1. Introduction
About 85% of patients with high-grade serous ovarian cancer (HGSOC) will achieve a clinical remission with a combination of surgery and platinum-based chemotherapy despite presenting with advanced stage disease [
1]. Though many will respond to additional rounds of treatment and may experience prolonged remission, disease-free intervals generally become progressively shorter, culminating in platinum and ultimately chemotherapy resistance, suggesting a fundamental and progressive change in the biology of the tumor.
The development of chemotherapy resistance is hypothesized to occur through multiple routes, most notably selection of resistant clones and upregulation of tumor-protective pathways. To elucidate the mechanisms underlying platinum resistance several groups have generated chemoresistant versions of established ovarian cancer cell lines and compared gene expression between the chemoresistant line and its chemosensitive parental line [
2,
3,
4,
5]. These studies provide insight into changes that occur as cells adapt to high levels of chemotherapy, but the genes identified in these studies are inconsistent between studies and the mechanisms of in vitro resistance could very likely be different from those arising in human tumors [
6].
Similarly, comparisons of gene expression between patients who achieved clinical remission versus those who were refractory identified expression signatures of chemoresistance, but patient heterogeneity was a noted confounding factor in these comparisons [
5,
7]. To eliminate interpatient heterogeneity, Patch et al. compared matched samples taken from patients at initial debulking and at recurrence [
8], suggesting that gene profiles in the latter may represent a chemoresistant gene signature.
A large systematic review of 42 studies attempting to define molecular signatures that predict resistance to chemotherapy in ovarian cancer found that gene signatures were not consistent between studies and concluded that there are no gene signatures currently appropriate for clinical use [
9]. With larger datasets becoming available, it may be possible to improve upon previous studies. For example, Yin et al. used TCGA data to devise a 131-gene signature correlating with platinum resistance [
10].
Another approach for predicting the response to chemotherapy is to perform in vitro growth assays using fresh tumor samples exposed to chemotherapy [
11]. The sensitivity and specificity of these in vitro assays vary, but they do show promise as a possible means to predict patient response, although they have not yet had widespread use in a clinical setting [
12,
13,
14].
A goal of precision medicine is to classify patients based on the genomic and transcriptomic characteristics of their tumor and use these classifications to guide treatment decisions. This approach has been successful in breast cancer, where gene expression signatures can be used for prognosis and to predict chemotherapy response [
15,
16]. There are now gene-expression-based lab diagnostics that are routinely used to stratify breast cancer patients into molecular subtypes and guide treatment options [
17,
18]. In ovarian cancer, several large-scale datasets have been used to stratify ovarian cancer patients into molecular subtypes based on gene expression [
19,
20,
21,
22]; this stratification has prognostic and therapeutic relevance [
23,
24], but has not yet been prospectively validated for clinical use [
25].
The aim of this study was to compare gene expression profiles for individuals before and early into the course of treatment in hopes of identifying early changes in expression that may herald either up-front chemoresistance or provide insights into the sequence of events involved in the development of resistance. Patients undergoing neoadjuvant chemotherapy (NACT), who typically require a biopsy for diagnosis followed by an interval cytoreduction, were felt to be ideal to address these questions.
