Transcriptomic Analysis Reveals Priming of The Host Antiviral Interferon Signaling Pathway by Bronchobini® Resulting in Balanced Immune Response to Rhinovirus Infection in Mouse Lung Tissue Slices

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
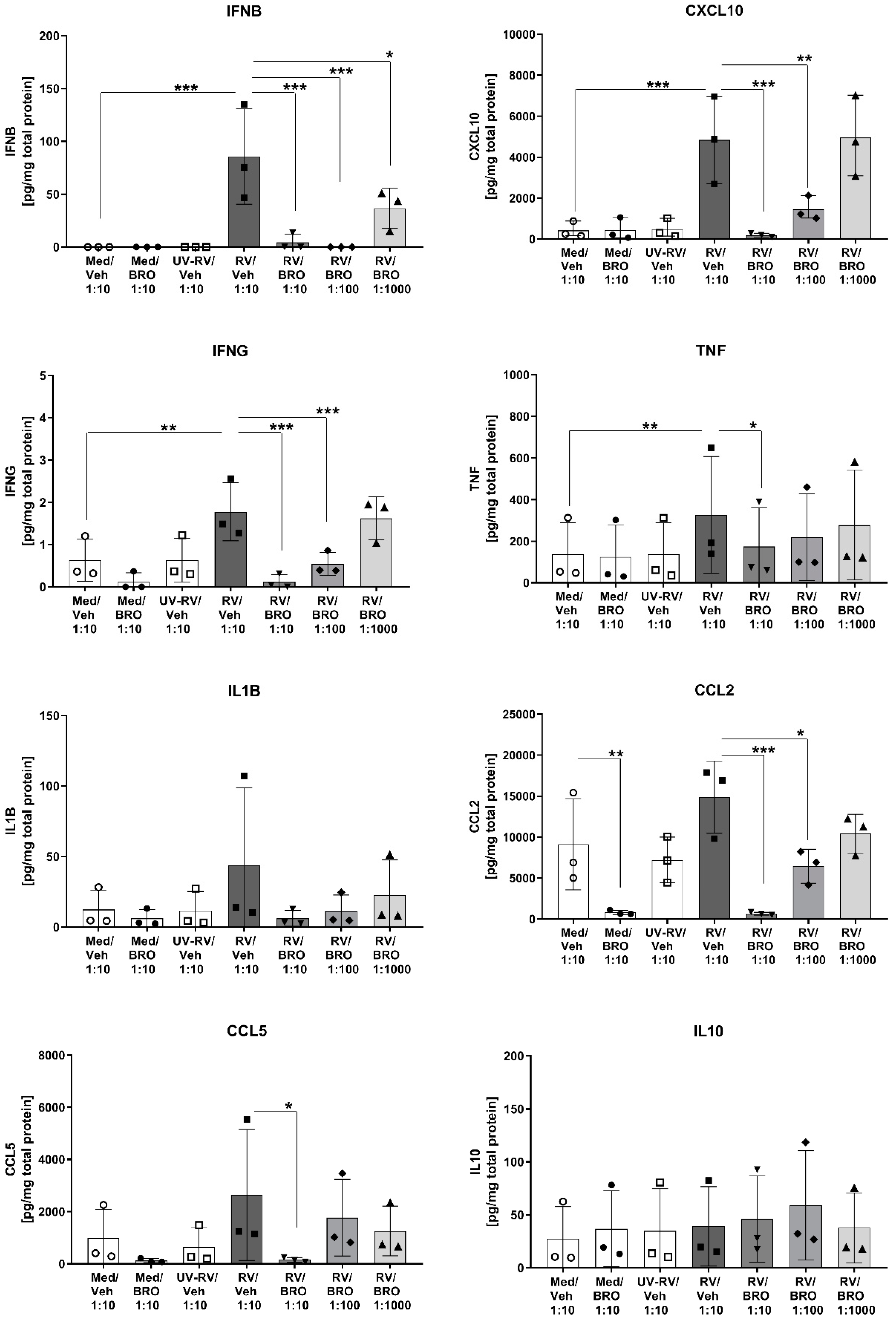
2.1. BRO Reduced RV-Induced Release of Pro-Inflammatory and Antiviral Cytokines
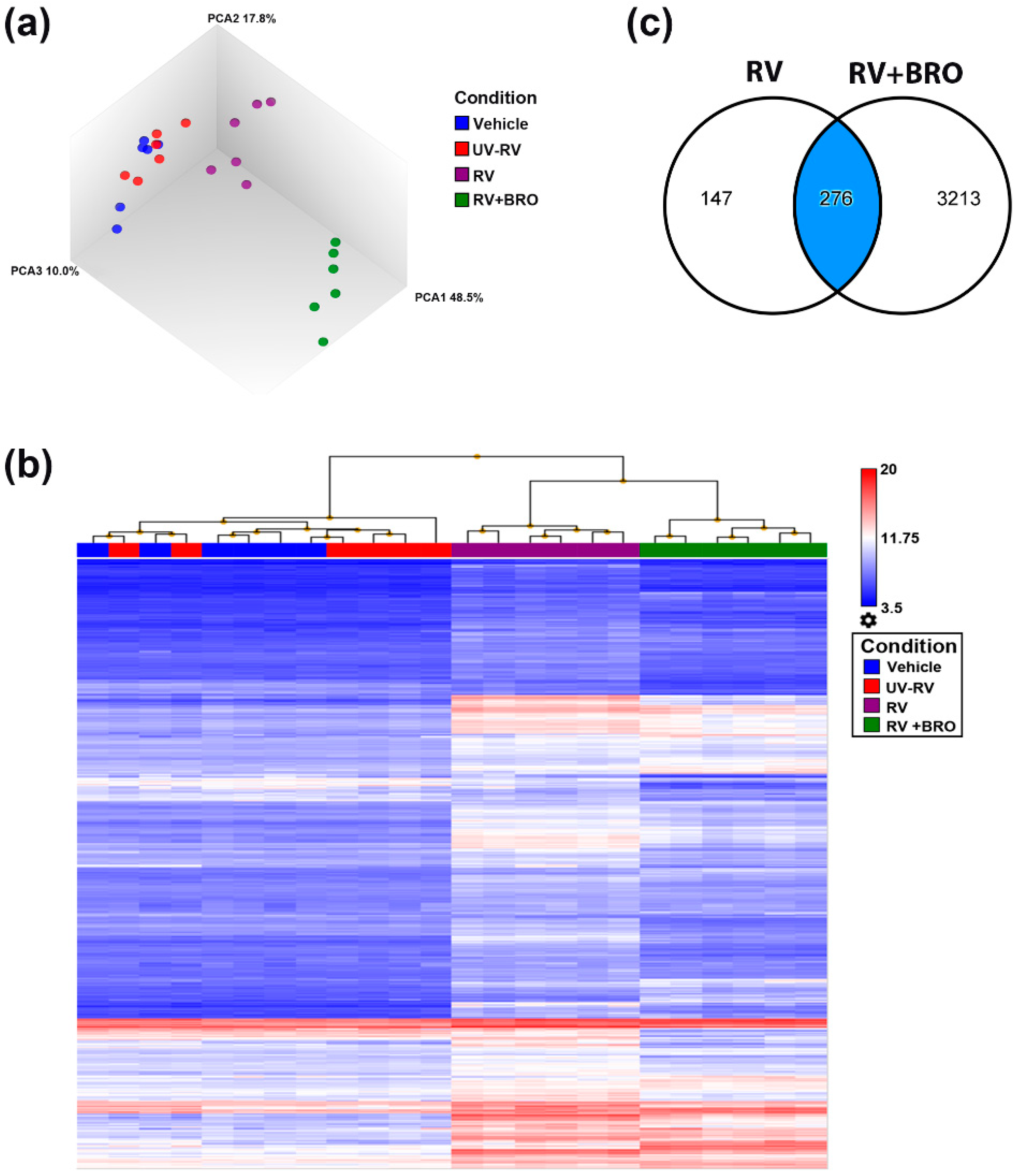
2.2. Analysis of BRO Mode-of-Action Using Whole Transcriptome Analysis
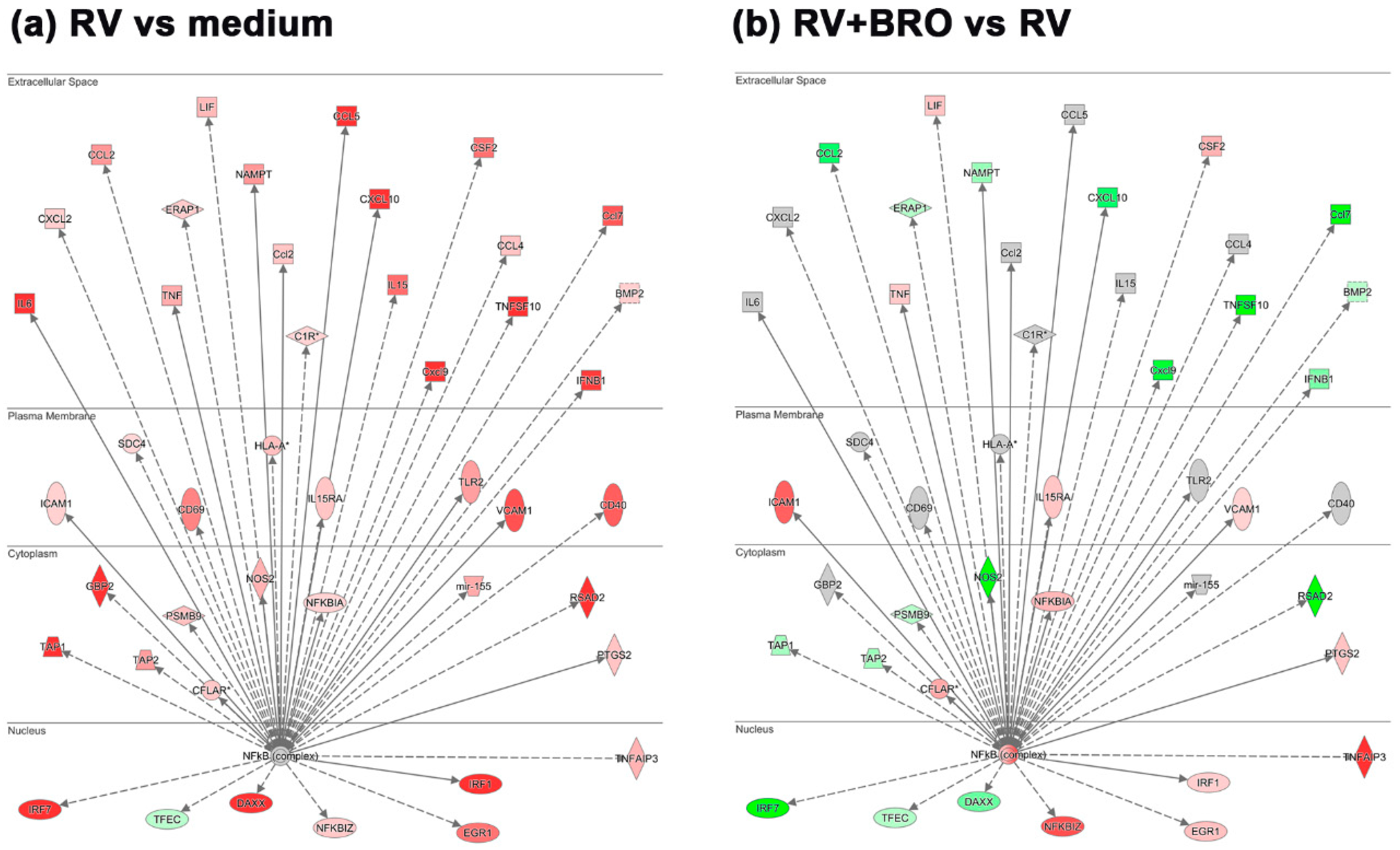
2.2.1. BRO Modulation of RV-Induced Interferon Signaling Pathway
2.2.2. BRO Modulation of NFkB-Mediated Pro-Inflammatory Response to Rhinovirus
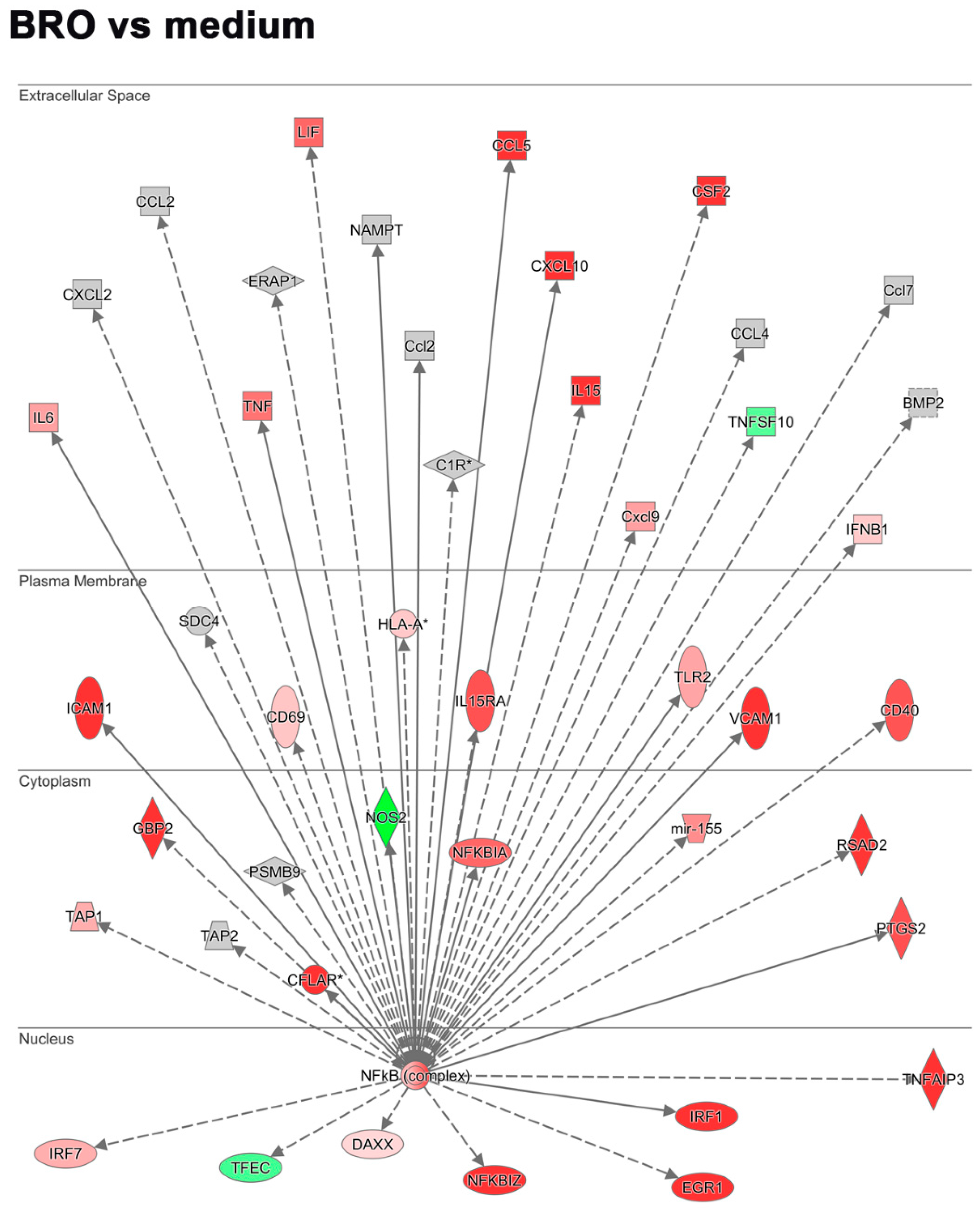
2.2.3. BRO Primes Antiviral and Pro-Inflammatory Host Signaling Pathways in Absence of Virus
3. Discussion
4. Materials and Methods
4.1. Media, Chemicals, and Reagents
4.2. Test Item
4.3. Animals
4.4. Preparation of Precision-Cut Lung Slices (PCLS)
4.5. Virus
4.6. Rhinovirus Infection and Pharmacological Treatment of PCLS
4.7. Analysis of Tissue Viability
