PMN-MDSCs Enhance CTC Metastatic Properties through Reciprocal Interactions via ROS/Notch/Nodal Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
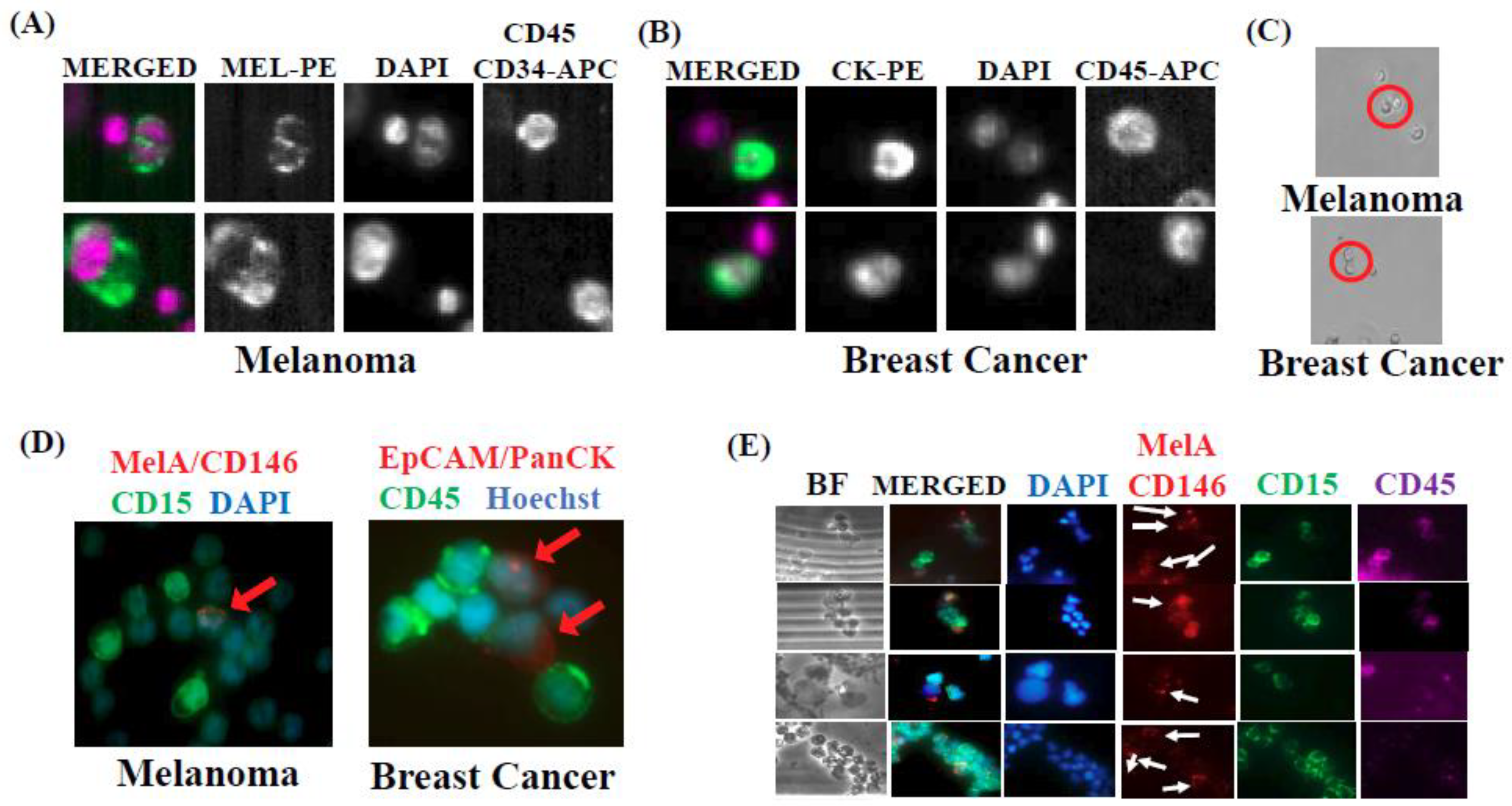
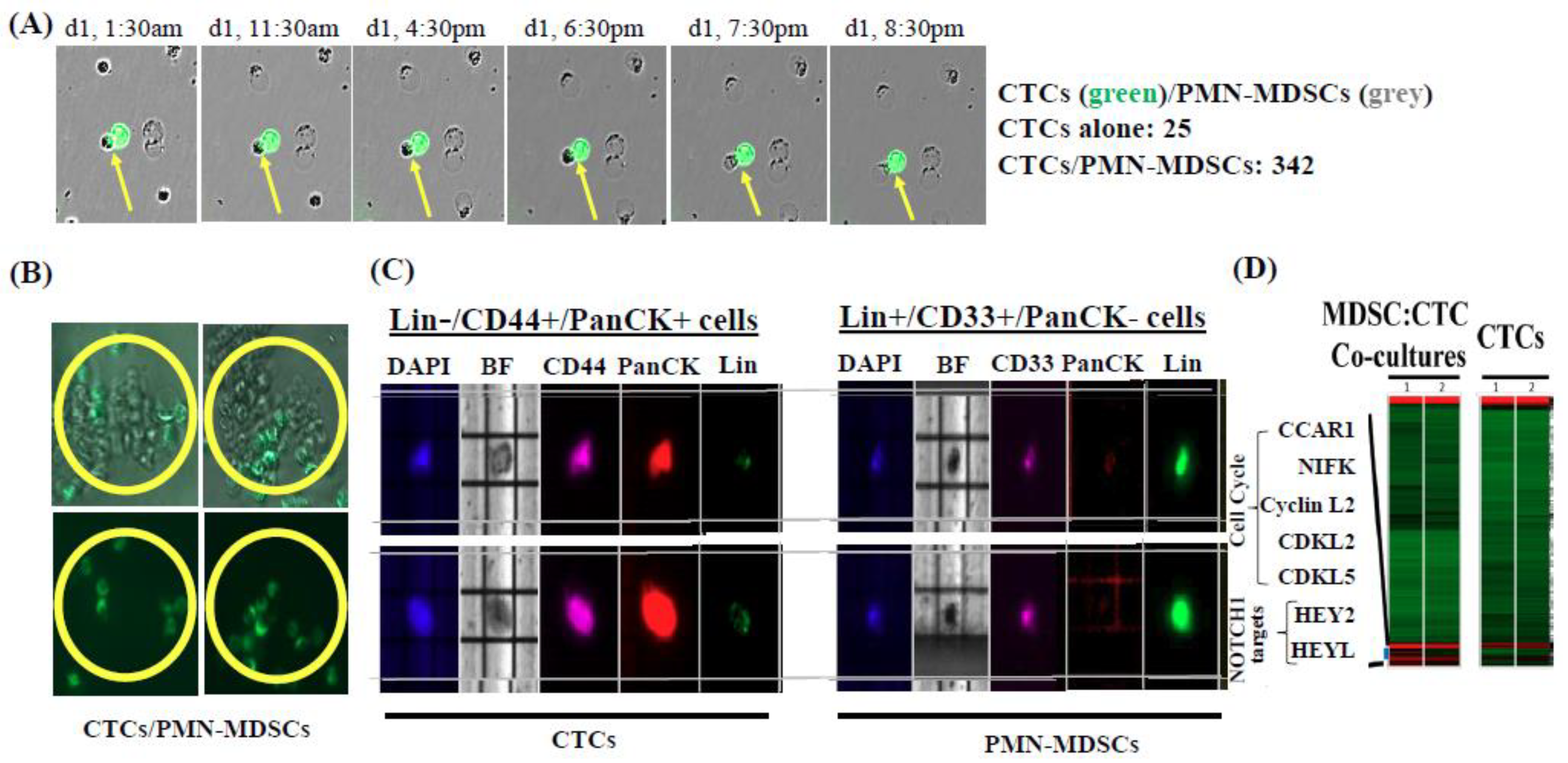
2.1. Isolation and Characterization of CTC/PMN-MDSC Clusters
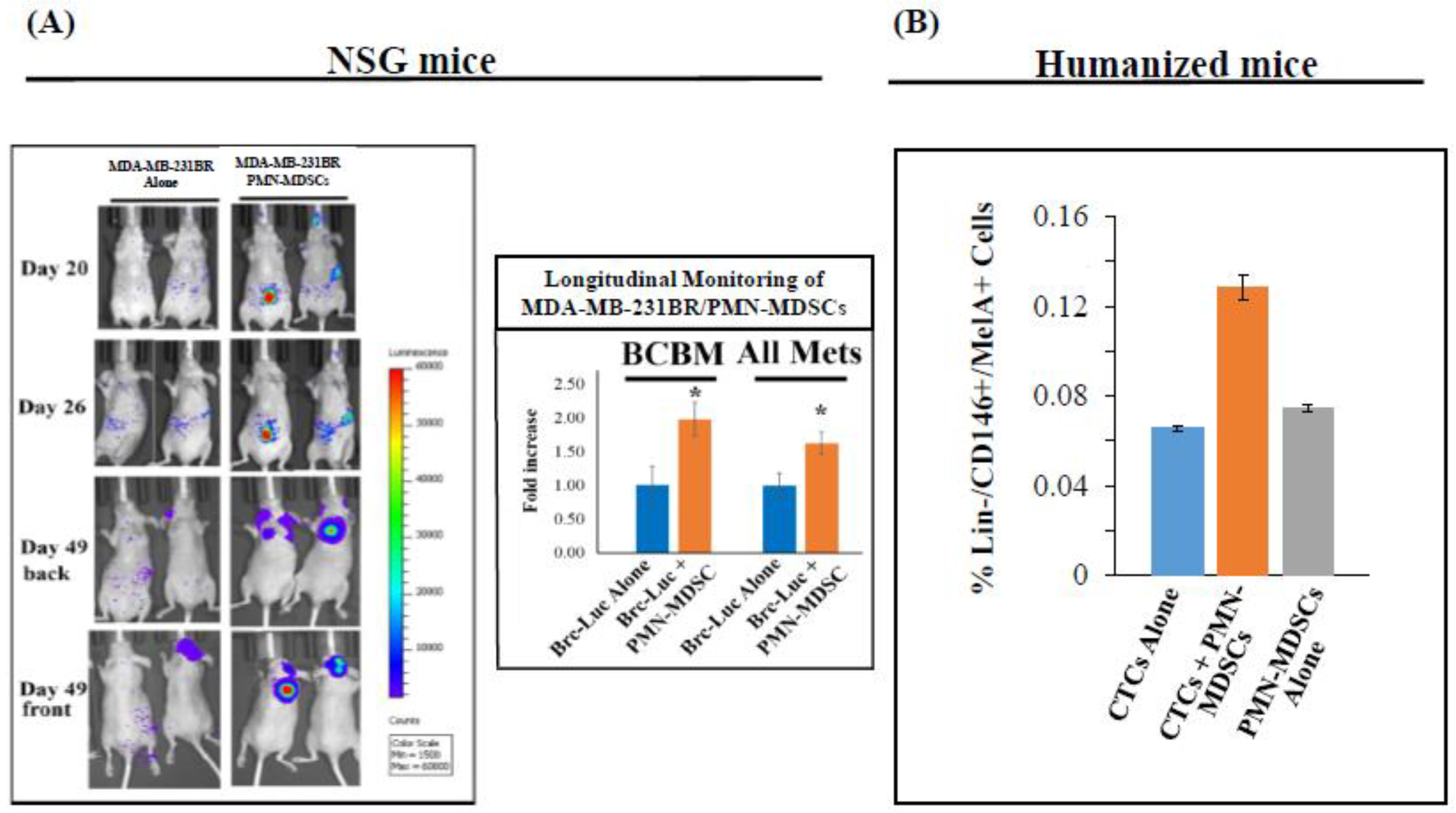
2.2. CTC-Driven Metastatic Dissemination is Enhanced by PMN-MDSCs
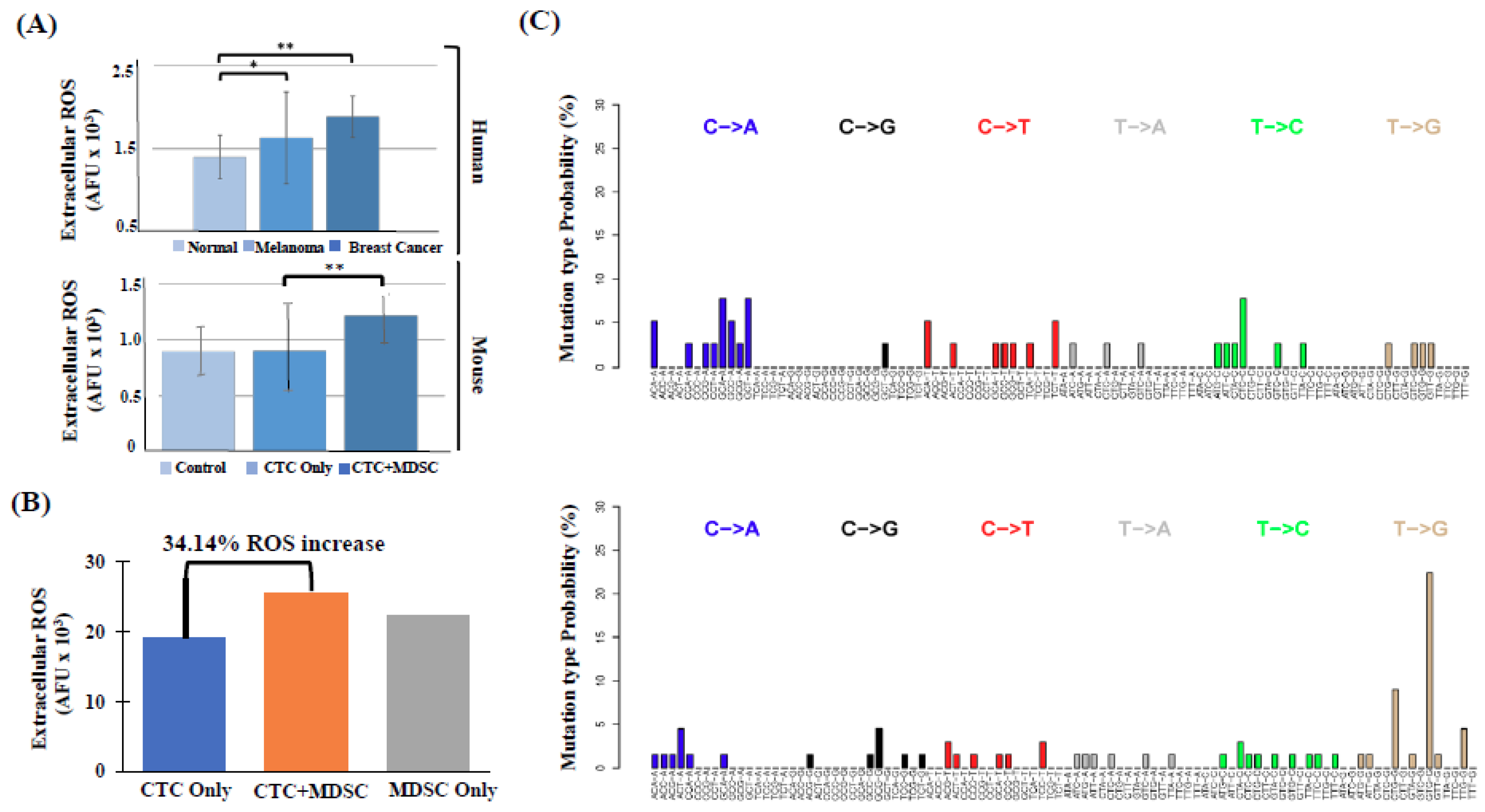
2.3. High Number of Circulatory PMN-MDSCs and Plasma ROS Levels are Linked to Metastasis
2.4. PMN-MDSCs Form Clusters with CTCs and Promote CTC Proliferation
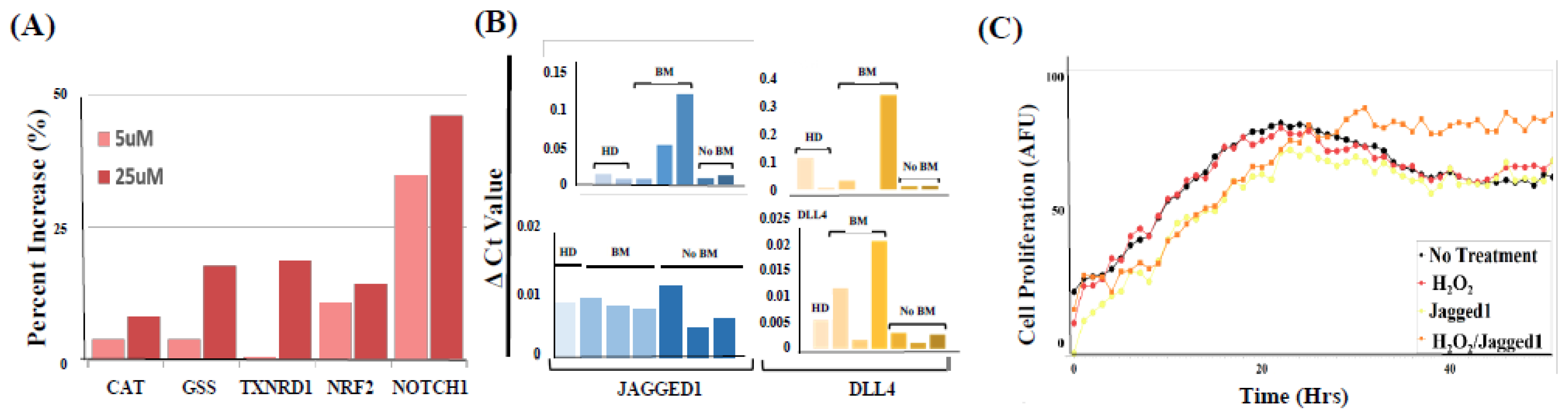
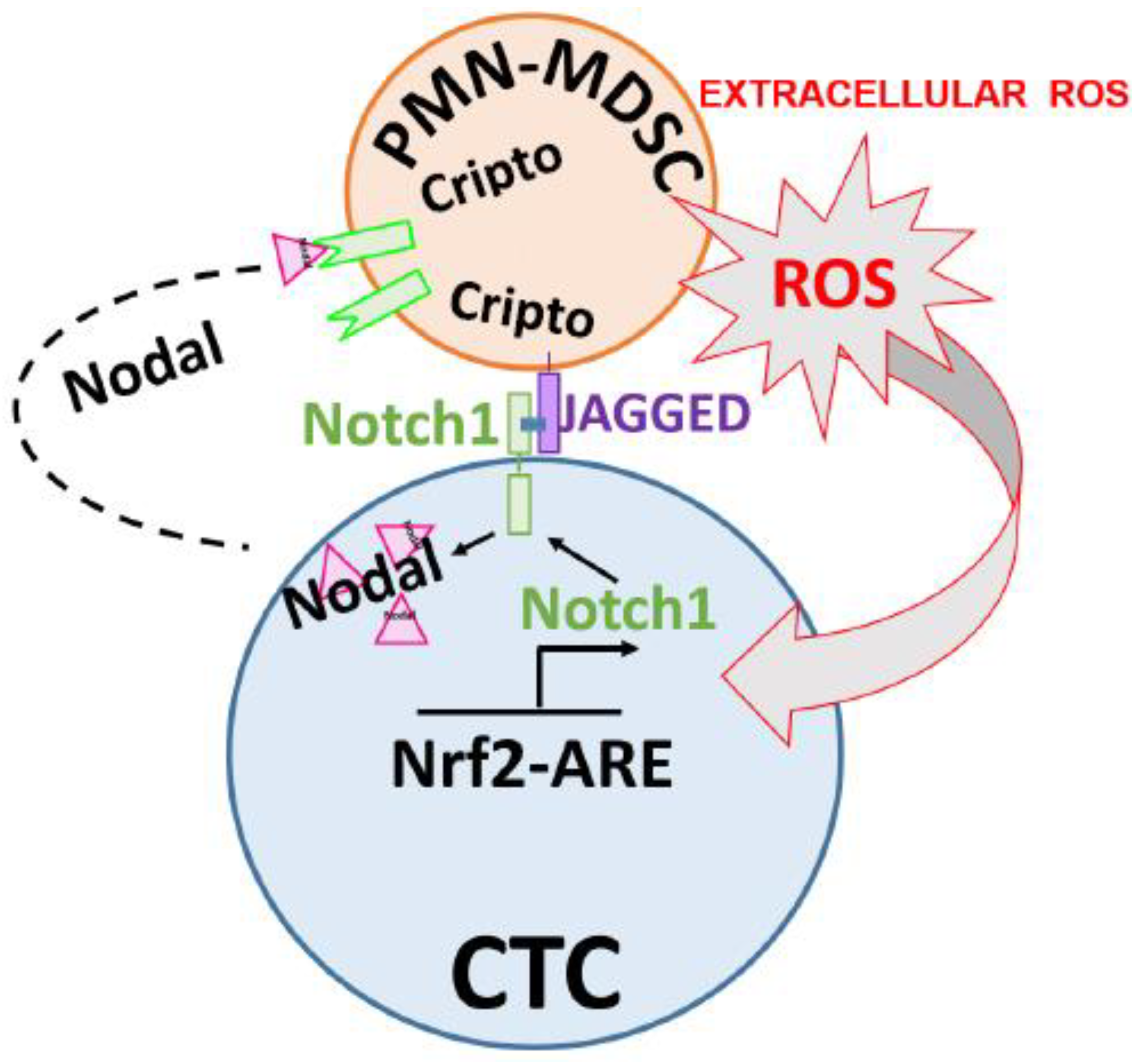
2.5. PMN-MDSCs Facilitate CTC Survival through Dual Activation of ROS-NRF2-ARE Axis and Notch Signaling Pathways
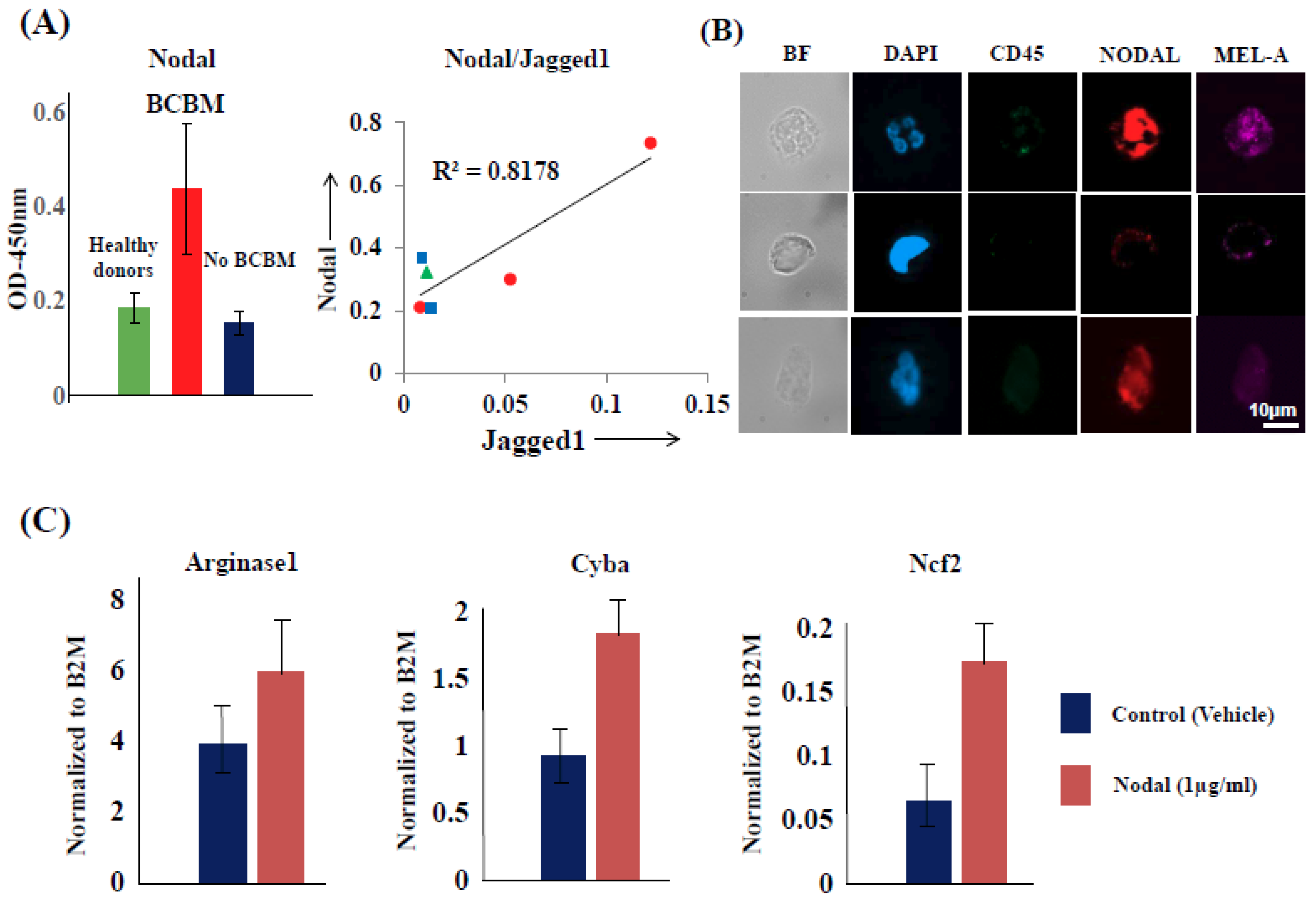
2.6. CTCs Induce the Pro-Tumorigenic Differentiation of PMN-MDSCs through Nodal-Cripto Axis
3. Discussion
4. Materials and Methods
4.1. Antibodies and Inhibitors
4.2. Patient Blood Collection, PBMC Isolation, and Multi Parametric Flow Cytometry (FACS)
4.3. CTC/PMN-MDSC Cluster Capture
4.4. Cell Culture and Cell Analyses
4.5. Live-Cell Microscopy
4.6. Experimental Animals and Mouse Models
4.7. Immunofluorescence and Immunohistochemistry
4.8. Extracellular ROS Determination
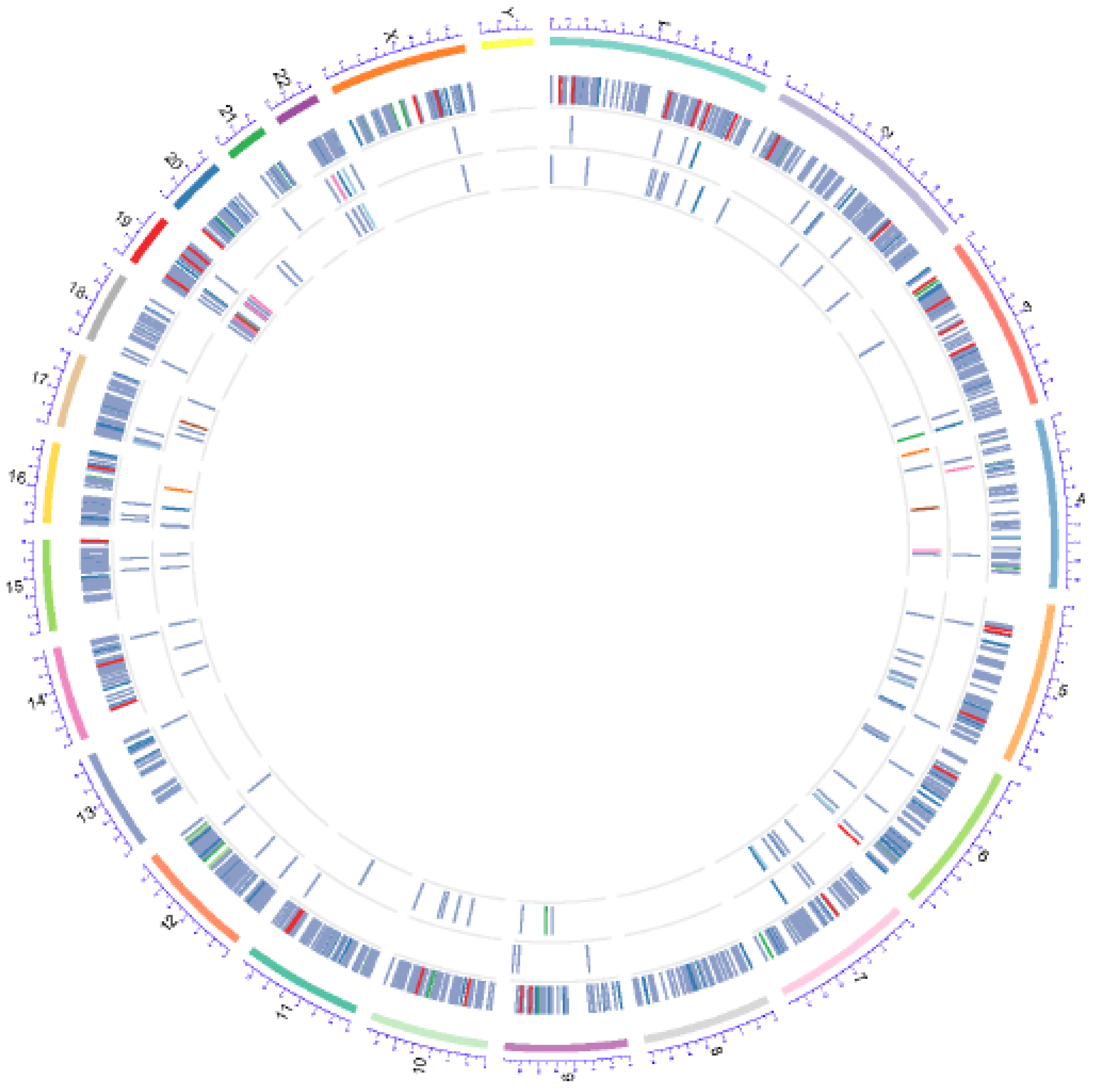
4.9. Whole Exome Sequencing (WES)
4.10. Sequence Mapping and Statistical Analyses