3. Discussion
Our data demonstrate that there are multiple changes in gene expression profiles following exposure of HGSOC to platinum and taxane therapy and that these appear to develop early in the treatment of cancer. These changes are sufficiently similar between patients to distinguish pre- and post-treatment specimens, suggesting that biological pressure may be more relevant to the initial response to chemotherapy than the expansion of minor resistant subclones. A recent study by Arend et al. measured pre- and post-NACT expression of 770 cancer genes using the Nanostring platform [
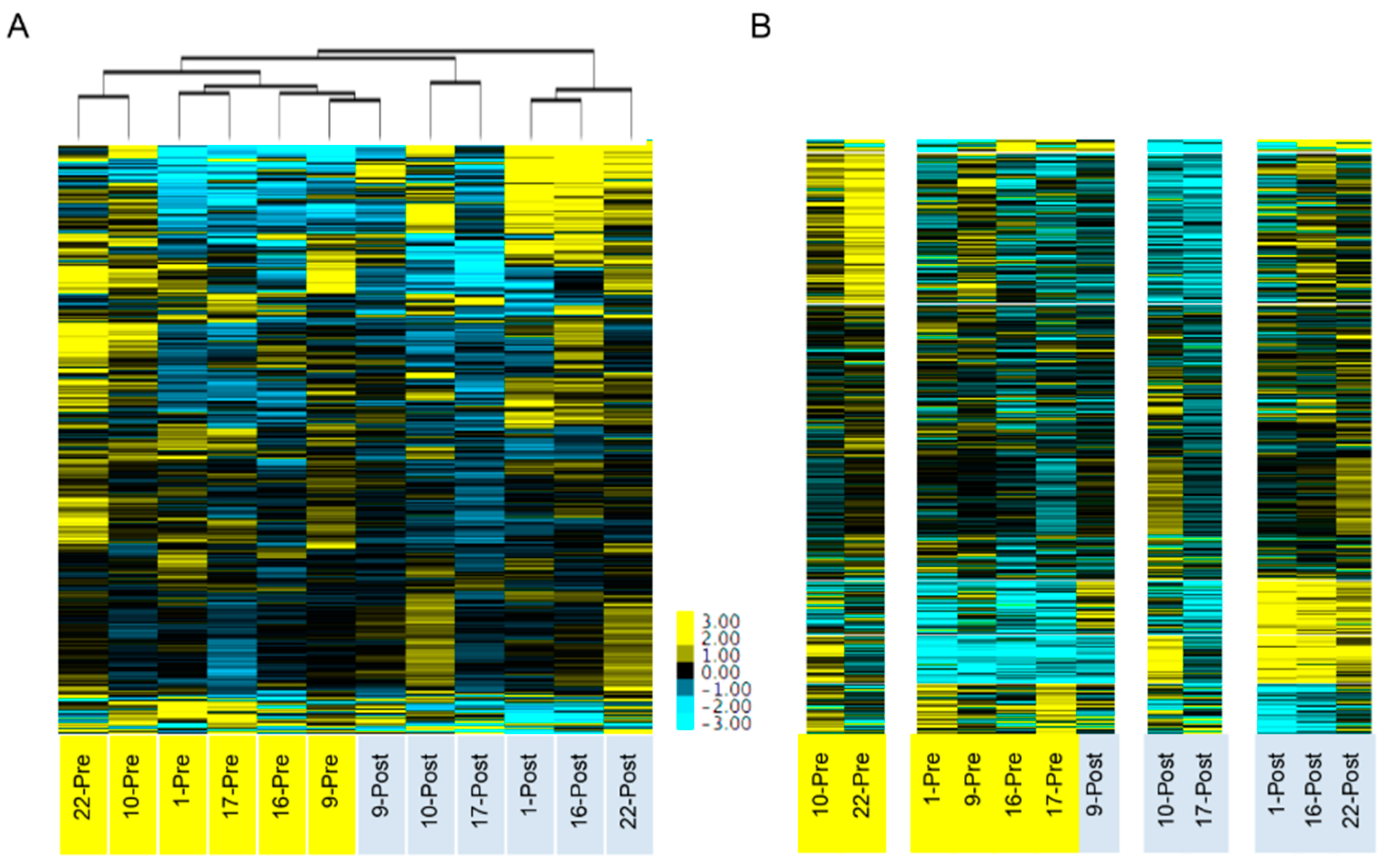
38]. Unsupervised clustering using the 86 differentially expressed genes from this panel of 770 genes resulted in a similar separation between pre- and post-NACT samples. Interestingly, when using all 770 genes, the samples did not cluster based on pre- and post-NACT status in the Arend study, while in our study, clustering using an expanded set of 6748 genes did result in separation (
Figure 2). This highlights the differences between using an unbiased whole transcriptome approach versus a targeted panel analysis. There was strong concordance between the two studies, with 19 of the top 20 differentially expressed genes identified in the Arend study also significantly changed in our study.
