Rosmarinic Acid Attenuates Cadmium-Induced Nephrotoxicity via Inhibition of Oxidative Stress, Apoptosis, Inflammation and Fibrosis

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results
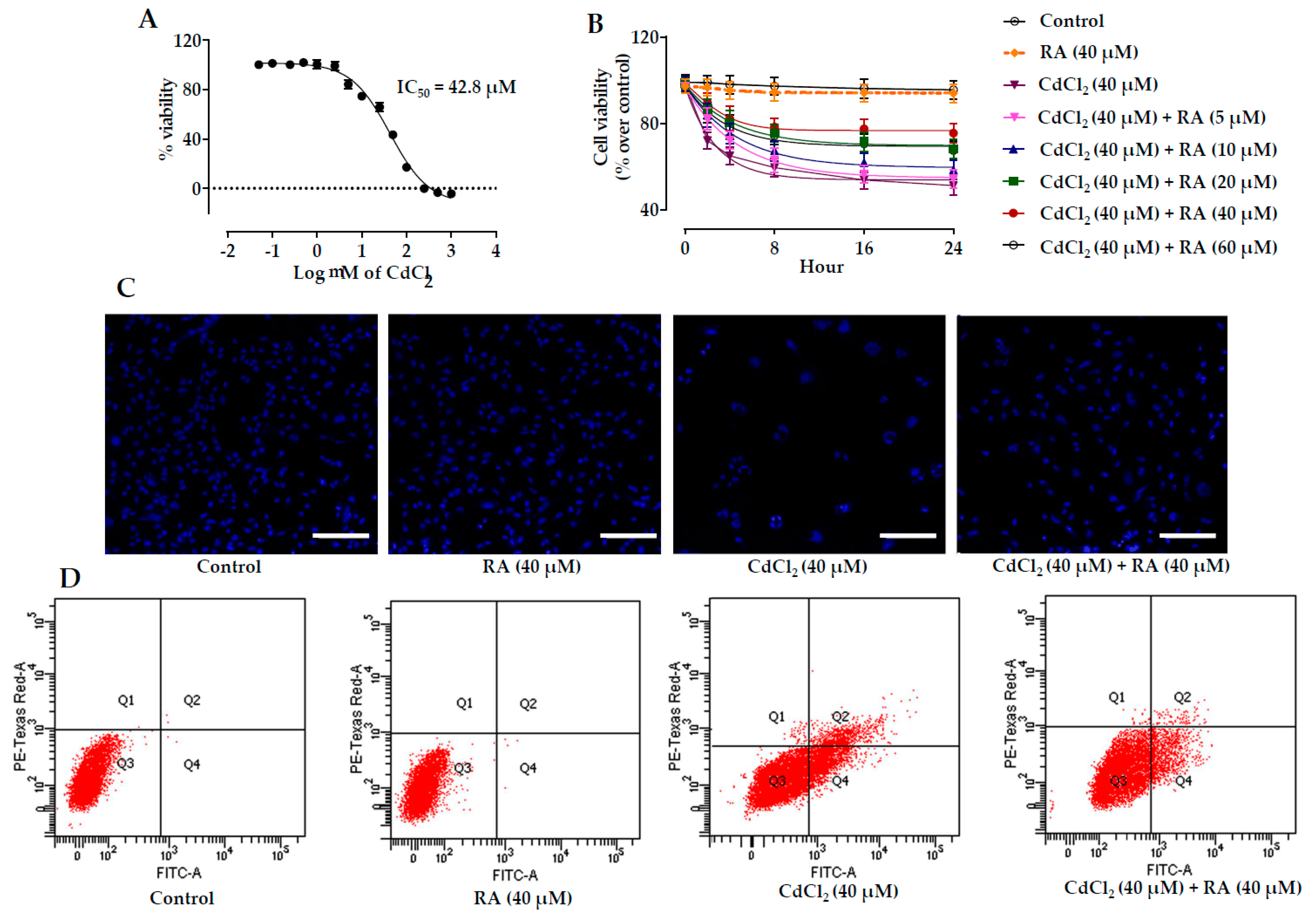
2.1. Effect of RA on CdCl2-Mediated Toxicity in Vitro
2.1.1. Dose-Dependent Cytotoxic Effect of CdCl2
2.1.2. Cytoprotective Effects of RA
2.1.3. Effects on Hoechst Staining
2.1.4. Flow Cytometric Analysis
2.1.5. Effects on Redox Status In Vitro
2.1.6. Effects on Signal Proteins In Vitro
2.2. Effect on CdCl2-Mediated Nephrotoxicity In Vivo
2.2.1. Effect on Serum Biochemical Parameters
2.2.2. Effect on Kidney Mass, Cd Accumulation in Kidneys, and Urine Parameters
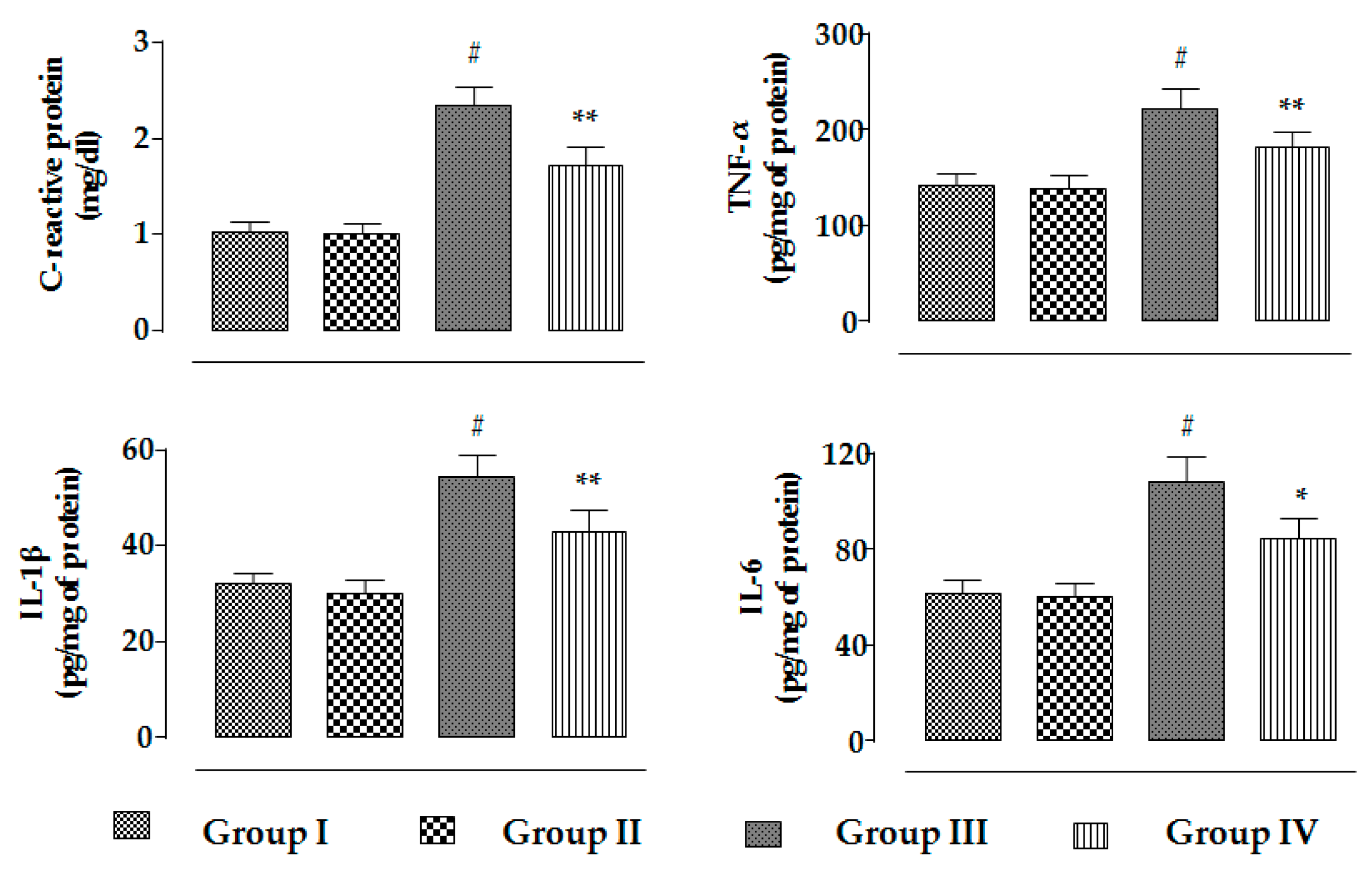
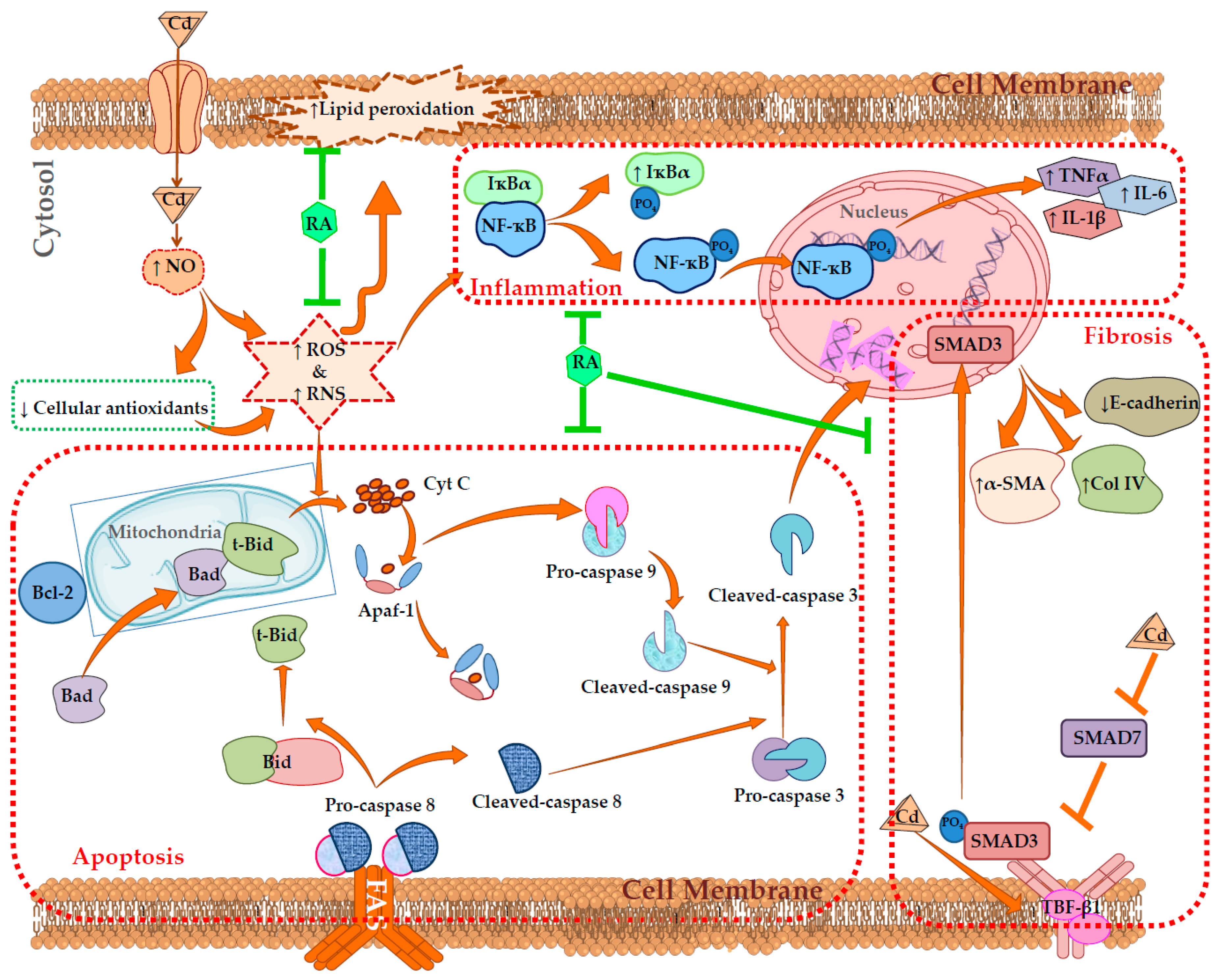
2.2.3. Effects on Inflammatory Mediators
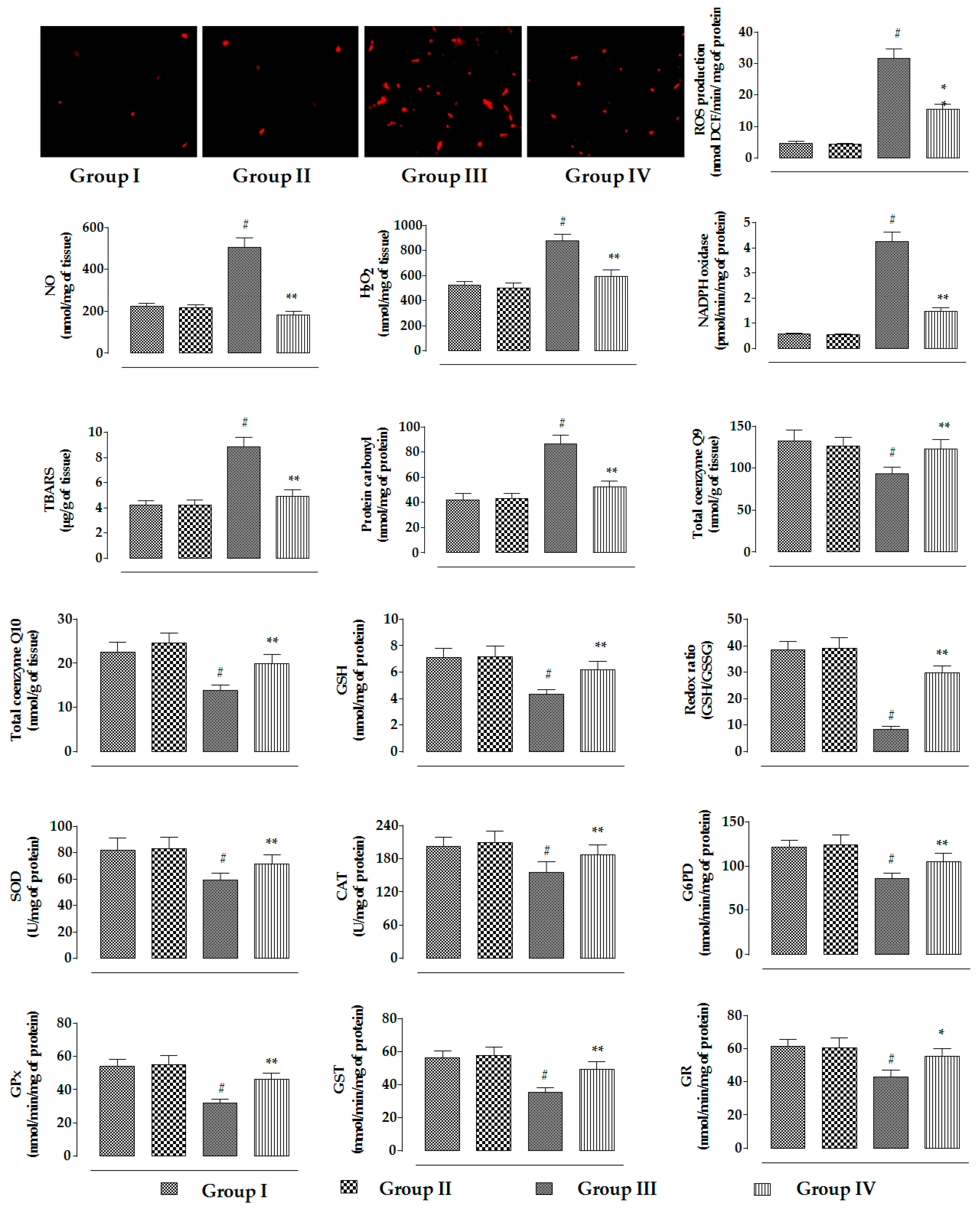
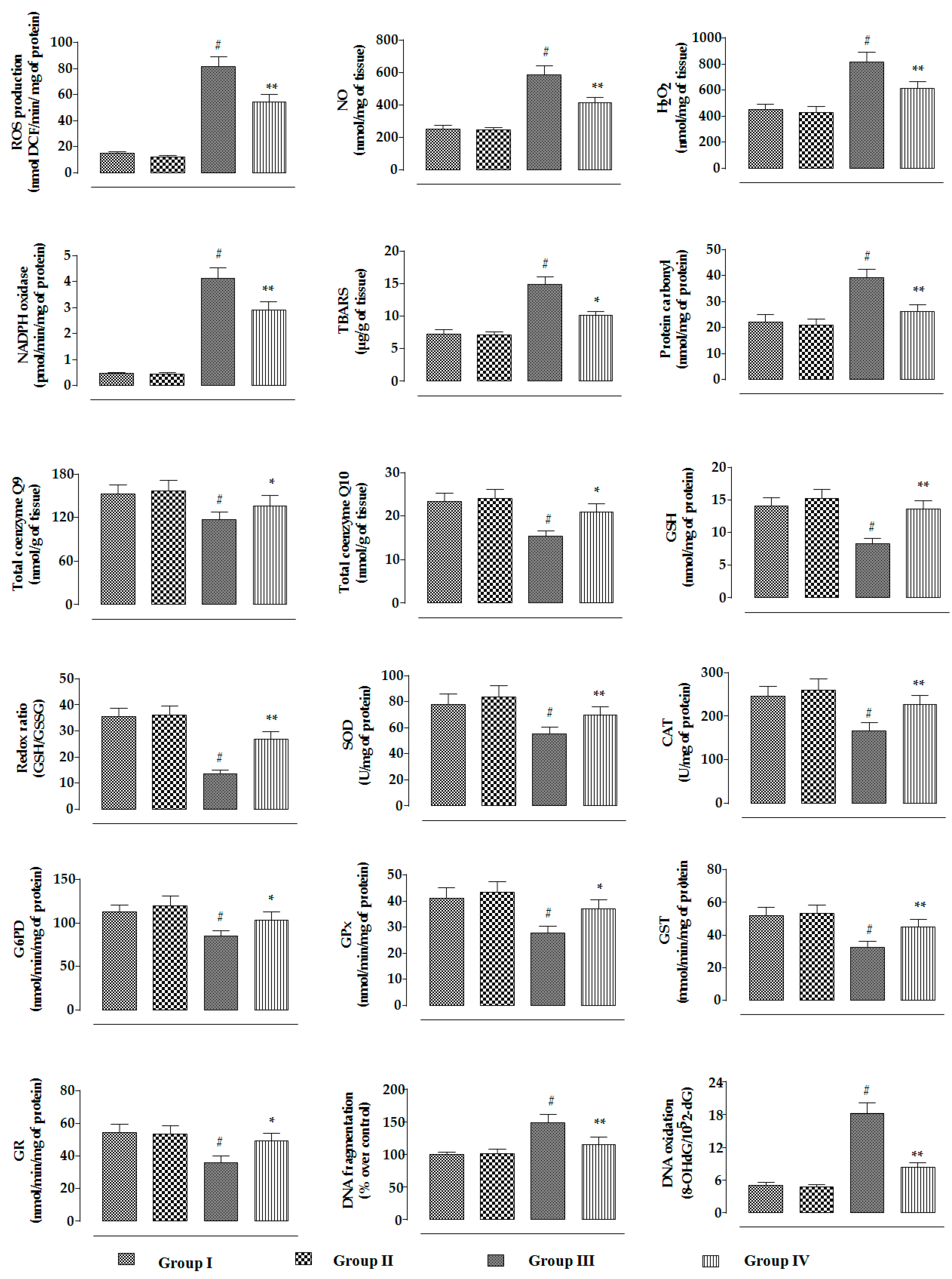
2.2.4. Effects on Redox Status In Vivo
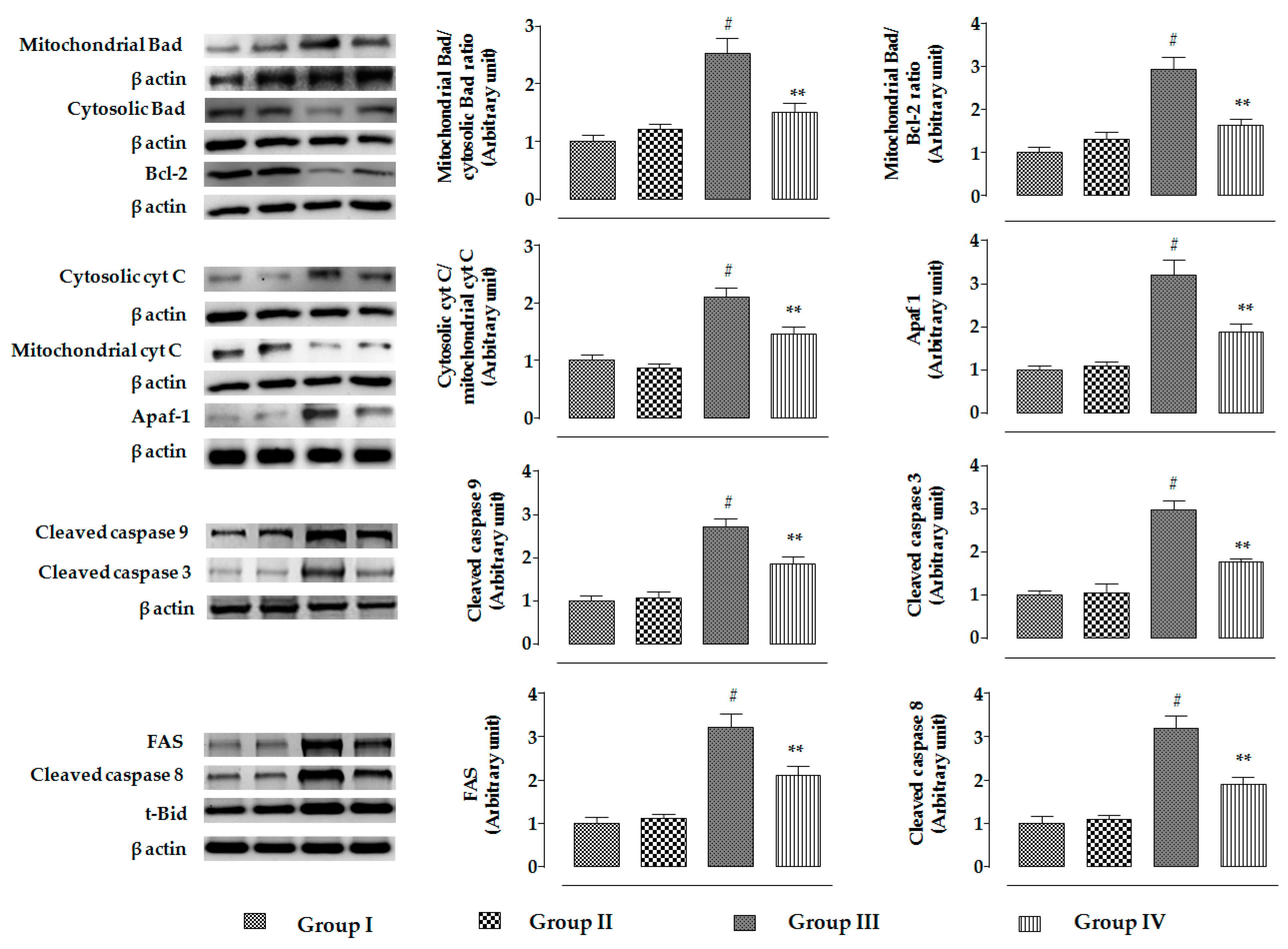
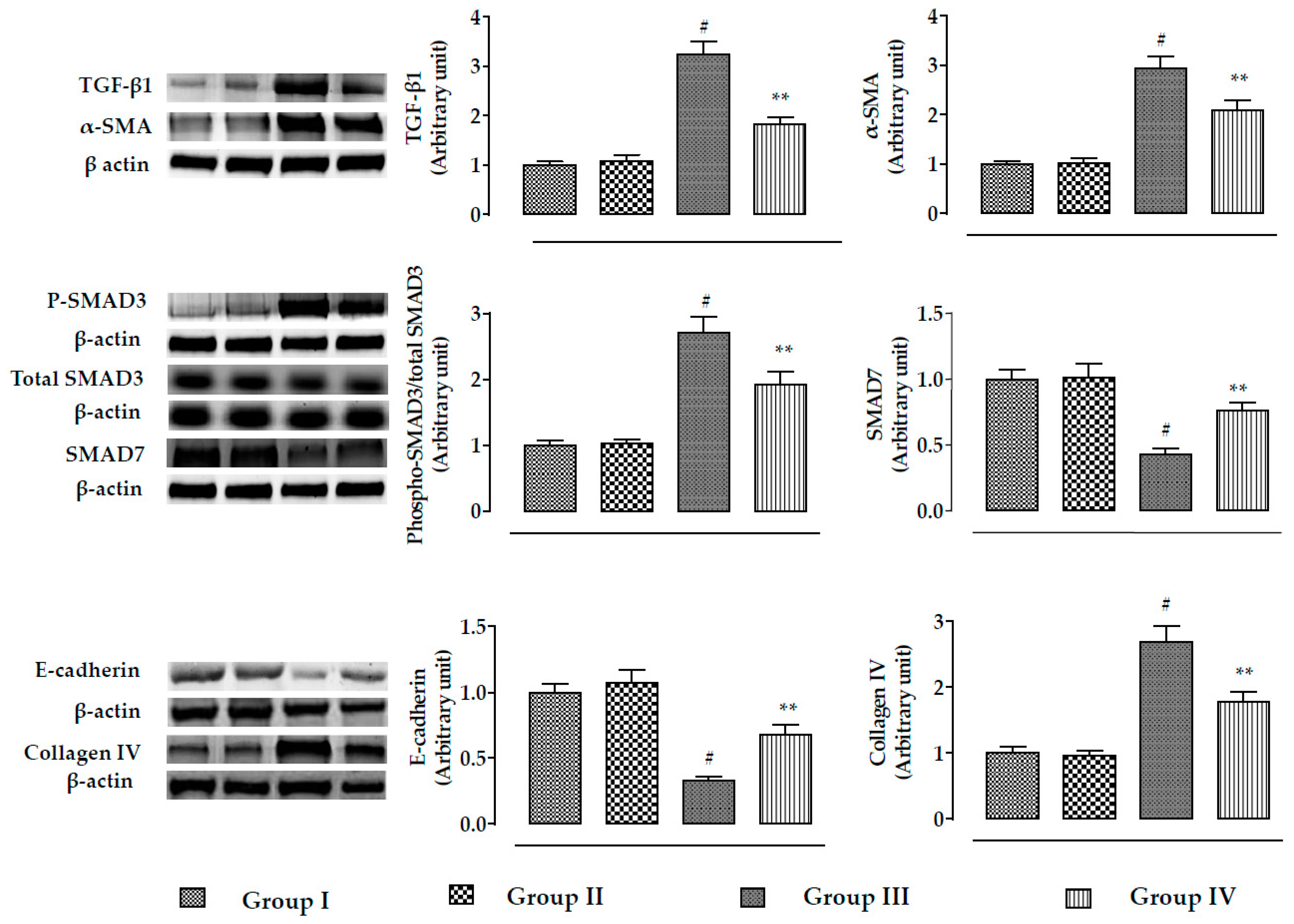
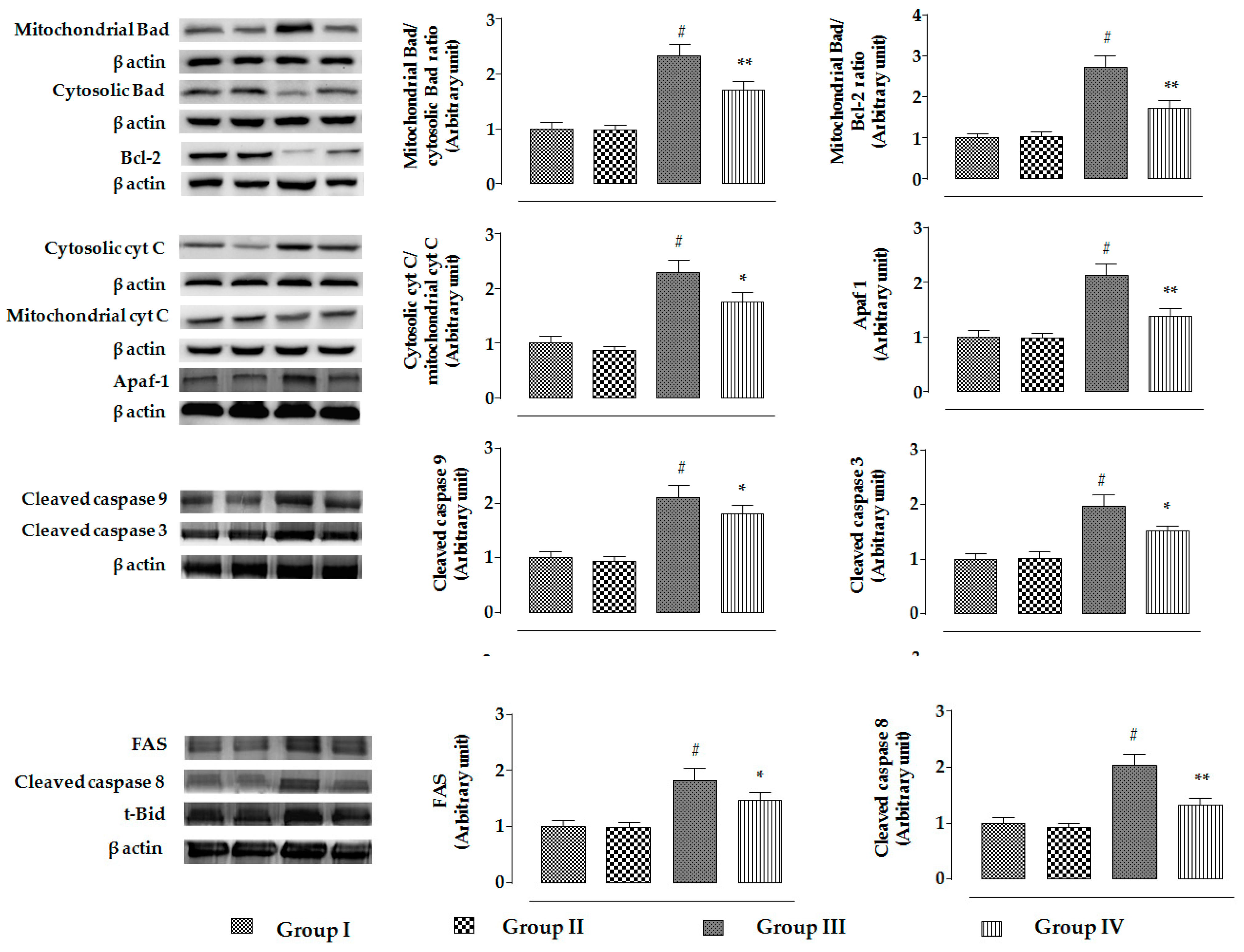
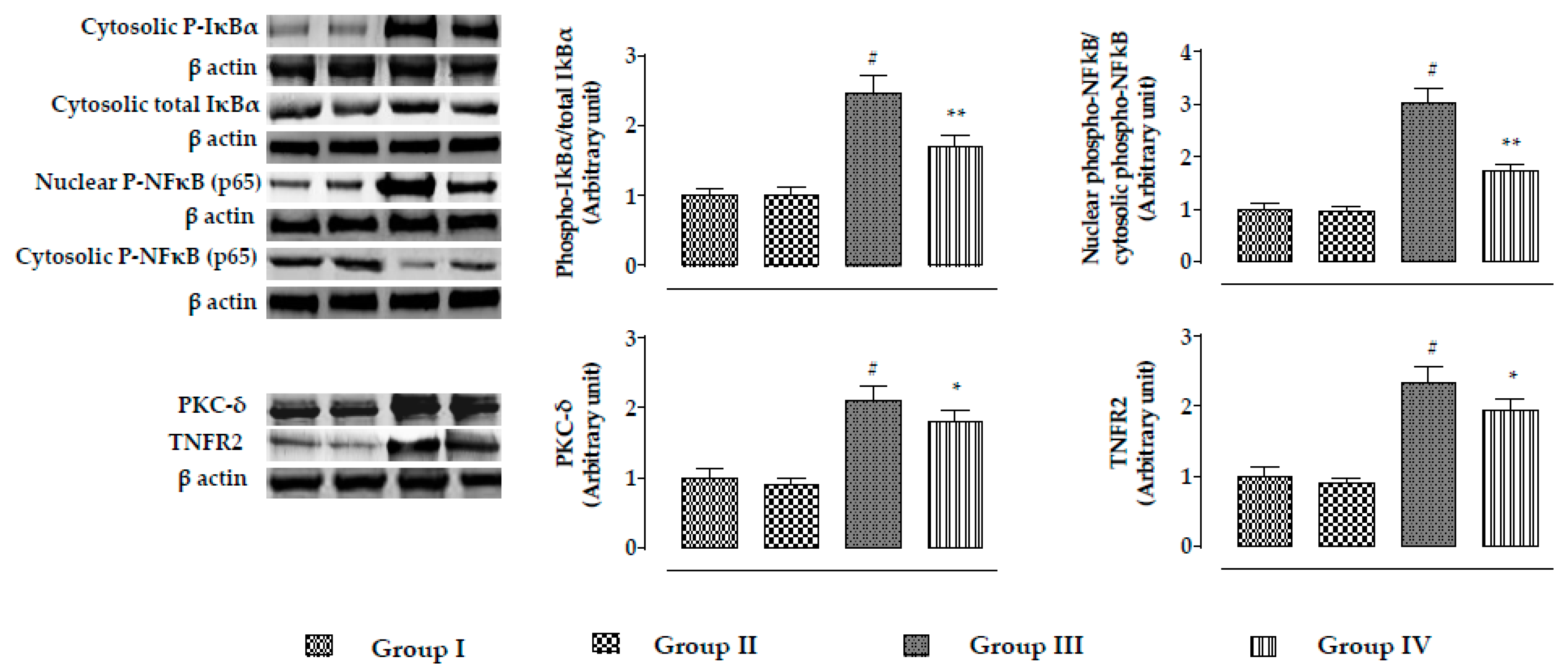
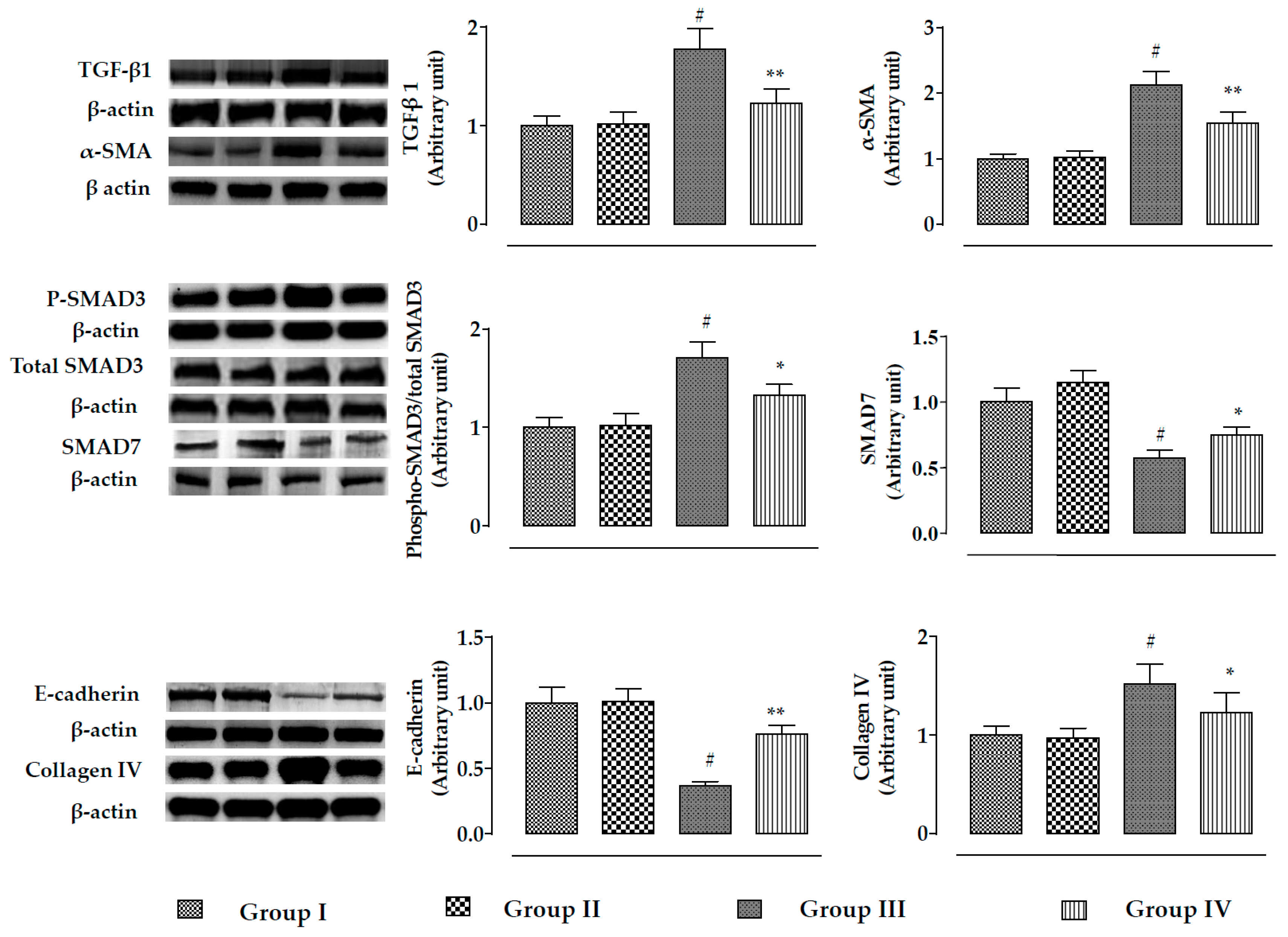
2.2.5. Effects on Signal Proteins In Vivo
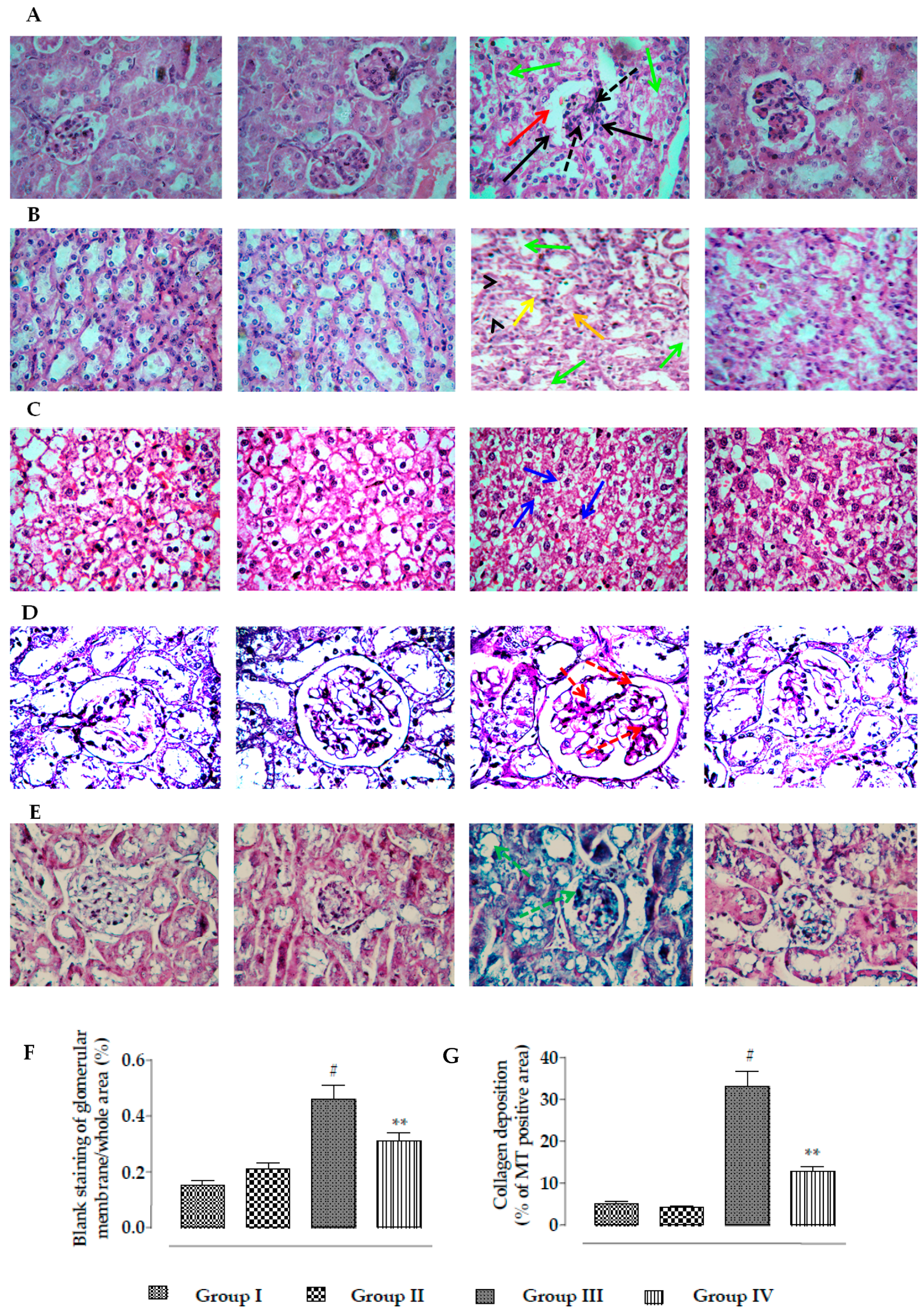
2.2.6. Effects on Histology of Kidneys
2.3. Predictive Pharmacology In Silico
2.3.1. Analyses of ADMET and Drug-Likeness Prediction
2.3.2. In Silico Binding Interaction Analysis through Molecular Docking Study
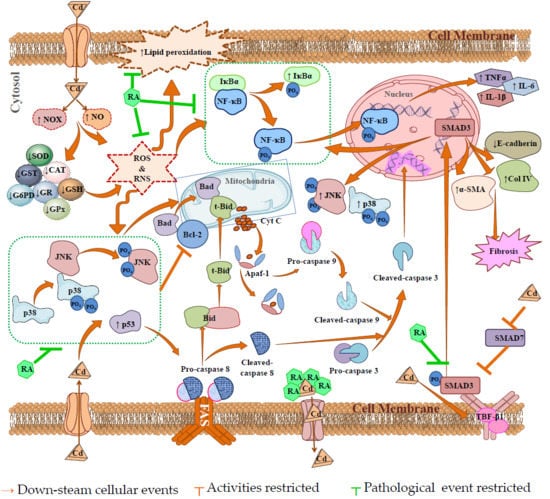
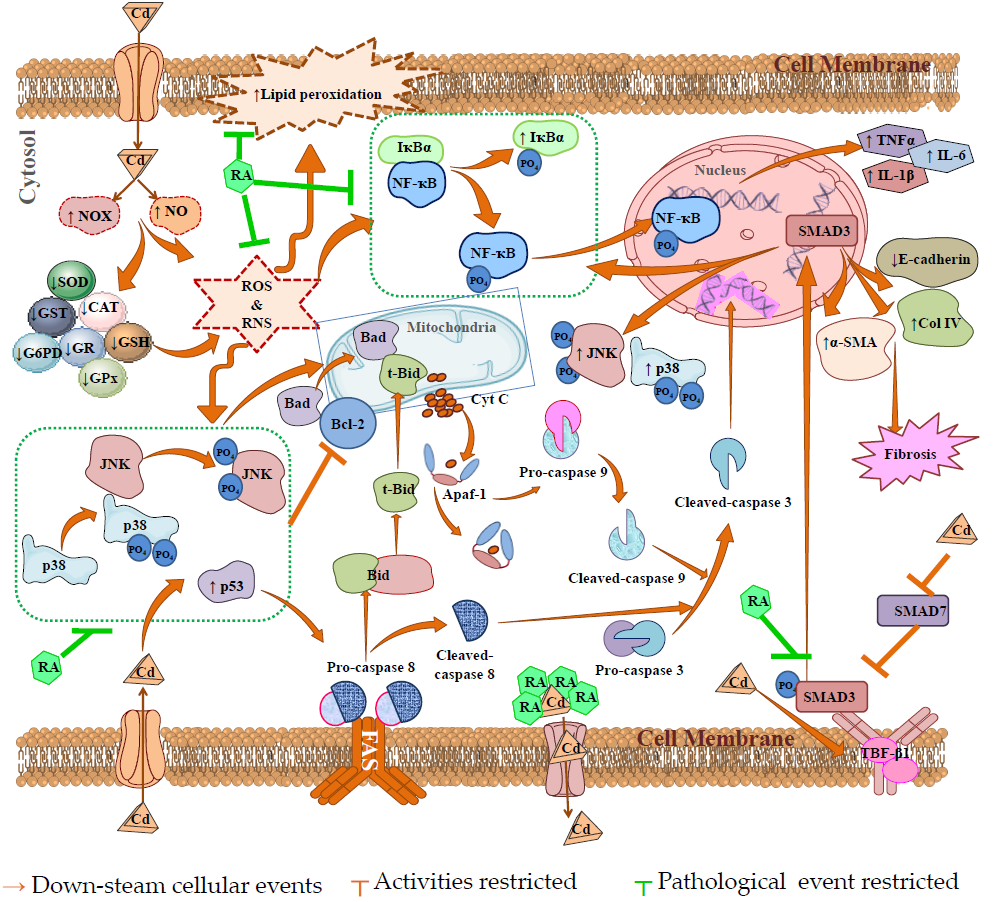
3. Discussion
4. Materials and Methods
4.1. Chemicals, Reagents, and Solvents
4.2. Animals
4.3. In Vitro Bioassays
4.3.1. Isolation of Mouse Proximal Tubular Epithelial Cells
4.3.2. Determination of Cytotoxic Effect of CdCl2 on Mouse Proximal Tubular Epithelial Cells
4.3.3. Determination of Cytoprotective Role of RA Against CdCl2
4.3.4. Hoechst Nuclear Staining