4.11. Transcriptome Analyses of CTCs Co-Cultured with PMN-MDSCs
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Giuliano, M.; Shaikh, A.; Lo, H.C.; Arpino, G.; de Placido, S.; Xiang, H.; Zhang, X.H.; Cristofanilli, M.; Schiff, R.; Trivedi, M.V. Perspective on Circulating Tumor Cell Clusters: Why it takes a village to metastasize. Cancer Res. 2018, 78, 845–852. [Google Scholar] [CrossRef]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H.; et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Boral, D.; Vishnoi, M.; Liu, H.N.; Yin, W.; Sprouse, M.L.; Scamardo, A.; Hong, D.S.; Tan, T.Z.; Thiery, J.-P.; Chang, J.C.; et al. Molecular characterization of breast cancer CTCs associated with brain metastasis. Nat. Commun. 2017, 8, 196–206. [Google Scholar] [CrossRef]
- Gabrilovich, D.I. Myeloid-derived suppressor cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Welte, T.; Kim, I.S.; Tian, L.; Gao, X.; Wang, H.; Li, J.; Herschkowitz, J.I.; Pond, A.; Dobrolecki, C.E.; Mo, Q.; et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat. Cell Biol. 2016, 18, 632–644. [Google Scholar] [CrossRef]
- Peng, D.; Tanikawa, T.; Li, W.; Zhao, L.; Vatan, L.; Szeliga, W.; Wan, S.; Wei, S.; Wang, Y.; Liu, Y.; et al. Myeloid-derived suppressor cells endow stem-like qualities to breast cancer cells through IL6/STAT3 and NO/NOTCH cross-talk signaling. Cancer Res. 2016, 76, 3156–3165. [Google Scholar] [CrossRef]
