The Impact of Tumor Heterogeneity on Diagnostics and Novel Therapeutic Strategies in Multiple Myeloma


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Inter-Patient Versus Intra-Tumor Heterogeneity
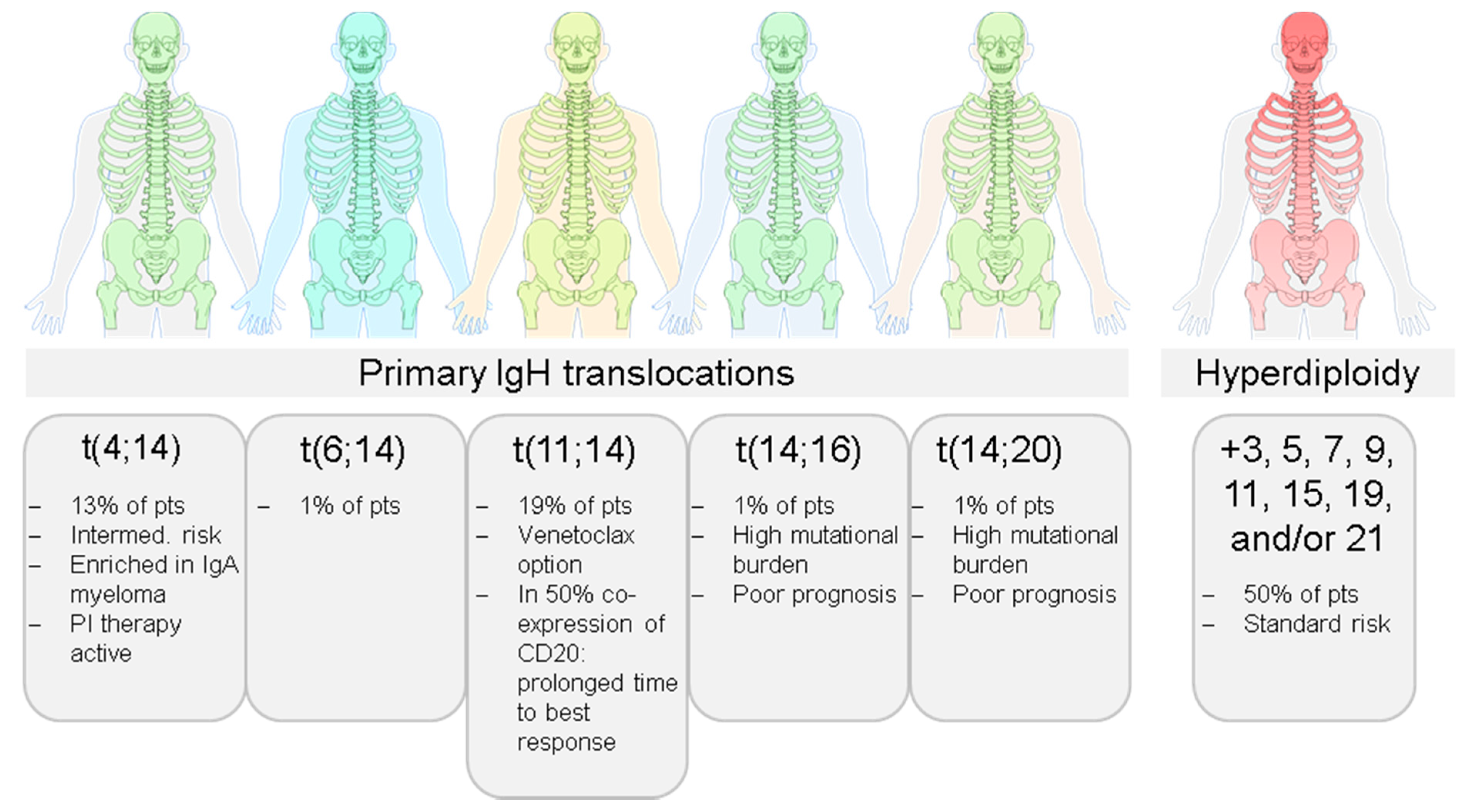
2.1. Inter-Patient Tumor Heterogeneity
2.2. Intra-Tumor Heterogeneity
3. Impact of Heterogeneity on Treatment Strategies
3.1. Targeted Therapy
3.2. Treatment Decisions Based on Disease Risk Status
3.3. Treatment Decisions Based on the Response and the Level of Minimal Residual Disease
4. Overcoming Tumor Heterogeneity
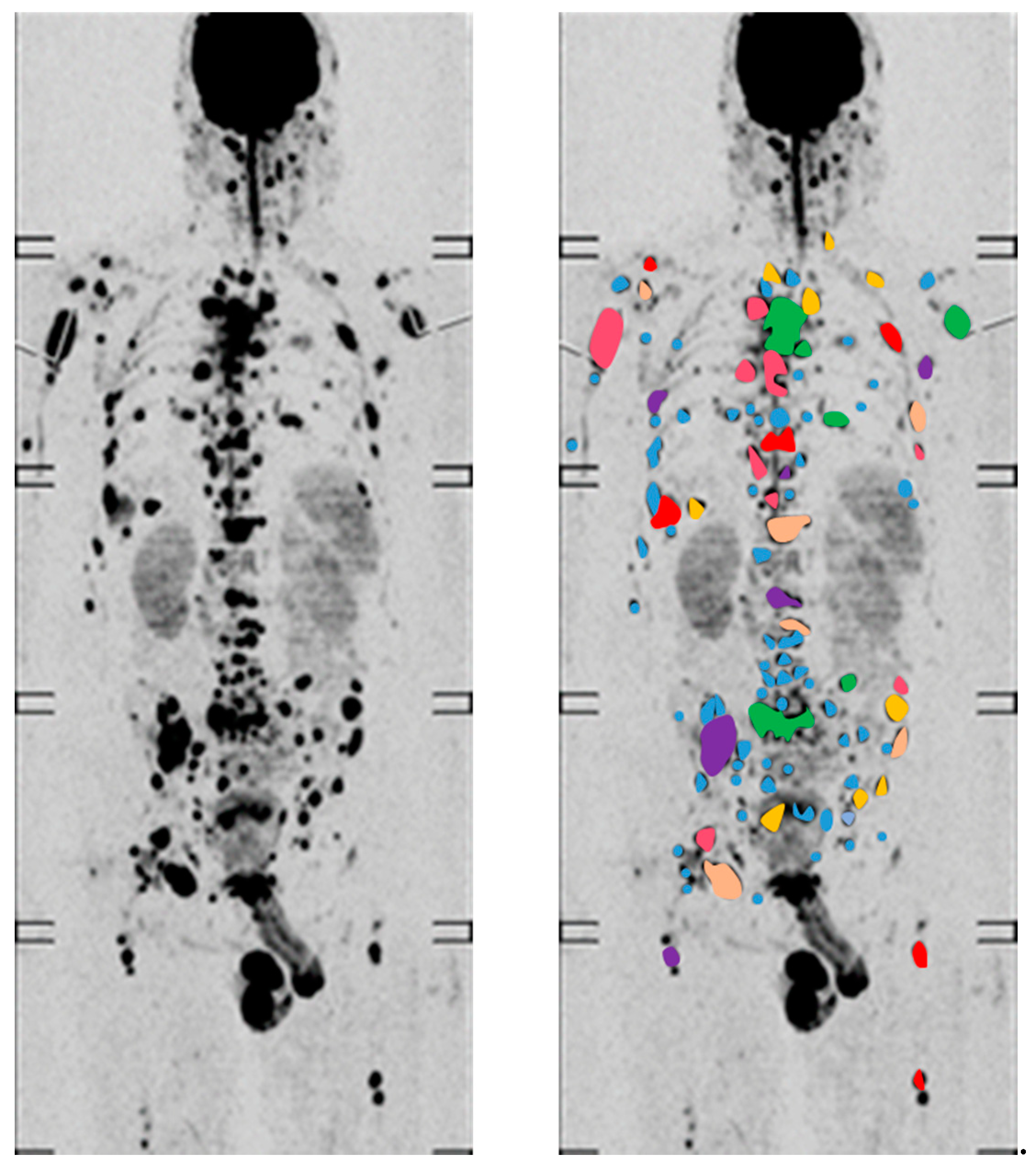
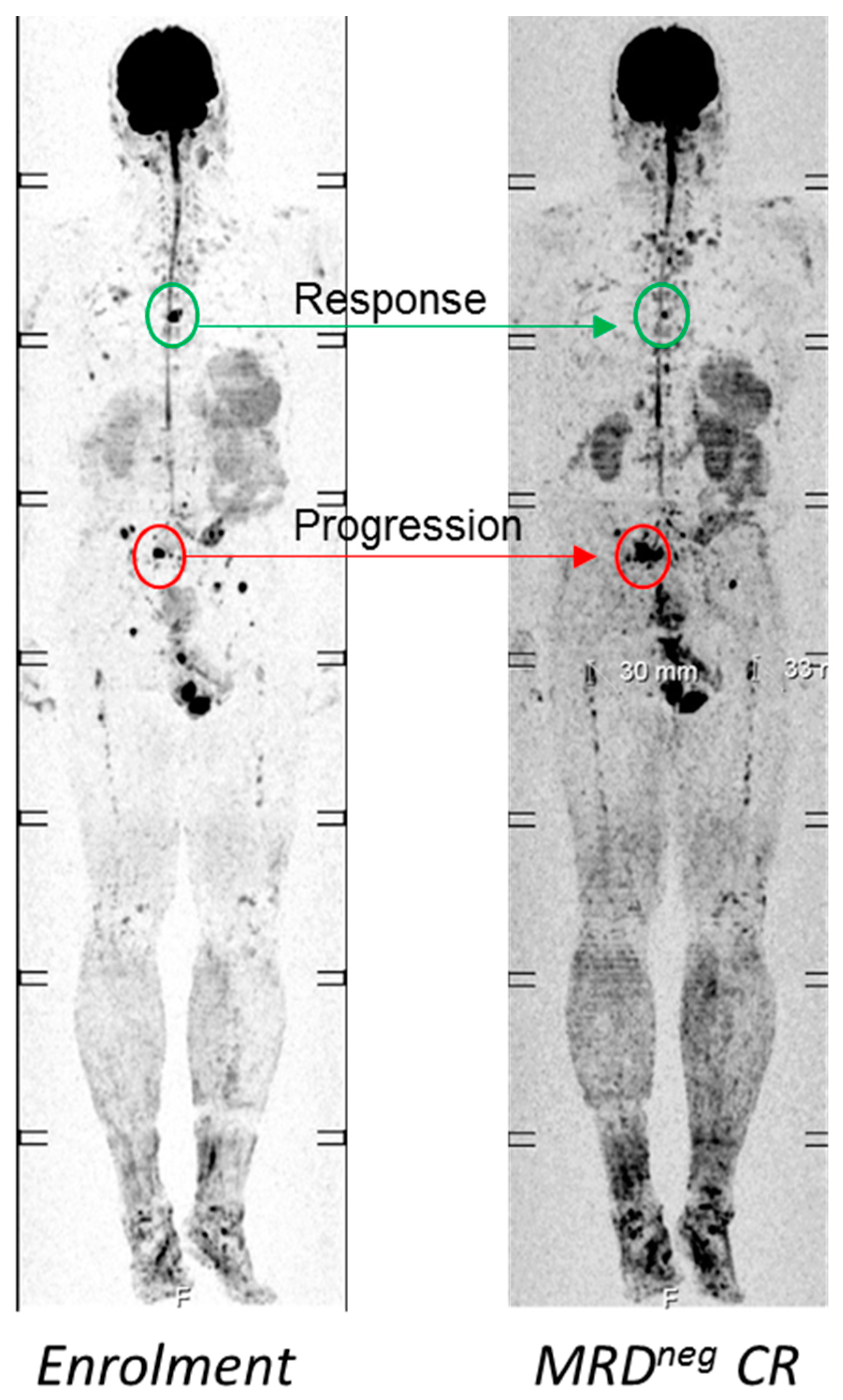
4.1. Functional Imaging to Decode Intra-Tumor Heterogeneity in MM
4.2. Immunotherapy to Target All Tumor Subclones
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Laubach, J.; Richardson, P.; Anderson, K. Multiple myeloma. Annu. Rev. Med. 2011, 62, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, L.M.; Hippe, E.; Hjorth, M.; Holmberg, E.; Westin, J. Renal function in newly diagnosed multiple myeloma--a demographic study of 1353 patients. Eur.J. Haematol. 1994, 53, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.W.; Lokhorst, H.M.; Anderson, K.C.; Richardson, P.G. How I treat plasma cell leukemia. Blood 2012, 120, 2376–2389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasche, L.; Buros, A.; Weinhold, N.; Stein, C.K.; McDonald, J.E.; Chavan, S.S.; Angtuaco, E.; Thanendrarajan, S.; Schinke, C.; Yaccoby, S.; et al. The Clinical Impact of Macrofocal Disease in Multiple Myeloma Differs Between Presentation and Relapse. Blood 2016, 128, 4431. [Google Scholar]
- Barlogie, B.; Mitchell, A.; van Rhee, F.; Epstein, J.; Morgan, G.J.; Crowley, J. Curing myeloma at last: Defining criteria and providing the evidence. Blood 2014, 124, 3043–3051. [Google Scholar] [CrossRef] [PubMed]
- Pawlyn, C.; Morgan, G.J. Evolutionary biology of high-risk multiple myeloma. Nat. Rev. Cancer. 2017, 17, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.J.; Walker, B.A.; Davies, F.E. The genetic architecture of multiple myeloma. Nat. Rev. Cancer. 2012, 12, 335–348. [Google Scholar] [CrossRef]
- Fox, E.J.; Loeb, L.A. Cancer: One cell at a time. Nature 2014, 512, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, J.R. The prognostic significance of cytogenetics and molecular profiling in multiple myeloma. Cancer Genet. 2011, 204, 3–12. [Google Scholar] [CrossRef]
- Went, M.; Sud, A.; Forsti, A.; Halvarsson, B.M.; Weinhold, N.; Kimber, S.; van Duin, M.; Thorleifsson, G.; Holroyd, A.; Johnson, D.C.; et al. Identification of multiple risk loci and regulatory mechanisms influencing susceptibility to multiple myeloma. Nat. Commun. 2018, 9, 3707. [Google Scholar] [CrossRef]
