Distinct Binding Dynamics, Sites and Interactions of Fullerene and Fullerenols with Amyloid-β Peptides Revealed by Molecular Dynamics Simulations


Abstract
:1. Introduction
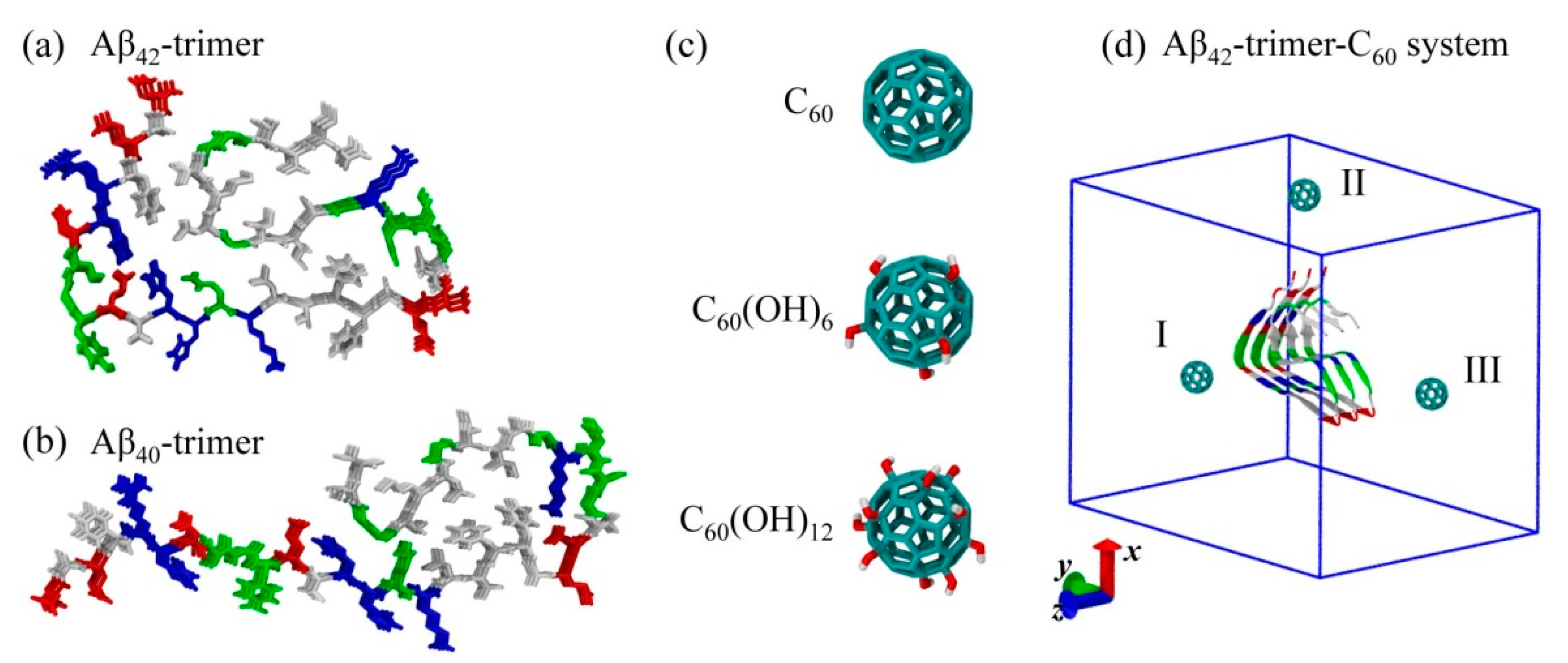
2. Results and Discussion
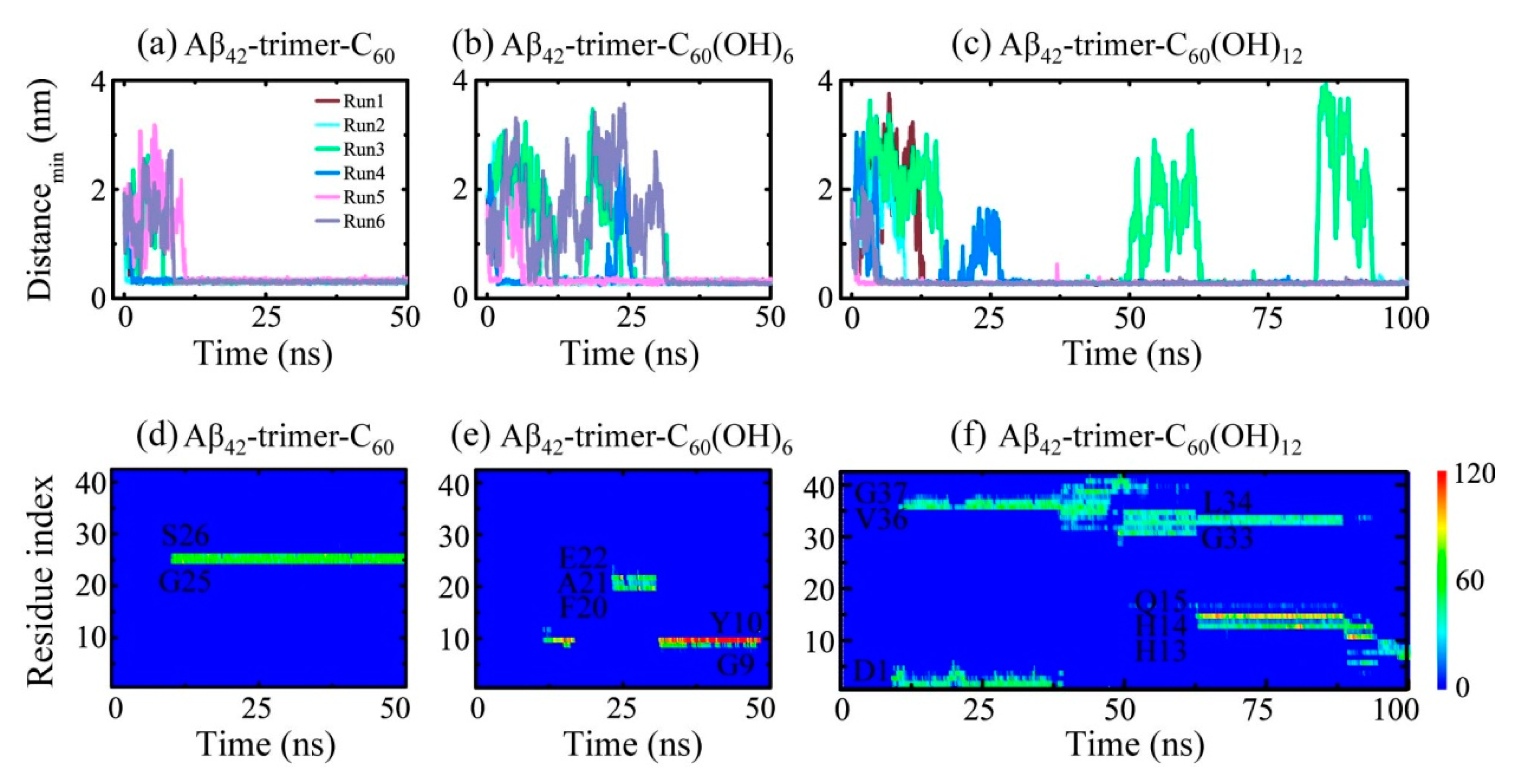
2.1. Dynamics of the Fullerene/Fullerenol Molecule Binding to Aβ42-Trimer
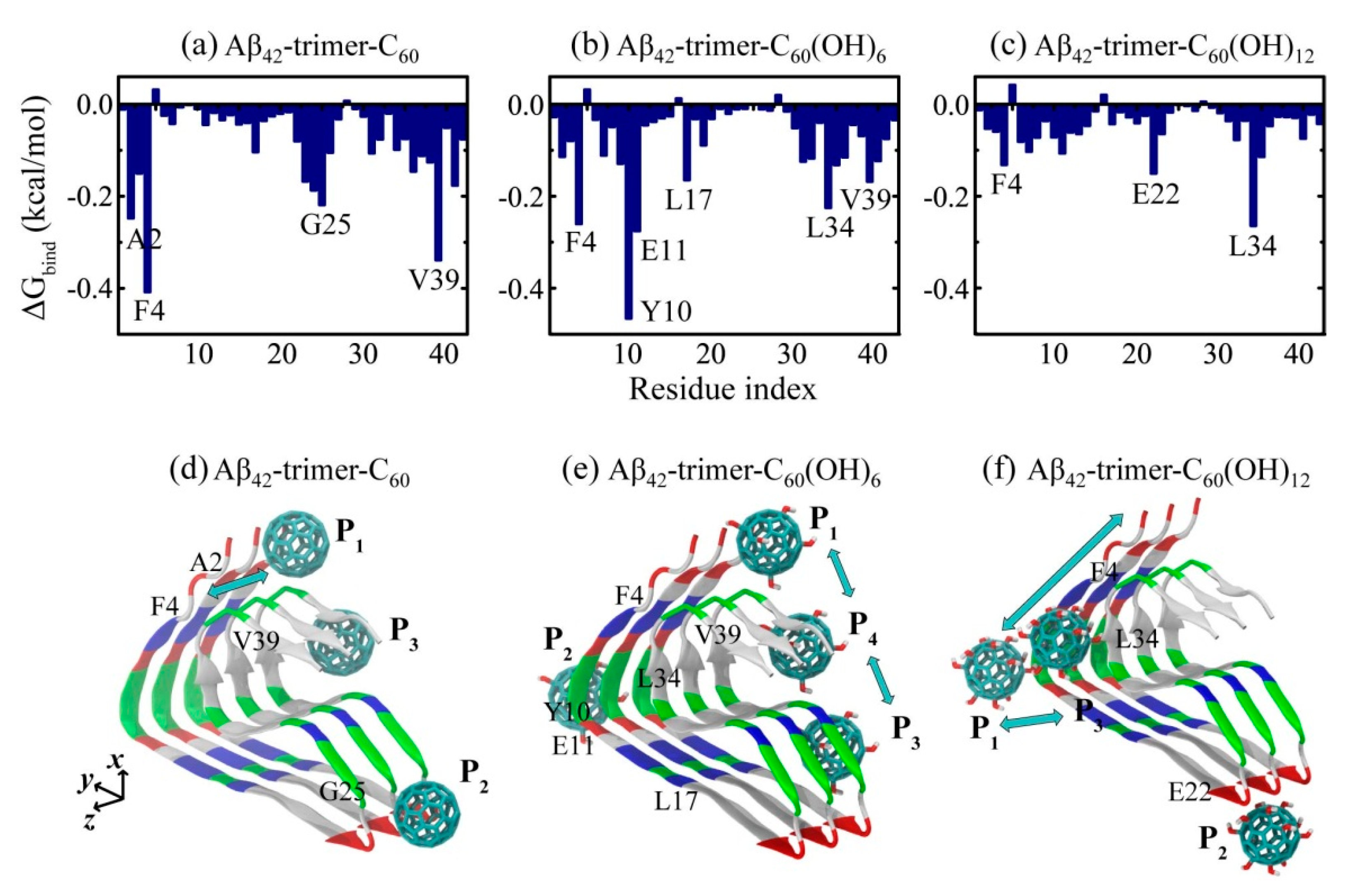
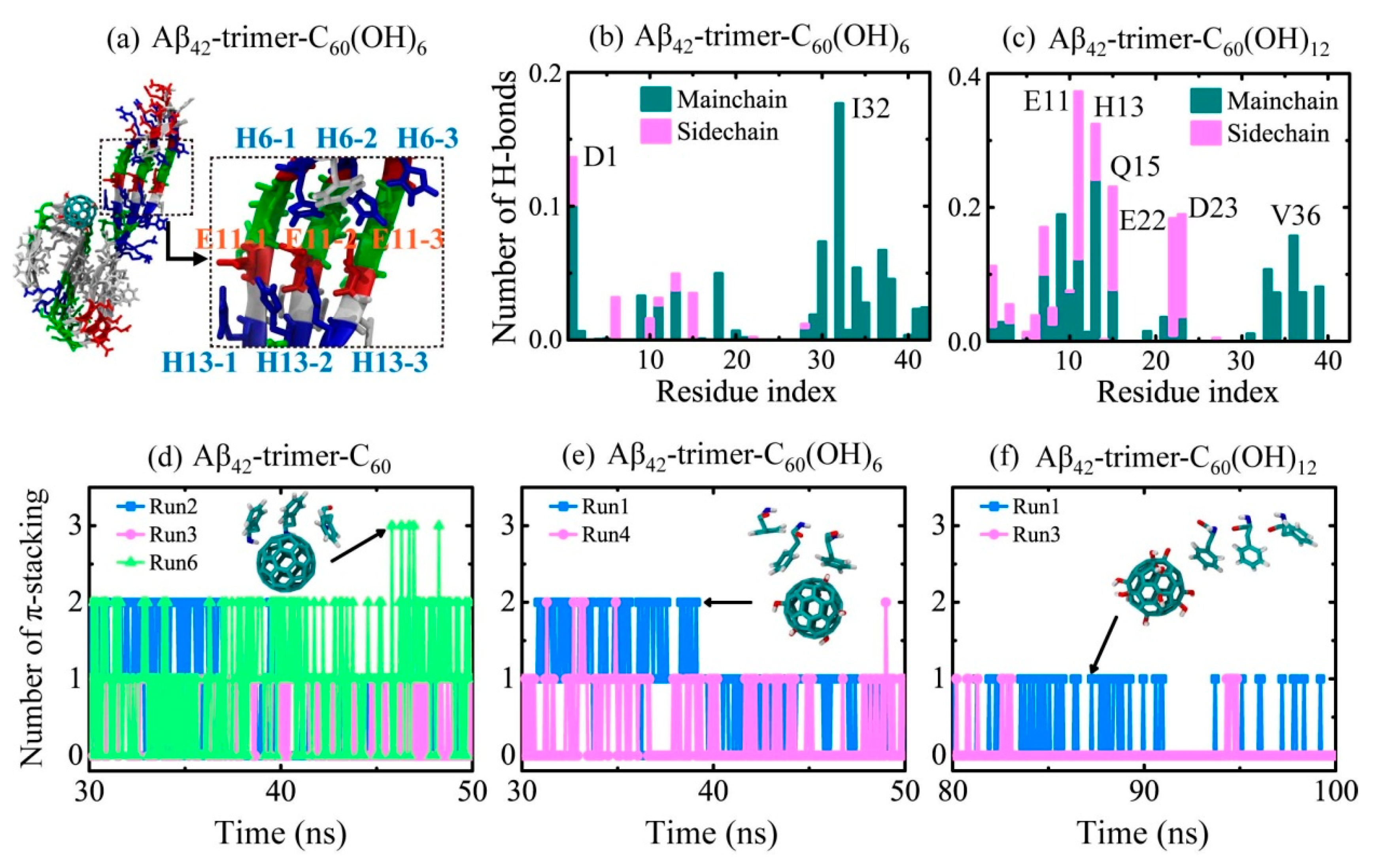
2.2. Binding Sites of The Fullerene/Fullerenol Molecule to Aβ42-Trimer
2.3. Structural Influence of The Fullerene/Fullerenol Molecule on Aβ42-Trimer
2.4. Dynamics, Sites and Interactions of The Fullerene/Fullerenol Molecule Binding to Aβ40-Trimer
3. Materials and Methods
3.1. Aβ40/42 Protofibrillar Trimer and C60/C60(OH)6 /C60(OH)12 Molecules
3.2. Details of MD Simulations
3.3. Analysis Methods
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Riek, R.; Eisenberg, D.S. The activities of amyloids from a structural perspective. Nature 2016, 539, 227–235. [Google Scholar] [CrossRef]
- Lu, J.X.; Qiang, W.; Yau, W.M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular structure of beta-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Gremer, L.; Schölzel, D.; Schenk, C.; Reinartz, E.; Labahn, J.; Ravelli, R.B.G.; Tusche, M.; Lopez-Iglesias, C.; Hoyer, W.; Heise, H.; et al. Fibril structure of amyloid-β(1–42) by cryo–electron microscopy. Science 2017, 358, 116–119. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, B.; Ge, X.; Ding, F. Distinct oligomerization and fibrillization dynamics of amyloid core sequences of amyloid-beta and islet amyloid polypeptide. Phys. Chem. Chem. Phys. 2017, 19, 28414–28423. [Google Scholar] [CrossRef]