- Cui, T.X.; Kryczek, I.; Zhao, L.; Zhao, E.; Kuick, R.; Roh, M.H.; Vatan, L.; Szeliga, W.; Mao, Y.; Thomas, D.G.; et al. Myeloid-derived suppressor cells enhance stemness of cancer cells by inducing microRNA101 and suppressing the corepressor CtBP2. Immunity 2013, 39, 611–621. [Google Scholar] [CrossRef]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, J.P. Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Ogrunc, M.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gougolis, U.G.; D’Adda Di Fagagna, F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014, 21, 998–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox regulation of PI 3-kinase signalling via inactivation of PTEN. EMBO J. 2003, 22, 5501–5510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, K.H.; Lemke, G.; Kim, J.W. The PI3K-PTEN tug-of-war, oxidative stress and retinal degeneration. Trends Mol. Med. 2009, 15, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Leite de Oliveira, R.; Huijberts, S.; Bosdriesz, E.; Pencheva, N.; Brunen, D.; Bosma, A.; Song, J.Y.; Zevehoven, J.; Los-de Vries, G.T.; et al. An acquired vulnerability of drug-resistant melanoma with therapeutic potential. Cell 2018, 173, 1413–1425. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Jo Harris, P.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: An update. Nat. Rev. Clin. Onc. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Zhang, L.; Ridgway, L.; Wetzel, M.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.; Marchetti, D. The identification and characterization of breast cancer CTCs with brain metastatic competence. Sci. Transl. Med. 2013, 5, 84–93. [Google Scholar] [CrossRef]