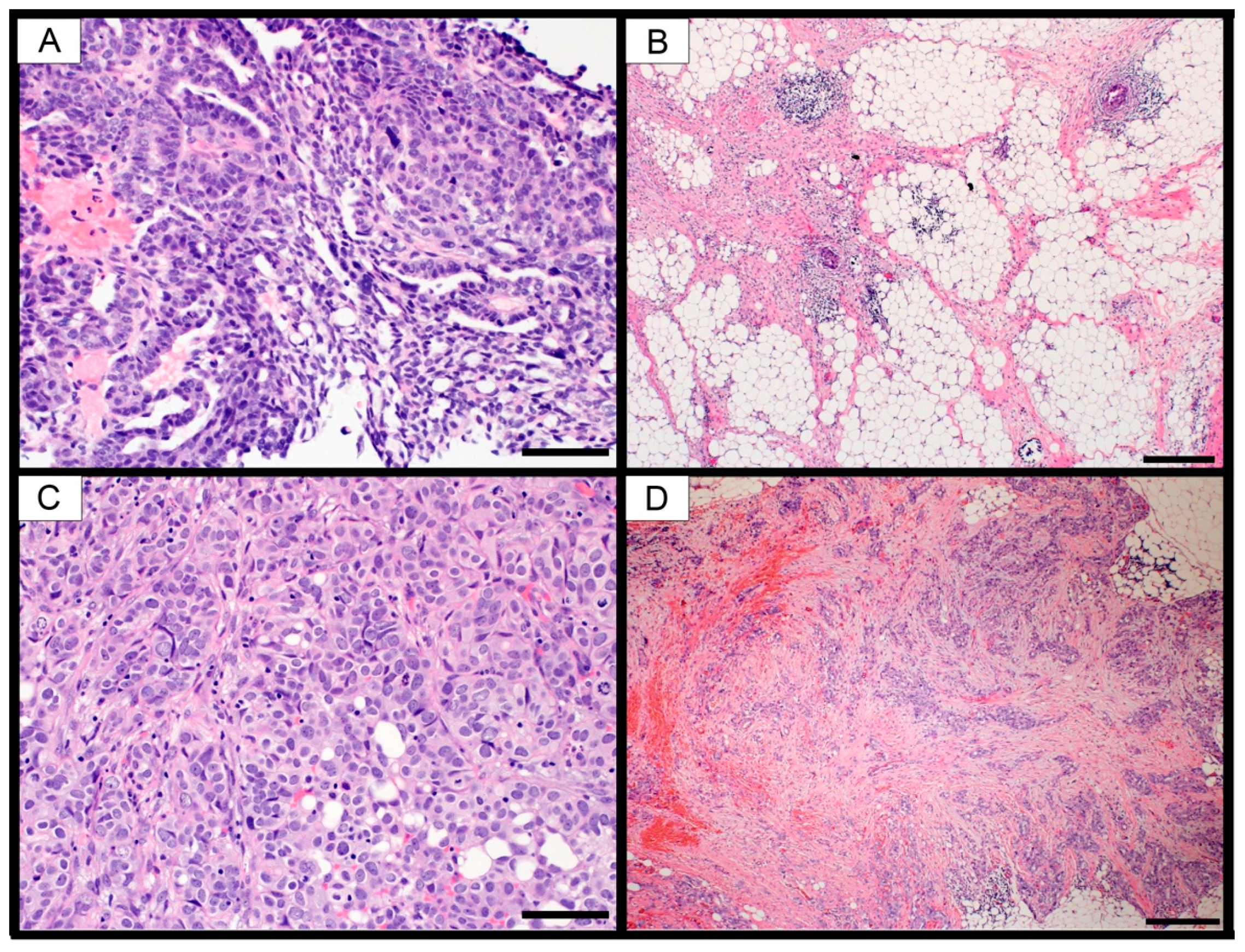
A caveat to gene expression analysis using bulk tissue samples is the contribution of non-cancer cell types within the tumor microenvironment. Gene transcripts from infiltrating stromal, vascular and immune cells are inevitably mixed in with the cancer cell mRNA, producing a “bulk” gene expression signature. Several groups have demonstrated that small numbers of infiltrating cells, comprising as little as 5% of the total sample, will affect the gene expression signature [
39,
40], which can heavily influence the molecular subtype assigned to the patient [
41]. Our samples had a similar range of tumor purity based on pathologic analysis and the ESTIMATEScore (
Table 1,
Figure S1) compared to TCGA samples [
28]. It is highly likely that these infiltrating cells contributed to the gene expression signatures. Future studies using microdissected tissue and/or single cell analyses will be required to determine the effect of these infiltrating cells on the molecular signatures.