- Weinhold, N.; Johnson, D.C.; Chubb, D.; Chen, B.; Forsti, A.; Hosking, F.J.; Broderick, P.; Ma, Y.P.; Dobbins, S.E.; Hose, D.; et al. The CCND1 c.870G>A polymorphism is a risk factor for t(11;14)(q13;q32) multiple myeloma. Nat. Genet. 2013, 45, 522–525. [Google Scholar] [CrossRef]
- Morgan, G.J.; Rasche, L. Maintaining therapeutic progress in multiple myeloma by integrating genetic and biological advances into the clinic. Expert Rev. Hematol. 2018, 11, 513–523. [Google Scholar] [CrossRef]
- Bolli, N.; Avet-Loiseau, H.; Wedge, D.C.; Van Loo, P.; Alexandrov, L.B.; Martincorena, I.; Dawson, K.J.; Iorio, F.; Nik-Zainal, S.; Bignell, G.R.; et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat. Commun. 2014, 5, 2997. [Google Scholar] [CrossRef] [Green Version]
- Lohr, J.G.; Kim, S.; Gould, J.; Knoechel, B.; Drier, Y.; Cotton, M.J.; Gray, D.; Birrer, N.; Wong, B.; Ha, G.; et al. Genetic interrogation of circulating multiple myeloma cells at single-cell resolution. Sci. Transl. Med. 2016, 8, 363ra147. [Google Scholar] [CrossRef]
- Walker, B.A.; Boyle, E.M.; Wardell, C.P.; Murison, A.; Begum, D.B.; Dahir, N.M.; Proszek, P.Z.; Johnson, D.C.; Kaiser, M.F.; Melchor, L.; et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients with Newly Diagnosed Myeloma. J. Clin. Oncol. 2015, 33, 3911–3920. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.E.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood 2018, 132, 587–597. [Google Scholar] [CrossRef]
- Stein, C.K.; Pawlyn, C.; Chavan, S.; Rasche, L.; Weinhold, N.; Corken, A.; Buros, A.; Sonneveld, P.; Jackson, G.H.; Landgren, O. The varied distribution and impact of RAS codon and other key DNA alterations across the translocation cyclin D subgroups in multiple myeloma. Oncotarget 2017, 8, 27854. [Google Scholar] [CrossRef]
- Bergsagel, P.L.; Kuehl, W.M. Molecular pathogenesis and a consequent classification of multiple myeloma. J. Clin. Oncol. 2005, 23, 6333–6338. [Google Scholar] [CrossRef]
- Zhan, F.; Huang, Y.; Colla, S.; Stewart, J.P.; Hanamura, I.; Gupta, S.; Epstein, J.; Yaccoby, S.; Sawyer, J.; Burington, B.; et al. The molecular classification of multiple myeloma. Blood 2006, 108, 2020–2028. [Google Scholar] [CrossRef] [Green Version]
- Shaughnessy, J.D.; Zhan, F.; Burington, B.E.; Huang, Y.; Colla, S.; Hanamura, I.; Stewart, J.P.; Kordsmeier, B.; Randolph, C.; Williams, D.R. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood 2007, 109, 2276–2284. [Google Scholar] [CrossRef] [Green Version]