- Strizzi, L.; Hardy, K.M.; Seftor, E.A.; Costa, F.F.; Kirschmann, D.A.; Seftor, R.E.; Postovit, L.M.; Hendrix, M.J. Development and cancer: at the crossroads of Nodal and Notch signaling. Cancer Res. 2009, 69, 7131–7134. [Google Scholar] [CrossRef]
- Bodenstine, T.M.; Chandler, G.S.; Reed, D.W.; Margaryan, N.V.; Gilgur, A.; Atkinson, J.; Ahmed, N.; Hyser, M.; Seftor, E.A.; Strizzi, L.; et al. Nodal expression in triple-negative breast cancer: Cellular effects of its inhibition following doxorubicin treatment. Cell Cycle 2016, 15, 1295–1302. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.F.; Wang, H.S.; Zhang, F.; Guo, Q.; Wang, H.; Wang, K.F.; Zhang, G.; Bu, X.Z.; Cai, S.H.; Du, J. Nodal promotes the generation of M2-like macrophages and downregulates the expression of IL-12. Eur. J. Immunol. 2014, 44, 173–183. [Google Scholar] [CrossRef]
- Vishnoi, M.V.; Boral, D.; Liu, H.N.; Sprouse, M.L.; Yin, W.; Goswami-Sewell, D.; Tetzlaff, M.T.; Davies, M.A.; Glitza-Oliva, I.C.; Marchetti, D. Targeting USP7 identifies a metastasis-competent state within BM-resident melanoma CTCs. Cancer Res. 2018, 78, 5349–5362. [Google Scholar] [CrossRef] [PubMed]
- Vishnoi, M.; Peddibohtla, S.; Yin, W.; Scamardo, A.; Goldy, G.; Hong, D.S.; Marchetti, D. The characterization of CTC subsets relating to markers of breast cancer dormancy. Sci. Rep. 2015, 5, 17533. [Google Scholar] [CrossRef]