- Michaels, T.C.T.; Šarić, A.; Habchi, J.; Chia, S.; Meisl, G.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P.J. Chemical kinetics for bridging molecular mechanisms and macroscopic measurements of amyloid fibril formation. Annu. Rev. Phys. Chem. 2018, 69, 273–298. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Straub, J.E.; Thirumalai, D. Toward a molecular theory of early and late events in monomer to amyloid fibril formation. Annu. Rev. Phys. Chem. 2011, 62, 437–463. [Google Scholar] [CrossRef]
- Nguyen, P.; Derreumaux, P. Understanding amyloid fibril nucleation and Aβ oligomer/drug interactions from computer simulations. Acc. Chem. Res. 2014, 47, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Mao, X.; Yu, Y.; Wang, C.-X.; Yang, Y.-L.; Wang, C. Nanomaterials for reducing amyloid cytotoxicity. Adv. Mater. 2013, 25, 3780–3801. [Google Scholar] [CrossRef]
- Radic, S.; Davis, T.P.; Ke, P.C.; Ding, F. Contrasting effects of nanoparticle-protein attraction on amyloid aggregation. RSC Adv. 2015, 5, 105498. [Google Scholar] [CrossRef]
- Wang, B.; Pilkington, E.H.; Sun, Y.; Davis, T.P.; Ke, P.C.; Ding, F. Modulating protein amyloid aggregation with nanomaterials. Environ. Sci. Nano 2017, 4, 1772–1783. [Google Scholar] [CrossRef]
- Young, L.M.; Saunders, J.C.; Mahood, R.A.; Revill, C.H.; Foster, R.J.; Tu, L.-H.; Raleigh, D.P.; Radford, S.E.; Ashcroft, A.E. Screening and classifying small-molecule inhibitors of amyloid formation using ion mobility spectrometry–mass spectrometry. Nat. Chem. 2014, 7, 73–81. [Google Scholar] [CrossRef]
- Saunders, J.C.; Young, L.M.; Mahood, R.A.; Jackson, M.P.; Revill, C.H.; Foster, R.J.; Smith, D.A.; Ashcroft, A.E.; Brockwell, D.J.; Radford, S.E. An in vivo platform for identifying inhibitors of protein aggregation. Nat. Chem. Biol. 2016, 12, 94–101. [Google Scholar] [CrossRef]
