Cyclooxygenase-2 Activity Regulates Recruitment of VEGF-Secreting Ly6Chigh Monocytes in Ventilator-Induced Lung Injury

 ,
,
Abstract
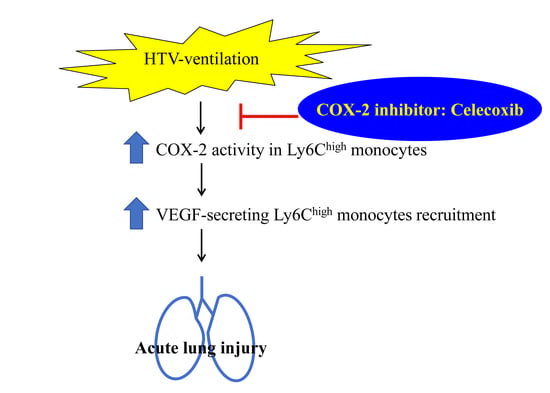
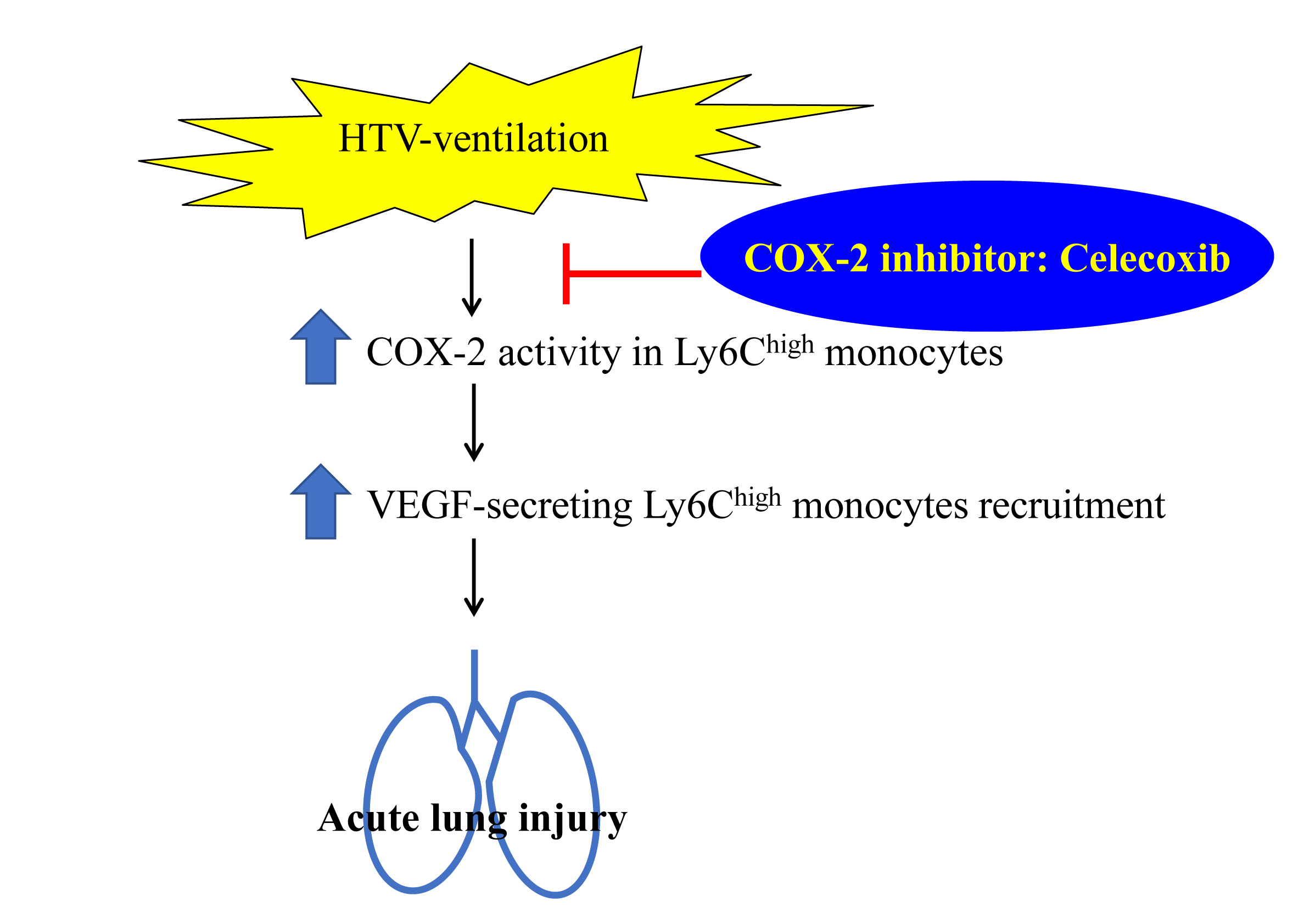
:
1. Introduction
2. Results
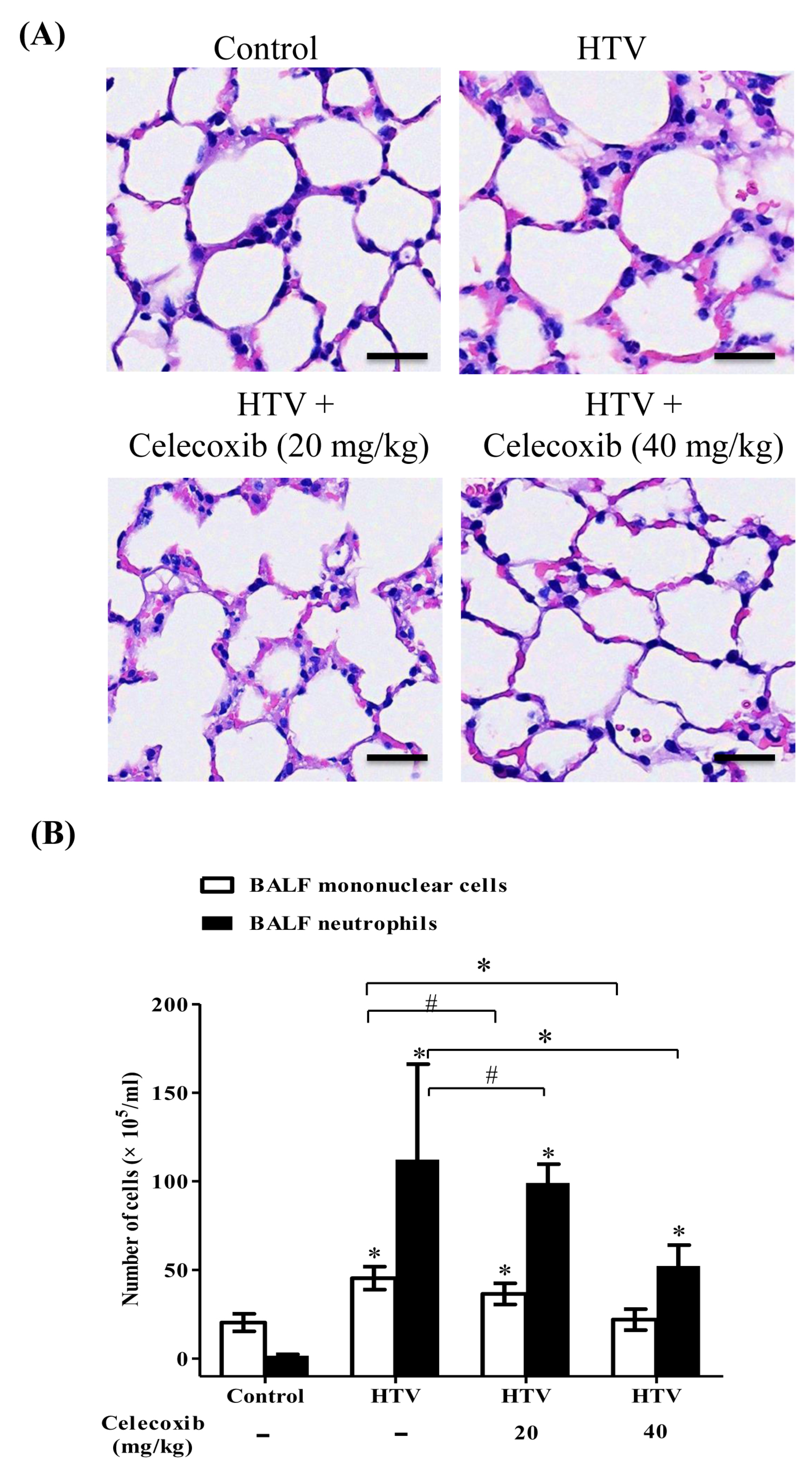
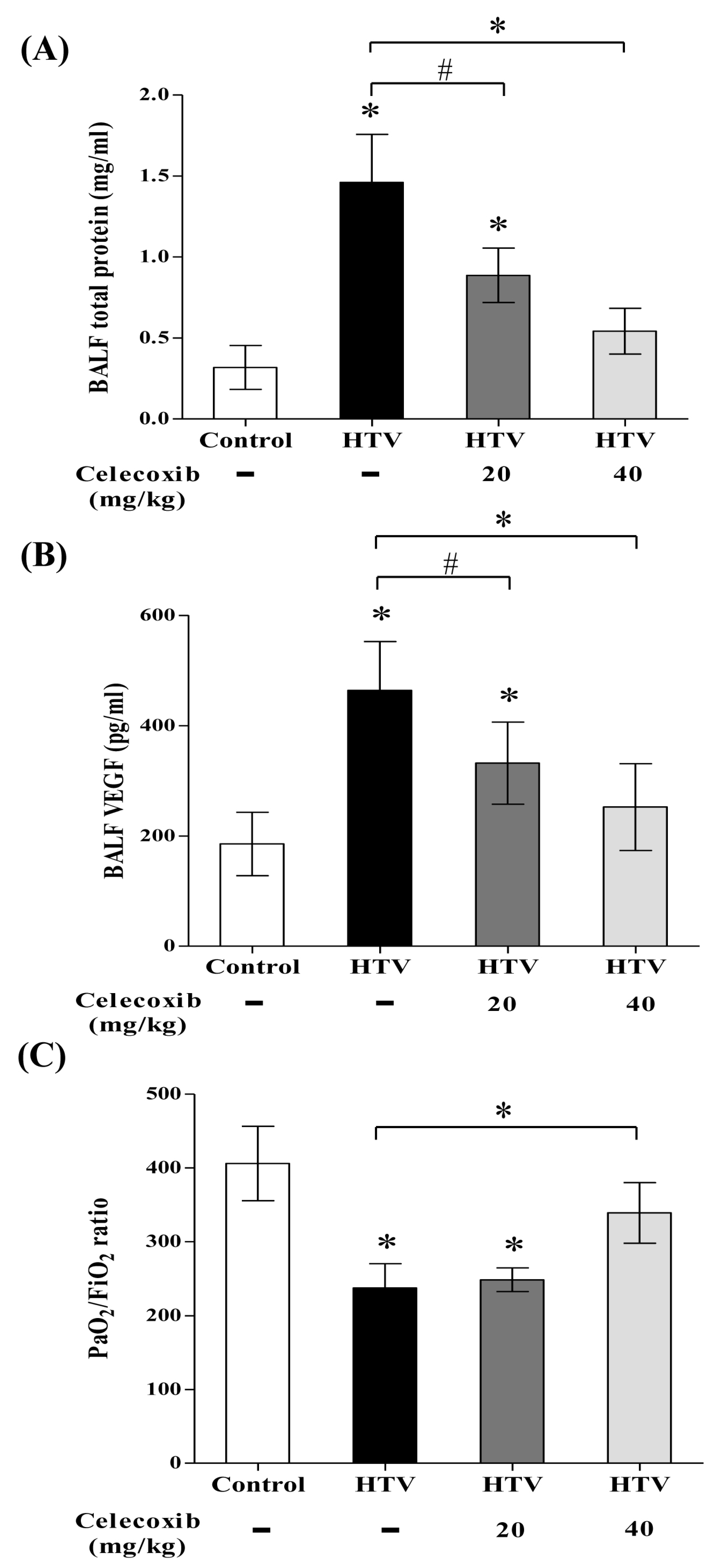
2.1. Celecoxib, a Clinical COX-2 Inhibitor, Attenuates VILI
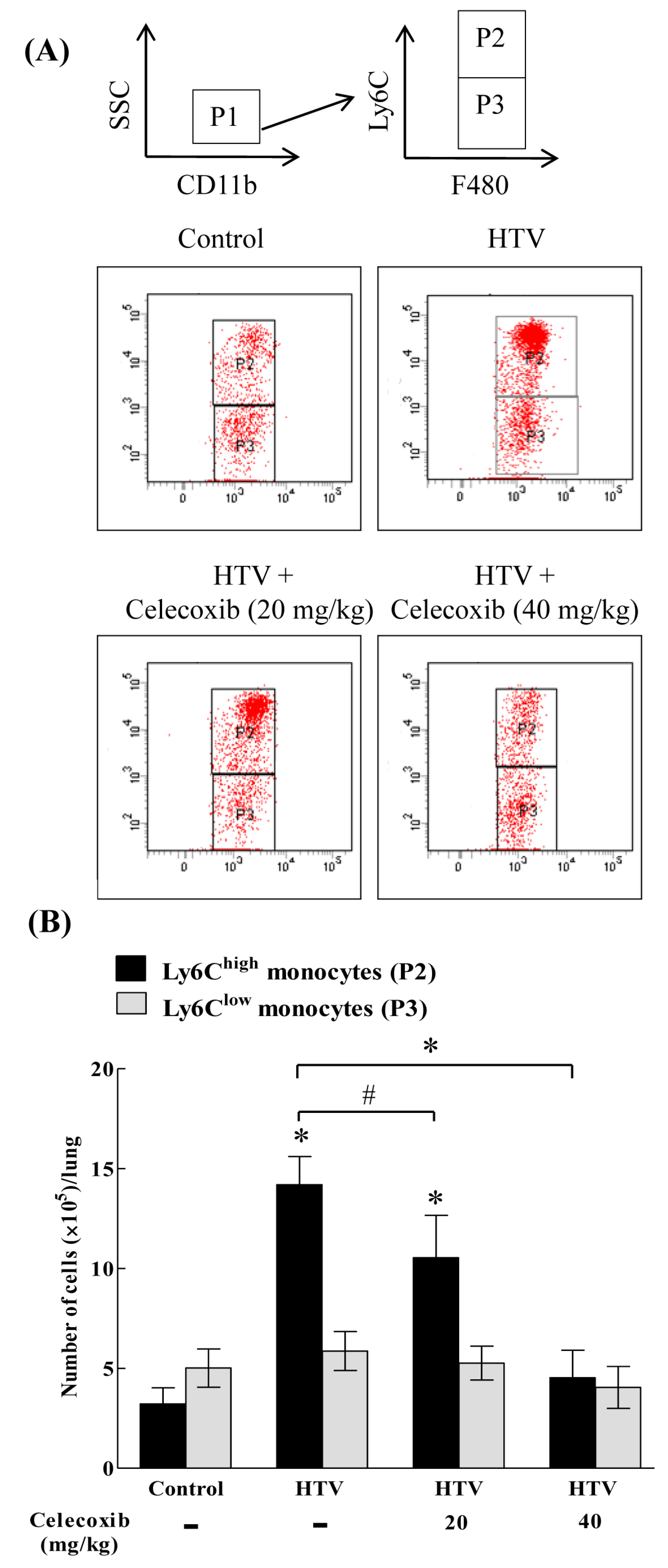
2.2. The Recruitment of COX-2-Expressing Neutrophils and Ly6Chigh, but Not Ly6Clow, Monocytes Is Enhanced during VILI
2.3. Celecoxib Aignificantly Mitigates the Recruitment of Ly6Chigh, but Not Ly6Clow, Monocytes in VILI
2.4. Celecoxib Reduces Alveolar Protein Outflow and VEGF Secretion into Bronchoalveolar Lavage Fluid (BALF) and Improves Pulmonary Oxygenation in VILI
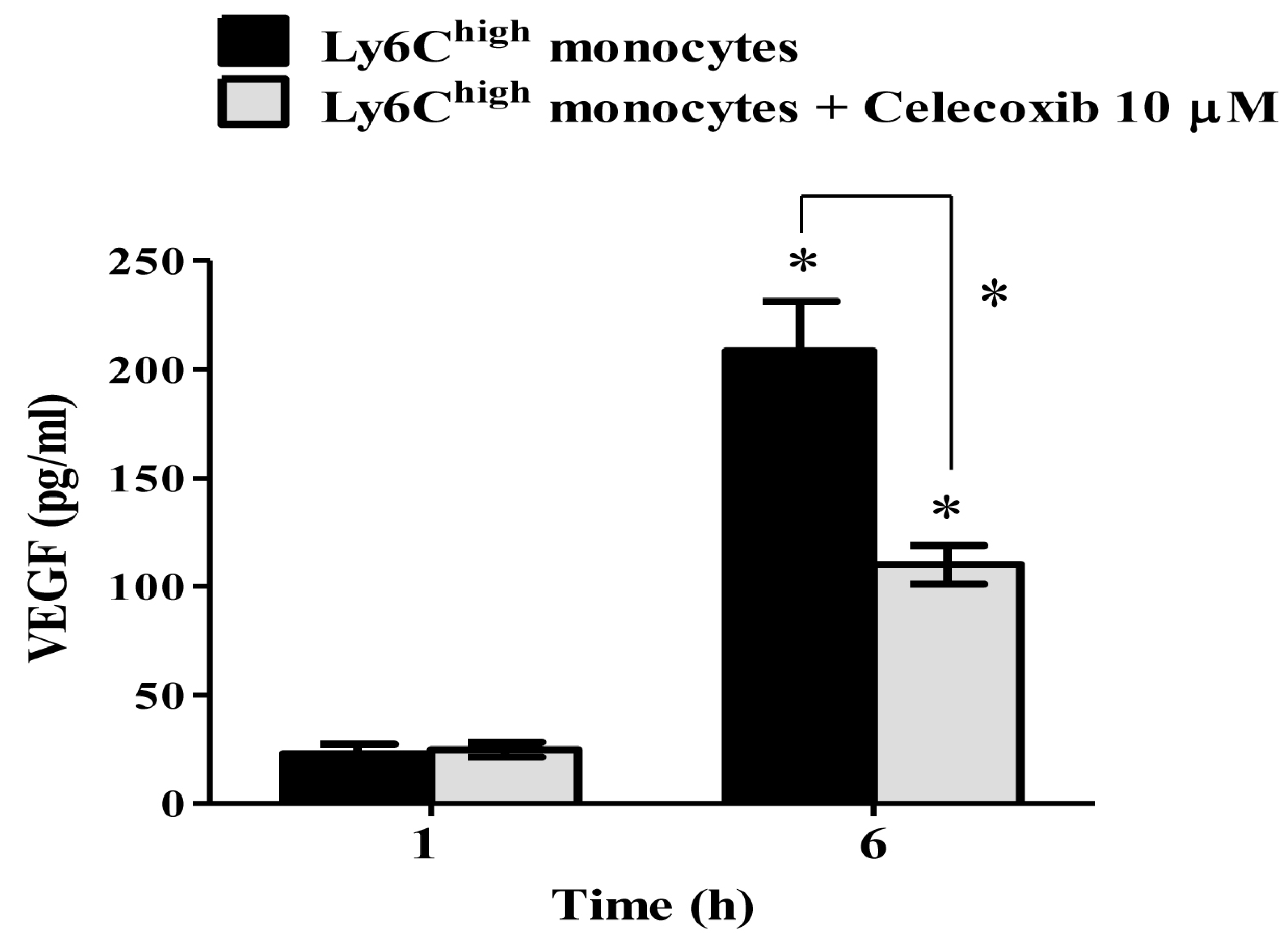
2.5. VEGF Secretion in Ly6Chigh Monocytes Is COX-2-Dependent
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Experimental Model of Ventilation-Induced Lung Injury
4.3. Analysis of BALF
4.4. Classification Strategy of Flow Cytometry
4.5. Ex vivo VEGF Secretion Assay
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| VILI | ventilator-induced lung injury |
| HTV | high tidal volume |
| BALF | bronchoalveolar lavage fluid |
| VEGF | vascular endothelial growth factor |
References
- Ware, L.B.; Matthay, M.A. The acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1334–1349. [Google Scholar] [CrossRef]
- Lee, H.Y.; Kim, S.D.; Shim, J.W.; Lee, S.Y.; Lee, H.; Cho, K.H.; Yun, J.; Bae, Y.S. Serum amyloid A induces CCL2 production via formyl peptide receptor-like 1-mediated signaling in human monocytes. J. Immunol. 2008, 181, 4332–4339. [Google Scholar] [CrossRef] [PubMed]