- Alix-Panabieres, C.; Pantel, K. Challenges in circulating tumour cell research. Nat. Rev. Cancer 2014, 14, 623–631. [Google Scholar] [CrossRef]
- Miller, M.C.; Robinson, P.S.; Wagner, C.; O’Shannessy, D.J. The Parsortix® cell separation system—A versatile liquid biopsy platform. Cytometry 2018. [Google Scholar] [CrossRef] [PubMed]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Singh, A.; Venkannagan, S.; Oh, K.H.; Zhang, Y.Q.; Rohde, J.M.; Liu, L.; Nimmagadda, S.; Sudini, K.; Brimacombe, K.R.; Gajbate, S.; et al. Small molecule inhibitor of NRF2 selectively intervenes therapeutic resistance in KEAP1-deficient NSCLC tumors. ACS Chem. Biol. 2016, 11, 3214–3225. [Google Scholar] [CrossRef] [Green Version]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Surnimann, C.U.; et al. Circulating tumor cell clustering shapes DNA methylation to enable metastasis seeding. Cell 2019, 176, 98–112. [Google Scholar] [CrossRef]
- Pearson, T.; Greiner, D.L.; Shultz, L.D. Creation of “Humanized” Mice to Study Human Immunity. Curr. Protocols Immunol. 2008. [Google Scholar] [CrossRef]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 7, 1195–1214. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Chartoumpekis, D.V.; Kensler, T.W. Crosstalk between Nrf2 and Notch signaling. Free Radic. Biol. Med. 2015, 88, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, M.J.; Kandela, I.; Mazar, A.P.; Seftor, E.A.; Seftor, R.E.; Margaryan, N.V.; Strizzi, L.; Murphy, G.F.; Long, G.V.; Scolyer, R.A. Targeting melanoma with front-line therapy does not abrogate Nodal-expressing tumor cells. Lab Investig. 