- Chen, Z.J.; Krause, G.; Reif, B. Structure and orientation of peptide inhibitors bound to β-amyloid fibrils. J. Mol. Biol. 2005, 354, 760–776. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, T.; Mihara, H. Peptide and protein mimetics inhibiting amyloid β-peptide aggregation. Acc. Chem. Res. 2008, 41, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussière, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef]
- Liao, Q.; Owen, M.C.; Bali, S.; Barz, B.; Strodel, B. Aβ under stress: The effects of acidosis, Cu2+-binding, and oxidation on amyloid β-peptide dimers. Chem. Commun. 2018, 54, 7766–7769. [Google Scholar] [CrossRef]
- Xiao, L.; Aoshima, H.; Saitoh, Y.; Miwa, N. Highly hydroxylated fullerene localizes at the cytoskeleton and inhibits oxidative stress in adipocytes and a subcutaneous adipose-tissue equivalent. Free Radical Biol. Med. 2011, 51, 1376–1389. [Google Scholar] [CrossRef]
- Grebowski, J.; Kazmierska, P.; Krokosz, A. Fullerenols as a new therapeutic approach in nanomedicine. BioMed Res. Int. 2013, 2013, 9. [Google Scholar] [CrossRef]
- Bosi, S.; Da Ros, T.; Spalluto, G.; Prato, M. Fullerene derivatives: An attractive tool for biological applications. Eur. J. Med. Chem. 2003, 38, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Dugan, L.L.; Turetsky, D.M.; Du, C.; Lobner, D.; Wheeler, M.; Almli, C.R.; Shen, C.K.F.; Luh, T.Y.; Choi, D.W.; Lin, T.S. Carboxyfullerenes as neuroprotective agents. Proc. Natl. Acad. Sci. USA 1997, 94, 9434–9439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Lee, M. Fullerene inhibits β-amyloid peptide aggregation. Biochem. Biophys. Res. Commun. 2003, 303, 576–579. [Google Scholar] [CrossRef]
- Podolski, I.Y.; Podlubnaya, Z.A.; Kosenko, E.A.; Mugantseva, E.A.; Makarova, E.G.; Marsagishvili, L.G.; Shpagina, M.D.; Kaminsky, Y.G.; Andrievsky, G.V.; Klochkov, V.K. Effects of hydrated forms of C60 fullerene on amyloid 1-peptide fibrillization in vitro and performance of the cognitive task. J. Nanosci. Nanotechnol. 2007, 7, 1479–1485. [Google Scholar] [CrossRef]