- Melchor, L.; Brioli, A.; Wardell, C.P.; Murison, A.; Potter, N.E.; Kaiser, M.F.; Fryer, R.A.; Johnson, D.C.; Begum, D.B.; Hulkki Wilson, S.; et al. Single-cell genetic analysis reveals the composition of initiating clones and phylogenetic patterns of branching and parallel evolution in myeloma. Leukemia 2014, 28, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Bahlis, N.J. Darwinian evolution and tiding clones in multiple myeloma. Blood 2012, 120, 927–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinhold, N.; Ashby, C.; Rasche, L.; Chavan, S.S.; Stein, C.; Stephens, O.W.; Tytarenko, R.; Bauer, M.A.; Meissner, T.; Deshpande, S.; et al. Clonal selection and double-hit events involving tumor suppressor genes underlie relapse in myeloma. Blood 2016, 128, 1735–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keats, J.J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; Van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortum, K.M.; Langer, C.; Monge, J.; Bruins, L.; Zhu, Y.X.; Shi, C.X.; Jedlowski, P.; Egan, J.B.; Ojha, J.; Bullinger, L.; et al. Longitudinal analysis of 25 sequential sample-pairs using a custom multiple myeloma mutation sequencing panel (M(3)P). Ann. Hematol. 2015, 94, 1205–1211. [Google Scholar] [CrossRef]
- Jones, J.R.; Weinhold, N.; Ashby, C.; Walker, B.A.; Wardell, C.; Pawlyn, C.; Rasche, L.; Melchor, L.; Cairns, D.A.; Gregory, W.M.; et al. Clonal evolution in myeloma: The impact of maintenance lenalidomide and depth of response on the genetics and sub-clonal structure of relapsed disease in uniformly treated newly diagnosed patients. Haematologica 2019. [Google Scholar] [CrossRef]
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef] [Green Version]
- Heuck, C.J.; Jethava, Y.; Khan, R.; van Rhee, F.; Zangari, M.; Chavan, S.; Robbins, K.; Miller, S.E.; Matin, A.; Mohan, M.; et al. Inhibiting MEK in MAPK pathway-activated myeloma. Leukemia 2016, 30, 976–980. [Google Scholar] [CrossRef]
- Kortuem, K.M.; Braggio, E.; Bruins, L.; Barrio, S.; Shi, C.S.; Zhu, Y.X.; Tibes, R.; Viswanatha, D.; Votruba, P.; Ahmann, G.; et al. Panel sequencing for clinically oriented variant screening and copy number detection in 142 untreated multiple myeloma patients. Blood Cancer J. 2016, 6, e397. [Google Scholar] [CrossRef]
- Andrulis, M.; Lehners, N.; Capper, D.; Penzel, R.; Heining, C.; Huellein, J.; Zenz, T.; von Deimling, A.; Schirmacher, P.; Ho, A.D.; et al. Targeting the BRAF V600E mutation in multiple myeloma. Cancer Discov. 2013, 3, 862–869. [Google Scholar] [CrossRef]
- Sharman, J.P.; Chmielecki, J.; Morosini, D.; Palmer, G.A.; Ross, J.S.; Stephens, P.J.; Stafl, J.; Miller, V.A.; Ali, S.M. Vemurafenib response in 2 patients with posttransplant refractory BRAF V600E-mutated multiple myeloma. Clin. Lymphoma Myeloma Leuk. 2014, 14, e161–e163. [Google Scholar] [CrossRef]
- Raab, M.S.; Lehners, N.; Xu, J.; Ho, A.D.; Schirmacher, P.; Goldschmidt, H.; Andrulis, M. Spatially divergent clonal evolution in multiple myeloma: Overcoming resistance to BRAF inhibition. Blood 2016, 127, 2155–2157. [Google Scholar] [CrossRef]