2017, 97, 176–186. [Google Scholar] [CrossRef]
- Kalyan, A.; Carneiro, B.A.; Chandra, S.; Kaplan, J.; Chae, Y.K.; Matsangou, M.; Strizzi, L.; Murphy, G.F.; Long, G.V.; Scolyer, R.A. Nodal signaling as a developmental therapeutics target in oncology. Mol. Cancer Ther. 2017, 16, 787–792. [Google Scholar] [CrossRef]
- Cavallari, C.; Fonsato, V.; Herrera, M.B.; Bruno, S.; Tetta, C.; Camussi, G. Role of Lefty in the anti tumor activity of human adult liver stem cells. Oncogene 2013, 32, 819–826. [Google Scholar] [CrossRef]
- Husemann, Y.; Geigl, J.B.; Schubert, F.; Musiani, P.; Meyer, M.; Burghart, E.; Forni, G.; Eils, R.; Fehm, T.; Riethmuller, G.; et al. Systemic spread is an early step in breast cancer. Cancer Cell 2008, 13, 58–68. [Google Scholar] [CrossRef]
- Chen, J.F.; Ho, H.; Lichterman, J.; Lu, Y.T.; Zhang, Y.; Garcia, M.A.; Chen, S.F.; Liang, A.J.; Hodara, E.; Zhau, H.E.; et al. Subclassification of prostate cancer circulating tumor cells by nuclear size reveals very small nuclear circulating tumor cells in patients with visceral metastases. Cancer 2015, 121, 3240–3251. [Google Scholar] [CrossRef]
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Rev. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef]