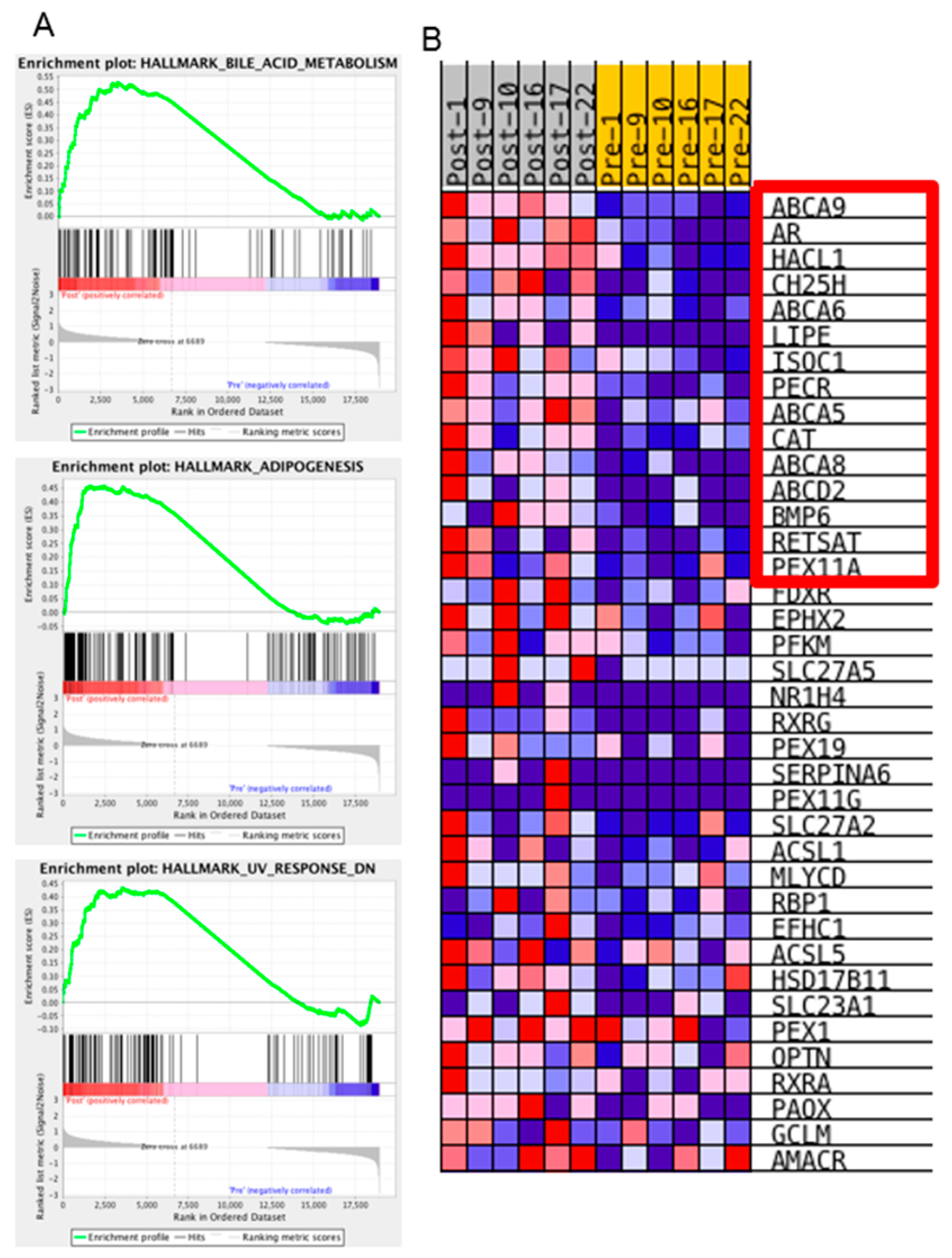
We were most interested in the pathways or biological states that were enriched in the post-NACT samples, as these could represent potential therapeutic windows if targeting those pathways could block the ovarian cancer cells’ ability to resist chemotherapy treatment. Interestingly, only three hallmark gene sets were enriched in the post-NACT samples compared to the pre-NACT samples: Bile acid metabolism, Adipogenesis, and Ultraviolet-response-down (
Figure 5). Tumor purity may have affected these results, although changes in tumor purity comparing pre- to post-NACT samples were varied, with half of the samples showing increased purity and half showing decreased purity (
Table 1, ESTIMATEScore change). However, three of the four samples from the omentum had decreased purity, which could indicate increased infiltration of adipocytes, which could account for the enriched adipogenesis gene set.
