Metabotropic Glutamate Receptor 5 and 8 Modulate the Ameliorative Effect of Ultramicronized Palmitoylethanolamide on Cognitive Decline Associated with Neuropathic Pain

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
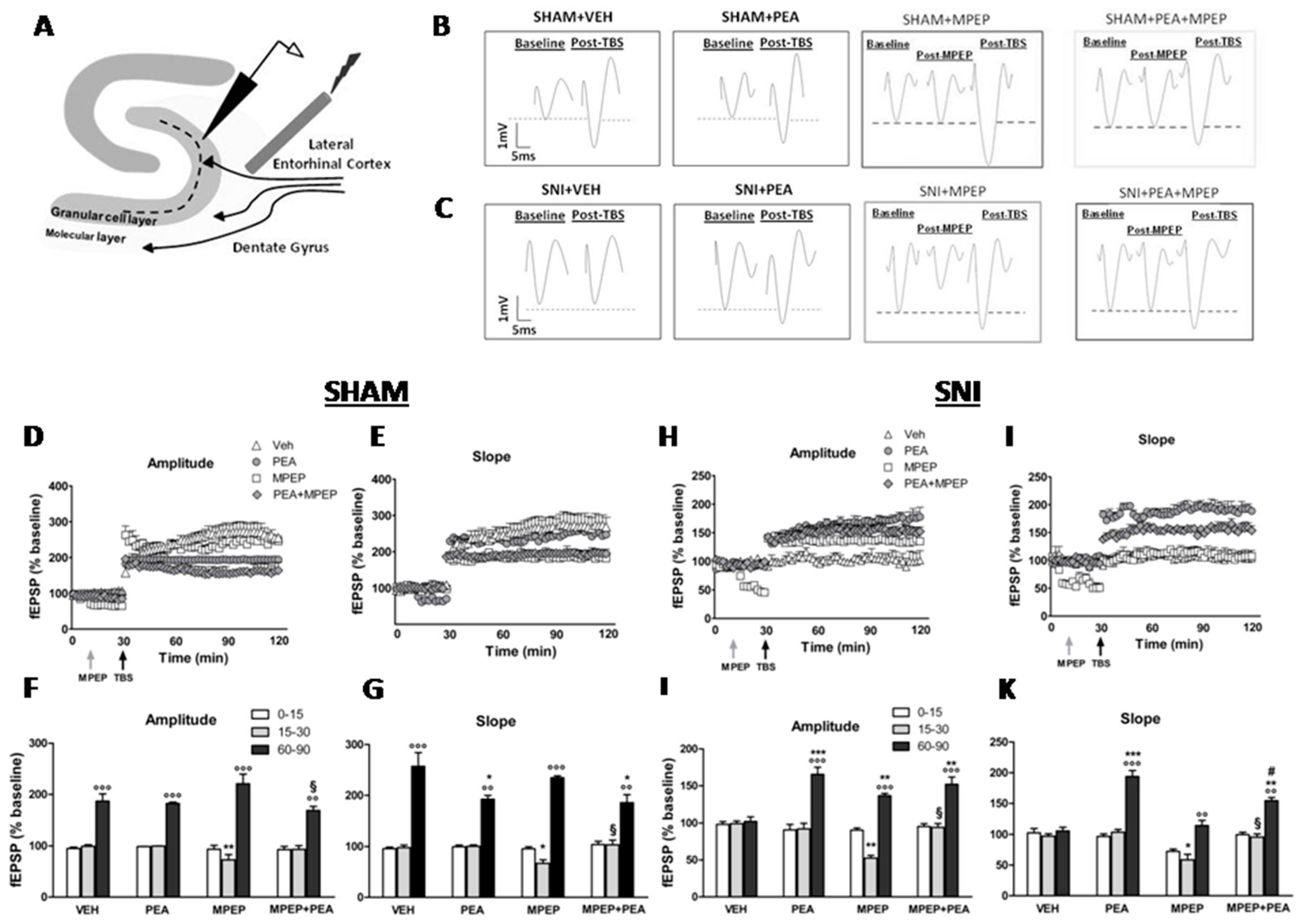
2. Results
2.1. Discriminative Memory
2.2. Effect of MPEP on LTP at the LEC-DG Pathway in Sham Mice Treated with Vehicle or PEA
2.3. Effect of MPEP on LTP at the LEC-DG Pathway in SNI Mice Treated with Vehicle or with PEA
2.4. Effect of MDCPG on LTP at the LEC-DG Pathway in Sham Mice Treated with Vehicle or with PEA
2.5. Effect of MDCPG on LTP at the LEC-DG Pathway in SNI Mice Treated with Vehicle or PEA
3. Discussion
3.1. Discriminative Memory and LTP at the LEC-DG Pathway in SNI Mice
3.2. The Effect of a Chronic Treatment with PEA on Discriminative Memory and LTP at the LEC-DG Pathway in Sham and SNI Mice
3.3. The Role of mGluR5 on the Effect of PEA on Discriminative Memory and LTP at the LEC-DG Pathway in Sham and SNI Mice
3.4. The Role of mGluR8 on the Effect of PEA on Discriminative Memory and LTP at the LEC-DG Pathway in Sham and SNI Mice
4. Materials and Methods
4.1. Animals
4.2. Spared Nerve Injury
4.3. Treatment
4.4. Novel Object Recognition
4.5. Surgical Preparation for In Vivo Field Potential Recordings at LEC-DG Pathway
4.6. Drugs
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Moriarty, O.; McGuire, B.E.; Finn, D.P. The effect of pain on cognitive function: A review of clinical and preclinical research. Prog. Neurobiol. 2011, 93, 385–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuner, R. Spinal excitatory mechanisms of pathological pain. Pain 2015, 156, S11–S17. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, O.; Gorman, C.L.; McGowan, F.; Ford, G.K.; Roche, M.; Thompson, K.; Dockery, P.; McGuire, B.E.; Finn, D.P. Impaired recognition memory and cognitive flexibility in the rat L5–L6 spinal nerve ligation model of neuropathic pain. Scand. J. Pain 2016, 10, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Gol, A.; Faibish, G.M. Hippocampectomy for relief of intractable pain. Tex. Med. 1966, 62, 76–79. [Google Scholar]
- Grilli, M. Chronic pain and adult hippocampal neurogenesis: Translational implications from preclinical studies. J. Pain Res. 2017, 10, 2281–2286. [Google Scholar] [CrossRef] [PubMed]
- Mutso, A.A.; Radzicki, D.; Baliki, M.N.; Huang, L.; Banisadr, G.; Centeno, M.V.; Radulovic, J.; Martina, M.; Miller, R.J.; Apkarian, A.V. Abnormalities in hippocampal functioning with persistent pain. J. Neurosci. 2012, 32, 5747–5756. [Google Scholar] [CrossRef]