- Brower, R.G.; Matthay, M.A.; Morris, A.; Schoenfeld, D.; Thompson, B.T.; Wheeler, A. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 2000, 342, 1301–1308. [Google Scholar]
- International consensus conferences in intensive care medicine: Ventilator-associated Lung Injury in ARDS. This official conference report was cosponsored by the American Thoracic Society, The European Society of Intensive Care Medicine, and The Societe de Reanimation de Langue Francaise, and was approved by the ATS Board of Directors, July 1999. Am. J. Respir. Crit. Care Med. 1999, 160, 2118–2124. [CrossRef]
- Dreyfuss, D.; Basset, G.; Soler, P.; Saumon, G. Intermittent positive-pressure hyperventilation with high inflation pressures produces pulmonary microvascular injury in rats. Am. Rev. Respir. Dis. 1985, 132, 880–884. [Google Scholar]
- Maniatis, N.A.; Kotanidou, A.; Catravas, J.D.; Orfanos, S.E. Endothelial pathomechanisms in acute lung injury. Vasc. Pharmacol. 2008, 49, 119–133. [Google Scholar] [CrossRef]
- Parker, J.C.; Hernandez, L.A.; Peevy, K.J. Mechanisms of ventilator-induced lung injury. Crit. Care Med. 1993, 21, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.A.; Wray, C.M.; McAuley, D.F.; Schwendener, R.; Matthay, M.A. Alveolar macrophages contribute to alveolar barrier dysfunction in ventilator-induced lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1191–L1198. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, S.; Wilson, M.R.; Goddard, M.E.; O’Dea, K.P.; Takata, M. Mechanisms of early pulmonary neutrophil sequestration in ventilator-induced lung injury in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L902–L910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, E. Neutrophils and acute lung injury. Crit. Care Med. 2003, 31, S195–S199. [Google Scholar] [CrossRef] [PubMed]
- Wilson, M.R.; O’Dea, K.P.; Zhang, D.; Shearman, A.D.; van Rooijen, N.; Takata, M. Role of lung-marginated monocytes in an in vivo mouse model of ventilator-induced lung injury. Am. J. Respir. Crit. Care Med. 2009, 179, 914–922. [Google Scholar] [CrossRef]
- Muller, H.C.; Hellwig, K.; Rosseau, S.; Tschernig, T.; Schmiedl, A.; Gutbier, B.; Schmeck, B.; Hippenstiel, S.; Peters, H.; Morawietz, L.; et al. Simvastatin attenuates ventilator-induced lung injury in mice. Crit. Care 2010, 14, R143. [Google Scholar] [CrossRef]
- Shi, C.S.; Huang, T.H.; Lin, C.K.; Li, J.M.; Chen, M.H.; Tsai, M.L.; Chang, C.C. VEGF Production by Ly6C+high Monocytes Contributes to Ventilator-Induced Lung Injury. PLoS ONE 2016, 11, e0165317. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.; Taylor, P.R. Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef]
- Rose, S.; Misharin, A.; Perlman, H. A novel Ly6C/Ly6G-based strategy to analyze the mouse splenic myeloid compartment. Cytom. Part A 2012, 81, 343–350. [Google Scholar] [CrossRef]
- O’Dea, K.P.; Young, A.J.; Yamamoto, H.; Robotham, J.L.; Brennan, F.M.; Takata, M. Lung-marginated monocytes modulate pulmonary microvascular injury during early endotoxemia. Am. J. Respir. Crit. Care Med. 2005, 172, 1119–1127. [Google Scholar] [CrossRef]