- Chavan, S.S.; He, J.; Tytarenko, R.; Deshpande, S.; Patel, P.; Bailey, M.; Stein, C.K.; Stephens, O.; Weinhold, N.; Petty, N.; et al. Bi-allelic inactivation is more prevalent at relapse in multiple myeloma, identifying RB1 as an independent prognostic marker. Blood Cancer J. 2017, 7, e535. [Google Scholar] [CrossRef] [Green Version]
- Kortum, K.M.; Mai, E.K.; Hanafiah, N.H.; Shi, C.X.; Zhu, Y.X.; Bruins, L.; Barrio, S.; Jedlowski, P.; Merz, M.; Xu, J.; et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 2016, 128, 1226–1233. [Google Scholar] [CrossRef] [Green Version]
- Barrio, S.; Stuhmer, T.; Da-Via, M.; Barrio-Garcia, C.; Lehners, N.; Besse, A.; Cuenca, I.; Garitano-Trojaola, A.; Fink, S.; Leich, E.; et al. Spectrum and functional validation of PSMB5 mutations in multiple myeloma. Leukemia 2019, 33, 447–456. [Google Scholar] [CrossRef]
- Jethava, Y.; Mitchell, A.; Epstein, J.; Zangari, M.; Yaccoby, S.; Tian, E.; Waheed, S.; Khan, R.; Papanikolaou, X.; Grazziutti, M.; et al. Adverse metaphase cytogenetics can be overcome by adding bortezomib and thalidomide to fractionated melphalan transplants. Clin. Cancer Res. 2017, 23, 2665–2672. [Google Scholar] [CrossRef]
- Jethava, Y.; Mitchell, A.; Zangari, M.; Waheed, S.; Schinke, C.; Thanendrarajan, S.; Sawyer, J.; Alapat, D.; Tian, E.; Stein, C.; et al. Dose-dense and less dose-intense Total Therapy 5 for gene expression profiling-defined high-risk multiple myeloma. Blood Cancer J. 2016, 6, e453. [Google Scholar] [CrossRef] [Green Version]
- Knop, S.; Liebisch, P.; Hebart, H.; Holler, E.; Engelhardt, M.; Metzner, B.; Peest, D.; Aulitzky, W.; Bunjes, D.W.; Straka, C.; et al. Autologous Followed by Allogeneic Versus TandemAutologous Stem Cell Transplant in Newly Diagnosed FISH-del13q Myeloma. Blood 2014, 124, 43. [Google Scholar]
- Mikhael, J.R.; Dingli, D.; Roy, V.; Reeder, C.B.; Buadi, F.K.; Hayman, S.R.; Dispenzieri, A.; Fonseca, R.; Sher, T.; Kyle, R.A.; et al. Management of newly diagnosed symptomatic multiple myeloma: Updated Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) consensus guidelines 2013. Mayo Clin Proc. 2013, 88, 360–376. [Google Scholar] [CrossRef]
- Meissner, T.; Seckinger, A.; Hemminki, K.; Bertsch, U.; Foersti, A.; Haenel, M.; Duering, J.; Salwender, H.; Goldschmidt, H.; Morgan, G.J.; et al. Profound impact of sample processing delay on gene expression of multiple myeloma plasma cells. BMC Med. Genom. 2015, 8, 85. [Google Scholar] [CrossRef]
- Palumbo, A.; Avet-Loiseau, H.; Oliva, S.; Lokhorst, H.M.; Goldschmidt, H.; Rosinol, L.; Richardson, P.; Caltagirone, S.; Lahuerta, J.J.; Facon, T.; et al. Revised International Staging System for Multiple Myeloma: A Report from International Myeloma Working Group. J. Clin. Oncol. 2015, 33, 2863–2869. [Google Scholar] [CrossRef]