4.3.5. Flow Cytometric Analysis
4.3.6. Oxidative Stress Analyses In Vitro
4.3.7. Immunoblotting of Signal Proteins in Vitro
4.4. In Vivo Bioassay
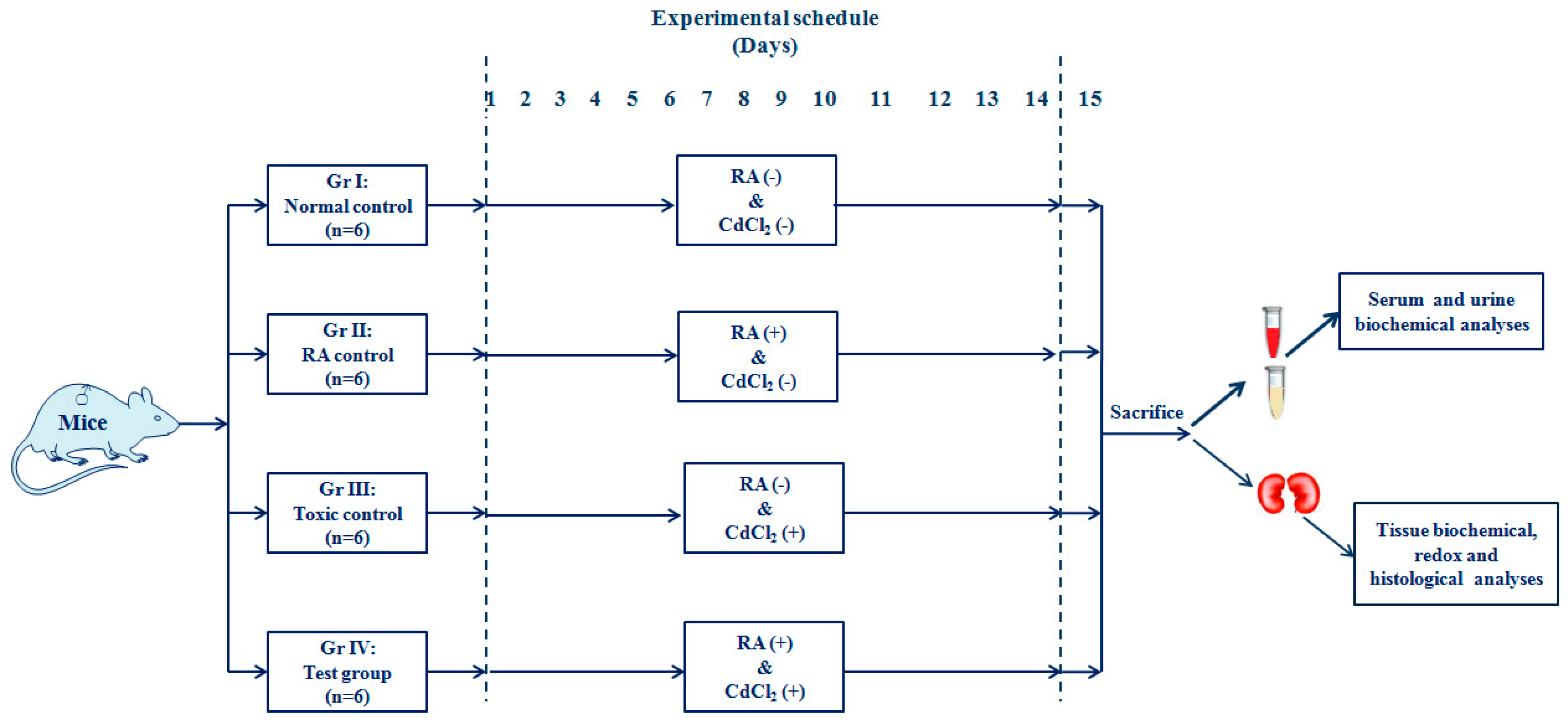
4.4.1. Experimental Set-Up
4.4.2. Estimation of Serum and Urine Biochemical Parameters
4.4.3. Estimation of Cd Contents in Renal Tissue
4.4.4. Oxidative Stress Analyses In Vivo
4.4.5. Immunoblotting of Signal Proteins in Vivo
4.4.6. Histological Analyses
4.5. Statistical Analysis
4.6. In Silico Analyses
4.6.1. In Silico ADMET and Drug-Likeness Prediction
4.6.2. In Silico Molecular Docking Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dua, T.K.; Dewanjee, S.; Khanra, R.; Bhattacharya, N.; Bhaskar, B.; Zia-Ul-Haq, M.; De Feo, V. The effects of two common edible herbs, Ipomoea aquatica and Enhydra fluctuans, on cadmium-induced pathophysiology: A focus on oxidative defence and anti-apoptotic mechanism. J. Transl. Med. 2015, 13, 245. [Google Scholar] [CrossRef]
- Grigholm, B.; Mayewski, P.A.; Aizen, V.; Kreutz, K.; Wake, C.P.; Aizen, E.; Kang, S.; Maasch, K.A.; Handley, M.J.; Sneed, S.B. Mid-twentieth century increases in anthropogenic Pb, Cd and Cu in central Asia set in hemispheric perspective using Tien Shan ice core. Atmos. Environ. 2016, 131, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Rafati-Rahimzadeh, M.; Rafati-Rahimzadeh, M.; Kazemi, S.; Moghadamnia, A. Cadmium toxicity and treatment: An update. Casp. J. Intern. Med. 2017, 8, 135–145. [Google Scholar]
- Wu, H.; Liao, Q.; Chillrud, S.N.; Yang, Q.; Huang, L.; Bi, J.; Yan, B. Environmental Exposure to Cadmium: Health Risk Assessment and its Associations with Hypertension and Impaired Kidney Function. Sci Rep. 2016, 6, 29989. [Google Scholar] [CrossRef] [Green Version]