- Ye, S.; Zhou, T.; Pan, D.; Lai, Y.; Yang, P.; Chen, M.; Wang, Y.; Hou, Z.; Ren, L.; Jiang, Y. Fullerene C60 derivatives attenuated microglia-mediated prion peptide neurotoxicity. J. Biomed. Nanotechnol. 2016, 12, 1820–1833. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, F.-Y.; Zhilenkov, A.V.; Voronov, I.I.; Khakina, E.A.; Mischenko, D.V.; Troshin, P.A.; Hsu, S.-h. Water-soluble fullerene derivatives as brain medicine: Surface chemistry determines if they are neuroprotective and antitumor. ACS Appl. Mat. Interfaces 2017, 9, 11482–11492. [Google Scholar] [CrossRef]
- Huy, P.D.Q.; Li, M.S. Binding of fullerenes to amyloid beta fibrils: Size matters. Phys. Chem. Chem. Phys. 2014, 16, 20030–20040. [Google Scholar] [CrossRef]
- Bednarikova, Z.; Huy, P.D.Q.; Mocanu, M.-M.; Fedunova, D.; Li, M.S.; Gazova, Z. Fullerenol C60(OH)16 prevents amyloid fibrillization of Aβ40 – in vitro and in silico approach. Phys. Chem. Chem. Phys. 2016, 18, 18855–18867. [Google Scholar] [CrossRef]
- Zhou, X.Y.; Xi, W.H.; Luo, Y.; Cao, S.Q.; Wei, G.H. Interactions of a water-soluble fullerene derivative with amyloid-beta protofibrils: Dynamics, binding mechanism, and the resulting salt-bridge disruption. J. Phys. Chem. B 2014, 118, 6733–6741. [Google Scholar] [CrossRef] [PubMed]
- Radic, S.; Nedumpully-Govindan, P.; Chen, R.; Salonen, E.; Brown, J.M.; Ke, P.C.; Ding, F. Effect of fullerenol surface chemistry on nanoparticle binding-induced protein misfolding. Nanoscale 2014, 6, 8340–8349. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Luo, Y.; Lin, D.; Xi, W.; Yang, X.; Wei, G. The molecular mechanism of fullerene-inhibited aggregation of Alzheimer’s β-amyloid peptide fragment. Nanoscale 2014, 6, 9752–9762. [Google Scholar] [CrossRef]