The hallmark gene set with the strongest normalized enrichment score was the bile acid metabolism gene set. This gene set is based on 28 founder gene sets, including several datasets relating to transporters, peroxisome, and response to drug. To better understand this enrichment, we identified the specific genes that were upregulated in our samples that caused gene set enrichment. Of the 38 genes that contributed to this enrichment, the top 15 were characterized by drug transport, fatty acid metabolism, and peroxisome production genes (
Table 6 and
Figure 5B). Both the Adipogenesis hallmark enrichment and the fatty acid metabolism enrichment could be the result of changes in the cancer epithelial cells or, alternatively, infiltration of fat cells within the sample taken during interval debulking surgery. Five of the 15 genes contributing to the Bile Acid Metabolism hallmark are drug transporters. Although we did not identify upregulation of
ABCB1 in our analysis, there is evidence that upregulation of
ABCB1 occurs via fusion with upstream promoters [
8]. It is possible that we did not detect upregulation of
ABCB1 because our sequence analysis pipeline rejected these transcripts due to non-alignment with annotated transcripts, or the upregulation did not change during the first three cycles of chemotherapy administration. Our data indicate that, in addition to
ABCB1, other transporters, especially those in the ABC1 family of transporters, may be playing an important role in the ovarian cancer response to chemotherapy treatment. Finally, our data indicate that cancer cells respond to chemotherapy by increasing peroxisome activity. Peroxisomes are key hubs within the cell for controlling reactive oxygen species [
42,
43]. Together these results suggest that inhibitors of drug transport, not just limited to ABCB1/MDR, and inhibitors of peroxisomes and fatty acid metabolism may be effective in blocking cancer cells’ response to chemotherapy.
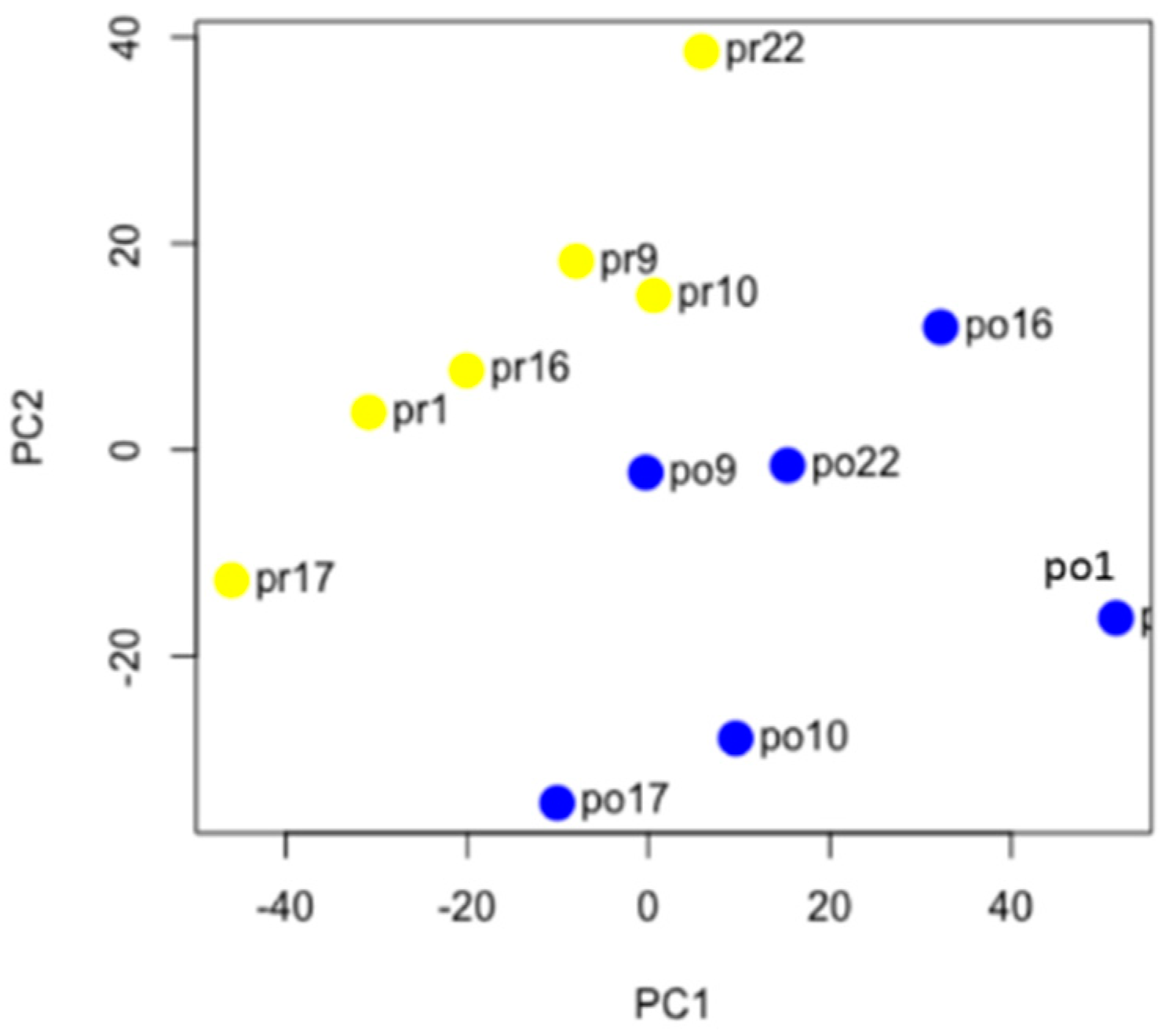
Numerous studies have demonstrated a large degree of heterogeneity in gene expression patterns found in HGSOC patients [
8,