- Ren, W.J.; Liu, Y.; Zhou, L.J.; Li, W.; Zhong, Y.; Pang, R.P.; Xin, W.J.; Wei, X.H.; Wang, J.; Zhu, H.Q.; et al. Peripheral Nerve Injury Leads to Working Memory Deficits and Dysfunction of the Hippocampus by Upregulation of TNF-α in Rodents. Neuropsychopharmacology 2011, 36, 979–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.M.; Neugebauer, V. Amygdala Plasticity and Pain. Pain Res. Manag. 2017, 2017, 8296501. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, S.; Riekkinen, P.J. Effects of apamin on memory processing of hippocampal-lesioned mice. Eur. J. Pharmacol. 1999, 382, 151–156. [Google Scholar] [CrossRef]
- Van der Staay, F.J.; Fanelli, R.J.; Blokland, A.; Schmidt, B. Behavioral effects of apamin, a selective inhibitor of the SK(Ca)-channel, in mice and rats. Neurosci. Biobehav. Rev. 2000, 23, 1087–1110. [Google Scholar] [CrossRef]
- Kwon, M.; Han, J.; Kim, U.J.; Cha, M.; Um, S.W.; Bai, S.J.; Hong, S.K.; Lee, B.H. Inhibition of Mammalian Target of Rapamycin (mTOR) Signaling in the Insular Cortex Alleviates Neuropathic Pain after Peripheral Nerve Injury. Front. Mol. Neurosci. 2017, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Kodama, D.; Ono, H.; Tanabe, M. Altered hippocampal long-term potentiation after peripheral nerve injury in mice. Eur. J. Pharmacol. 2007, 574, 127–132. [Google Scholar] [CrossRef]
- Dellarole, A.; Morton, P.; Brambilla, R.; Walters, W.; Summers, S.; Bernardes, D.; Grilli, M.; Bethea, J.R. Neuropathic pain-induced depressive-like behavior and hippocampal neurogenesis and plasticity are dependent on TNFR1 signaling. BrainBehav. Immun. 2014, 41, 65–81. [Google Scholar] [CrossRef]
- Witter, M.P. The perforant path: Projections from the entorhinal cortex to the dentate gyrus. Prog. Brain Res. 2007, 163, 43–61. [Google Scholar]
- Bush, D.; Caswell, B.; Burgess, N. What do grid cells contribute to place cell firing? Trends Neurosci. 2014, 37, 136–145. [Google Scholar] [CrossRef] [Green Version]
- Boccella, S.; Cristiano, C.; Romano, R.; Iannotta, M.; Belardo, C.; Farina, A.; Guida, F.; Piscitelli, F.; Palazzo, E.; Imperatore, R.; et al. Ultra-micronized palmitoylethanolamide rescues the cognitive decline-associated loss of neural plasticity in the neuropathic mouse entorhinal cortex-dentate gyrus pathway. Neurobiol. Dis. 2019, 121, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Guida, F.; Luongo, L.; Marmo, F.; Romano, R.; Iannotta, M.; Napolitano, F.; Belardo, C.; Marabese, I.; D’Aniello, A.; De Gregorio, D.; et al. Palmitoylethanolamide reduces pain-related behaviors and restores glutamatergic synapses homeostasis in the medial prefrontal cortex of neuropathic mice. Mol. Brain 2015, 8, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guida, F.; Boccella, S.; Iannotta, M.; De Gregorio, D.; Giordano, C.; Belardo, C.; Romano, R.; Palazzo, E.; Scafuro, M.A.; Serra, N.; et al. Palmitoylethanolamide Reduces Neuropsychiatric Behaviors by Restoring Cortical Electrophysiological Activity in a Mouse Model of Mild Traumatic Brain Injury. Front. Pharmacol. 