- Sun, Y.; Qian, Z.; Wei, G. The inhibitory mechanism of a fullerene derivative against amyloid-β peptide aggregation: An atomistic simulation study. Phys. Chem. Chem. Phys. 2016, 18, 12582–12591. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, Q.; Liang, G.; Zheng, J. Molecular dynamics simulations of low-ordered Alzheimer β-amyloid oligomers from dimer to hexamer on self-assembled monolayers. Langmuir 2011, 27, 14876–14887. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xi, W.; Wei, G. Atomic-level study of the effects of O4 molecules on the structural properties of protofibrillar Abeta trimer: Beta-sheet stabilization, salt bridge protection, and binding mechanism. J. Phys. Chem. B 2015, 119, 2786–2794. [Google Scholar] [CrossRef] [PubMed]
- Ngo, S.T.; Li, M.S. Curcumin binds to Aβ1–40 peptides and fibrils stronger than ibuprofen and naproxen. J. Phys. Chem. B 2012, 116, 10165–10175. [Google Scholar] [CrossRef] [PubMed]
- Thai, N.Q.; Nguyen, H.L.; Linh, H.Q.; Li, M.S. Protocol for fast screening of multi-target drug candidates: Application to Alzheimer’s disease. J. Mol. Graphics Modell. 2017, 77, 121–129. [Google Scholar] [CrossRef]
- Andujar, S.A.; Lugli, F.; Hofinger, S.; Enriz, R.D.; Zerbetto, F. Amyloid-beta fibril disruption by C60-molecular guidance for rational drug design. Phys. Chem. Chem. Phys. 2012, 14, 8599–8607. [Google Scholar] [CrossRef] [PubMed]
- Benyamini, H.; Shulman-Peleg, A.; Wolfson, H.J.; Belgorodsky, B.; Fadeev, L.; Gozin, M. Interaction of C60-fullerene and carboxyfullerene with proteins: Docking and binding site alignment. Bioconjugate Chem. 2006, 17, 378–386. [Google Scholar] [CrossRef]
- Do, T.D.; LaPointe, N.E.; Nelson, R.; Krotee, P.; Hayden, E.Y.; Ulrich, B.; Quan, S.; Feinstein, S.C.; Teplow, D.B.; Eisenberg, D.; et al. Amyloid β-protein C-terminal fragments: Formation of cylindrins and β-barrels. J. Am. Chem. Soc. 2016, 138, 549–557. [Google Scholar] [CrossRef]