20], which presumably would result in pre-NACT and post-NACT samples from the same patient clustering together due to their unique pattern of copy number changes. In contrast to this prediction, unsupervised hierarchical clustering and k-means clustering, using the most variably expressed genes, indicated that the effects of chemotherapy on gene expression appear to outweigh the effects of inter-patient variability (
Figure 2). Arend et al. performed a similar study using a targeted gene panel and also found that pre- and post-NACT samples cluster together [
38]. A previous study comparing the change in chromosomal alterations between matched pre-NACT and post-NACT samples found that there were no significant changes in the chromosomal architecture during the first three cycles of chemotherapy [
44]. These findings combined with our findings of extensive gene expression changes indicate that chemotherapy has a stronger effect on gene expression than clonal evolution within the short time frame of NACT.
Currently there are no clinically useful biomarkers or gene expression patterns that predict response to carboplatin and paclitaxel [
7,
45]. Analyses of gene expression in large cohorts of HGSOC patients has led to classification of patients into molecular subtypes based on various unsupervised clustering algorithms. Several groups have proposed between four and nine molecular subtypes for ovarian cancer based on gene expression and other omics datasets in hope of improving patient outcomes through tumor-directed therapy selection [
8,
19,
20,
21,
23,
24,
25]. While appealing, especially for the treatment of patients who are categorized as likely to be platinum-resistant, one problem with this approach is that the patient cohort and the platform for measuring gene expression can affect the robustness and applicability to other datasets. Our group has previously demonstrated that the technology used to measure gene expression can affect placement of patients into their molecular subtype. When we re-analyzed the original TCGA cohort using RNASeq gene expression data, instead of the original microarray gene expression data, a large percentage of the patients were classified into different molecular subtypes [