- Weinhold, N.; Heuck, C.J.; Rosenthal, A.; Thanendrarajan, S.; Stein, C.K.; Van Rhee, F.; Zangari, M.; Hoering, A.; Tian, E.; Davies, F.E.; et al. Clinical value of molecular subtyping multiple myeloma using gene expression profiling. Leukemia 2016, 30, 423–430. [Google Scholar] [CrossRef]
- Thanendrarajan, S.; Tian, E.; Qu, P.; Mathur, P.; Schinke, C.; van Rhee, F.; Zangari, M.; Rasche, L.; Weinhold, N.; Alapat, D.; et al. The level of deletion 17p and bi-allelic inactivation of TP53 has a significant impact on clinical outcome in multiple myeloma. Haematologica 2017, 102, e364–e367. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019, 33, 159–170. [Google Scholar] [CrossRef]
- Shah, V.; Johnson, D.C.; Sherborne, A.L.; Ellis, S.; Aldridge, F.M.; Howard-Reeves, J.; Begum, F.; Price, A.; Kendall, J.; Chiecchio, L.; et al. Subclonal TP53 copy number is associated with prognosis in multiple myeloma. Blood 2018, 132, 2465–2469. [Google Scholar] [CrossRef]
- Perrot, A.; Lauwers-Cances, V.; Corre, J.; Robillard, N.; Hulin, C.; Chretien, M.L.; Dejoie, T.; Maheo, S.; Stoppa, A.M.; Pegourie, B.; et al. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood 2018, 132, 2456–2464. [Google Scholar] [CrossRef]
- Landgren, O.; Lu, S.X.; Hultcrantz, M. MRD Testing in Multiple Myeloma: The Main Future Driver for Modern Tailored Treatment. Semin. Hematol. 2018, 55, 44–50. [Google Scholar] [CrossRef]
- Schinke, C.; Hoering, A.; Wang, H.; Carlton, V.; Thanandrarajan, S.; Deshpande, S.; Patel, P.; Molnar, G.; Susanibar, S.; Mohan, M.; et al. The prognostic value of the depth of response in multiple myeloma depends on the time of assessment, risk status and molecular subtype. Haematologica 2017, 102, e313–e316. [Google Scholar] [CrossRef] [Green Version]
- Rasche, L.; Alapat, D.; Kumar, M.; Gershner, G.; McDonald, J.; Wardell, C.P.; Samant, R.; Van Hemert, R.; Epstein, J.; Williams, A.F.; et al. Combination of flow cytometry and functional imaging for monitoring of residual disease in myeloma. Leukemia 2018. [Google Scholar] [CrossRef]
- Bartel, T.B.; Haessler, J.; Brown, T.L.; Shaughnessy, J.D., Jr.; van Rhee, F.; Anaissie, E.; Alpe, T.; Angtuaco, E.; Walker, R.; Epstein, J.; et al. F18-fluorodeoxyglucose positron emission tomography in the context of other imaging techniques and prognostic factors in multiple myeloma. Blood 2009, 114, 2068–2076. [Google Scholar] [CrossRef] [Green Version]