2017, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghoul, W.M.; Volsi, G.L.; Weinberg, R.J.; Rustioni, A. Glutamate immunocytochemistry in the dorsal horn after injury or stimulation of the sciatic nerve of rats. Brain Res. Bull. 1993, 30, 453–459. [Google Scholar] [CrossRef]
- De Novellis, V.; Vita, D.; Gatta, L.; Luongo, L.; Bellini, G.; De Chiaro, M.; Marabese, I.; Siniscalco, D.; Boccella, S.; Piscitelli, F.; et al. The blockade of the transient receptor potential vanilloid type 1 and fatty acid amide hydrolase decreases symptoms and central sequelae in the medial prefrontal cortex of neuropathic rats. Mol. Pain 2011, 7, 7–25. [Google Scholar] [CrossRef]
- Lin, T.Y.; Lu, C.W.; Wu, C.C.; Huang, S.K.; Wang, S.J. Palmitoylethanolamide inhibits glutamate release in rat cerebrocortical nerve terminals. Int. J. Mol. Sci. 2015, 16, 5555–5571. [Google Scholar] [CrossRef]
- Marcello, L.; Cavaliere, C.; Colangelo, A.M.; Bianco, M.R.; Cirillo, G.; Alberghina, L.; Papa, M. Remodelling of supraspinal neuroglial network in neuropathic pain is featured by a reactive gliosis of the nociceptive amygdala. Eur. J. Pain 2013, 17, 799–810. [Google Scholar] [CrossRef]
- Santangelo, R.M.; Acker, T.M.; Zimmerman, S.S.; Katzman, B.M.; Strong, K.L.; Traynelis, S.F.; Liotta, D.C. Novel NMDA receptor modulators: An update. Exp. Opin. Ther. Pat. 2012, 22, 1337–1352. [Google Scholar] [CrossRef]
- Goudet, C.; Chapuy, E.; Alloui, A.; Acher, F.; Pin, J.P.; Eschalier, A. Group III metabotropic glutamate receptors inhibit hyperalgesia in animal models of inflammation and neuropathic pain. Pain 2008, 137, 112–124. [Google Scholar] [CrossRef] [Green Version]
- Montana, M.C.; Cavallone, L.F.; Stubbert, K.K.; Stefanescu, A.D.; Kharasch, E.D.; Gereau, R.W. The metabotropic glutamate receptor subtype 5 antagonist fenobam is analgesic and has improved in vivo selectivity compared with the prototypical antagonist 2-methyl-6-(phenylethynyl)-pyridine. J. Pharmacol. Exp. Ther. 2009, 330, 834–843. [Google Scholar] [CrossRef]
- Osikowicz, M.; Mika, J.; Makuch, W.; Przewlocka, B. Glutamate receptor ligands attenuate allodynia and hyperalgesia and potentiate morphine effects in a mouse model of neuropathic pain. Pain 2008, 139, 117–126. [Google Scholar] [CrossRef]
- Palazzo, E.; Genovese, R.; Mariani, L.; Siniscalco, D.; Marabese, I.; de Novellis, V.; Rossi, F.; Maione, S. Metabotropic glutamate receptor 5 and dorsal raphe serotonin release in inflammatory pain in rat. Eur. J. Pharmacol. 2004, 492, 169–176. [Google Scholar] [CrossRef]
