Complementary Role of P2 and Adenosine Receptors in ATP Induced-Anti-Apoptotic Effects Against Hypoxic Injury of HUVECs

 , , , , ,
, , , , ,
Abstract
:
1. Introduction
2. Results
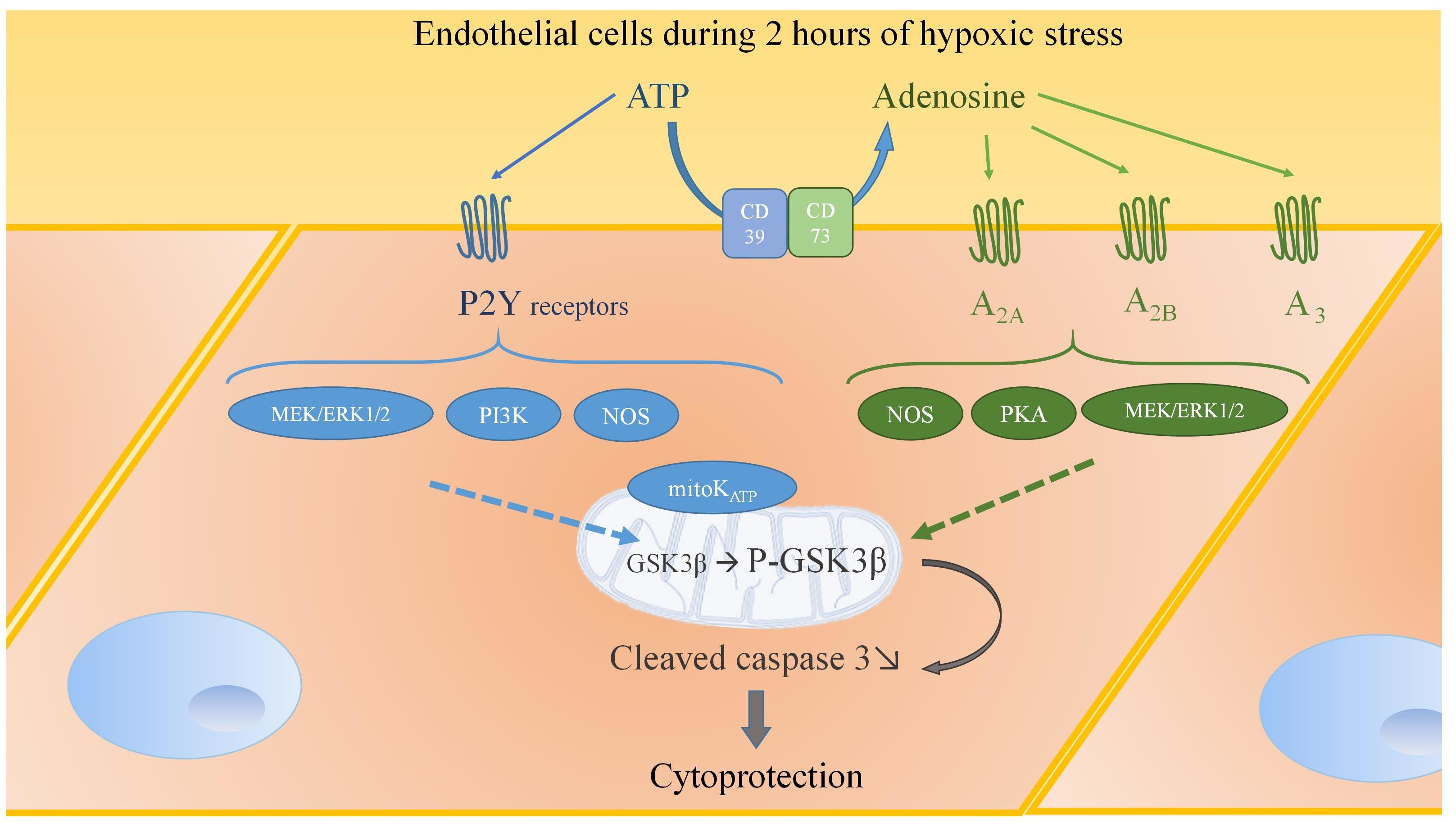
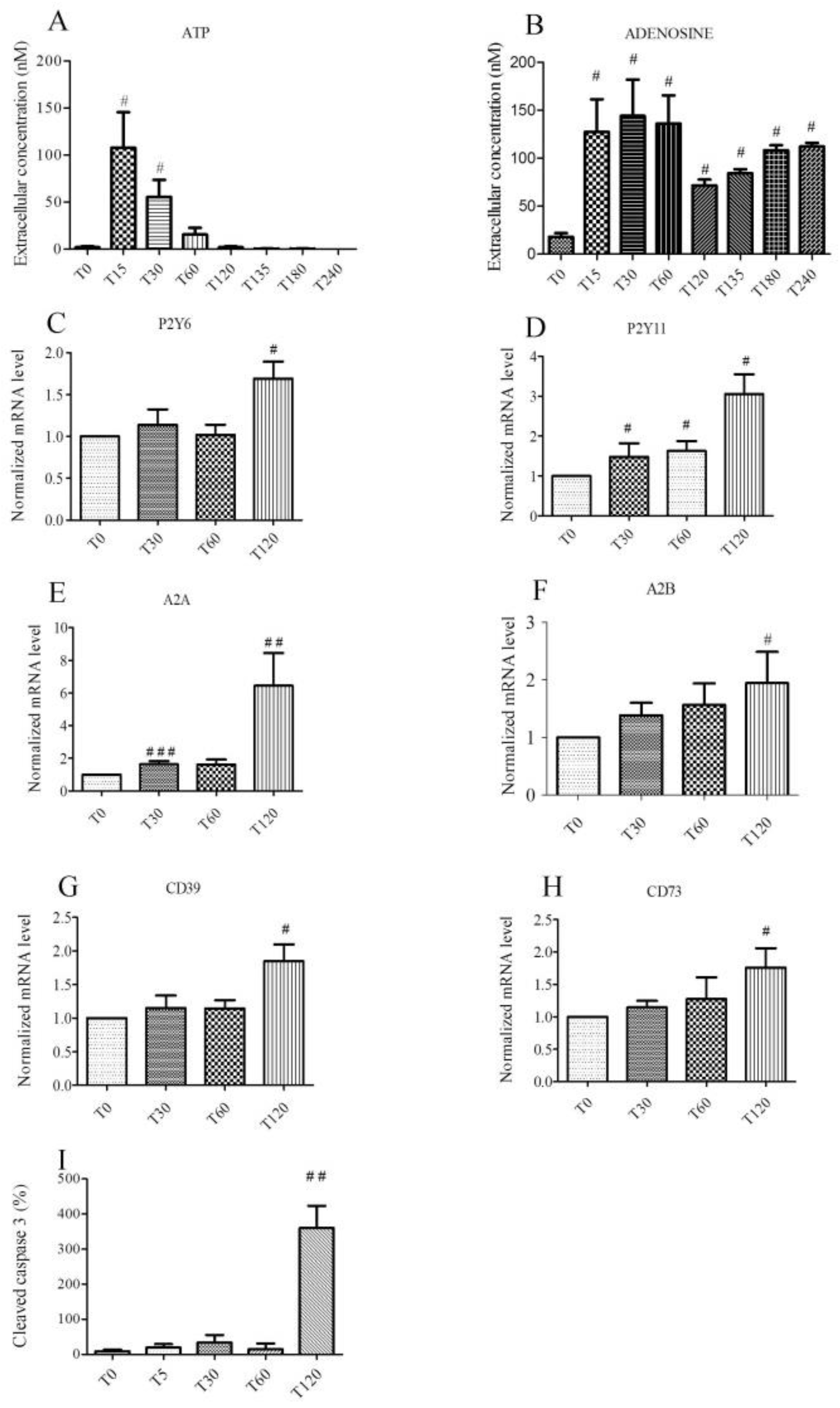
2.1. Hypoxic Stress Induces both ATP and Adenosine Release from Endothelial Cells
2.2. Hypoxic Stress Induces Overexpression of P2Y6, P2Y11, A2A, A2B, CD 39, and CD 73 mRNAs
2.3. Hypoxia Promotes Apoptosis in Endothelial Cells
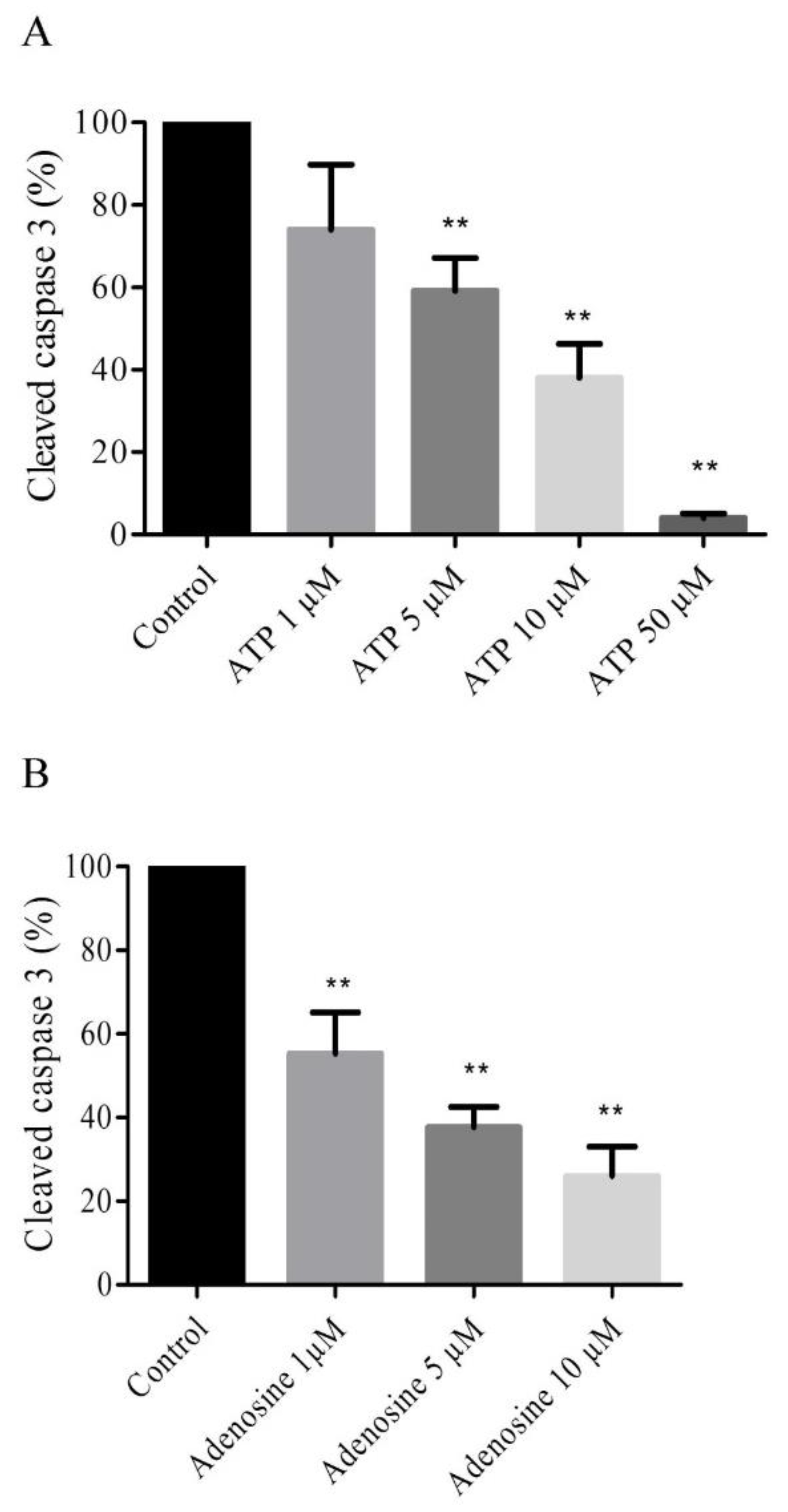
2.4. Extracellular ATP and Adenosine Induced an Anti-Apoptotic Effect
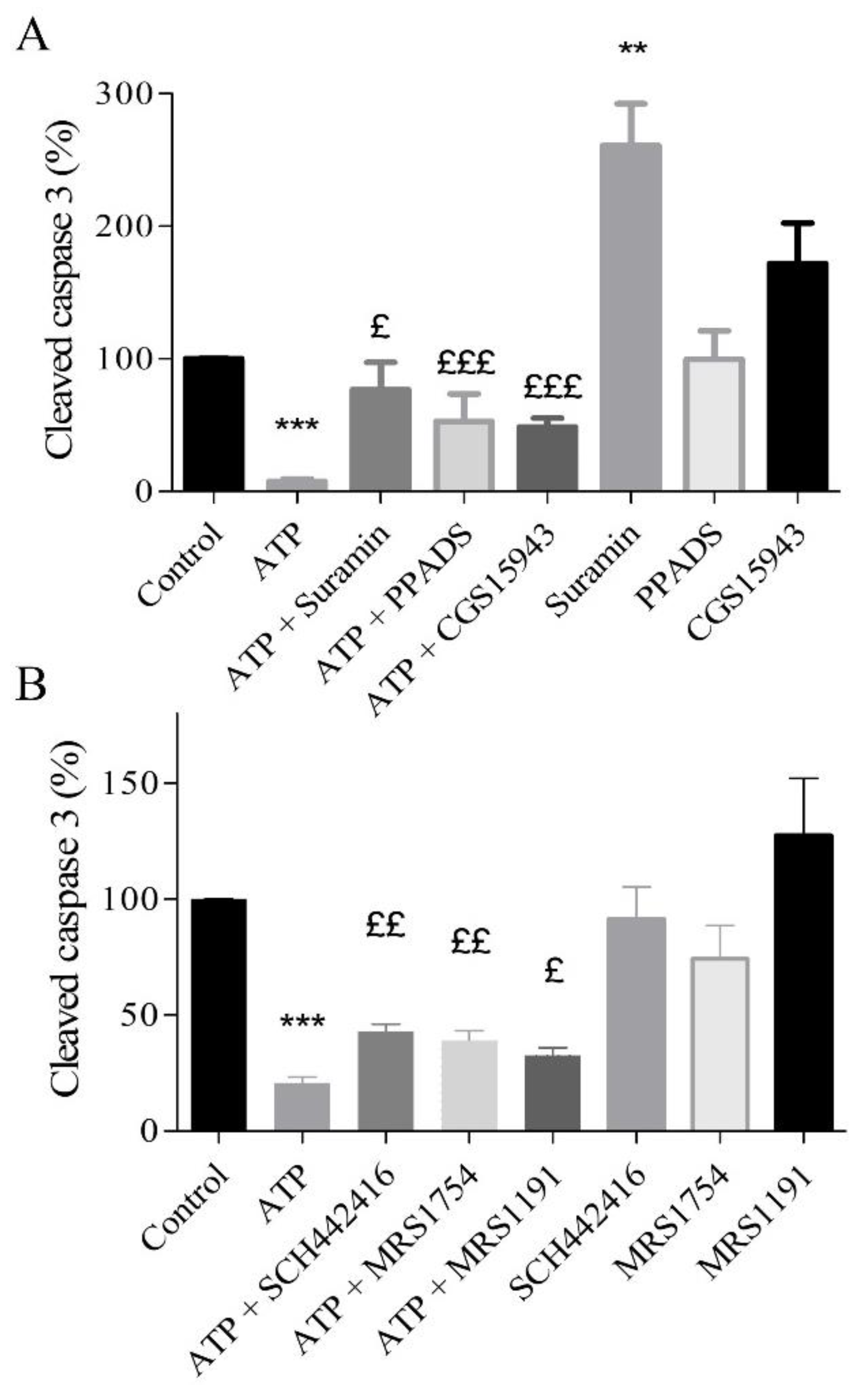
2.5. Protective Effects of Extracellular ATP Are Mediated by P2 and Adenosine Receptors
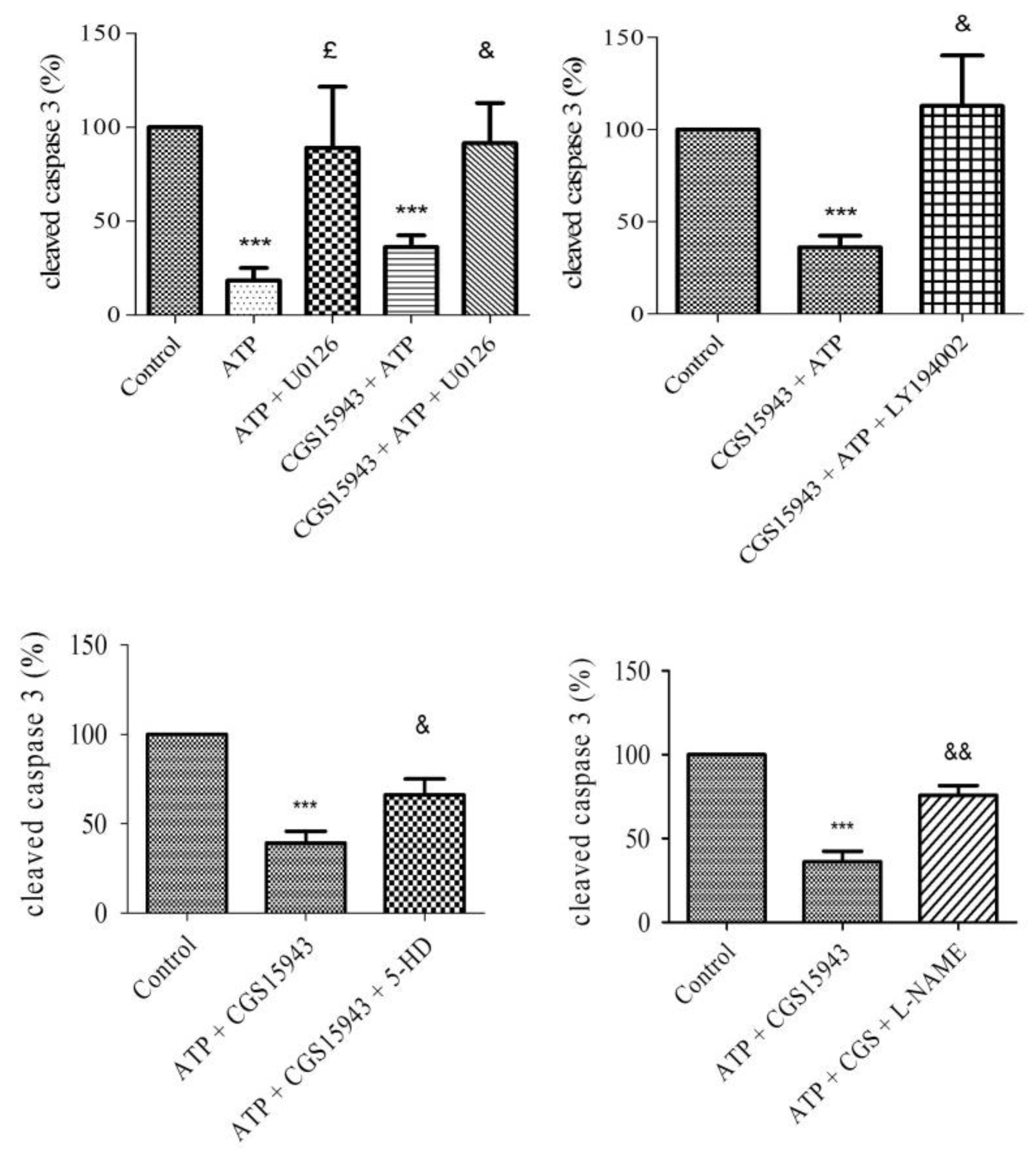
2.6. P2 Receptor-Mediated Anti-Apoptotic Effect of ATP Involves PI3K, MEK/ERK1/2, mitoKATP, and NOS Pathways
2.7. A2A, A2B, and A3 Receptors are Involved in Endothelial Protection Induced by Extracellular ATP
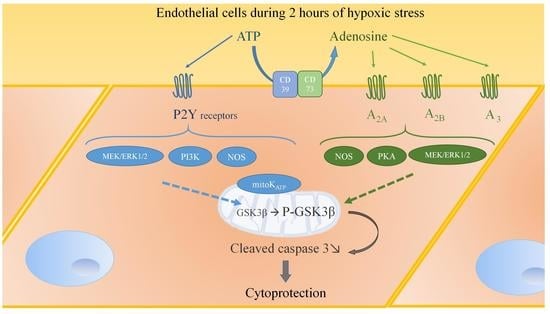
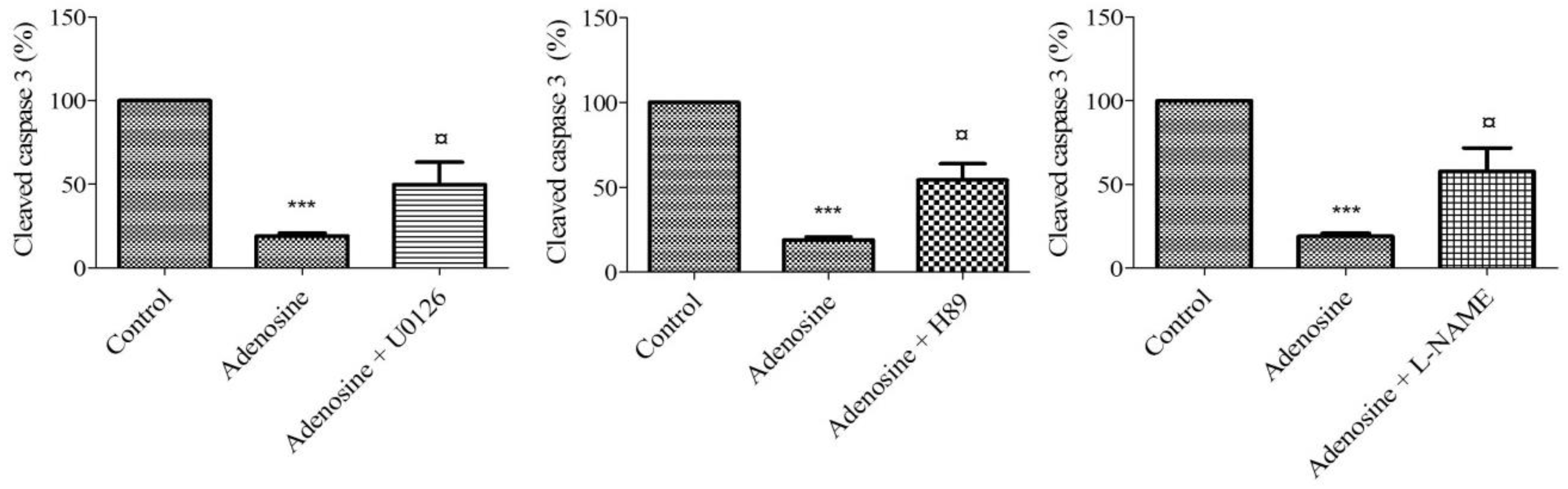
2.8. Adenosine Receptor-Mediated Anti-Apoptotic Effect Involves MEK/ERK1/2, PKA, and NOS
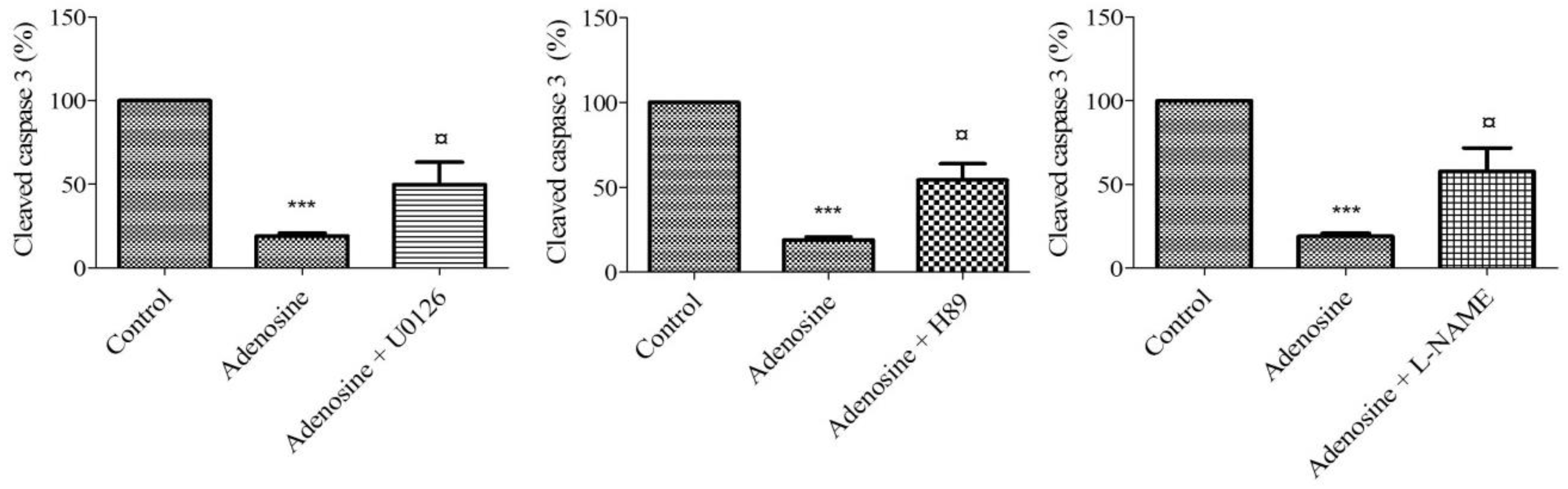
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Experimental Protocols
4.3. Quantification of Nucleotides in Extracellular Medium
4.4. RT-PCR
4.5. Immunoblotting
4.6. LDH Activity
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgements
Conflicts of Interest
References
- Mason, J.C. Cytoprotective pathways in the vascular endothelium. Do they represent a viable therapeutic target? Vascul. Pharmacol. 2016, 86, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Förstermann, U. Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014, 237, 208–219. [Google Scholar] [CrossRef]
- Fisher, M. Injuries to the vascular endothelium: Vascular wall and endothelial dysfunction. Rev. Neurol. Dis. 2008, 5 (Suppl. 1), S4–S11. [Google Scholar]
- Singhal, A.K.; Symons, J.D.; Boudina, S.; Jaishy, B.; Shiu, Y.-T. Role of Endothelial Cells in Myocardial Ischemia-Reperfusion Injury. Vasc. Dis. Prev. 2010, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Scarabelli, T.; Stephanou, A.; Rayment, N.; Pasini, E.; Comini, L.; Curello, S.; Ferrari, R.; Knight, R.; Latchman, D. Apoptosis of endothelial cells precedes myocyte cell apoptosis in ischemia/reperfusion injury. Circulation 2001, 104, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Parolari, A.; Rubini, P.; Cannata, A.; Bonati, L.; Alamanni, F.; Tremoli, E.; Biglioli, P. Endothelial damage during myocardial preservation and storage. Ann. Thorac. Surg. 2002, 73, 682–690. [Google Scholar] [CrossRef]