- Liu, X.; Taftaf, R.; Kawaguchi, M.; Chang, Y.-F.; Chen, W.; Entenberg, D.; Zhang, Y.; Gerratana, L.; Huang, S.; Patel, D.B.; et al. Homophilic CD44 interactions mediate tumor cell aggregation and polyclonal metastasis in patient-derived breast cancer models. Cancer Discov. 2019, 1, 96–113. [Google Scholar] [CrossRef]
- Arnoletti, J.P.; Zhu, X.; Almodovar, A.J.; Veldhuis, P.P.; Sause, R.; Griffith, E.; Corpus, G.; Chang, J.C.; Fanaian, N.; Litherland, S.A. Portal venous blood circulation supports immunosuppressive environment and pancreatic cancer circulating tumor cell activation. Pancreas 2017, 46, 116–123. [Google Scholar] [CrossRef]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Hong, Y.; Fang, F.; Zhang, Q. Circulating tumor cell clusters: What we know and what we expect. Int. J. Oncol. 2016, 49, 2206–2216. [Google Scholar] [CrossRef]
- Palmieri, D.; Bronder, J.L.; Herring, J.M.; Yoneda, T.; Weil, R.J.; Stark, A.M.; Kurek, R.; Vega-Valle, E.; Feigenbaum, L.; Halverson, D.; et al. Her-2 overexpression increases the metastatic outgrowth of breast cancer cells in the brain. Cancer Res. 2017, 67, 4190–4198. [Google Scholar] [CrossRef]
- Roy, M.; Reiland, J.; Murry, B.P.; Chouljenko, V.; Kousoulas, K.G.; Marchetti, D. Antisense-mediated suppression of heparanase gene inhibits melanoma cell invasion. Neoplasia 2005, 3, 253–262. [Google Scholar] [CrossRef]