- Prozialeck, W.C.; Edwards, J.R. Mechanisms of cadmium-induced proximal tubule injury: New insights with implications for biomonitoring and therapeutic interventions. J. Pharmacol. Exp. Ther. 2012, 343, 2–12. [Google Scholar] [CrossRef]
- Dewanjee, S.; Gangopadhyay, M.; Sahu, R.; Karmakar, S. Cadmium induced pathophysiology: Prophylactic role of edible jute (Corchorus olitorius) leaves with special emphasis on oxidative stress and mitochondrial involvement. Food Chem. Toxicol. 2013, 60, 188–198. [Google Scholar] [CrossRef]
- Luo, T.; Liu, G.; Long, M.; Yang, J.; Song, R.; Wang, Y.; Yuan, Y.; Bian, J.; Liu, X.; Gu, J.; et al. Treatment of cadmium-induced renal oxidative damage in rats by administration of alpha-lipoic acid. Environ. Sci. Pollut. Res. Int. 2017, 24, 1832–1844. [Google Scholar] [CrossRef]
- Liu, J.; Qian, S.Y.; Guo, Q.; Jiang, J.; Waalkes, M.P.; Mason, R.P.; Kadiiska, M.B. Cadmium generates reactive oxygen- and carbon-centered radical species in rats: Insights from in vivo spin-trapping studies. Free Radic. Biol. Med. 2008, 45, 475–481. [Google Scholar] [CrossRef]
- Angeli, J.K.; Cruz Pereira, C.A.; de Oliveira Faria, T.; Stefanon, I.; Padilha, A.S.; Vassallo, D.V. Cadmium exposure induces vascular injury due to endothelial oxidative stress: The role of local angiotensin II and COX-2. Free Radic. Biol. Med. 2013, 65, 838–848. [Google Scholar] [CrossRef]
- Zhang, H.; Li, L.; Wang, Y.; Dong, F.; Chen, X.; Liu, F.; Xu, D.; Yi, F.; Kapron, C.M.; Liu, J. NF-kappaB signaling maintains the survival of cadmium-exposed human renal glomerular endothelial cells. Int. J. Mol. Med. 2016, 38, 417–422. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Du, L.; Li, J.; Song, H. Epigallocatechin-3-gallate attenuates cadmium-induced chronic renal injury and fibrosis. Food Chem. Toxicol. 2016, 96, 70–78. [Google Scholar] [CrossRef]
