The Role of Tyrosine Phosphorylation of Protein Kinase C Delta in Infection and Inflammation


Abstract
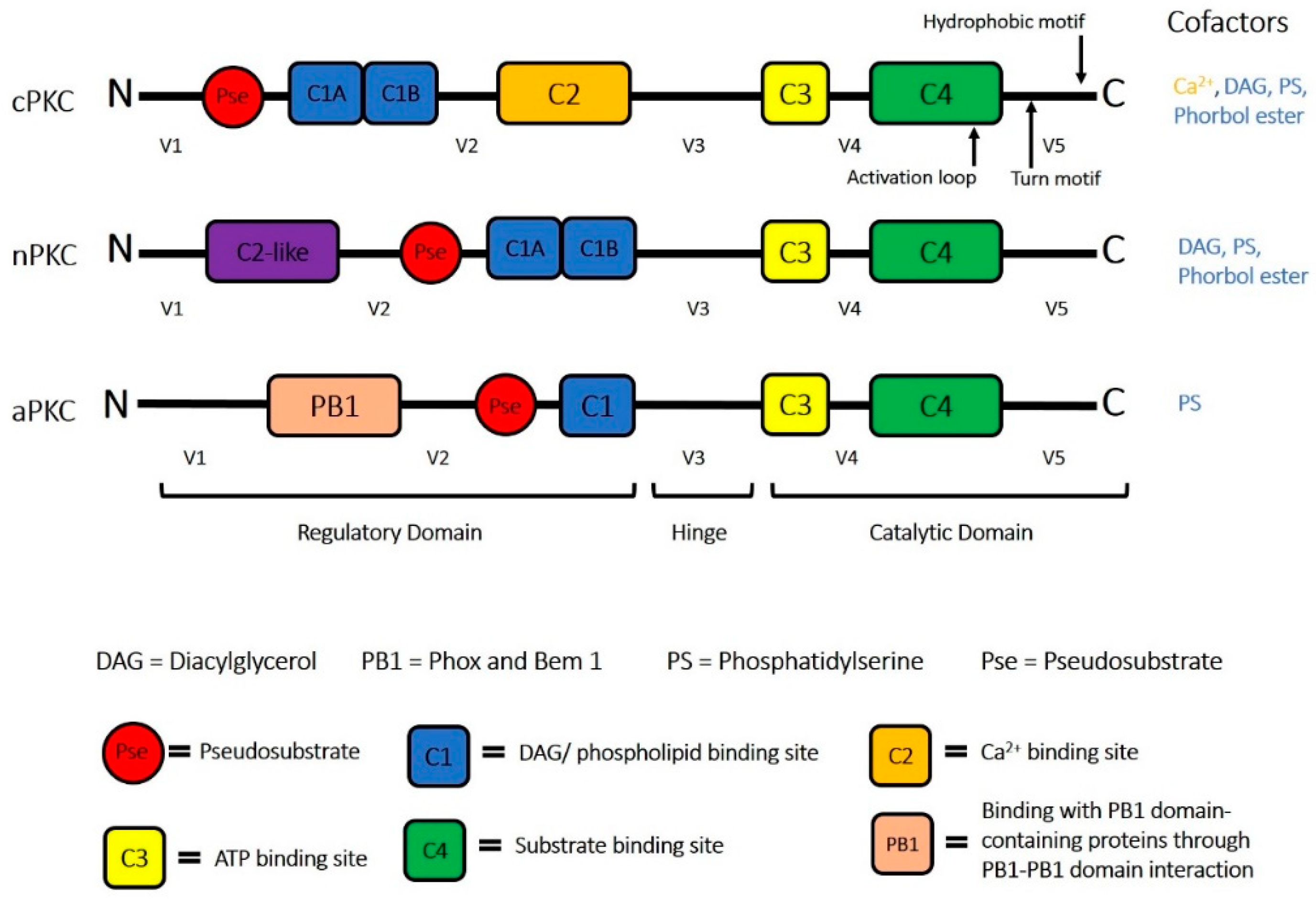
:1. Protein Kinase C (PKC) Superfamily
2. PKCδ and Its Unique Role in Health and Disease
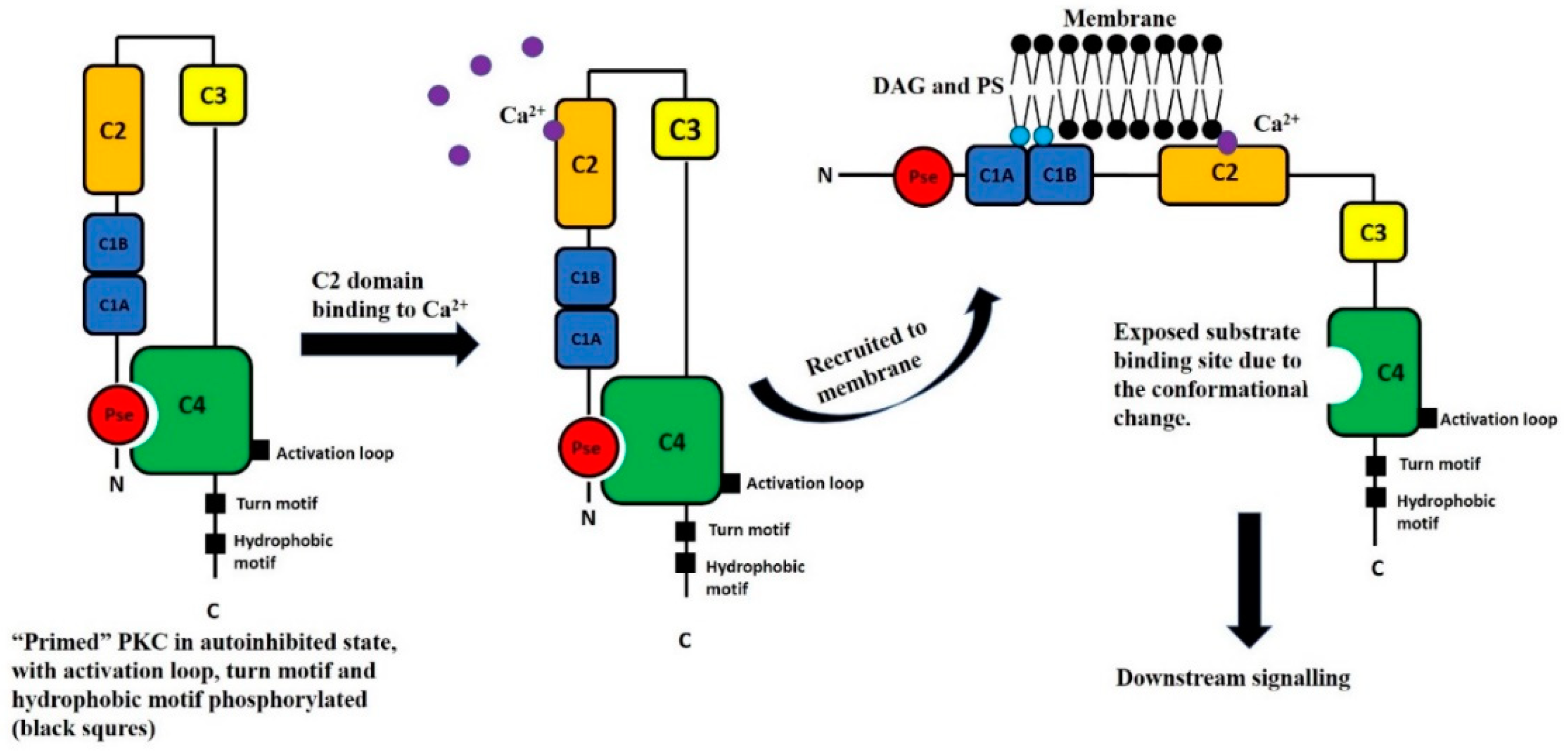
2.1. PKCδ Activation
2.1.1. PKCδ Phosphorylation
2.1.2. PKCδ Translocation and Subcellular Localization
2.2. PKCδ in Inflammatory Diseases
2.2.1. Role of PKCδ in Sepsis—Animal Studies
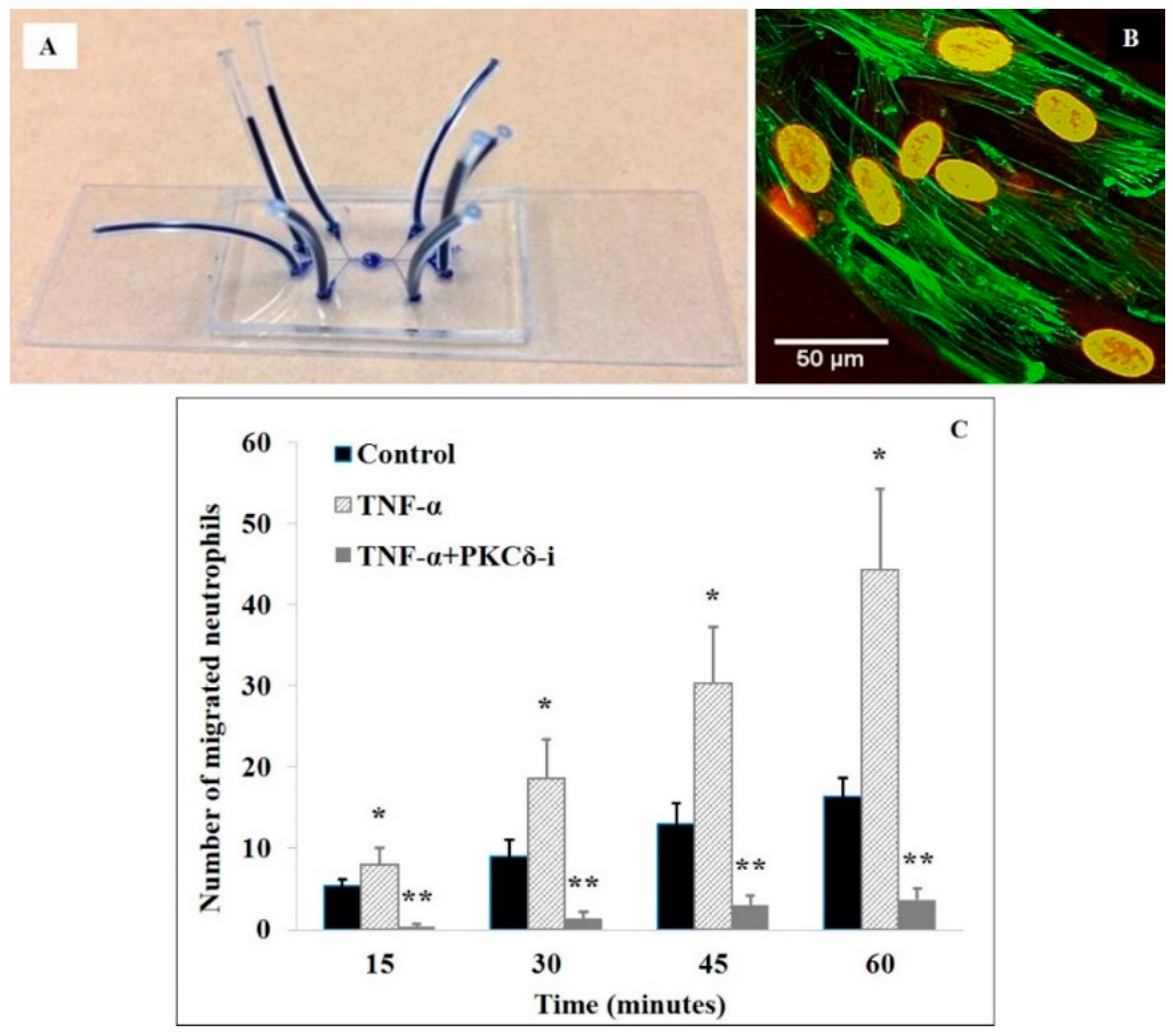
2.2.2. Role of PKCδ in Neutrophil-Endothelial Cell Interactions—In Vitro Studies Using Microfluidics-Based Biomimetic Assays
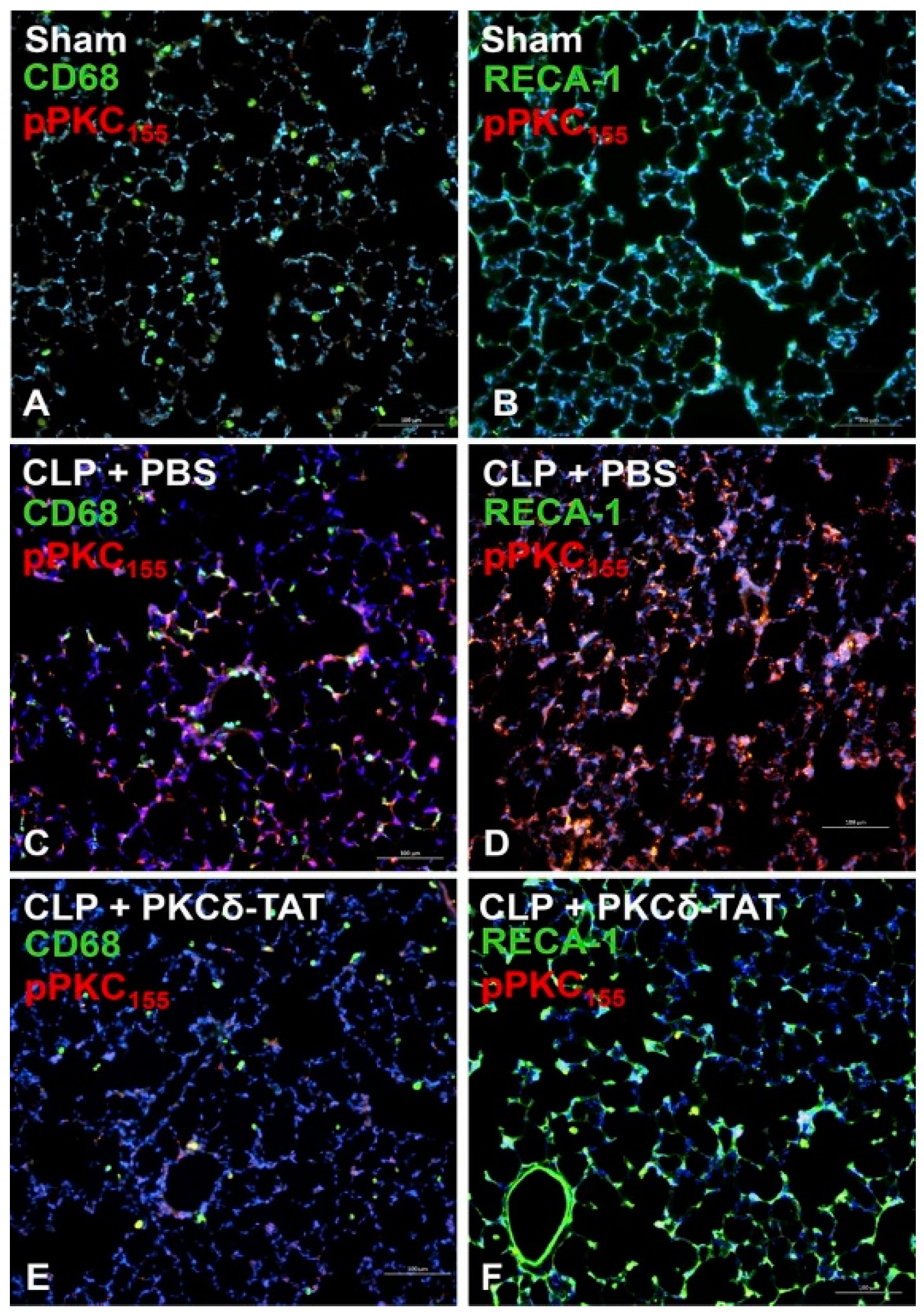
2.2.3. PKCδ Phosphorylation in Sepsis and Inflammation—In Vivo Studies
2.2.4. PKCδ Phosphorylation in Sepsis and Inflammation—In Vitro Studies
3. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Inoue, M.; Kishimoto, A.; Takai, Y.; Nishizuka, Y. Studies on a cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. II. Proenzyme and its activation by calcium-dependent protease from rat brain. J. Biol. Chem. 1977, 252, 7610–7616. [Google Scholar]
- Steinberg, S.F. Structural Basis of Protein Kinase C Isoform Function. Physiol. Rev. 2008, 88, 1341–1378. [Google Scholar] [CrossRef] [Green Version]
- Dempsey, E.C.; Newton, A.C.; Mochly-Rosen, D.; Fields, A.P.; Reyland, M.E.; Insel, P.A.; Messing, R.O. Protein kinase C isozymes and the regulation of diverse cell responses. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 279, L429–L438. [Google Scholar] [CrossRef]
- Sabri, A.; Steinberg, S.F. Protein kinase C isoform-selective signals that lead to cardiac hypertrophy and the progression of heart failure. In Biochemistry of Hypertrophy and Heart Failure; Springer: New York, NY, USA, 2003; pp. 97–101. [Google Scholar]
- Mellor, H.; Parker, P.J. The extended protein kinase C superfamily. Biochem. J. 1998, 332, 281–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, A.; Seki, N.; Hattori, A.; Kozuma, S.; Saito, T. PKCν, a new member of the protein kinase C family, composes a fourth subfamily with PKCμ1. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 1999, 1450, 99–106. [Google Scholar] [CrossRef]
- Lenz, J.C.; Reusch, H.P.; Albrecht, N.; Schultz, G.; Schaefer, M. Ca2+-controlled competitive diacylglycerol binding of protein kinase C isoenzymes in living cells. J. Cell Biol. 2002, 159, 291–302. [Google Scholar] [CrossRef]
- Mondrinos, M.J.; Kennedy, P.A.; Lyons, M.; Deutschman, C.S.; Kilpatrick, L.E. Protein Kinase C and Acute Respiratory Distress Syndrome. Shock 2013, 39, 467–479. [Google Scholar] [CrossRef] [Green Version]