- Truex, N.L.; Wang, Y.; Nowick, J.S. Assembly of peptides derived from β-sheet regions of β-amyloid. J. Am. Chem. Soc. 2016, 138, 13882–13890. [Google Scholar] [CrossRef]
- Qian, Z.; Zhang, Q.; Liu, Y.; Chen, P. Assemblies of amyloid-β30–36 hexamer and its G33V/L34T mutants by replica-exchange molecular dynamics simulation. PLoS ONE 2017, 12, e0188794. [Google Scholar] [CrossRef]
- Kar, R.K.; Brender, J.R.; Ghosh, A.; Bhunia, A. Nonproductive binding modes as a prominent feature of Aβ40 fiber elongation: Insights from molecular dynamics simulation. J. Chem. Inf. Model. 2018, 58, 1576–1586. [Google Scholar] [CrossRef]
- Brender, J.R.; Ghosh, A.; Kotler, S.A.; Krishnamoorthy, J.; Bera, S.; Morris, V.; Sil, T.B.; Garai, K.; Reif, B.; Bhunia, A.; et al. Probing transient non-native states in amyloid beta fiber elongation by NMR. Chem. Commun. 2019, 55, 4483–4486. [Google Scholar] [CrossRef] [PubMed]
- Ferkinghoff-Borg, J.; Fonslet, J.; Andersen, C.B.; Krishna, S.; Pigolotti, S.; Yagi, H.; Goto, Y.; Otzen, D.; Jensen, M.H. Stop-and-go kinetics in amyloid fibrillation. Phys. Rev. E 2010, 82, 010901. [Google Scholar] [CrossRef] [PubMed]
- Melchor, M.-H.; Susana, F.-G.; Francisco, G.-S.; Hiram, I.B.; Norma, R.-F.; Jorge, A.L.-R.; Perla, Y.L.-C.; Gustavo, B.-I. Fullerenemalonates inhibit amyloid beta aggregation, in vitro and in silico evaluation. RSC Adv. 2018, 8, 39667–39677. [Google Scholar] [CrossRef]
- Gazit, E. A possible role for π-stacking in the self-assembly of amyloid fibrils. FASEB J. 2002, 16, 77–83. [Google Scholar] [CrossRef]
- Xie, L.G.; Lin, D.D.; Luo, Y.; Li, H.Y.; Yang, X.J.; Wei, G.H. Effects of Hydroxylated Carbon Nanotubes on the Aggregation of A beta(16-22) Peptides: A Combined Simulation and Experimental Study. Biophys. J. 2014, 107, 1930–1938. [Google Scholar] [CrossRef]