- O’Dea, K.P.; Wilson, M.R.; Dokpesi, J.O.; Wakabayashi, K.; Tatton, L.; van Rooijen, N.; Takata, M. Mobilization and margination of bone marrow Gr-1high monocytes during subclinical endotoxemia predisposes the lungs toward acute injury. J. Immunol. 2009, 182, 1155–1166. [Google Scholar] [CrossRef]
- Ermert, L.; Ermert, M.; Merkle, M.; Goppelt-Struebe, M.; Duncker, H.R.; Grimminger, F.; Seeger, W. Rat pulmonary cyclooxygenase-2 expression in response to endotoxin challenge: Differential regulation in the various types of cells in the lung. Am. J. Pathol. 2000, 156, 1275–1287. [Google Scholar] [CrossRef]
- Nonas, S.A.; Moreno-Vinasco, L.; Ma, S.F.; Jacobson, J.R.; Desai, A.A.; Dudek, S.M.; Flores, C.; Hassoun, P.M.; Sam, L.; Ye, S.Q.; et al. Use of consomic rats for genomic insights into ventilator-associated lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L292–L302. [Google Scholar] [CrossRef] [Green Version]
- Robertson, J.A.; Sauer, D.; Gold, J.A.; Nonas, S.A. The role of cyclooxygenase-2 in mechanical ventilation-induced lung injury. Am. J. Respir. Cell Mol. Biol. 2012, 47, 387–394. [Google Scholar] [CrossRef]
- Li, L.-F.; Huang, C.-C.; Liu, Y.-Y.; Lin, H.-C.; Kao, K.-C.; Yang, C.-T.; Liao, S.-K. Hydroxyethyl starch reduces high stretch ventilation-augmented lung injury via vascular endothelial growth factor. Transl. Res. 2011, 157, 293–305. [Google Scholar] [CrossRef]