- Mochly-Rosen, D.; Das, K.; Grimes, K.V. Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Discov. 2012, 11, 937. [Google Scholar] [CrossRef] [PubMed]
- Ono, Y.; Fujii, T.; Ogita, K.; Kikkawa, U.; Igarashi, K.; Nishizuka, Y. The structure, expression, and properties of additional members of the protein kinase C family. J. Biol. Chem. 1988, 263, 6927–6932. [Google Scholar]
- Kim, J.; Koyanagi, T.; Mochly-Rosen, D. PKCδ activation mediates angiogenesis via NADPH oxidase activity in PC-3 prostate cancer cells. Prostate 2011, 71, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Bright, R.; Raval, A.P.; Dembner, J.M.; Perez-Pinzon, M.A.; Steinberg, G.K.; Yenari, M.A.; Mochly-Rosen, D. Protein kinase C δ mediates cerebral reperfusion injury in vivo. J. Neurosci. 2004, 24, 6880–6888. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Hahn, H.; Wu, G.; Chen, C.H.; Liron, T.; Schechtman, D.; Cavallaro, G.; Banci, L.; Guo, Y.; Bolli, R.; et al. Opposing cardioprotective actions and parallel hypertrophic effects of δ PKC and epsilon PKC. Proc. Natl. Acad. Sci. USA 2001, 98, 11114–11119. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, K.; Chen, L.; Ikeno, F.; Lee, F.H.; Imahashi, K.; Bouley, D.M.; Rezaee, M.; Yock, P.G.; Murphy, E.; Mochly-Rosen, D. Inhibition of δ-protein kinase C protects against reperfusion injury of the ischemic heart in vivo. Circulation 2003, 108, 2304–2307. [Google Scholar] [CrossRef]
- Pereira, S.; Park, E.; Mori, Y.; Haber, C.A.; Han, P.; Uchida, T.; Stavar, L.; Oprescu, A.I.; Koulajian, K.; Ivovic, A. FFA-induced hepatic insulin resistance in vivo is mediated by PKCδ, NADPH oxidase, and oxidative stress. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E34–E46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geraldes, P.; Hiraoka-Yamamoto, J.; Matsumoto, M.; Clermont, A.; Leitges, M.; Marette, A.; Aiello, L.P.; Kern, T.S.; King, G.L. Activation of PKC-δ and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat. Med. 2009, 15, 1298. [Google Scholar] [CrossRef]
- Qi, X.; Disatnik, M.-H.; Shen, N.; Sobel, R.A.; Mochly-Rosen, D. Aberrant mitochondrial fission in neurons induced by protein kinase Cδ under oxidative stress conditions in vivo. Mol. Biol. Cell 2011, 22, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Qi, X.; Inagaki, K.; Sobel, R.A.; Mochly-Rosen, D. Sustained pharmacological inhibition of δPKC protects against hypertensive encephalopathy through prevention of blood-brain barrier breakdown in rats. J. Clin. Investig. 