- Palazzo, E.; de Novellis, V.; Rossi, F.; Maione, S. Supraspinal metabotropic glutamate receptor subtype 8: A switch to turn off pain. Amino Acids 2014, 46, 1441–1448. [Google Scholar] [CrossRef]
- Palazzo, E.; Marabese, I.; de Novellis, V.; Rossi, F.; Maione, S. Supraspinal metabotropic glutamate receptors: A target for pain relief and beyond. Eur. J. Neurosci. 2014, 39, 444–454. [Google Scholar] [CrossRef]
- Palazzo, E.; Marabese, I.; Luongo, L.; Guida, F.; de Novellis, V.; Maione, S. Nociception modulation by supraspinal group III metabotropic glutamate receptors. J. Neurochem. 2017, 141, 507–519. [Google Scholar] [CrossRef]
- Almeida-Santos, A.F.; Moreira, F.A.; Guimaraes, F.S.; Aguiar, D.C. 2-Arachidonoylglycerol endocannabinoid signaling coupled to metabotropic glutamate receptor type-5 modulates anxiety-like behavior in the rat ventromedial prefrontal cortex. J. Psychopharmacol. 2017, 31, 740–749. [Google Scholar] [CrossRef]
- Olmo, I.G.; Ferreira-Vieira, T.H.; Ribeiro, F.M. Dissecting the Signaling Pathways Involved in the Crosstalk between Metabotropic Glutamate 5 and Cannabinoid Type 1 Receptors. Mol. Pharmacol. 2016, 90, 609–619. [Google Scholar] [CrossRef]
- Palazzo, E.; Marabese, I.; de Novellis, V.; Oliva, P.; sca Rossi, F.; Berrino, L.; Rossi, F.; Maione, S. Metabotropic and NMDA glutamate receptors participate in the cannabinoid-induced antinociception. Neuropharmacology 2001, 40, 319–326. [Google Scholar] [CrossRef]
- Palazzo, E.; de Novellis, V.; Petrosino, S.; Marabese, I.; Vita, D.; Giordano, C.; Di Marzo, V.; Mangoni, G.S.; Rossi, F.; Maione, S. Neuropathic pain and the endocannabinoid system in the dorsal raphe: Pharmacological treatment and interactions with the serotonergic system. Eur. J. Neurosci. 2006, 24, 2011–2020. [Google Scholar] [CrossRef]
- Palazzo, E.; Luongo, L.; Bellini, G.; Guida, F.; Marabese, I.; Boccella, S.; Rossi, F.; Maione, S.; de Novellis, V. Changes in cannabinoid receptor subtype 1 activity and interaction with metabotropic glutamate subtype 5 receptors in the periaqueductal gray-rostral ventromedial medulla pathway in a rodent neuropathic pain model. CNS Neurol. Dis. Drug Targets 2012, 11, 148–161. [Google Scholar] [CrossRef]
- Yalcin, I.; Bohren, Y.; Waltisperger, E.; Sage-Ciocca, D.; Yin, J.C.; Freund-Mercier, M.J.; Barrot, M. A time-dependent history of mood disorders in a murine model of neuropathic pain. Biol. Psychiatry 2011, 70, 946–953. [Google Scholar] [CrossRef]
- Wilson, D.I.; Langston, R.F.; Schlesiger, M.I.; Wagner, M.; Watanabe, S.; Ainge, J.A. Lateral entorhinal cortex is critical for novel object-context recognition. Hippocampus 2013, 23, 352–366. [Google Scholar] [CrossRef]