4.8. Total Protein Determination
4.9. Determination of Cytokines
4.10. Analysis of Virus Load
4.11. RNA Isolation and Quality Analysis
4.12. Transcriptome Arrays
4.13. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BRO | Bronchobini®’s ingredients |
| COPD | Chronic obstructive pulmonary disease |
| DEG | Differential expressed genes |
| DMEM | Dulbecco’s modified Eagle’s medium |
| DPBS | Dulbecco’s phosphate buffered salt solution |
| EBSS | Earle’s balanced salt solution |
| FAM | Carboxyfluorescein |
| FC | Fold change |
| IPA | Ingenuity pathway analysis software |
| IU | Infectious units |
| LDH | Lactate dehydrogenase |
| LDLR | Low density lipoprotein receptor |
| LPS | Lipopolysaccharides |
| MEM | Minimal essential medium |
| mPCLS | Mouse precision-cut lung slices |
| MSD | Mesoscale Discovery |
| MTA | Mouse transcriptome arrays |
| NEAA | Non-essential amino acids |
| PCA | Principal Component Analysis |
| qPCR | Quantitative polymerase chain reaction |
| RIN | RNA integrity number |
| RV | Rhinovirus |
| TAC | Transcriptome analysis console software |
| TCID50 | Tissue culture infective dose 50 |
References
- Passioti, M.; Maggina, P.; Megremis, S.; Papadopoulos, N.G. The common cold: Potential for future prevention or cure. Curr. Allergy Asthma Rep. 2014, 14, 413. [Google Scholar] [CrossRef] [PubMed]
- Heikkinen, T.; Järvinen, A. The common cold. Lancet 2003, 361, 51–59. [Google Scholar] [CrossRef]
- Fendrick, A.M.; Monto, A.S.; Nightengale, B.; Sarnes, M. The economic burden of non-influenza-related viral respiratory tract infection in the United States. Arch. Intern. Med. 2003, 163, 487–494. [Google Scholar] [CrossRef]
- Oppong, R.; Coast, J.; Hood, K.; Nuttall, J.; Smith, R.D.; Butler, C.C. Resource use and costs of treating acute cough/lower respiratory tract infections in 13 European countries: Results and challenges. Eur. J. Health Econ. 2011, 12, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Monto, A.S. Epidemiology of viral respiratory infections. Am. J. Med. 2002, 112 (Suppl. 6A), 4S–12S. [Google Scholar] [CrossRef]