- Puybasset, L.; Gusman, P.; Muller, J.C.; Cluzel, P.; Coriat, P.; Rouby, J.J. Regional distribution of gas and tissue in acute respiratory distress syndrome. III. Consequences for the effects of positive end-expiratory pressure. CT Scan ARDS Study Group. Adult Respiratory Distress Syndrome. Intensive Care Med. 2000, 26, 1215–1227. [Google Scholar] [CrossRef]
- Meng, F.-Y.; Gao, W.; Ju, Y.-N. Parecoxib reduced ventilation induced lung injury in acute respiratory distress syndrome. BMC Pharm. Toxicol. 2017, 18, 25. [Google Scholar] [CrossRef]
- Shi, C.; Pamer, E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011, 11, 762–774. [Google Scholar] [CrossRef] [Green Version]
- Pflucke, D.; Hackel, D.; Mousa, S.A.; Partheil, A.; Neumann, A.; Brack, A.; Rittner, H.L. The molecular link between C-C-chemokine ligand 2-induced leukocyte recruitment and hyperalgesia. J. Pain 2013, 14, 897–910. [Google Scholar] [CrossRef]
- Altemeier, W.A.; Matute-Bello, G.; Frevert, C.W.; Kawata, Y.; Kajikawa, O.; Martin, T.R.; Glenny, R.W. Mechanical ventilation with moderate tidal volumes synergistically increases lung cytokine response to systemic endotoxin. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L533–L542. [Google Scholar] [CrossRef] [Green Version]
- Osterholzer, J.J.; Olszewski, M.A.; Murdock, B.J.; Chen, G.H.; Erb-Downward, J.R.; Subbotina, N.; Browning, K.; Lin, Y.; Morey, R.E.; Dayrit, J.K.; et al. Implicating exudate macrophages and Ly-6C(high) monocytes in CCR2-dependent lung fibrosis following gene-targeted alveolar injury. J. Immunol. 2013, 190, 3447–3457. [Google Scholar] [CrossRef]
- Zhang, G.S.; Liu, D.S.; Dai, C.W.; Li, R.J. Antitumor effects of celecoxib on K562 leukemia cells are mediated by cell-cycle arrest, caspase-3 activation, and downregulation of Cox-2 expression and are synergistic with hydroxyurea or imatinib. Am. J. Hematol. 2006, 81, 242–255. [Google Scholar] [CrossRef] [Green Version]