- Lyu, D.; Yu, W.; Tang, N.; Wang, R.; Zhao, Z.; Xie, F.; He, Y.; Du, H.; Chen, J. The mTOR signaling pathway regulates pain-related synaptic plasticity in rat entorhinal-hippocampal pathways. Mol. Pain 2013, 9, 64. [Google Scholar] [CrossRef]
- Sestito, R.S.; Trindade, L.B.; de Souza, R.G.; Kerbauy, L.N.; Iyomasa, M.M.; Rosa, M.L. Effect of isolation rearing on the expression of AMPA glutamate receptors in the hippocampal formation. J. Psychopharmacol. 2011, 25, 1720–1729. [Google Scholar] [CrossRef]
- Barnes, C.A.; Jung, M.W.; McNaughton, B.L.; Korol, D.L.; Andreasson, K.; Worley, P.F. LTP saturation and spatial learning disruption: Effects of task variables and saturation levels. J. Neurosci. 1994, 14, 5793–5806. [Google Scholar] [CrossRef]
- Kanno, T.; Nagata, T.; Yamamoto, S.; Okamura, H.; Nishizaki, T. Interleukin-18 stimulates synaptically released glutamate and enhances postsynaptic AMPA receptor responses in the CA1 region of mouse hippocampal slices. Brain Res. 2004, 1012, 190–193. [Google Scholar] [CrossRef]
- Giordano, C.; Cristino, L.; Luongo, L.; Siniscalco, D.; Petrosino, S.; Piscitelli, F.; Marabese, I.; Gatta, L.; Rossi, F.; Imperatore, R.; et al. TRPV1-dependent and -independent alterations in the limbic cortex of neuropathic mice: Impact on glial caspases and pain perception. Cereb. Cortex 2012, 22, 2495–2518. [Google Scholar] [CrossRef]
- Hudson, L.J.; Bevan, S.; McNair, K.; Gentry, C.; Fox, A.; Kuhn, R.; Winter, J. Metabotropic glutamate receptor 5 upregulation in A-fibers after spinal nerve injury: 2-methyl-6-(phenylethynyl)-pyridine (MPEP) reverses the induced thermal hyperalgesia. J. Neurosci. 2002, 22, 2660–2668. [Google Scholar] [CrossRef]
- Chaise, T.N.; Oh, J.D. Striatal dopamine- and glutamate-mediated dysregulation in experimental parkinsonism. Trends Neurosci. 2000, 23, S86–S91. [Google Scholar] [CrossRef]
- Greenamyre, J.T. Glutamatergic influences on the basal ganglia. Clin. Neuropharmacol. 2001, 24, 65–70. [Google Scholar] [CrossRef]
- Cartmell, J.; Schoepp, D.D. Regulation of neurotransmitter release by metabotropic glutamate receptors. J. Neurochem. 2000, 75, 889–907. [Google Scholar] [CrossRef]
- Palazzo, E.; Marabese, I.; Soukupova, M.; Luongo, L.; Boccella, S.; Giordano, C.; de Novellis, V.; Rossi, F.; Maione, S. Metabotropic glutamate receptor subtype 8 in the amygdala modulates thermal threshold, neurotransmitter release, and rostral ventromedial medulla cell activity in inflammatory pain. J. Neurosci. 2011, 31, 4687–4697. [Google Scholar] [CrossRef]
- Rossi, F.; Marabese, I.; De Chiaro, M.; Boccella, S.; Luongo, L.; Guida, F.; De Gregorio, D.; Giordano, C.; de Novellis, V.; Palazzo, E.; et al. Dorsal striatum metabotropic glutamate receptor 8 affects nocifensive responses and rostral ventromedial medulla cell activity in neuropathic pain conditions. J. Neurophysiol. 2014, 111, 2196–2209. [Google Scholar] [CrossRef] [Green Version]