- Peltola, V.; Waris, M.; Osterback, R.; Susi, P.; Hyypia, T.; Ruuskanen, O. Clinical effects of rhinovirus infections. J. Clin. Virol. 2008, 43, 411–414. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, J.L.; Turner, R.B.; Braciale, T.; Heymann, P.W.; Borish, L. Pathogenesis of rhinovirus infection. Curr. Opin. Virol. 2012, 2, 287–293. [Google Scholar] [CrossRef]
- Henquell, C.; Mirand, A.; Deusebis, A.-L.; Regagnon, C.; Archimbaud, C.; Chambon, M.; Bailly, J.-L.; Gourdon, F.; Hermet, E.; Dauphin, J.-B.; et al. Prospective genotyping of human rhinoviruses in children and adults during the winter of 2009–2010. J. Clin. Virol. 2012, 53, 280–284. [Google Scholar] [CrossRef]
- Palmenberg, A.C.; Spiro, D.; Kuzmickas, R.; Wang, S.; Djikeng, A.; Rathe, J.A.; Fraser-Liggett, C.M.; Liggett, S.B. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science 2009, 324, 55–59. [Google Scholar] [CrossRef]
- Bochkov, Y.A.; Gern, J.E. Clinical and molecular features of human rhinovirus C. Microbes Infect. 2012, 14, 485–494. [Google Scholar] [CrossRef]