- Dua, T.K.; Dewanjee, S.; Khanra, R.; Joardar, S.; Barma, S.; Das, S.; Zia-Ul-Haq, M.; De Feo, V. Cytoprotective and Antioxidant Effects of an Edible Herb, Enhydra fluctuans Lour. (Asteraceae), against Experimentally Induced Lead Acetate Intoxication. PLoS ONE 2016, 11, e0148757. [Google Scholar] [CrossRef] [PubMed]
- Psotová, J.; Lasovský, J.; Vičar, J. Metal-chelating properties, electrochemical behavior, scavenging and cytoprotective activities of six natural phenolics. Biomed. Pap. 2003, 147, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Dewanjee, S.; Gangopadhyay, M.; Das, U.; Sahu, R.; Samanta, A.; Banerjee, P. Signal transducer and oxidative stress mediated modulation of phenylpropanoid pathway to enhance rosmarinic acid biosynthesis in fungi elicited whole plant culture of Solenostemon scutellarioides. Enzym. Microb. Technol. 2014, 66, 1–9. [Google Scholar] [CrossRef]
- Nunes, S.; Madureira, A.R.; Campos, D.; Sarmento, B.; Gomes, A.M.; Pintado, M.; Reis, F. Therapeutic and nutraceutical potential of rosmarinic acid-Cytoprotective properties and pharmacokinetic profile. Crit. Rev. Food Sci. Nutr. 2017, 57, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Malgorzata, M.B.; Sylwia, B.; Michal, T. Antioxidants as a Potential Preventive and Therapeutic Strategy for Cadmium. Curr. Drug Targets 2016, 17, 1350–1384. [Google Scholar]
- Micali, A.; Pallio, G.; Irrera, N.; Marini, H.; Trichilo, V.; Puzzolo, D.; Pisani, A.; Malta, C.; Santoro, G.; Laura, R.; et al. Flavocoxid, a natural antioxidant, protects mouse kidney from cadmium-induced toxicity. Oxid Med. Cell. Longev. 2018, 2018, 9162946. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Sadhukhan, P.; Sinha, K.; Agarwal, N.; Sil, P.C. Mangiferin attenuates oxidative stress induced renal cell damage through activation of PI3K induced Akt and Nrf-2 mediated signaling pathways. Biochem. Biophys. Rep. 2016, 5, 313–327. [Google Scholar] [CrossRef] [Green Version]