2008, 118, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, L.E.; Standage, S.W.; Li, H.; Raj, N.R.; Korchak, H.M.; Wolfson, M.R.; Deutschman, C.S. Protection against sepsis-induced lung injury by selective inhibition of protein kinase C-δ (δ-PKC). J. Leuk. Biol. 2011, 89, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Liverani, E.; Mondrinos, M.J.; Sun, S.; Kunapuli, S.P.; Kilpatrick, L.E. Role of Protein Kinase C-δ in regulating platelet activation and platelet-leukocyte interaction during sepsis. PLoS ONE 2018, 13, e0195379. [Google Scholar] [CrossRef]
- Soroush, F.; Tang, Y.; Guglielmo, K.; Engelmann, A.; Liverani, E.; Langston, J.; Sun, S.; Kunapuli, S.; Kiani, M.F.; Kilpatrick, L.E. Protein Kinase C-δ (PKCδ) Tyrosine Phosphorylation is a Critical Regulator of Neutrophil-Endothelial Cell Interaction in Inflammation. Shock 2018. [Google Scholar] [CrossRef]
- Kilpatrick, L.E.; Sun, S.; Li, H.; Vary, T.C.; Korchak, H.M. Regulation of TNF-induced oxygen radical production in human neutrophils: Role of δ-PKC. J. Leuk. Biol. 2010, 87, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Soroush, F.; Zhang, T.; King, D.J.; Tang, Y.; Deosarkar, S.; Prabhakarpandian, B.; Kilpatrick, L.E.; Kiani, M.F. A novel microfluidic assay reveals a key role for protein kinase C δ in regulating human neutrophil–endothelium interaction. J. Leuk. Biol. 2016, 100, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Mondrinos, M.J.; Knight, L.C.; Kennedy, P.A.; Wu, J.; Kauffman, M.; Baker, S.T.; Wolfson, M.R.; Kilpatrick, L.E. Biodistribution and efficacy of targeted pulmonary delivery of a protein kinase C-δ inhibitory peptide: Impact on indirect lung injury. J. Pharmacol. Exp. Ther. 2015, 355, 86–98. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Soroush, F.; Sun, S.; Liverani, E.; Langston, J.C.; Yang, Q.; Kilpatrick, L.E.; Kiani, M.F. Protein kinase C-δ inhibition protects blood-brain barrier from sepsis-induced vascular damage. J. Neuroinflamm. 2018, 15, 309. [Google Scholar] [CrossRef]
- Soroush, F.; Tang, Y.; Zaidi, J.; Sheffield, L.; Kilpatrick, L.E.; Kiani, M.F. PKCδ inhibition as a novel medical countermeasure for radiation-induced vascular damage. FASEB J. 2018, 32, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- Malavez, Y.; Gonzalez-Mejia, M.E.; Doseff, A.I. PRKCD (Protein Kinase C, Delta). Available online: http://atlasgeneticsoncology.org/Genes/GC_PRKCD.html (accessed on 24 March 2009).