- Frank, J.A.; Pittet, J.F.; Wray, C.; Matthay, M.A. Protection from experimental ventilator-induced acute lung injury by IL-1 receptor blockade. Thorax 2008, 63, 147–153. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhou, Q.; Gu, C.; Li, D.; Zhu, L. Depletion of circulating monocytes suppresses IL-17 and HMGB1 expression in mice with LPS-induced acute lung injury. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2017, 312, L231–L242. [Google Scholar] [CrossRef]
- Zhu, Y.M.; Azahri, N.S.; Yu, D.C.; Woll, P.J. Effects of COX-2 inhibition on expression of vascular endothelial growth factor and interleukin-8 in lung cancer cells. BMC Cancer 2008, 8, 218. [Google Scholar] [CrossRef]
- Wei, D.; Wang, L.; He, Y.; Xiong, H.Q.; Abbruzzese, J.L.; Xie, K. Celecoxib inhibits vascular endothelial growth factor expression in and reduces angiogenesis and metastasis of human pancreatic cancer via suppression of Sp1 transcription factor activity. Cancer Res. 2004, 64, 2030–2038. [Google Scholar] [CrossRef]
- Gust, R.; Kozlowski, J.K.; Stephenson, A.H.; Schuster, D.P. Role of cyclooxygenase-2 in oleic acid-induced acute lung injury. Am. J. Respir. Crit. Care Med. 1999, 160, 1165–1170. [Google Scholar] [CrossRef]
- Hodges, R.J.; Jenkins, R.G.; Wheeler-Jones, C.P.D.; Copeman, D.M.; Bottoms, S.E.; Bellingan, G.J.; Nanthakumar, C.B.; Laurent, G.J.; Hart, S.L.; Foster, M.L.; et al. Severity of Lung Injury in Cyclooxygenase-2-Deficient Mice Is Dependent on Reduced Prostaglandin E2 Production. Am. J. Pathol. 2004, 165, 1663–1676. [Google Scholar] [CrossRef]