- Yang, Q.; He, G.-W.; Underwood, M.J.; Yu, C.-M. Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: Perspectives and implications for postischemic myocardial protection. Am. J. Transl. Res. 2016, 8, 765–777. [Google Scholar]
- Djerada, Z.; Feliu, C.; Richard, V.; Millart, H. Current knowledge on the role of P2Y receptors in cardioprotection against ischemia-reperfusion. Pharmacol. Res. 2016, 118, 5–18. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef] [Green Version]
- Erlinge, D.; Burnstock, G. P2 receptors in cardiovascular regulation and disease. Purinergic Signal. 2008, 4, 1–20. [Google Scholar] [CrossRef]
- Djerada, Z.; Millart, H. Intracellular NAADP increase induced by extracellular NAADP via the P2Y11-like receptor. Biochem. Biophys. Res. Commun. 2013, 436, 199–203. [Google Scholar] [CrossRef]
- Wee, S.; Peart, J.N.; Headrick, J.P. P2 purinoceptor-mediated cardioprotection in ischemic-reperfused mouse heart. J. Pharmacol. Exp. Ther. 2007, 323, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, H.; Otani, H.; Lu, K.; Uchiyama, T.; Kido, M.; Imamura, H. Complementary role of extracellular ATP and adenosine in ischemic preconditioning in the rat heart. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1810–H1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, M.V.; Downey, J.M. Adenosine: Trigger and mediator of cardioprotection. Basic Res. Cardiol. 2008, 103, 203–215. [Google Scholar] [CrossRef]
- Djerada, Z.; Peyret, H.; Dukic, S.; Millart, H. Extracellular NAADP affords cardioprotection against ischemia and reperfusion injury and involves the P2Y11-like receptor. Biochem. Biophys. Res. Commun. 2013, 434, 428–433. [Google Scholar] [CrossRef] [PubMed]
- Avanzato, D.; Genova, T.; Fiorio Pla, A.; Bernardini, M.; Bianco, S.; Bussolati, B.; Mancardi, D.; Giraudo, E.; Maione, F.; Cassoni, P.; et al. Activation of P2X7 and P2Y11 purinergic receptors inhibits migration and normalizes tumor-derived endothelial cells via cAMP signaling. Sci. Rep. 2016, 6, 32602. [Google Scholar] [CrossRef] [Green Version]