46]. Due to this and other factors, it is difficult to make robust molecular classifications based on gene expression [
47]. In this present study, we demonstrate that classification of patients into specific molecular subtypes changes after administration of chemotherapy (
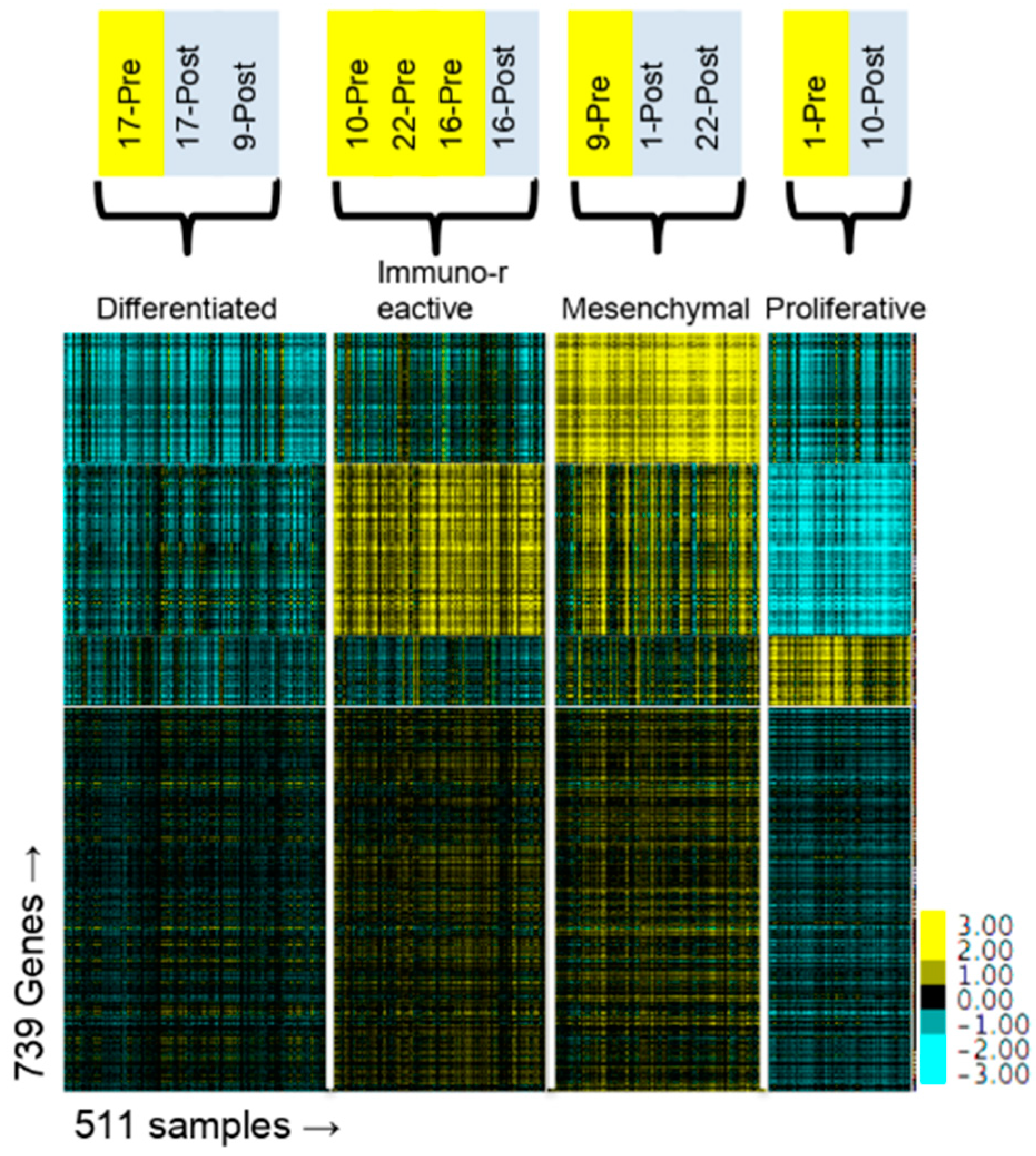
Figure 4 and
Table 2). One caveat to this analysis is that our samples were from metastatic sites (
Table 1) and not from the primary ovarian tumor. The TCGA molecular subtypes were based on gene expression in the primary ovarian tumor and it has not been established that gene expression from metastatic sites or from post-NACT samples can be applied to the molecular subtype stratification. Nevertheless, our data suggest that the subtypes may not be as useful as has been proposed because a patient’s classification into a subtype might change after exposure to chemotherapy. Furthermore, the change is not predictable based on the initial classification, as the three patients classified in the Immunoreactive subtype were all assigned to different subtypes after NACT.
Unsurprisingly, pre-NACT samples were notably enriched for multiple pathways involving cell cycle progression (
Table 3,
Tables S3 and S4) consistent with the concept that HGSOC is a highly proliferative disease and that platinum and taxane therapy reduces proliferation dramatically. This is further supported by the finding that common proliferation markers,
MKI67 and
PCNA were both significantly downregulated in the post-NACT samples (
Tables S2 and S5). A subset of ovarian cancer is known to overexpress
CCNE1 or
CCND1 [
8], suggesting that cell-cycle-targeted drugs such as palbociclib, a CDK4/6 inhibitor, may be effective in specific subsets of ovarian cancer [
48]. In our study, the levels of
CCND1,
CCND2,
CCNE1 and
CCNE2 were relatively unchanged after treating with chemotherapy, while
CCNB and
CCNA were significantly downregulated (
Table S12), suggesting cell cycle inhibitors may be effective even in patients without elevated
CCNE or
CCND.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}