- Usmani, S.Z.; Mitchell, A.; Waheed, S.; Crowley, J.; Hoering, A.; Petty, N.; Brown, T.; Bartel, T.; Anaissie, E.; van Rhee, F.; et al. Prognostic implications of serial 18-fluoro-deoxyglucose emission tomography in multiple myeloma treated with total therapy 3. Blood 2013, 121, 1819–1823. [Google Scholar] [CrossRef] [Green Version]
- Waheed, S.; Mitchell, A.; Usmani, S.; Epstein, J.; Yaccoby, S.; Nair, B.; van Hemert, R.; Angtuaco, E.; Brown, T.; Bartel, T.; et al. Standard and novel imaging methods for multiple myeloma: Correlates with prognostic laboratory variables including gene expression profiling data. Haematologica 2013, 98, 71–78. [Google Scholar] [CrossRef]
- Rasche, L.; Angtuaco, E.J.; Alpe, T.L.; Gershner, G.H.; McDonald, J.E.; Samant, R.S.; Kumar, M.; Van Hemert, R.; Epstein, J.; Deshpande, S.; et al. The presence of large focal lesions is a strong independent prognostic factor in multiple myeloma. Blood 2018, 132, 59–66. [Google Scholar] [CrossRef]
- Davies, F.E.; Rosenthal, A.; Rasche, L.; Petty, N.M.; McDonald, J.E.; Ntambi, J.A.; Steward, D.M.; Panozzo, S.B.; van Rhee, F.; Zangari, M.; et al. Treatment to suppression of focal lesions on positron emission tomography-computed tomography is a therapeutic goal in newly diagnosed multiple myeloma. Haematologica 2018, 103, 1047–1053. [Google Scholar] [CrossRef] [Green Version]
- Moreau, P.; Attal, M.; Caillot, D.; Macro, M.; Karlin, L.; Garderet, L.; Facon, T.; Benboubker, L.; Escoffre-Barbe, M.; Stoppa, A.M.; et al. Prospective Evaluation of Magnetic Resonance Imaging and [(18)F]Fluorodeoxyglucose Positron Emission Tomography-Computed Tomography at Diagnosis and Before Maintenance Therapy in Symptomatic Patients with Multiple Myeloma Included in the IFM/DFCI 2009 Trial: Results of the IMAJEM Study. J. Clin. Oncol. 2017, 35, 2911–2918. [Google Scholar]
- Rasche, L.; Angtuaco, E.; McDonald, J.E.; Buros, A.; Stein, C.; Pawlyn, C.; Thanendrarajan, S.; Schinke, C.; Samant, R.; Yaccoby, S.; et al. Low expression of hexokinase-2 is associated with false-negative FDG-positron emission tomography in multiple myeloma. Blood 2017, 130, 30–34. [Google Scholar] [CrossRef]
- Mishima, Y.; Paiva, B.; Shi, J.; Park, J.; Manier, S.; Takagi, S.; Massoud, M.; Perilla-Glen, A.; Aljawai, Y.; Huynh, D.; et al. The Mutational Landscape of Circulating Tumor Cells in Multiple Myeloma. Cell Rep. 2017, 19, 218–224. [Google Scholar] [CrossRef] [Green Version]
- Kis, O.; Kaedbey, R.; Chow, S.; Danesh, A.; Dowar, M.; Li, T.; Li, Z.; Liu, J.; Mansour, M.; Masih-Khan, E.; et al. Circulating tumour DNA sequence analysis as an alternative to multiple myeloma bone marrow aspirates. Nat. Commun. 2017, 8, 15086. [Google Scholar] [CrossRef] [Green Version]
- Mithraprabhu, S.; Khong, T.; Ramachandran, M.; Chow, A.; Klarica, D.; Mai, L.; Walsh, S.; Broemeling, D.; Marziali, A.; Wiggin, M.; et al. Circulating tumour DNA analysis demonstrates spatial mutational heterogeneity that coincides with disease relapse in myeloma. Leukemia 2017, 31, 1695–1705. [Google Scholar] [CrossRef]