- YU, J.; HUANG, X.; WU, Q.; WANG, J.; YU, X.; ZHAO, P. Effect of A2A receptor antagonist (SCH 442416) on the mRNA expression of glutamate aspartate transporter and glutamine synthetase in rat retinal Müller cells under hypoxic conditions in vitro. Exp. Ther. Med. 2012, 3, 803–806. [Google Scholar] [CrossRef] [Green Version]
- Salie, R.; Moolman, J.A.; Lochner, A. The mechanism of beta-adrenergic preconditioning: Roles for adenosine and ROS during triggering and mediation. Basic Res. Cardiol. 2012, 107, 281. [Google Scholar] [CrossRef]
- Urban, D.; Härtel, F.V.; Gadiraju, K.; Gündüz, D.; Aslam, M.; Piper, H.M.; Noll, T. Extracellular ATP attenuates ischemia-induced caspase-3 cleavage in human endothelial cells. Biochem. Biophys. Res. Commun. 2012, 425, 230–236. [Google Scholar] [CrossRef]
- Millart, H.; Alouane, L.; Oszust, F.; Chevallier, S.; Robinet, A. Involvement of P2Y receptors in pyridoxal-5′-phosphate-induced cardiac preconditioning. Fundam. Clin. Pharmacol. 2009, 23, 279–292. [Google Scholar] [CrossRef] [Green Version]
- Alm, R.; Edvinsson, L.; Malmsjö, M. Organ culture: A new model for vascular endothelium dysfunction. BMC Cardiovasc. Disord. 2002, 2, 8. [Google Scholar] [CrossRef]
- Casanello, P.; Torres, A.; Sanhueza, F.; González, M.; Farías, M.; Gallardo, V.; Pastor-Anglada, M.; San Martín, R.; Sobrevia, L. Equilibrative nucleoside transporter 1 expression is downregulated by hypoxia in human umbilical vein endothelium. Circ. Res. 2005, 97, 16–24. [Google Scholar] [CrossRef]
- Kumar, S.; Reusch, H.P.; Ladilov, Y. Acidic pre-conditioning suppresses apoptosis and increases expression of Bcl-xL in coronary endothelial cells under simulated ischaemia. J. Cell. Mol. Med. 2008, 12, 1584–1592. [Google Scholar] [CrossRef]
- Gerasimovskaya, E.V.; Ahmad, S.; White, C.W.; Jones, P.L.; Carpenter, T.C.; Stenmark, K.R. Extracellular ATP is an autocrine/paracrine regulator of hypoxia-induced adventitial fibroblast growth. Signaling through extracellular signal-regulated kinase-1/2 and the Egr-1 transcription factor. J. Biol. Chem. 2002, 277, 44638–44650. [Google Scholar] [CrossRef]
- Bodin, P.; Burnstock, G. Synergistic effect of acute hypoxia on flow-induced release of ATP from cultured endothelial cells. Experientia 1995, 51, 256–259. [Google Scholar] [CrossRef]