- Qiao, S.; Li, W.; Tsubouchi, R.; Haneda, M.; Murakami, K.; Takeuchi, F.; Nisimoto, Y.; Yoshino, M. Rosmarinic acid inhibits the formation of reactive oxygen and nitrogen species in RAW264.7 macrophages. Free Radic. Res. 2005, 39, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Revoltella, S.; Baraldo, G.; Waltenberger, B.; Schwaiger, S.; Kofler, P.; Moesslacher, J.; Huber-Seidel, A.; Pagitz, K.; Kohl, R.; Jansen-Duerr, P.; et al. Identification of the NADPH Oxidase 4 Inhibiting Principle of Lycopus europaeus. Molecules 2018, 23, 653. [Google Scholar] [CrossRef]
- Adomako-Bonsu, A.G.; Chan, S.L.; Pratten, M.; Fry, J.R. Antioxidant activity of rosmarinic acid and its principal metabolites in chemical and cellular systems: Importance of physico-chemical characteristics. Toxicol. In Vitro 2017, 40, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Dewanjee, S.; Das, A.K.; Sahu, R.; Gangopadhyay, M. Antidiabetic activity of Diospyros peregrina fruit: Effect on hyperglycemia, hyperlipidemia and augmented oxidative stress in experimental type 2 diabetes. Food Chem. Toxicol. 2009, 47, 2679–2685. [Google Scholar] [CrossRef]
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012, 64, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Fernando, P.M.; Piao, M.J.; Kang, K.A.; Ryu, Y.S.; Hewage, S.R.; Chae, S.W.; Hyun, J.W. Rosmarinic acid attenuates cell damage against UVB radiation-induced oxidative stress via enhancing antioxidant effects in human HaCaT cells. Biomol. Ther. 2016, 24, 75–84. [Google Scholar] [CrossRef]
- Ding, Y.; Zhang, Z.; Yue, Z.; Ding, L.; Zhou, Y.; Huang, Z.; Huang, H. Rosmarinic acid ameliorates H2O2-induced oxidative stress in L02 cells through MAPK and Nrf2 pathways. Rejuvenation Res. 2018. [Google Scholar] [CrossRef]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef]