- Mailankody, S.; Devlin, S.M.; Korde, N.; Lendvai, N.; Lesokhin, A.; Landau, H.; Hassoun, H.; Ballagi, A.; Ekman, D.; Chung, D.J.; et al. Proteomic profiling in plasma cell disorders: a feasibility study. Leuk Lymphoma 2017, 58, 1757–1759. [Google Scholar] [CrossRef]
- Rasche, L.; Weinhold, N.; Morgan, G.J.; van Rhee, F.; Davies, F.E. Immunologic approaches for the treatment of multiple myeloma. Cancer Treat. Rev. 2017, 55, 190–199. [Google Scholar] [CrossRef]
- Rasche, L.; Rollig, C.; Stuhler, G.; Danhof, S.; Mielke, S.; Grigoleit, G.U.; Dissen, L.; Schemmel, L.; Middeke, J.M.; Rucker, V.; et al. Allogeneic Hematopoietic Cell Transplantation in Multiple Myeloma: Focus on Longitudinal Assessment of Donor Chimerism, Extramedullary Disease, and High-Risk Cytogenetic Features. Biol. Blood Marrow Transplant. 2016, 22, 1988–1996. [Google Scholar] [CrossRef]
- Rasche, L.; Bernard, C.; Topp, M.S.; Kapp, M.; Duell, J.; Wesemeier, C.; Haralambieva, E.; Maeder, U.; Einsele, H.; Knop, S. Features of extramedullary myeloma relapse: high proliferation, minimal marrow involvement, adverse cytogenetics: a retrospective single-center study of 24 cases. Ann. Hematol. 2012, 91, 1031–1037. [Google Scholar] [CrossRef]
- Weinstock, M.; Aljawai, Y.; Morgan, E.A.; Laubach, J.; Gannon, M.; Roccaro, A.M.; Varga, C.; Mitsiades, C.S.; Paba-Prada, C.; Schlossman, R.; et al. Incidence and clinical features of extramedullary multiple myeloma in patients who underwent stem cell transplantation. Br. J. Haematol. 2015, 169, 851–858. [Google Scholar] [CrossRef] [Green Version]
- Usmani, S.Z.; Heuck, C.; Mitchell, A.; Szymonifka, J.; Nair, B.; Hoering, A.; Alsayed, Y.; Waheed, S.; Haider, S.; Restrepo, A.; et al. Extramedullary disease portends poor prognosis in multiple myeloma and is over-represented in high-risk disease even in the era of novel agents. Haematologica 2012, 97, 1761–1767. [Google Scholar] [CrossRef] [Green Version]
- Alegre, A.; Granda, A.; Martinez-Chamorro, C.; Diaz-Mediavilla, J.; Martinez, R.; Garcia-Larana, J.; Lahuerta, J.J.; Sureda, A.; Blade, J.; de la Rubia, J.; et al. Spanish Registry of Transplants in Multiple, M.; Spanish Group of Hemopoietic, T.; Pethema, Different patterns of relapse after autologous peripheral blood stem cell transplantation in multiple myeloma: clinical results of 280 cases from the Spanish Registry. Haematologica 2002, 87, 609–614. [Google Scholar]
- Vincent, L.; Ceballos, P.; Plassot, C.; Meniane, J.C.; Quittet, P.; Navarro, R.; Cyteval, C.; Szablewski, V.; Lu, Z.Y.; Kanouni, T.; et al. Factors influencing extramedullary relapse after allogeneic transplantation for multiple myeloma. Blood Cancer J. 2015, 5, e341. [Google Scholar] [CrossRef]