- O’Brien, G.; Shields, C.J.; Winter, D.C.; Dillon, J.P.; Kirwan, W.O.; Redmond, H.P. Cyclooxygenase-2 plays a central role in the genesis of pancreatitis and associated lung injury. Hepatobiliary Pancreat. Dis. Int. 2005, 4, 126–129. [Google Scholar]
- Smith, W.L.; Garavito, R.M.; DeWitt, D.L. Prostaglandin Endoperoxide H Synthases (Cyclooxygenases)-1 and −2. J. Boil. Chem. 1996, 271, 33157–33160. [Google Scholar] [CrossRef] [Green Version]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arter. Thromb. Vasc. Boil. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- Wong, D.; Wang, M.; Cheng, Y.; FitzGerald, G.A. Cardiovascular hazard and non-steroidal anti-inflammatory drugs. Curr. Opin. Pharm. 2005, 5, 204–210. [Google Scholar] [CrossRef]
- Patrono, C.; Baigent, C. Nonsteroidal Anti-Inflammatory Drugs and the Heart. Circulation 2014, 129, 907–916. [Google Scholar] [CrossRef] [Green Version]
- Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: Meta-analyses of individual participant data from randomised trials. Lancet 2013, 382, 769–779. [CrossRef]
- Patrono, C. Cardiovascular effects of cyclooxygenase-2 inhibitors: A mechanistic and clinical perspective. Br. J. Clin. Pharm. 2016, 82, 957–964. [Google Scholar] [CrossRef]
- Vidal Melo, M.F. Effect of cardiac output on pulmonary gas exchange: Role of diffusion limitation with Va/Q mismatch. Respir. Physiol. 1998, 113, 23–32. [Google Scholar] [CrossRef]