- Bodin, P.; Burnstock, G. Purinergic signalling: ATP release. Neurochem. Res. 2001, 26, 959–969. [Google Scholar] [CrossRef]
- Gorman, M.W.; Feigl, E.O.; Buffington, C.W. Human Plasma ATP Concentration. Clin. Chem. 2007, 53, 318–325. [Google Scholar] [CrossRef]
- Picher, M.; Burch, L.H.; Boucher, R.C. Metabolism of P2 receptor agonists in human airways: Implications for mucociliary clearance and cystic fibrosis. J. Biol. Chem. 2004, 279, 20234–20241. [Google Scholar] [CrossRef]
- Yegutkin, G.G. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim. Biophys. Acta 2008, 1783, 673–694. [Google Scholar] [CrossRef] [Green Version]
- Koeppen, M.; Eckle, T.; Eltzschig, H.K. Chapter 6—Interplay of Hypoxia and A2B Adenosine Receptors in Tissue Protection. In Advances in Pharmacology; Pharmacology of Purine and Pyrimidine Receptors; Jacobson, K.A., Linden, J., Eds.; Academic Press: New York, NY, USA, 2011; Volume 61, pp. 145–186. [Google Scholar]
- Conde, S.V.; Monteiro, E.C. Hypoxia induces adenosine release from the rat carotid body. J. Neurochem. 2004, 89, 1148–1156. [Google Scholar] [CrossRef] [Green Version]
- Harrison, G.J.; Willis, R.J.; Headrick, J.P. Extracellular adenosine levels and cellular energy metabolism in ischemically preconditioned rat heart. Cardiovasc. Res. 1998, 40, 74–87. [Google Scholar] [CrossRef] [Green Version]
- Chaudary, N.; Naydenova, Z.; Shuralyova, I.; Coe, I.R. Hypoxia regulates the adenosine transporter, mENT1, in the murine cardiomyocyte cell line, HL-1. Cardiovasc. Res. 2004, 61, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Löffler, M.; Morote-Garcia, J.C.; Eltzschig, S.A.; Coe, I.R.; Eltzschig, H.K. Physiological roles of vascular nucleoside transporters. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1004–1013. [Google Scholar] [CrossRef]
- Morote-Garcia, J.C.; Rosenberger, P.; Nivillac, N.M.I.; Coe, I.R.; Eltzschig, H.K. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology 2009, 136, 607–618. [Google Scholar] [CrossRef]
