 479–486. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Carl, M.N.; Lakshmanan, M.R.; Porter, J.W. Fatty acid synthase from rat liver. Methods. Enzymol. 1975, 35, 37–44. [Google Scholar]
- Ochoa, S.; Mehler, A.H.; Kornberg, A. Biosynthesis of dicarboxylic acids by carbon dioxide fixation; isolation and properties of an enzyme from pigeon liver catalyzing the reversible oxidative decarboxylation of 1-malic acid. J. Biol. Chem. 1948, 174, 979–1000. [Google Scholar] [PubMed]
- Pitkänen, E.; Pitkänen, O.; Uotila, L. Enzymatic determination of unbound D-mannose in serum. Eur. J. Clin. Chem. Clin. Biochem. 1997, 35, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Lazarow, P.B. Assay of peroxisomal β-oxidation of fatty acids. Methods. Enzymol. 1981, 72, 315–319. [Google Scholar] [PubMed]
- Markwell, M.A.; McGroarty, E.J.; Bieber, L.L.; Tolbert, N.E. The subcellular distribution of carnitine acyltransferases in mammalian liver and kidney. A new peroxisomal enzyme. J. Biol. Chem. 1973, 248, 3426–3432. [Google Scholar] [PubMed]
- Walton, P.A.; Possmayer, F. The role of Mg2+-dependent phosphatidate phosphohydrolase in pulmonary glycerolipid biosynthesis. Biochim. Biophys. Acta. 1984, 796, 346–372. [Google Scholar] [CrossRef]
- Davidson, A.L.; Arion, W.J. Factors underlying significant underestimations of glucokinase activity in crude liver extracts: Physiological implications of higher cellular activity. Arch. Biochem. Biophys. 1987, 253, 156–167. [Google Scholar] [CrossRef]
- Bentle, L.A.; Lardy, H.A. Interaction of anions and divalent metal ions with phosphoenolpyruvate carboxykinase. J. Biol. Chem. 1976, 251, 2916–2921. [Google Scholar] [PubMed]
- Alegre, M.; Ciudad, C.J.; Fillat, C.; Guinovart, J.J. Determination of glucose-6-phosphatase activity using the glucose dehydrogenase-coupled reaction. Anal. Biochem. 1988, 173, 185–189. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Direction | Primer Sequence |
|---|---|---|
| Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) | Forward | 5′-CAAGTTCAACGGCACAGTCAAGG-3′ |
| Reverse | 5′-ACATACTCAGCACCAGCATCACC-3′ | |
| Peroxisome proliferator-activated receptor alpha (PPARα) | Forward | 5′-CCTGAACATCGAGTGTCGAATAT-3′ |
| Reverse | 5′-GGTCTTCTTCTGAATCTTGCAGCT-3′ | |
| PPAR-gamma coactivator 1alpha (PGC1α) | Forward | 5′-AAGTGTGGAACTCTCTGGAACTG-3′ |
| Reverse | 5′-GGGTTATCTTGGTTGGCTTTATG-3′ | |
| PGC1β | Forward | 5′-GGTCCCTGGCTGACATTCAC-3′ |
| Reverse | 5′-GGCACATCGAGGGCAGAG-3′ | |
| Sterol regulatory element-binding transcription factor 1a (SREBP1a) | Forward | 5′-TAGTCCGAAGCCGGGTGGGCGCCGGCGCCAT-3′ |
| Reverse | 5′-GATGTCGTTCAAAACCGCTGTGTGTCCAGTTC-3′ | |
| Acetyl-CoA carboxylase (ACC) | Forward | 5′- GCCTCTTCCTGACAAACGAG-3′ |
| Reverse | 5′-TGACTGCCGAAACATCTCTG-3′ | |
| Fatty acid synthase (FAS) | Forward | 5′-GCTGCGGAAACTTCAGGAAAT-3′ |
| Reverse | 5′-AGAGACGTGTCACTCCTGGACTT-3′ | |
| Steroly-CoA desaturase 1 (SCD1) | Forward | 5′-CCCCTGCGGATCTTCCTTAT-3′ |
| Reverse | 5′-AGGGTCGGCGTGTGTTTCT-3′ | |
| Lipoprotein lipase (LPL) | Forward | 5′-GACTCGCTCTCAGATGCCCTAC-3′ |
| Reverse | 5′-GCCTGGTTGTGTTGCTTGCC-3′ | |
| CD antigen 36 (CD36) | Forward | 5′-TGGTGGATGGTTTCCTAGCCTTTC-3′ |
| Reverse | 5′-TCGCCAACTCCCAGGTACAATC-3′ | |
| Adrenoreceptor beta 3 (ADRB3) | Forward | 5′-ACCAACGTGTTCGTGACT-3′ |
| Reverse | 5′-ACAGCTAGGTAGCGGTCC-3′ | |
| CPT2 | Forward | 5′-GCCTGCTGTTGCGTGACTG-3′ |
| Reverse | 5′-TGGTGGGTACGATGCTGTGC-3′ | |
| Cytochrome c oxidase subunit 8B (COX8B) | Forward | 5′-TGTGGGGATCTCAGCCATAGT-3′ |
| Reverse | 5′-AGTGGGCTAAGACCCATCCTG-3′ | |
| Uncoupling protein 1 (UCP1) | Forward | 5′-AGATCTTCTCAGCCGGAGTTT-3′ |
| Reverse | 5′-CTGTACAGTTTCGGCAATCCT-3′ | |
| Insulin receptor substrate 2 (IRS2) | Forward | 5′-CCCATGTCCCGCCGTGAAG-3′ |
| Reverse | 5′-CTCCAGTGCCAAGGTCTGAAGG-3′ | |
| Phosphoenol pyruvate carboxykinase (PEPCK) | Forward | 5′-ATCATCTTTGGTGGCCGTAG-3′ |
| Reverse | 5′-ATCTTGCCCTTGTGTTCTGC-3′ | |
| Glucose-6-phosphatase (G6PC) | Forward | 5′-GGAGGAAGGATGGAGGAAGGAATG-3′ |
| Reverse | 5′-GGTCAGCAATCACAGACACAAGG-3′ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwon, E.-Y.; Choi, M.-S. Dietary Eriodictyol Alleviates Adiposity, Hepatic Steatosis, Insulin Resistance, and Inflammation in Diet-Induced Obese Mice. Int. J. Mol. Sci. 2019, 20, 1227. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051227
Kwon E-Y, Choi M-S. Dietary Eriodictyol Alleviates Adiposity, Hepatic Steatosis, Insulin Resistance, and Inflammation in Diet-Induced Obese Mice. International Journal of Molecular Sciences. 2019; 20(5):1227. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051227
Chicago/Turabian StyleKwon, Eun-Young, and Myung-Sook Choi. 2019. "Dietary Eriodictyol Alleviates Adiposity, Hepatic Steatosis, Insulin Resistance, and Inflammation in Diet-Induced Obese Mice" International Journal of Molecular Sciences 20, no. 5: 1227. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051227