- Qvit, N.; Mochly-Rosen, D. The many hats of protein kinase Cdelta: One enzyme with many functions. Biochem. Soc. Trans. 2014, 42, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, S.F. Distinctive activation mechanisms and functions for protein kinase Cδ. Biochem. J. 2004, 384, 449–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronfeld, I.; Kazimirsky, G.; Lorenzo, P.S.; Garfield, S.H.; Blumberg, P.M.; Brodie, C. Phosphorylation of Protein Kinase Cd on Distinct Tyrosine Residues Regulates Specific Cellular Functions. J. Biol. Chem. 2000, 275, 35491–35498. [Google Scholar] [CrossRef]
- Newton, A.C. Protein kinase C: Structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev. 2001, 101, 2353–2364. [Google Scholar] [CrossRef]
- Dempsey, E.C.; Badesch, D.B.; Dobyns, E.L.; Stenmark, K.R. Enhanced growth capacity of neonatal pulmonary artery smooth muscle cells in vitro: Dependence on cell size, time from birth, insulin-like growth factor I, and auto-activation of protein Kinase, C. J. Cell. Physiol. 1994, 160, 469–481. [Google Scholar] [CrossRef]
- Ono, Y.; FuJII, T.; Igarashi, K.; Kuno, T.; Tanaka, C.; Kikkawa, U.; Nishizuka, Y. Phorbol ester binding to protein kinase C requires a cysteine-rich zinc-finger-like sequence. Proc. Natl. Acad. Sci. USA 1989, 86, 4868–4871. [Google Scholar] [CrossRef]
- Hommel, U.; Zurini, M.; Luyten, M. Solution structure of a cysteine rich domain of rat protein kinase C. Nat. Struct. Mol. Biol. 1994, 1, 383. [Google Scholar] [CrossRef]
- Diaz-Meco, M.T.; Moscat, J. The atypical PKCs in inflammation: NF-κB and beyond. Immunol. Rev. 2012, 246, 154–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oancea, E.; Meyer, T. Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell 1998, 95, 307–318. [Google Scholar] [CrossRef]
- Shao, X.; Davletov, B.A.; Sutton, R.B.; Südhof, T.C.; Rizo, J. Bipartite Ca2+-Binding Motif in C2 Domains of Synaptotagmin and Protein Kinase C. Science 1996, 273, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Sutton, R.B.; Sprang, S.R. Structure of the protein kinase Cβ phospholipid-binding C2 domain complexed with Ca2+. Structure 1998, 6, 1395–1405. [Google Scholar] [CrossRef]
- Verdaguer, N.; Corbalan-Garcia, S.; Ochoa, W.F.; Fita, I.; Gómez-Fernández, J.C. Ca2+ bridges the C2 membrane-binding domain of protein kinase Cα directly to phosphatidylserine. EMBO J. 1999, 18, 6329–6338. [Google Scholar] [CrossRef] [PubMed]
- Rosse, C.; Linch, M.; Kermorgant, S.; Cameron, A.J.; Boeckeler, K.; Parker, P.J. PKC and the control of localized signal dynamics. Nat. Rev. Mol. Cell Biol. 2010, 11, 103. [Google Scholar] [CrossRef]
- Moscat, J.; Rennert, P.; Diaz-Meco, M. PKCζ at the crossroad of NF-κB and Jak1/Stat6 signaling pathways. Cell Death Differ. 2006, 13, 702. [Google Scholar] [CrossRef]
- Keranen, L.M.; Dutil, E.M.; Newton, A.C. Protein kinase C is regulated in vivo by three functionally distinct phosphorylations. Curr. Biol. 1995, 5, 1394–1403. [Google Scholar] [CrossRef] [Green Version]
- Kilpatrick, L.E.; Song, Y.H.; Rossi, M.W.; Korchak, H.M. Serine phosphorylation of p60 tumor necrosis factor receptor by PKC-δ in TNF-α-activated neutrophils. Am. J. Physiol. Cell Physiol. 2000, 279, C2011–C2018. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, L.E.; Lee, J.Y.; Haines, K.M.; Campbell, D.E.; Sullivan, K.E.; Korchak, H.M. A role for PKC-δ and PI 3-kinase in TNF-α-mediated antiapoptotic signaling in the human neutrophil. Am. J. Physiol. Cell Physiol. 2002, 283, C48–C57. [Google Scholar] [CrossRef] [PubMed]
- Kilpatrick, L.E.; Sun, S.; Korchak, H.M. Selective regulation by δ-PKC and PI 3-kinase in the assembly of the antiapoptotic TNFR-1 signaling complex in neutrophils. Am. J. Physiol. Cell Physiol. 2004, 287, C633–C642. [Google Scholar] [CrossRef] [Green Version]
- Kilpatrick, L.E.; Sun, S.; Mackie, D.; Baik, F.; Li, H.; Korchak, H.M. Regulation of TNF mediated antiapoptotic signaling in human neutrophils: Role of δ-PKC and ERK1/2. J. Leuk. Biol. 2006, 80, 1512–1521. [Google Scholar] [CrossRef]
- Mondrinos, M.J.; Zhang, T.; Sun, S.; Kennedy, P.A.; King, D.J.; Wolfson, M.R.; Knight, L.C.; Scalia, R.; Kilpatrick, L.E. Pulmonary Endothelial Protein Kinase C-δ (PKCd) Regulates Neutrophil Migration in Acute Lung Inflammation. Am. J. Pathol. 2014, 184, 200–213. [Google Scholar] [CrossRef]
- Page, K.; Li, J.; Zhou, L.; Iasvovskaia, S.; Corbit, K.C.; Soh, J.W.; Weinstein, I.B.; Brasier, A.R.; Lin, A.; Hershenson, M.B. Regulation of airway epithelial cell NF-κ B-dependent gene expression by protein kinase C δ. J. Immunol. 2003, 170, 5681–5689. [Google Scholar] [CrossRef]
- Stempka, L.; Girod, A.; Müller, H.-J.; Rincke, G.; Marks, F.; Gschwendt, M.; Bossemeyer, D. Phosphorylation of Protein Kinase Cδ (PKCδ) at Threonine 505 is not a prerequisite for enzymatic activity expression of rat pkcδ and an alanine 505 mutant in bacteria in a functional form. J. Biol. Chem. 1997, 272, 6805–6811. [Google Scholar] [CrossRef] [PubMed]
- Rybin, V.O.; Sabri, A.; Short, J.; Braz, J.C.; Molkentin, J.D.; Steinberg, S.F. Cross regulation of nPKC isoform function in cardiomyocytes: Role of PKCepsilon in activation loop phosphorylations and PKCδ in hydrophobic motif phosphorylations. J. Biol. Chem. 2003. [Google Scholar] [CrossRef]
- Le Good, J.A.; Ziegler, W.H.; Parekh, D.B.; Alessi, D.R.; Cohen, P.; Parker, P.J. Protein kinase C isotypes controlled by phosphoinositide 3-kinase through the protein kinase PDK1. Science 1998, 281, 2042–2045. [Google Scholar] [CrossRef]
- Stempka, L.; Schnölzer, M.; Radke, S.; Rincke, G.; Marks, F.; Gschwendt, M. Requirements of Protein Kinase Cδ for Catalytic Function Role of Glutamic Acid 500 and Autophosphorylation on Serine 643. J. Biol. Chem. 1999, 274, 8886–8892. [Google Scholar] [CrossRef]