- Gwak, Y.S.; Hulsebosch, C.E. Neuronal hyperexcitability: A substrate for central neuropathic pain after spinal cord injury. Curr. Pain Headache Rep. 2011, 15, 215–222. [Google Scholar] [CrossRef]
- Anwyl, R. Metabotropic glutamate receptor-dependent long-term potentiation. Neuropharmacology 2009, 56, 735–740. [Google Scholar] [CrossRef]
- Bashir, Z.I.; Bortolotto, Z.A.; Davies, C.H.; Berretta, N.; Irving, A.J.; Seal, A.J.; Henley, J.M.; Jane, D.E.; Watkins, J.C.; Collingridge, G.L. Induction of LTP in the hippocampus needs synaptic activation of glutamate metabotropic receptors. Nature 1993, 363, 347–350. [Google Scholar] [CrossRef]
- Cumiskey, D.; Pickering, M.; O’Connor, J.J. Interleukin-18 mediated inhibition of LTP in the rat dentate gyrus is attenuated in the presence of mGluR antagonists. Neurosci. Lett. 2007, 412, 206–210. [Google Scholar] [CrossRef]
- Richter-Levin, G.; Errington, M.L.; Maegawa, H.; Bliss, T.V. Activation of metabotropic glutamate receptors is necessary for long-term potentiation in the dentate gyrus and for spatial learning. Neuropharmacology 1994, 33, 853–857. [Google Scholar] [CrossRef]
- Riedel, G.; Casabona, G.; Reymann, K.G. Inhibition of long-term potentiation in the dentate gyrus of freely moving rats by the metabotropic glutamate receptor antagonist MCPG. J. Neurosci. 1995, 15, 87–98. [Google Scholar] [CrossRef]
- Artola, A.; Singer, W. Long-term depression of excitatory synaptic transmission and its relationship to long-term potentiation. Trends Neurosci. 1993, 16, 480–487. [Google Scholar] [CrossRef]
- Fernyhough, P.; Calcutt, N.A. Abnormal calcium homeostasis in peripheral neuropathies. Cell Calcium 2010, 47, 130–139. [Google Scholar] [CrossRef]
- Walker, K.R.; Tesco, G. Molecular mechanisms of cognitive dysfunction following traumatic brain injury. Front. Aging Neurosci. 2013, 5, 29. [Google Scholar] [CrossRef]
- Rodrigues, S.M.; Bauer, E.P.; Farb, C.R.; Schafe, G.E.; LeDoux, J.E. The group I metabotropic glutamate receptor mGluR5 is required for fear memory formation and long-term potentiation in the lateral amygdala. J. Neurosci. 2002, 22, 5219–5229. [Google Scholar] [CrossRef]
- Simonyi, A.; Serfozo, P.; Shelat, P.B.; Dopheide, M.M.; Coulibaly, A.P.; Schachtman, T.R. Differential roles of hippocampal metabotropic glutamate receptors 1 and 5 in inhibitory avoidance learning. Neurobiol. Learn. Mem. 2007, 88, 305–311. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Trieu, B.H.; Palmer, L.C.; Jia, Y.; Pham, D.T.; Jung, K.M.; Karsten, C.A.; Merrill, C.B.; Mackie, K.; Gall, C.M.; et al. A Primary Cortical Input to Hippocampus Expresses a Pathway-Specific and Endocannabinoid-Dependent Form of Long-Term Potentiation. eNeuro 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Fendt, M.; Bürki, H.; Imobersteg, S.; van der Putten, H.; McAllister, K.; Leslie, J.C.; Shaw, D.; Hölscher, C. The effect of mGlu8 deficiency in animal models of psychiatric diseases. Genes Brain Behav. 2010, 9, 33–44. [Google Scholar] [CrossRef]