- Lee, A.S.; Wu, J.C. Imaging of embryonic stem cell migration in vivo. Methods Mol. Biol. 2011, 750, 101–114. [Google Scholar]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Yan, C.; Hsu, C.H.; Chen, Q.R.; Niu, K.; Komatsoulis, G.A.; Meerzaman, D. OmicCircos: A simple-to-use R Package for the circular visualization of multidimensional omics data. Cancer Inform. 2014, 13, 13–20. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]










© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sprouse, M.L.; Welte, T.; Boral, D.; Liu, H.N.; Yin, W.; Vishnoi, M.; Goswami-Sewell, D.; Li, L.; Pei, G.; Jia, P.; et al. PMN-MDSCs Enhance CTC Metastatic Properties through Reciprocal Interactions via ROS/Notch/Nodal Signaling. Int. J. Mol. Sci. 2019, 20, 1916. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081916
Sprouse ML, Welte T, Boral D, Liu HN, Yin W, Vishnoi M, Goswami-Sewell D, Li L, Pei G, Jia P, et al. PMN-MDSCs Enhance CTC Metastatic Properties through Reciprocal Interactions via ROS/Notch/Nodal Signaling. International Journal of Molecular Sciences. 2019; 20(8):1916. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081916
Chicago/Turabian StyleSprouse, Marc L., Thomas Welte, Debasish Boral, Haowen N. Liu, Wei Yin, Monika Vishnoi, Debalina Goswami-Sewell, Lili Li, Guangsheng Pei, Peilin Jia, and et al. 2019. "PMN-MDSCs Enhance CTC Metastatic Properties through Reciprocal Interactions via ROS/Notch/Nodal Signaling" International Journal of Molecular Sciences 20, no. 8: 1916. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20081916