- Kikkawa, U.; Matsuzaki, H.; Yamamoto, T. Protein kinase Cδ (PKCδ): Activation mechanisms and functions. J. Biochem. 2002, 132, 831–839. [Google Scholar] [CrossRef]
- Zhao, M.; Xia, L.; Chen, G.-Q. Protein Kinase Cd in Apoptosis: A Brief Overview. Arch. Immunol. Ther. Exp. 2012, 60, 361–372. [Google Scholar] [CrossRef]
- Humphries, M.J.; Ohm, A.M.; Schaack, J.; Adwan, T.S.; Reyland, M.E. Tyrosine phosphorylation regulates nuclear translocation of PKCδ. Oncogene 2007, 27, 3045. [Google Scholar] [CrossRef]
- Pabla, N.; Dong, G.; Jiang, M.; Huang, S.; Kumar, M.V.; Messing, R.O.; Dong, Z. Inhibition of PKCd reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J. Clin. Investig. 2011, 121, 2709–2722. [Google Scholar] [CrossRef]
- Li, W.; Li, W.; Chen, X.-H.; Kelley, C.A.; Alimandi, M.; Zhang, J.; Chen, Q.; Bottaro, D.P.; Pierce, J.H. Identification of tyrosine 187 as a protein kinase C-δ phosphorylation site. J. Biol. Chem. 1996, 271, 26404–26409. [Google Scholar] [CrossRef]
- Jackson, D.N.; Foster, D.A. The enigmatic protein kinase Cδ: Complex roles in cell proliferation and survival. FASEB J. 2004, 18, 627–636. [Google Scholar] [CrossRef]
- Pappa, H.; Murray-Rust, J.; Dekker, L.; Parker, P.; McDonald, N. Crystal structure of the C2 domain from protein kinase C-δ. Structure 1998, 6, 885–894. [Google Scholar] [CrossRef] [Green Version]
- Yamanashi, Y.; Mori, S.; Yoshida, M.; Kishimoto, T.; Inoue, K.; Yamamoto, T.; ToYOSHIMA, K. Selective expression of a protein-tyrosine kinase, p56lyn, in hematopoietic cells and association with production of human T-cell lymphotropic virus type I. Proc. Natl. Acad. Sci. USA 1989, 86, 6538–6542. [Google Scholar] [CrossRef]
- Umemori, H.; Wanaka, A.; Kato, H.; Takeuchi, M.; Tohyama, M.; Yamamoto, T. Specific expressions of Fyn and Lyn, lymphocyte antigen receptor-associated tyrosine kinases, in the central nervous system. Mol. Brain Res. 1992, 16, 303–310. [Google Scholar] [CrossRef]
- Yamada, E.; Pessin, J.E.; Kurland, I.J.; Schwartz, G.J.; Bastie, C.C. Fyn-dependent regulation of energy expenditure and body weight is mediated by tyrosine phosphorylation of LKB1. Cell Metab. 2010, 11, 113–124. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.L.; Strehler, E.E. Plasma membrane calcium ATPases as critical regulators of calcium homeostasis during neuronal cell function. Front. Biosci. 1999, 4, D869–D882. [Google Scholar] [CrossRef]
- Ahn, B.K.; Jeong, S.K.; Lee, S.H. Role of PKC-δ as a signal mediator in epidermal barrier homeostasis. Arch. Dermatol. Res. 2007, 299, 53–57. [Google Scholar] [CrossRef]
- Voss, O.H.; Kim, S.; Wewers, M.D.; Doseff, A.I. Regulation of monocyte apoptosis by the protein kinase C δ (PKCδ)-dependent phosphorylation of caspase-3. J. Biol. Chem. 2005, 280, 17371–17379. [Google Scholar] [CrossRef]
- Yoshida, K.; Liu, H.; Miki, Y. Protein kinase C δ regulates Ser46 phosphorylation of p53 tumor suppressor in the apoptotic response to DNA damage. J. Biol. Chem. 2006, 281, 5734–5740. [Google Scholar] [CrossRef]
- Hu, Y.; Kang, C.; Philp, R.J.; Li, B. PKC δ phosphorylates p52ShcA at Ser29 to regulate ERK activation in response to H2O2. Cell. Signal. 2007, 19, 410–418. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Lee, D.-H.; Cho, C.-K.; Bae, S.; Jhon, G.-J.; Lee, S.-J.; Soh, J.-W.; Lee, Y.-S. HSP25 inhibits PKCδ-mediated cell death through direct interaction. J. Biol. Chem. 2005, 280, 18108–18119. [Google Scholar] [CrossRef]
- Disatnik, M.-H.; Boutet, S.C.; Lee, C.H.; Mochly-Rosen, D.; Rando, T.A. Sequential activation of individual PKC isozymes in integrin-mediated muscle cell spreading: A role for MARCKS in an integrin signaling pathway. J. Cell Sci. 2002, 115, 2151–2163. [Google Scholar]
- Jideama, N.M.; Noland, T.A.; Raynor, R.L.; Blobe, G.C.; Fabbro, D.; Kazanietz, M.G.; Blumberg, P.M.; Hannun, Y.A.; Kuo, J. Phosphorylation specificities of protein kinase C isozymes for bovine cardiac troponin I and troponin T and sites within these proteins and regulation of myofilament properties. J. Biol. Chem. 1996, 271, 23277–23283. [Google Scholar] [CrossRef]
- Churchill, E.N.; Murriel, C.L.; Chen, C.-H.; Mochly-Rosen, D.; Szweda, L.I. Reperfusion-induced translocation of δPKC to cardiac mitochondria prevents pyruvate dehydrogenase reactivation. Circ. Res. 2005, 97, 78–85. [Google Scholar] [CrossRef]
- Sawai, H.; Okazaki, T.; Takeda, Y.; Tashima, M.; Sawada, H.; Okuma, M.; Kishi, S.; Umehara, H.; Domae, N. Ceramide-induced Translocation of Protein Kinase C-δ and-ϵ to the Cytosol Implications in Apoptosis. J. Biol. Chem. 1997, 272, 2452–2458. [Google Scholar] [CrossRef]
- Rybin, V.O.; Xu, X.; Steinberg, S.F. Activated protein kinase C isoforms target to cardiomyocyte caveolae: Stimulation of local protein phosphorylation. Circ. Res. 1999, 84, 980–988. [Google Scholar] [CrossRef]
- Qi, X.; Mochly-Rosen, D. The PKCδ-Abl complex communicates ER stress to the mitochondria–an essential step in subsequent apoptosis. J. Cell Sci. 2008, 121, 804–813. [Google Scholar] [CrossRef]
- Kajimoto, T.; Ohmori, S.; Shirai, Y.; Sakai, N.; Saito, N. Subtype-specific translocation of the δ subtype of protein kinase C and its activation by tyrosine phosphorylation induced by ceramide in HeLa cells. Mol. Cell. Biol. 2001, 21, 1769–1783. [Google Scholar] [CrossRef]
- Gomel, R.; Xiang, C.; Finniss, S.; Lee, H.K.; Lu, W.; Okhrimenko, H.; Brodie, C. The localization of protein kinase Cδ in different subcellular sites affects its proapoptotic and antiapoptotic functions and the activation of distinct downstream signaling pathways. Mol. Cancer Res. 2007, 5, 627–639. [Google Scholar] [CrossRef]