- Duvoisin, R.M.; Zhang, C.; Pfankuch, T.F.; O’Connor, H.; Gayet-Primo, J.; Quraishi, S.; Raber, J. Increased measures of anxiety and weight gain in mice lacking the group III metabotropic glutamate receptor mGluR8. Eur. J. Neurosci. 2005, 22, 425–436. [Google Scholar] [CrossRef]
- Goddyn, H.; Callaerts-Vegh, Z.; D’Hooge, R. Functional Dissociation of Group III Metabotropic Glutamate Receptors Revealed by Direct Comparison between the Behavioral Profiles of Knockout Mouse Lines. Int. J. Neuropsychopharmacol. 2015, 18, 1–11. [Google Scholar] [CrossRef]
- Shigemoto, R.; Kinoshita, A.; Wada, E.; Nomura, S.; Ohishi, H.; Takada, M.; Flor, P.J.; Neki, A.; Abe, T.; Nakanishi, S.; et al. Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J. Neurosci. 1997, 17, 7503–7522. [Google Scholar] [CrossRef]
- Scanziani, M.; Salin, P.A.; Vogt, K.E.; Malenka, R.C.; Nicoll, R.A. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature 1997, 385, 630–634. [Google Scholar] [CrossRef]
- Ferraguti, F.; Klausberger, T.; Cobden, P.; Baude, A.; Roberts, J.D.; Szucs, P.; Kinoshita, A.; Shigemoto, R.; Somogyi, P.; Dalezios, Y. Metabotropic glutamate receptor 8-expressing nerve terminals target subsets of GABAergic neurons in the hippocampus. J. Neurosci. 2005, 25, 10520–10536. [Google Scholar] [CrossRef]
- Mercier, M.S.; Lodge, D.; Fang, G.; Nicolas, C.S.; Collett, V.J.; Jane, D.E.; Collingridge, G.L.; Bortolotto, Z.A. Characterisation of an mGlu8 receptor-selective agonist and antagonist in the lateral and medial perforant path inputs to the dentate gyrus. Neuropharmacology 2013, 67, 294–303. [Google Scholar] [CrossRef]
- D’Hooge, R.; Lüllmann-Rauch, R.; Beckers, T.; Balschun, D.; Schwake, M.; Reiss, K.; von Figura, K.; Saftig, P. Neurocognitive and psychotiform behavioral alterations and enhanced hippocampal long-term potentiation in transgenic mice displaying neuropathological features of human alpha-mannosidosis. J. Neurosci. 2005, 25, 6539–6549. [Google Scholar] [CrossRef]
- Gerlai, R.; Henderson, J.T.; Roder, J.C.; Jia, Z. Multiple behavioral anomalies in GluR2 mutant mice exhibiting enhanced LTP. Behav. Brain Res. 1998, 95, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Jolas, T.; Zhang, X.S.; Zhang, Q.; Wong, G.; Del Vecchio, R.; Gold, L.; Priestley, T. Long-term potentiation is increased in the CA1 area of the hippocampus of APP (swe/ind) CRND8 mice. Neurobiol. Dis. 2002, 11, 394–409. [Google Scholar] [CrossRef]