- Perez-Simon, J.A.; Sureda, A.; Fernandez-Aviles, F.; Sampol, A.; Cabrera, J.R.; Caballero, D.; Martino, R.; Petit, J.; Tomas, J.F.; Moraleda, J.M.; et al. Reduced-intensity conditioning allogeneic transplantation is associated with a high incidence of extramedullary relapses in multiple myeloma patients. Leukemia 2006, 20, 542–545. [Google Scholar] [CrossRef] [Green Version]
- Minnema, M.C.; van de Donk, N.W.; Zweegman, S.; Hegenbart, U.; Schonland, S.; Raymakers, R.; Zijlmans, J.M.; Kersten, M.J.; Bos, G.M.; Lokhorst, H.M. Extramedullary relapses after allogeneic non-myeloablative stem cell transplantation in multiple myeloma patients do not negatively affect treatment outcome. Bone Marrow Transplant 2008, 41, 779–784. [Google Scholar] [CrossRef] [Green Version]
- Zeiser, R.; Deschler, B.; Bertz, H.; Finke, J.; Engelhardt, M. Extramedullary vs medullary relapse after autologous or allogeneic hematopoietic stem cell transplantation (HSCT) in multiple myeloma (MM) and its correlation to clinical outcome. Bone Marrow Transplant 2004, 34, 1057–1065. [Google Scholar] [CrossRef] [Green Version]
- Pick, M.; Vainstein, V.; Goldschmidt, N.; Lavie, D.; Libster, D.; Gural, A.; Grisariu, S.; Avni, B.; Ben Yehuda, D.; Gatt, M.E.; et al. Daratumumab resistance is frequent in advanced-stage multiple myeloma patients irrespective of CD38 expression and is related to dismal prognosis. Eur.J. Haematol. 2018, 100, 494–501. [Google Scholar] [CrossRef]
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560. [Google Scholar] [CrossRef]
- Zhao, W.H.; Liu, J.; Wang, B.Y.; Chen, Y.X.; Cao, X.M.; Yang, Y.; Zhang, Y.L.; Wang, F.X.; Zhang, P.Y.; Lei, B.; et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J. Hematol. Oncol. 2018, 11, 141. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasche, L.; Kortüm, K.M.; Raab, M.S.; Weinhold, N. The Impact of Tumor Heterogeneity on Diagnostics and Novel Therapeutic Strategies in Multiple Myeloma. Int. J. Mol. Sci. 2019, 20, 1248. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051248
Rasche L, Kortüm KM, Raab MS, Weinhold N. The Impact of Tumor Heterogeneity on Diagnostics and Novel Therapeutic Strategies in Multiple Myeloma. International Journal of Molecular Sciences. 2019; 20(5):1248. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051248
Chicago/Turabian StyleRasche, Leo, K. Martin Kortüm, Marc S. Raab, and Niels Weinhold. 2019. "The Impact of Tumor Heterogeneity on Diagnostics and Novel Therapeutic Strategies in Multiple Myeloma" International Journal of Molecular Sciences 20, no. 5: 1248. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051248