- Bhavanasi, D.; Badolia, R.; Manne, B.K.; Janapati, S.; Dangelmaier, C.T.; Mazharian, A.; Jin, J.; Kim, S.; Zhang, X.; Chen, X.; et al. Cross talk between serine/threonine and tyrosine kinases regulates ADP-induced thromboxane generation in platelets. Thromb. Haem. 2015, 114, 558–568. [Google Scholar] [CrossRef]
- Song, J.S.; Swann, P.G.; Szallasi, Z.; Blank, U.; Blumberg, P.M.; Rivera, J. Tyrosine phosphorylation-dependent and-independent associations of protein kinase C-δ with Src family kinases in the RBL-2H3 mast cell line: Regulation of Src family kinase activity by protein kinase C-δ. Oncogene 1998, 16, 3357. [Google Scholar] [CrossRef]
- Zang, Q.; Lu, Z.; Curto, M.; Barile, N.; Shalloway, D.; Foster, D.A. Association between v-Src and protein kinase C δ in v-Src-transformed fibroblasts. J. Biol. Chem. 1997, 272, 13275–13280. [Google Scholar] [CrossRef]
- Sun, X.; Wu, F.; Datta, R.; Kharbanda, S.; Kufe, D. Interaction between protein kinase C δ and the c-Abl tyrosine kinase in the cellular response to oxidative stress. J. Biol. Chem. 2000, 275, 7470–7473. [Google Scholar] [CrossRef]
- Yoshida, K.; Kufe, D. Negative regulation of the SHPTP1 protein tyrosine phosphatase by protein kinase C δ in response to DNA damage. Mol. Pharmacol. 2001, 60, 1431–1438. [Google Scholar] [CrossRef]
- Brodie, C.; Steinhart, R.; Kazimirsky, G.; Rubinfeld, H.; Hyman, T.; Ayres, J.N.; Hur, G.M.; Toth, A.; Yang, D.; Garfield, S.H. PKCδ associates with and is involved in the phosphorylation of RasGRP3 in response to phorbol esters. Mol. Pharmacol. 2004, 66, 76–84. [Google Scholar] [CrossRef]
- Brandt, D.T.; Goerke, A.; Heuer, M.; Gimona, M.; Leitges, M.; Kremmer, E.; Lammers, R.; Haller, H.; Mischak, H. Protein kinase Cδ induces Src kinase activity via activation of the protein tyrosine phosphatase PTPα. J. Biol. Chem. 2003, 278, 34073–34078. [Google Scholar] [CrossRef]
- Uddin, S.; Sassano, A.; Deb, D.K.; Verma, A.; Majchrzak, B.; Rahman, A.; Malik, A.B.; Fish, E.N.; Platanias, L.C. Protein kinase C-δ (PKC-δ) is activated by type I interferons and mediates phosphorylation of Stat1 on serine 727. J. Biol. Chem. 2002, 277, 14408–14416. [Google Scholar] [CrossRef]
- Jain, N.; Zhang, T.; Kee, W.H.; Li, W.; Cao, X. Protein kinase C δ associates with and phosphorylates Stat3 in an interleukin-6-dependent manner. J. Biol. Chem. 1999, 274, 24392–24400. [Google Scholar] [CrossRef]
- Yuan, L.; Soh, J.-W.; Weinstein, I.B. Inhibition of histone acetyltransferase function of p300 by PKCδ. BBA Mol. Cell Res. 2002, 1592, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Hamaguchi, A.; Suzuki, E.; Murayama, K.; Fujimura, T.; Hikita, T.; Iwabuchi, K.; Handa, K.; Withers, D.A.; Masters, S.C.; Fu, H. Sphingosine-dependent protein kinase-1 (SDK1), directed to 14-3-3, is identified as the kinase domain of PKC δ. J. Biol. Chem. 2003, 278, 41557–41565. [Google Scholar] [CrossRef]
- Novotny-Diermayr, V.; Zhang, T.; Gu, L.; Cao, X. Protein kinase C δ associates with the interleukin-6 receptor subunit glycoprotein (gp) 130 via Stat3 and enhances Stat3-gp130 interaction. J. Biol. Chem. 2002, 130, 49134–49142. [Google Scholar] [CrossRef]
- Fontayne, A.; Dang, P.M.-C.; Gougerot-Pocidalo, M.-A.; El Benna, J. Phosphorylation of p47 p hox Sites by PKC α, βΙΙ, δ, and ζ: Effect on Binding to p22 p hox and on NADPH Oxidase Activation. Biochemistry 2002, 41, 7743–7750. [Google Scholar] [CrossRef]
- Alt, A.; Ohba, M.; Li, L.; Gartsbein, M.; Belanger, A.; Denning, M.F.; Kuroki, T.; Yuspa, S.H.; Tennenbaum, T. Protein kinase Cδ-mediated phosphorylation of α6β4 is associated with reduced integrin localization to the hemidesmosome and decreased keratinocyte attachment. Cancer Res. 2001, 61, 4591–4598. [Google Scholar]
- Siwko, S.; Mochly-Rosen, D. Use of a novel method to find substrates of protein kinase C δ identifies M2 pyruvate kinase. Int. J. Biochem. Cell Biol. 2007, 39, 978–987. [Google Scholar] [CrossRef]
- Vidyasagar, A.; Wilson, N.A.; Djamali, A. Heat shock protein 27 (HSP27): Biomarker of disease and therapeutic target. Fibrogene. Tissue Repair 2012, 5, 7. [Google Scholar] [CrossRef]
- Bharti, A.; Kraeft, S.-K.; Gounder, M.; Pandey, P.; Jin, S.; Yuan, Z.-M.; Lees-Miller, S.P.; Weichselbaum, R.; Weaver, D.; Chen, L.B. Inactivation of DNA-dependent protein kinase by protein kinase Cδ: Implications for apoptosis. Mol. Cell. Biol. 1998, 18, 6719–6728. [Google Scholar] [CrossRef]
- Murriel, C.L.; Churchill, E.; Inagaki, K.; Szweda, L.I.; Mochly-Rosen, D. Protein kinase Cδ activation induces apoptosis in response to cardiac ischemia and reperfusion damage a mechanism involving Bad and the mitochondria. J. Biol. Chem. 2004, 279, 47985–47991. [Google Scholar] [CrossRef]
- Yogalingam, G.; Hwang, S.; Ferreira, J.C.; Mochly-Rosen, D. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) phosphorylation by protein kinase Cδ (PKCδ) inhibits mitochondria elimination by lysosomal-like structures following ischemia and reoxygenation-induced injury. J. Biol. Chem. 2013, 288, 18947–18960. [Google Scholar] [CrossRef]
- Liu, J.; Chen, J.; Dai, Q.; Lee, R.M. Phospholipid scramblase 3 is the mitochondrial target of protein kinase C δ-induced apoptosis. Cancer Res. 2003, 63, 1153–1156. [Google Scholar]
- Cross, T.; Griffiths, G.; Deacon, E.; Sallis, R.; Gough, M.; Watters, D.; Lord, J.M. PKC-δ is an apoptotic lamin kinase. Oncogene 2000, 19, 2331. [Google Scholar] [CrossRef]
- Yoshida, K.; Wang, H.G.; Miki, Y.; Kufe, D. Protein kinase Cδ is responsible for constitutive and DNA damage-induced phosphorylation of Rad9. EMBO J. 2003, 22, 1431–1441. [Google Scholar] [CrossRef] [Green Version]