- Walther, T.; Balschun, D.; Voigt, J.P.; Fink, H.; Zuschratter, W.; Birchmeier, C.; Ganten, D.; Bader, M. Sustained long term potentiation and anxiety in mice lacking the Mas protooncogene. J. Biol. Chem. 1998, 273, 11867–11873. [Google Scholar] [CrossRef]
- Decosterd, I.; Woolf, C.J. Spared nerve injury: An animal model of persistent peripheral neuropathic pain. Pain 2000, 87, 149–158. [Google Scholar] [CrossRef]
- Paxinos, G.; Franklin, K. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates, 4th ed.; Academic Press: San Diego, CA, USA, 1997. [Google Scholar]
- Lodge, D.; Tidball, P.; Mercier, M.S.; Lucas, S.J.; Hanna, L.; Ceolin, L.; Kritikos, M.; Fitzjohn, S.M.; Sherwood, J.L.; Bannister, N.; et al. Antagonists reversibly reverse chemical LTD induced by group I, group II and group III metabotropic glutamate receptors. Neuropharmacology 2013, 74, 135–146. [Google Scholar] [CrossRef]
- Palazzo, E.; de Novellis, V.; Marabese, I.; Cuomo, D.; sca Rossi, F.; Berrino, L.; Rossi, F.; Maione, S. Interaction between vanilloid and glutamate receptors in the central modulation of nociception. Eur. J. Pharmacol. 2002, 439, 69–75. [Google Scholar] [CrossRef]
- Palazzo, E.; Fu, Y.; Ji, G.; Maione, S.; Neugebauer, V. Group III mGluR7 and mGluR8 in the amygdala differentially modulate nocifensive and affective pain behaviors. Neuropharmacology 2008, 55, 537–545. [Google Scholar] [CrossRef] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boccella, S.; Marabese, I.; Iannotta, M.; Belardo, C.; Neugebauer, V.; Mazzitelli, M.; Pieretti, G.; Maione, S.; Palazzo, E. Metabotropic Glutamate Receptor 5 and 8 Modulate the Ameliorative Effect of Ultramicronized Palmitoylethanolamide on Cognitive Decline Associated with Neuropathic Pain. Int. J. Mol. Sci. 2019, 20, 1757. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071757
Boccella S, Marabese I, Iannotta M, Belardo C, Neugebauer V, Mazzitelli M, Pieretti G, Maione S, Palazzo E. Metabotropic Glutamate Receptor 5 and 8 Modulate the Ameliorative Effect of Ultramicronized Palmitoylethanolamide on Cognitive Decline Associated with Neuropathic Pain. International Journal of Molecular Sciences. 2019; 20(7):1757. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071757
Chicago/Turabian StyleBoccella, Serena, Ida Marabese, Monica Iannotta, Carmela Belardo, Volker Neugebauer, Mariacristina Mazzitelli, Gorizio Pieretti, Sabatino Maione, and Enza Palazzo. 2019. "Metabotropic Glutamate Receptor 5 and 8 Modulate the Ameliorative Effect of Ultramicronized Palmitoylethanolamide on Cognitive Decline Associated with Neuropathic Pain" International Journal of Molecular Sciences 20, no. 7: 1757. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071757