- Bhattacharjee, N.; Dua, T.K.; Khanra, R.; Joardar, S.; Nandy, A.; Saha, A.; De Feo, V.; Dewanjee, S. Protocatechuic Acid, a phenolic from Sansevieria roxburghiana Leaves, suppresses diabetic cardiomyopathy via stimulating glucose metabolism, ameliorating oxidative stress, and inhibiting inflammation. Front. Pharmacol. 2017, 8, 251. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. BBA Mol. Cell Res. 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Khanra, R.; Bhattacharjee, N.; Dua, T.K.; Nandy, A.; Saha, A.; Kalita, J.; Manna, P.; Dewanjee, S. Taraxerol, a pentacyclic triterpenoid, from Abroma augusta leaf attenuates diabetic nephropathy in type 2 diabetic rats. Biomed. Pharmacother. 2017, 94, 726–741. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, G.; Marcocci, M.E.; Torcia, M.; Lucibello, M.; Rosini, P.; Bonini, P.; Higashimoto, Y.; Damonte, G.; Armirotti, A.; Amodei, S.; et al. Bcl-2 Phosphorylation by p38 MAPK: Identification of target sites and biologic consequences. J. Biol. Chem. 2006, 281, 21353–21361. [Google Scholar] [CrossRef] [PubMed]
- Blaser, H.; Dostert, C.; Mak, T.W.; Brenner, D. TNF and ROS Crosstalk in Inflammation. Trends Cell Biol. 2016, 26, 249–261. [Google Scholar] [CrossRef]
- Wang, L.; Azad, N.; Kongkaneramit, L.; Chen, F.; Lu, Y.; Jiang, B.-H.; Rojanasakul, Y. The Fas death signaling pathway connecting reactive oxygen species generation and flice inhibitory protein down-regulation. J. Immunol. 2008, 180, 3072–3080. [Google Scholar] [CrossRef] [PubMed]
- Kantari, C.; Walczak, H. Caspase-8 and bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta 2011, 1813, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Cabal-Hierro, L.; Rodriguez, M.; Artime, N.; Iglesias, J.; Ugarte, L.; Prado, M.A.; Lazo, P.S. TRAF-mediated modulation of NF-kB AND JNK activation by TNFR2. Cell Signal. 2014, 26, 2658–2666. [Google Scholar] [CrossRef]