- Rose, J.B.; Naydenova, Z.; Bang, A.; Eguchi, M.; Sweeney, G.; Choi, D.-S.; Hammond, J.R.; Coe, I.R. Equilibrative nucleoside transporter 1 plays an essential role in cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H771–H777. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Faigle, M.; Knapp, S.; Karhausen, J.; Ibla, J.; Rosenberger, P.; Odegard, K.C.; Laussen, P.C.; Thompson, L.F.; Colgan, S.P. Endothelial catabolism of extracellular adenosine during hypoxia: The role of surface adenosine deaminase and CD26. Blood 2006, 108, 1602–1610. [Google Scholar] [CrossRef]
- Wang, L.; Karlsson, L.; Moses, S.; Hultgårdh-Nilsson, A.; Andersson, M.; Borna, C.; Gudbjartsson, T.; Jern, S.; Erlinge, D. P2 receptor expression profiles in human vascular smooth muscle and endothelial cells. J. Cardiovasc. Pharmacol. 2002, 40, 841–853. [Google Scholar] [CrossRef]
- Burnstock, G.; Ralevic, V. Purinergic signaling and blood vessels in health and disease. Pharmacol. Rev. 2014, 66, 102–192. [Google Scholar] [CrossRef]
- Burnstock, G. Purine and pyrimidine receptors. Cell. Mol. Life Sci. 2007, 64, 1471–1483. [Google Scholar] [CrossRef]
- Beitner-Johnson, D.; Rust, R.T.; Hsieh, T.C.; Millhorn, D.E. Hypoxia activates Akt and induces phosphorylation of GSK-3 in PC12 cells. Cell. Signal. 2001, 13, 23–27. [Google Scholar] [CrossRef]
- Miura, T.; Tanno, M.; Sato, T. Mitochondrial kinase signalling pathways in myocardial protection from ischaemia/reperfusion-induced necrosis. Cardiovasc. Res. 2010, 88, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Hu, S.M.; Xie, H.; Qiao, S.G.; Liu, H.; Liu, C.F. Role of mitochondrial ATP-sensitive potassium channel-mediated PKC-ε in delayed protection against myocardial ischemia/reperfusion injury in isolated hearts of sevoflurane-preconditioned rats. Braz. J. Med. Biol. Res. Rev. Bras. Pesqui. Medicas E Biol. 2015, 48, 528–536. [Google Scholar] [CrossRef] [Green Version]
- Park, J.I.; Shin, C.Y.; Lee, Y.W.; Huh, I.H.; Sohn, U.D. Endothelium-dependent Sensory Non-adrenergic Non-cholinergic Vasodilatation in Rat Thoracic Aorta: Involvement of ATP and a Role for NO. J. Pharm. Pharmacol. 2000, 52, 409–416. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Yellon, D.M. Ischaemic conditioning and reperfusion injury. Nat. Rev. Cardiol. 2016, 13, 193–209. [Google Scholar] [CrossRef]
- Daiber, A.; Xia, N.; Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Münzel, T.; Li, H. New Therapeutic Implications of Endothelial Nitric Oxide Synthase (eNOS) Function/Dysfunction in Cardiovascular Disease. Int. J. Mol. Sci. 2019, 20, 187. [Google Scholar] [CrossRef]