- Berendsen, H.; Postma, J.; Van Gunsteren, W.; Hermans, J. Interaction models for water in relation to protein hydration. Intermol. Forces 1981, 11, 331–342. [Google Scholar]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Nguyen, P.H.; Li, M.S.; Stock, G.; Straub, J.E.; Thirumalai, D. Monomer adds to preformed structured oligomers of Aβ-peptides by a two-stage dock-lock mechanism. Proc. Natl. Acad. Sci. USA 2007, 104, 111–116. [Google Scholar] [CrossRef]
- Krone, M.G.; Hua, L.; Soto, P.; Zhou, R.; Berne, B.J.; Shea, J.E. Role of water in mediating the assembly of Alzheimer amyloid-β Aβ16-22 protofilaments. J. Am. Chem. Soc. 2008, 130, 11066–11072. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald - an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure - pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Xu, Z.; Lei, X.; Tu, Y.; Tan, Z.-J.; Song, B.; Fang, H. Dynamic cooperation of hydrogen binding and π stacking in ssDNA adsorption on graphene oxide. Chem.-Eur. J. 2017, 23, 13100–13104. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; David, L.; Gilson, M.K. Accelerated Poisson–Boltzmann calculations for static and dynamic systems. J. Comput. Chem. 2002, 23, 1244–1253. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Berhanu, W.M.; Hansmann, U.H.E. The stability of cylindrin β-barrel amyloid oligomer models—A molecular dynamics study. Proteins 2013, 81, 1542–1555. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Qian, Z.; Chen, Y.; Qian, H.; Wei, G.; Zhang, Q. Norepinephrine inhibits Alzheimer’s amyloid-β peptide aggregation and destabilizes amyloid-β protofibrils: A molecular dynamics simulation study. ACS Chem. Neurosci. 2019, 10, 1585–1594. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Systems | ΔEvdw | ΔEelec | ΔEMM | ΔGpolar | ΔGnonpolar | ΔGsolv | ΔGbind |