- Force, A.D.T.; Ranieri, V.M.; Rubenfeld, G.D.; Thompson, B.T.; Ferguson, N.D.; Caldwell, E.; Fan, E.; Camporota, L.; Slutsky, A.S. Acute respiratory distress syndrome: The Berlin Definition. JAMA 2012, 307, 2526–2533. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | HTV | HTV | HTV | |
|---|---|---|---|---|
| Celecoxib (mg/kg) | — | — | +20 | +40 |
| pH | 7.371 ± 0.031 | 7.322 ± 0.026 | 7.307 ± 0.02 | 7.37 ± 0.025 |
| PaCO2 (mm Hg) | 35.83 ± 5.56 | 30.5 ± 3.72 | 34.9 ± 4.23 | 36.23 ± 2.15 |
| HCO3 (mmol/l) | 21.48 ± 1.75 | 17.51 ± 1.30 | 18.8 ± 0.89 | 21.3 ± 1.45 |
| PaO2 (mm Hg) | 82.83 ± 11.28 | 51.0 ± 6.89 * | 52.3 ± 3.31 | 71.21 ± 8.6 # |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, T.-H.; Fang, P.-H.; Li, J.-M.; Ling, H.-Y.; Lin, C.-M.; Shi, C.-S. Cyclooxygenase-2 Activity Regulates Recruitment of VEGF-Secreting Ly6Chigh Monocytes in Ventilator-Induced Lung Injury. Int. J. Mol. Sci. 2019, 20, 1771. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071771
Huang T-H, Fang P-H, Li J-M, Ling H-Y, Lin C-M, Shi C-S. Cyclooxygenase-2 Activity Regulates Recruitment of VEGF-Secreting Ly6Chigh Monocytes in Ventilator-Induced Lung Injury. International Journal of Molecular Sciences. 2019; 20(7):1771. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071771
Chicago/Turabian StyleHuang, Tzu-Hsiung, Pin-Hui Fang, Jhy-Ming Li, Huan-Yuan Ling, Chieh-Mo Lin, and Chung-Sheng Shi. 2019. "Cyclooxygenase-2 Activity Regulates Recruitment of VEGF-Secreting Ly6Chigh Monocytes in Ventilator-Induced Lung Injury" International Journal of Molecular Sciences 20, no. 7: 1771. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20071771