- Vancurova, I.; Miskolci, V.; Davidson, D. NF-κB activation in tumor necrosis factor α-stimulated neutrophils is mediated by protein kinase Cδ. Correlation to nuclear IκBα. J. Biol. Chem. 2001, 276, 19746–19752. [Google Scholar] [CrossRef]
- Brown, K.A.; Brain, S.D.; Pearson, J.D.; Edgeworth, J.D.; Lewis, S.M.; Treacher, D.F. Neutrophils in development of multiple organ failure in sepsis. Lancet 2006, 368, 157–169. [Google Scholar] [CrossRef]
- Aldridge, A.J. Role of the neutrophil in septic shock and the adult respiratory distress syndrome. Eur. J. Surg. 2002, 168, 204–214. [Google Scholar] [CrossRef]
- Williams, A.E.; Chambers, R.C. The mercurial nature of neutrophils: Still an enigma in ARDS? Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 306, L217–L230. [Google Scholar] [CrossRef]
- Reutershan, J.; Ley, K. Bench-to-bedside review: Acute respiratory distress syndrome—How neutrophils migrate into the lung. Crit Care 2004, 8, 453–461. [Google Scholar] [CrossRef]
- Chou, W.H.; Choi, D.S.; Zhang, H.; Mu, D.; McMahon, T.; Kharazia, V.N.; Lowell, C.A.; Ferriero, D.M.; Messing, R.O. Neutrophil protein kinase Cδ as a mediator of stroke-reperfusion injury. J. Clin. Investig. 2004, 114, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Shukla, A.; Lounsbury, K.M.; Barrett, T.F.; Gell, J.; Rincon, M.; Butnor, K.J.; Taatjes, D.J.; Davis, G.S.; Vacek, P.; Nakayama, K.I.; et al. Asbestos-induced peribronchiolar cell proliferation and cytokine production are attenuated in lungs of protein kinase C-δ knockout mice. Am. J. Pathol. 2007, 170, 140–151. [Google Scholar] [CrossRef]
- Ramnath, R.; Sun, J.; Bhatia, M. PKC δ mediates pro-inflammatory responses in a mouse model of caerulein-induced acute pancreatitis. J. Mol. Med. 2010, 88, 1–9. [Google Scholar] [CrossRef]
- Chichger, H.; Grinnell, K.L.; Casserly, B.; Chung, C.S.; Braza, J.; Lomas-Neira, J.; Ayala, A.; Rounds, S.; Klinger, J.R.; Harrington, E.O. Genetic disruption of protein kinase Cδ reduces endotoxin-induced lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L880–L888. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Roller, J.; Menger, M.D.; Thorlacius, H. Sepsis-induced leukocyte adhesion in the pulmonary microvasculature in vivo is mediated by CD11a and CD11b. Eur. J. Pharmacol. 2013, 702, 135–141. [Google Scholar] [CrossRef]
- Roller, J.; Wang, Y.; Rahman, M.; Schramm, R.; Laschke, M.W.; Menger, M.D.; Jeppsson, B.; Thorlacius, H. Direct in vivo observations of P-selectin glycoprotein ligand-1-mediated leukocyte-endothelial cell interactions in the pulmonary microvasculature in abdominal sepsis in mice. Inflamm. Res. 2013, 62, 275–282. [Google Scholar] [CrossRef]
- Reinhart, K.; Bayer, O.; Brunkhorst, F.; Meisner, M. Markers of endothelial damage in organ dysfunction and sepsis. Crit. Care Med. 2002, 30, S302–S312. [Google Scholar] [CrossRef]
- Craciun, F.L.; Iskander, K.N.; Chiswick, E.L.; Stepien, D.M.; Henderson, J.M.; Remick, D.G. Early murine polymicrobial sepsis predominantly causes renal injury. Shock 2014, 41, 97–103. [Google Scholar] [CrossRef]
- Soltoff, S.P. Rottlerin: An inappropriate and ineffective inhibitor of PKCδ. Trends Pharmacol. Sci. 2007, 28, 453–458. [Google Scholar] [CrossRef]
- Davies, S.P.; Reddy, H.; Caivano, M.; Cohen, P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J. 2000, 351, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Moschos, S.A.; Jones, S.W.; Perry, M.M.; Williams, A.E.; Erjefalt, J.S.; Turner, J.J.; Barnes, P.J.; Sproat, B.S.; Gait, M.J.; Lindsay, M.A. Lung Delivery Studies Using siRNA Conjugated to TAT(48–60) and Penetratin Reveal Peptide Induced Reduction in Gene Expression and Induction of Innate Immunity. Bioconj. Chem. 2007, 18, 1450–1459. [Google Scholar] [CrossRef]
- Begley, R.; Liron, T.; Baryza, J.; Mochly-Rosen, D. Biodistribution of intracellularly acting peptides conjugated reversibly to Tat. Biochem. Biophys. Res. Commun. 2004, 318, 949–954. [Google Scholar] [CrossRef]
- Goldenberg, N.M.; Steinberg, B.E.; Slutsky, A.S.; Lee, W.L. Broken Barriers: A New Take on Sepsis Pathogenesis. Sci. Transl. Med. 2011, 3, 88ps25. [Google Scholar] [CrossRef]
- Maniatis, N.A.; Orfanos, S.E. The endothelium in acute lung injury/acute respiratory distress syndrome. Curr. Opin. Crit. Care 2008, 14, 22–30. [Google Scholar] [CrossRef]
- Ley, K.; Reutershan, J. Leucocyte-endothelial interactions in health and disease. Handb. Exp. Pharmacol. 2006, 97–133. [Google Scholar]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef]
- Guo, R.-F.; Riedemann, N.C.; Laudes, I.J.; Sarma, V.J.; Kunkel, R.G.; Dilley, K.A.; Paulauskis, J.D.; Ward, P.A. Altered Neutrophil Trafficking During Sepsis. J. Immunol. 2002, 169, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Roth, N.M.; Kiani, M.F. A geographic information systems based technique for the study of microvascular networks. Ann. Biomed. Eng. 1999, 27, 42–47. [Google Scholar] [CrossRef]
- Prabhakarpandian, B.; Wang, Y.I.; Rea-Ramsey, A.; Sundaram, S.; Kiani, M.F.; Pant, K. Bifurcations: Focal Points of Particle Adhesion in Microvascular Networks. Microcirculation 2011, 18, 380–389. [Google Scholar] [CrossRef]
- Prabhakarpandian, B.; Pant, K.; Scott, R.; Patillo, C.; Irimia, D.; Kiani, M.; Sundaram, S. Synthetic microvascular networks for quantitative analysis of particle adhesion. Biomed. Microd. 2008, 10, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Rosano, J.; Tousi, N.; Scott, R.; Krynska, B.; Rizzo, V.; Prabhakarpandian, B.; Pant, K.; Sundaram, S.; Kiani, M. A physiologically realistic in vitro model of microvascular networks. Biomed. Microd. 2009, 11, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Tousi, N.; Wang, B.; Pant, K.; Kiani, M.F.; Prabhakarpandian, B. Preferential adhesion of leukocytes near bifurcations is endothelium independent. Microvasc. Res. 2010, 80, 384–388. [Google Scholar] [CrossRef]