- Cosentino-Gomes, D.; Rocco-Machado, N.; Meyer-Fernandes, J.R. Cell signaling through protein kinase C oxidation and activation. Int. J. Mol. Sci. 2012, 13, 10697–10721. [Google Scholar] [CrossRef] [PubMed]
- Dewanjee, S.; Bhattacharjee, N. MicroRNA: A new generation therapeutic target in diabetic nephropathy. Biochem. Pharmacol. 2018, 155, 32–47. [Google Scholar] [CrossRef]
- Yan, X.; Liu, Z.; Chen, Y. Regulation of TGF-β signaling by Smad7. Acta Biochim. Biophys. Sin. 2009, 41, 263–272. [Google Scholar] [CrossRef]
- Bhattacharjee, N.; Barma, S.; Konwar, N.; Dewanjee, S.; Manna, P. Mechanistic insight of diabetic nephropathy and its pharmacotherapeutic targets: An update. Eur. J. Pharmacol. 2016, 791, 8–24. [Google Scholar] [CrossRef]
- Wei, Q.; Liu, Q.; Ren, C.; Liu, J.; Cai, W.; Zhu, M.; Jin, H.; He, M.; Yu, J. Effects of bradykinin on TGFbeta1induced epithelialmesenchymal transition in ARPE19 cells. Mol. Med. Rep. 2018, 17, 5878–5886. [Google Scholar] [PubMed]
- Cutroneo, K.R.; White, S.L.; Phan, S.H.; Ehrlich, H.P. Therapies for bleomycin induced lung fibrosis through regulation of TGF-beta1 induced collagen gene expression. J. Cell Physiol. 2007, 211, 585–589. [Google Scholar] [CrossRef]
- Pandya, V.; Rao, A.; Chaudhary, K. Lipid abnormalities in kidney disease and management strategies. World J. Nephrol. 2015, 4, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Bulbul, M.C.; Dagel, T.; Afsar, B.; Ulusu, N.N.; Kuwabara, M.; Covic, A.; Kanbay, M. Disorders of Lipid Metabolism in Chronic Kidney Disease. Blood Purif. 2018, 46, 144–152. [Google Scholar] [CrossRef]
- Giordano, C.; Karasik, O.; King-Morris, K.; Asmar, A. Uric Acid as a Marker of Kidney Disease: Review of the Current Literature. Dis. Markers 2015, 2015, 382918. [Google Scholar] [CrossRef]
- Higgins, C. Urea and Creatinine Concentration, the Urea: Creatinine Ratio. Available online: www.acutecaretesting.org (accessed on 10 October 2016).