|---|---|---|---|---|---|---|---|
| Aβ42-trimer-C60 | −24.44 ± 0.69 | 0 | −24.44 ± 0.69 | 0 | −3.92 ± 0.16 | −3.92 ± 0.16 | −28.36 ± 0.71 |
| Aβ42-trimer-C60(OH)6 | −24.02 ± 0.74 | −5.16 ± 0.69 | −29.18 ± 0.25 | 15.27 ± 1.68 | −3.61 ± 0.16 | 11.66 ± 1.69 | −17.52 ± 1.71 |
| Aβ42-trimer-C60(OH)12 | −18.20 ± 1.02 | −14.60 ± 1.45 | −32.80 ± 1.77 | 27.06 ± 2.52 | −3.30 ± 0.17 | 23.77 ± 2.53 | −9.03 ± 3.09 |
| System | Aβ42-trimer-C60 | Aβ42-trimer-C60(OH)6 | Aβ42-trimer-C60(OH)12 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Binding site | 2–4 | 23–25 | 31–41 | 2–4 | 9–11 | 17–19 | 31–41 | 4–14 | 22–23 | 34–35 |
| ΔGbind | −0.80 | −0.57 | −1.32 | −0.45 | −0.87 | −0.28 | −1.23 | −0.73 | −0.21 | −0.38 |
| Deviation | 0.09 | 0.02 | 0.01 | 0.06 | 0.03 | 0.04 | 0.05 | 0.07 | 0.04 | 0.05 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Zou, Y.; Zhang, Q.; Chen, P.; Liu, Y.; Qian, Z. Distinct Binding Dynamics, Sites and Interactions of Fullerene and Fullerenols with Amyloid-β Peptides Revealed by Molecular Dynamics Simulations. Int. J. Mol. Sci. 2019, 20, 2048. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20082048
Liu Z, Zou Y, Zhang Q, Chen P, Liu Y, Qian Z. Distinct Binding Dynamics, Sites and Interactions of Fullerene and Fullerenols with Amyloid-β Peptides Revealed by Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2019; 20(8):2048. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20082048
Chicago/Turabian StyleLiu, Zhiwei, Yu Zou, Qingwen Zhang, Peijie Chen, Yu Liu, and Zhenyu Qian. 2019. "Distinct Binding Dynamics, Sites and Interactions of Fullerene and Fullerenols with Amyloid-β Peptides Revealed by Molecular Dynamics Simulations" International Journal of Molecular Sciences 20, no. 8: 2048. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20082048