- Jacobson, K.A.; Jarvis, M.F.; Williams, M. Purine and Pyrimidine (P2) Receptors as Drug Targets. J. Med. Chem. 2002, 45, 4057–4093. [Google Scholar] [CrossRef]
- Ralevic, V.; Burnstock, G. Receptors for Purines and Pyrimidines. Pharmacol. Rev. 1998, 50, 413–492. [Google Scholar]
- Coddou, C.; Yan, Z.; Obsil, T.; Huidobro-Toro, J.P.; Stojilkovic, S.S. Activation and Regulation of Purinergic P2X Receptor Channels. Pharmacol. Rev. 2011, 63, 641–683. [Google Scholar] [CrossRef] [Green Version]
- Peart, J.N.; Headrick, J.P. Adenosinergic cardioprotection: Multiple receptors, multiple pathways. Pharmacol. Ther. 2007, 114, 208–221. [Google Scholar] [CrossRef]
- Maddock, H.L.; Broadley, K.J.; Bril, A.; Khandoudi, N. Role of endothelium in ischaemia-induced myocardial dysfunction of isolated working hearts: Cardioprotection by activation of adenosine A(2A) receptors. J. Auton. Pharmacol. 2001, 21, 263–271. [Google Scholar] [CrossRef]
- Day, Y.-J.; Huang, L.; McDuffie, M.J.; Rosin, D.L.; Ye, H.; Chen, J.-F.; Schwarzschild, M.A.; Fink, J.S.; Linden, J.; Okusa, M.D. Renal protection from ischemia mediated by A2A adenosine receptors on bone marrow-derived cells. J. Clin. Investig. 2003, 112, 883–891. [Google Scholar] [CrossRef]
- Eckle, T.; Krahn, T.; Grenz, A.; Köhler, D.; Mittelbronn, M.; Ledent, C.; Jacobson, M.A.; Osswald, H.; Thompson, L.F.; Unertl, K.; et al. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation 2007, 115, 1581–1590. [Google Scholar] [CrossRef]
- Peart, J.; Flood, A.; Linden, J.; Matherne, G.P.; Headrick, J.P. Adenosine-mediated cardioprotection in ischemic-reperfused mouse heart. J. Cardiovasc. Pharmacol. 2002, 39, 117–129. [Google Scholar] [CrossRef]
- Maddock, H.L.; Mocanu, M.M.; Yellon, D.M. Adenosine A(3) receptor activation protects the myocardium from reperfusion/reoxygenation injury. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1307–H1313. [Google Scholar] [CrossRef]
- Nanhwan, M.K.; Ling, S.; Kodakandla, M.; Nylander, S.; Ye, Y.; Birnbaum, Y. Chronic treatment with ticagrelor limits myocardial infarct size: An adenosine and cyclooxygenase-2-dependent effect. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2078–2085. [Google Scholar] [CrossRef]