- Prabhakarpandian, B.; Shen, M.-C.; Pant, K.; Kiani, M.F. Microfluidic devices for modeling cell-cell and particle-cell interactions in the microvasculature. Microvasc. Res. 2011, 82, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Molla, M.; Panes, J.; Casadevall, M.; Salas, A.; Conill, C.; Biete, A.; Anderson, D.C.; Granger, D.N.; Pique, J.M. Influence of dose-rate on inflammatory damage and adhesion molecule expression after abdominal radiation in the rat. Int. J. Radiat. Oncol. Biol. Phys. 1999, 45, 1011–1018. [Google Scholar] [CrossRef]
- Yuan, H.; Gaber, M.W.; McColgan, T.; Naimark, M.D.; Kiani, M.F.; Merchant, T.E. Radiation-induced permeability and leukocyte adhesion in the rat blood-brain barrier: Modulation with anti-ICAM-1 antibodies. Brain Res. 2003, 969, 59–69. [Google Scholar] [CrossRef]
- Yuan, H.; Gaber, M.W.; Boyd, K.; Wilson, C.M.; Kiani, M.F.; Merchant, T.E. Effects of fractionated radiation on the brain vasculature in a murine model: Blood-brain barrier permeability, astrocyte proliferation, and ultrastructural changes. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Goetz, D.J.; Gaber, M.W.; Issekutz, A.C.; Merchant, T.E.; Kiani, M.F. Radiation-induced up-regulation of adhesion molecules in brain microvasculature and their modulation by dexamethasone. Radiat. Res. 2005, 163, 544–551. [Google Scholar] [CrossRef]
- Flanders, K.C.; Ho, B.M.; Arany, P.R.; Stuelten, C.; Mamura, M.; Paterniti, M.O.; Sowers, A.; Mitchell, J.B.; Roberts, A.B. Absence of Smad3 induces neutrophil migration after cutaneous irradiation: Possible contribution to subsequent radioprotection. Am. J. Pathol. 2008, 173, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Francois, A.; Milliat, F.; Guipaud, O.; Benderitter, M. Inflammation and immunity in radiation damage to the gut mucosa. BioMed Res. Int. 2013, 2013, 123241. [Google Scholar] [CrossRef]
- Wong, S.L.; Lau, C.W.; Wong, W.T.; Xu, A.; Au, C.L.; Ng, C.F.; Ng, S.S.M.; Gollasch, M.; Yao, X.; Huang, Y. Pivotal Role of Protein Kinase Cd in Angiotensin II-Induced Endothelial Cyclooxygenase-2 Expression: A Link to Vascular Inflammation. Arterioscl. Thromb. Vasc. Biol. 2011, 31, 1169–1176. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | Effects |
|---|---|
| c-Abl | Increased activity [78,79] |
| SFKs | Variable [80] |
| SHPTP1 (protein tyrosine phosphatase) (SHP1) | Decreased phosphatase activity [81] |
| RasGRP | Uncertain [82] |
| Protein tyrosine phosphatase PTPα | Increased phosphatase activity [83] |
| PKCε (hydrophobic motif) | Yields release from membranes [50] |
| STAT1 (Ser-727) | Interferon gene expression [84] |
| STAT3 (Ser-727) | Reduced DNA binding and transcription [85] |
| p300 | HAT activity lowered, decreased transcriptional function [86] |
| 14-3-3 | Interfere with 14-3-3 polymerization and interactions with partners [87] |
| gp130 | Increased gp130-STAT3 interaction [88] |
| p47(pbox) unit of NADPH | Activity enhancement [89] |
| β4-integrin | Cell-laminin attachment decreases [90] |
| Caspase-3 | Promote the apoptotic activity of caspase-3 in monocytes both in vitro and in vivo [65] |
| MARCKS | Cell attachment and spreading in skeletal muscle cells [69] |
| M2 Pyruvate Kinase | Tumor metabolism; uncertain [91] |
| Heat Shock Protein 27 (HSP27) | Protein chaperone, antioxidant, apoptosis inhibition [92] |
| Plasma membrane calcium ATPase (PMCA) | Regulate calcium levels in skin [30,65] |
| Heat Shock Protein 25 | Inhibition of apoptosis [92] |
| p52Shc protein | Positively regulates H2O2-induced ERK activation [67] |
| p66Shc protein | Negatively regulates H2O2-induced ERK activation [67] |
| Troponin | Decreased Calcium sensitivity of actomyosin [70] |
| Pyruvate Dehydrogenase Kinase | Inhibition of PDH resulting in necrosis and blocking ATP regeneration [71] |
| DNA-dependent protein kinase | Inhibition of p53 phosphorylation [93] |
| Bcl-2-associated death promoter (BAD) | Promotes apoptosis post-reperfusion after cardiac ischemia [94] |
| Dynamin-related protein 1 (Drp1) | Induction of mitochondrial fission and dysfunction following cardiac ischemia [17] |
| Glyceraldehyde-3-phosphoate dehydrogenase (GADPH) | Removal of injured mitochondria following ischemic damage [95] |
| PLS3 | Higher phospholipid movement [96] |
| DNA-PK | Increase apoptosis due to malfunctional DNA [93] |
| Lamin B | Apoptosis [97] |
| hRad4 | Increased hRad9-Bcl-2 interactions/apoptosis [98] |
| p73β(Ser-289) | p73β activation; apoptosis [66] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Langston, J.C.; Tang, Y.; Kiani, M.F.; Kilpatrick, L.E. The Role of Tyrosine Phosphorylation of Protein Kinase C Delta in Infection and Inflammation. Int. J. Mol. Sci. 2019, 20, 1498. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061498
Yang Q, Langston JC, Tang Y, Kiani MF, Kilpatrick LE. The Role of Tyrosine Phosphorylation of Protein Kinase C Delta in Infection and Inflammation. International Journal of Molecular Sciences. 2019; 20(6):1498. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061498
Chicago/Turabian StyleYang, Qingliang, Jordan C. Langston, Yuan Tang, Mohammad F. Kiani, and Laurie E. Kilpatrick. 2019. "The Role of Tyrosine Phosphorylation of Protein Kinase C Delta in Infection and Inflammation" International Journal of Molecular Sciences 20, no. 6: 1498. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20061498