- Tojo, A.; Kinugasa, S. Mechanisms of glomerular albumin filtration and tubular reabsorption. Int. J. Nephrol. 2012, 481520. [Google Scholar] [CrossRef]
- Xu, Z.; Jin, X.; Pan, T.; Liu, T.; Wan, N.; Li, S. Antagonistic effects of selenium on cadmium-induced apoptosis by restoring the mitochondrial dynamic equilibrium and energy metabolism in chicken spleens. Oncotarget 2017, 8, 52629–52641. [Google Scholar] [CrossRef] [Green Version]
- Dewanjee, S.; Dua, T.K.; Khanra, R.; Das, S.; Barma, S.; Joardar, S.; Bhattacharjee, N.; Zia-Ul-Haq, M.; Jaafar, H.Z. Water Spinach, Ipomoea aquatica (Convolvulaceae), Ameliorates Lead Toxicity by Inhibiting Oxidative Stress and Apoptosis. PLoS ONE 2015, 10, e0139831. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target Ther. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Joardar, S.; Manna, P.; Dua, T.K.; Bhattacharjee, N.; Khanra, R.; Bhowmick, S.; Kalita, J.; Saha, A.; Ray, S.; et al. Carnosic Acid, a natural diterpene, attenuates arsenic-induced hepatotoxicity via reducing oxidative stress, MAPK activation, and apoptotic cell death pathway. Oxid. Med. Cell Longev. 2018, 2018, 1421438. [Google Scholar] [CrossRef]
- Public Health Service. Public Health Service Policy on Human Care and Use of Laboratory Animals; National Institute of Health: Washington, DC, USA, 2015. [Google Scholar]
- Breggia, A.C.; Himmelfarb, J. Primary mouse renal tubular epithelial cells have variable injury tolerance to ischemic and chemical mediators of oxidative stress. Oxid. Med. Cell. Longev. 2008, 1, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, J.; Das, J.; Manna, P.; Sil, P.C. The protective role of arjunolic acid against doxorubicin induced intracellular ROS dependent JNK-p38 and p53-mediated cardiac apoptosis. Biomaterials 2011, 32, 4857–4866. [Google Scholar] [CrossRef] [PubMed]
- Stinson, L.J.; Darmon, A.J.; Dagnino, L.; D’Souza, S.J. Delayed apoptosis post-cadmium injury in renal proximal tubule epithelial cells. Am. J. Nephrol. 2003, 23, 27–37. [Google Scholar] [CrossRef]
- Mansoor, T.A.; Borralho, P.M.; Dewanjee, S.; Mulhovo, S.; Rodrigues, C.M.; Ferreira, M.J. Monoterpene bisindole alkaloids, from the African medicinal plant Tabernaemontana elegans, induce apoptosis in HCT116 human colon carcinoma cells. J. Ethnopharmacol. 2013, 149, 463–470. [Google Scholar] [CrossRef]
- Dewanjee, S.; Joardar, S.; Bhattacharjee, N.; Dua, T.K.; Das, S.; Kalita, J.; Manna, P. Edible leaf extract of Ipomoea aquatica Forssk. (Convolvulaceae) attenuates doxorubicin-induced liver injury via inhibiting oxidative impairment, MAPK activation and intrinsic pathway of apoptosis. Food Chem. Toxicol. 2017, 105, 322–336. [Google Scholar] [CrossRef]
- Manna, P.; Jain, S.K. L-cysteine and hydrogen sulfide increase PIP3 and AMPK/PPARgamma expression and decrease ROS and vascular inflammation markers in high glucose treated human U937 monocytes. J. Cell Biochem. 2013, 114, 2334–2345. [Google Scholar] [CrossRef]
- Fraga, C.G.; Leibovitz, B.E.; Tappel, A.L. Lipid peroxidation measured as thiobarbituric acid-reactive substances in tissue slices: Characterization and comparison with homogenates and microsomes. Free Radic. Biol. Med. 1988, 4, 155–161. [Google Scholar] [CrossRef]
- Herrera, B.; Murillo, M.M.; Alvarez-Barrientos, A.; Beltran, J.; Fernandez, M.; Fabregat, I. Source of early reactive oxygen species in the apoptosis induced by transforming growth factor-beta in fetal rat hepatocytes. Free Radic. Biol. Med. 2004, 36, 16–26. [Google Scholar] [CrossRef]
- Pal, S.; Pal, P.B.; Das, J.; Sil, P.C. Involvement of both intrinsic and extrinsic pathways in hepatoprotection of arjunolic acid against cadmium induced acute damage in vitro. Toxicology 2011, 283, 129–139. [Google Scholar] [CrossRef]
- Zhang, Y.; Aberg, F.; Appelkvist, E.L.; Dallner, G.; Ernster, L. Uptake of dietary coenzyme Q supplement is limited in rats. J. Nutr. 1995, 125, 446–453. [Google Scholar] [PubMed]
- Manna, P.; Sinha, M.; Sil, P.C. Protective role of arjunolic acid in response to streptozotocin-induced type-I diabetes via the mitochondrial dependent and independent pathways. Toxicology 2009, 257, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Hissin, P.J.; Hilf, R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. Anal. Biochem. 1976, 74, 214–226. [Google Scholar] [CrossRef]
- Baghirova, S.; Hughes, B.G.; Hendzel, M.J.; Schulz, R. Sequential fractionation and isolation of subcellular proteins from tissue or cultured cells. MethodsX 2015, 2, 440–445. [Google Scholar] [CrossRef] [PubMed]
- Khanra, R.; Dewanjee, S.; K Dua, T.; Sahu, R.; Gangopadhyay, M.; De Feo, V.; Zia-Ul-Haq, M. Abroma augusta L. (Malvaceae) leaf extract attenuates diabetes induced nephropathy and cardiomyopathy via inhibition of oxidative stress and inflammatory response. J. Transl. Med. 2015, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Pari, L.; Murugavel, P.; Sitasawad, S.L.; Kumar, K.S. Cytoprotective and antioxidant role of diallyl tetrasulfide on cadmium induced renal injury: An in vivo and in vitro study. Life Sci. 2007, 80, 650–658. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- OSIRIS Property Explorer. Available online: www.organic-chemistry.org/prog/peo/ (accessed on 10 October 2018).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protein Data Bank. Available online: http://www.rcsb.org/ (accessed on 10 October 2018).
- Maestro, Schrödinger, LLC, New York, USA, 2017. Available online: www.schrodinger.com (accessed on 10 October 2018).















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Group I | Group II | Group III | Group IV |
|---|---|---|---|---|
| Total cholesterol (mg/dL) | 82.12 ± 7.54 | 81.33 ± 8.92 | 138.76 ± 12.50 # | 118.22 ± 10.67 ** |
| HDL cholesterol (mg/dL) | 31.58 ± 2.87 | 31.12 ± 3.24 | 23.16 ± 2.33 # | 28.22 ± 2.41 * |
| Triglycerides (mg/dL) | 91.15 ± 8.67 | 87.58 ± 8.52 | 118.21 ± 10.45 # | 104.11 ± 9.89 * |
| LDH (U/L) | 161.48 ± 14.23 | 155.23 ± 15.92 | 238.21 ± 22.78 # | 184.55 ± 17.24 ** |
| CK (IU/ mg of protein) | 27.28 ± 2.44 | 26.02 ± 2.72 | 36.42 ± 3.38 # | 31.57 ± 2.98 * |
| Urea (mg/dL) | 22.13 ± 1.78 | 23.02 ± 2.25 | 34.42 ± 3.08 # | 28.72 ± 2.77 ** |
| Uric acid (mg/dL) | 2.23 ± 0.25 | 2.03 ± 0.19 | 3.87 ± 0.29 # | 3.12 ± 0.31 ** |
| Creatinine (mg/dL) | 0.47 ± 0.05 | 0.45 ± 0.04 | 0.71 ± 0.08 # | 0.62 ± 0.06 * |
| Parameters | Group I | Group II | Group III | Group IV |
|---|---|---|---|---|
| Kidney mass (mg) | 47.24 ± 4.54 | 45.67 ± 3.92 | 55.67 ± 6.12 $ | 48.22 ± 5.01 * |
| Kidney mass/body mass (× 103) | 15.28 ± 1.48 | 13.94 ± 1.24 | 20.33 ± 2.17 # | 17.89 ± 1.31 * |
| Cd burden in kidney (ppm of wet tissue) | 0.02 ± 0.001 | 0.007 ± 0.0008 | 20.33 ± 2.37 # | 17.89 ± 1.31 * |
| Urinary creatinine (mg/dL) | 65.52 ± 6.33 | 64.33 ± 7.12 | 31.50 ± 3.21# | 41.57 ± 3.98 * |
| Urinary albumin (mg/dL) | 2.98 ± 0.24 | 2.78 ± 0.19 | 6.74 ± 0.75 # | 5.98 ± 0.49 * |
| Urinary nitrate/nitrite (nmol/g creatinine) | 0.85 ± 0.09 | 0.82 ± 0.08 | 1.45 ± 0.16 # | 1.17 ± 0.12 ** |
| Properties | Profile | Values |
|---|---|---|
| Properties under Lipinski’s rule of five | Molecular weight | 360.31 |
| Hydrogen bond acceptor | 8 | |
| Hydrogen bond donor | 5 | |
| Octanol water coefficient (LogP) | 2.36 | |
| Other physico-chemical properties | Number of rotatable bonds | 7 |
| Molecular refractivity | 91.4 | |
| Topological polar surface area | 144.52 | |
| Water solubility | Log S | Soluble |
| Pharmacokinetics profiles | GI absorption | Low |
| log Kp (cm/s) | −6.82 | |
| CYP1A2 inhibitor | No | |
| CYP2C19 inhibitor | No | |
| CYP2C9 inhibitor | No | |
| CYP2D6 inhibitor | No | |
| CYP3A4 inhibitor | No | |
| Drug-likeness profiles | Bioavailability score | 0.55 |
| Toxicity risk assessment | Mutagenicity risk | Low |
| Irritating effect | Low | |
| Reproductive toxicity effect | Low | |
| Tumorigenicity risk | Low |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joardar, S.; Dewanjee, S.; Bhowmick, S.; Dua, T.K.; Das, S.; Saha, A.; De Feo, V. Rosmarinic Acid Attenuates Cadmium-Induced Nephrotoxicity via Inhibition of Oxidative Stress, Apoptosis, Inflammation and Fibrosis. Int. J. Mol. Sci. 2019, 20, 2027. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20082027
Joardar S, Dewanjee S, Bhowmick S, Dua TK, Das S, Saha A, De Feo V. Rosmarinic Acid Attenuates Cadmium-Induced Nephrotoxicity via Inhibition of Oxidative Stress, Apoptosis, Inflammation and Fibrosis. International Journal of Molecular Sciences. 2019; 20(8):2027. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20082027
Chicago/Turabian StyleJoardar, Swarnalata, Saikat Dewanjee, Shovonlal Bhowmick, Tarun K. Dua, Sonjit Das, Achintya Saha, and Vincenzo De Feo. 2019. "Rosmarinic Acid Attenuates Cadmium-Induced Nephrotoxicity via Inhibition of Oxidative Stress, Apoptosis, Inflammation and Fibrosis" International Journal of Molecular Sciences 20, no. 8: 2027. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20082027