- Yang, X.-M.; Liu, Y.; Cui, L.; Yang, X.; Liu, Y.; Tandon, N.; Kambayashi, J.; Downey, J.M.; Cohen, M.V. Platelet P2Y12 blockers confer direct postconditioning-like protection in reperfused rabbit hearts. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 251–262. [Google Scholar] [CrossRef]
- Lochner, A.; Marais, E.; Genade, S.; Huisamen, B.; du Toit, E.F.; Moolman, J.A. Protection of the ischaemic heart: Investigations into the phenomenon of ischaemic preconditioning. Cardiovasc. J. Afr. 2009, 20, 43–51. [Google Scholar]
- Cohen, S.; Megherbi, M.; Jordheim, L.P.; Lefebvre, I.; Perigaud, C.; Dumontet, C.; Guitton, J. Simultaneous analysis of eight nucleoside triphosphates in cell lines by liquid chromatography coupled with tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 3831–3840. [Google Scholar] [CrossRef]
- Canelas, A.B.; ten Pierick, A.; Ras, C.; Seifar, R.M.; van Dam, J.C.; van Gulik, W.M.; Heijnen, J.J. Quantitative evaluation of intracellular metabolite extraction techniques for yeast metabolomics. Anal. Chem. 2009, 81, 7379–7389. [Google Scholar] [CrossRef]
- Zhang, G.; Walker, A.D.; Lin, Z.; Han, X.; Blatnik, M.; Steenwyk, R.C.; Groeber, E.A. Strategies for quantitation of endogenous adenine nucleotides in human plasma using novel ion-pair hydrophilic interaction chromatography coupled with tandem mass spectrometry. J. Chromatogr. A 2014, 1325, 129–136. [Google Scholar] [CrossRef]
- Feliu, C.; Millart, H.; Guillemin, H.; Vautier, D.; Binet, L.; Fouley, A.; Djerada, Z. Validation of a fast UPLC-MS/MS method for quantitative analysis of opioids, cocaine, amphetamines (and their derivatives) in human whole blood. Bioanalysis 2015, 7, 2685–2700. [Google Scholar] [CrossRef]
- Djerada, Z.; Feliu, C.; Tournois, C.; Vautier, D.; Binet, L.; Robinet, A.; Marty, H.; Gozalo, C.; Lamiable, D.; Millart, H. Validation of a fast method for quantitative analysis of elvitegravir, raltegravir, maraviroc, etravirine, tenofovir, boceprevir and 10 other antiretroviral agents in human plasma samples with a new UPLC-MS/MS technology. J. Pharm. Biomed. Anal. 2013, 86, 100–111. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Djerada, Z.; Feliu, C.; Cazaubon, Y.; Smati, F.; Gomis, P.; Guerrot, D.; Charbit, B.; Fernandes, O.; Malinovsky, J.-M. Population Pharmacokinetic-Pharmacodynamic Modeling of Ropivacaine in Spinal Anesthesia. Clin. Pharmacokinet. 2018, 57, 1135–1147. [Google Scholar] [CrossRef]
- Djerada, Z.; Fournet-Fayard, A.; Gozalo, C.; Lelarge, C.; Lamiable, D.; Millart, H.; Malinovsky, J.-M. Population pharmacokinetics of nefopam in elderly, with or without renal impairment, and its link to treatment response. Br. J. Clin. Pharmacol. 2014, 77, 1027–1038. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds Name | Target | Concentration | Reference |
|---|---|---|---|
| Suramin | P2 receptors antagonist | 10 µM | Wee et al. [12] |
| PPADS | P2 receptors antagonist | 10 µM | Wee et al. [12] |
| CGS 15943 | Adenosine receptors antagonist | 1 µM | Avanzato et al. [16] |
| SCH442416 | selective receptor antagonist A2A | 10 µM | Yu et al. [17] |
| MRS1754 | selective receptor antagonist A2B | 0.1 µM | Salie et al. [18] |
| MRS1191 | selective receptor antagonist A3 | 10 µM | Salie et al. [18] |
| U0126 | ERK1/2 inhibitor | 10 µM | Urban et al. [19] |
| LY294002 | PI3K inhibitor | 10 µM | Urban et al. [19] |
| 5-HD | mitoK+ATP inhibitor | 100 µM | Millart et al. [20] |
| L-NAME | NOS inhibitor | 10 µM | Millart et al. [20] |
| H89 | PKA inhibitor | 20 µM | Millart et al. [20] |
| indomethacin | COX inhibitor | 5 µM | Alm et al. [21] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feliu, C.; Peyret, H.; Poitevin, G.; Cazaubon, Y.; Oszust, F.; Nguyen, P.; Millart, H.; Djerada, Z. Complementary Role of P2 and Adenosine Receptors in ATP Induced-Anti-Apoptotic Effects Against Hypoxic Injury of HUVECs. Int. J. Mol. Sci. 2019, 20, 1446. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061446
Feliu C, Peyret H, Poitevin G, Cazaubon Y, Oszust F, Nguyen P, Millart H, Djerada Z. Complementary Role of P2 and Adenosine Receptors in ATP Induced-Anti-Apoptotic Effects Against Hypoxic Injury of HUVECs. International Journal of Molecular Sciences. 2019; 20(6):1446. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061446
Chicago/Turabian StyleFeliu, Catherine, Hélène Peyret, Gael Poitevin, Yoann Cazaubon, Floriane Oszust, Philippe Nguyen, Hervé Millart, and Zoubir Djerada. 2019. "Complementary Role of P2 and Adenosine Receptors in ATP Induced-Anti-Apoptotic Effects Against Hypoxic Injury of HUVECs" International Journal of Molecular Sciences 20, no. 6: 1446. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061446