- Makris, S.; Johnston, S. Recent advances in understanding rhinovirus immunity. F1000Research 2018, 7. [Google Scholar] [CrossRef]
- Fuchs, R.; Blaas, D. Productive entry pathways of human rhinoviruses. Adv. Virol. 2012, 2012, 826301. [Google Scholar] [CrossRef]
- Uncapher, C.R.; DeWitt, C.M.; Colonno, R.J. The major and minor group receptor families contain all but one human rhinovirus serotype. Virology 1991, 180, 814–817. [Google Scholar] [CrossRef]
- Hofer, F.; Gruenberger, M.; Kowalski, H.; Machat, H.; Huettinger, M.; Kuechler, E.; Blaas, D. Members of the low density lipoprotein receptor family mediate cell entry of a minor-group common cold virus. Proc. Natl. Acad. Sci. USA 1994, 91, 1839–1842. [Google Scholar] [CrossRef]
- Rankl, C.; Kienberger, F.; Wildling, L.; Wruss, J.; Gruber, H.J.; Blaas, D.; Hinterdorfer, P. Multiple receptors involved in human rhinovirus attachment to live cells. Proc. Natl. Acad. Sci. USA 2008, 105, 17778–17783. [Google Scholar] [CrossRef]
- Bochkov, Y.A.; Watters, K.; Ashraf, S.; Griggs, T.F.; Devries, M.K.; Jackson, D.J.; Palmenberg, A.C.; Gern, J.E. Cadherin-related family member 3, a childhood asthma susceptibility gene product, mediates rhinovirus C binding and replication. Proc. Natl. Acad. Sci. USA 2015, 112, 5485–5490. [Google Scholar] [CrossRef]
- Mosser, A.G.; Brockman-Schneider, R.; Amineva, S.; Burchell, L.; Sedgwick, J.B.; Busse, W.W.; Gern, J.E. Similar frequency of rhinovirus-infectible cells in upper and lower airway epithelium. J. Infect. Dis. 2002, 185, 734–743. [Google Scholar] [CrossRef]
- Papadopoulos, N.G.; Sanderson, G.; Hunter, J.; Johnston, S.L. Rhinoviruses replicate effectively at lower airway temperatures. J. Med. Virol. 1999, 58, 100–104. [Google Scholar] [CrossRef]
- Bartlett, N.W.; Walton, R.P.; Edwards, M.R.; Aniscenko, J.; Caramori, G.; Zhu, J.; Glanville, N.; Choy, K.J.; Jourdan, P.; Burnet, J.; et al. Mouse models of rhinovirus-induced disease and exacerbation of allergic airway inflammation. Nat. Med. 2008, 14, 199–204. [Google Scholar] [CrossRef]
- Newcomb, D.C.; Sajjan, U.S.; Nagarkar, D.R.; Wang, Q.; Nanua, S.; Zhou, Y.; McHenry, C.L.; Hennrick, K.T.; Tsai, W.C.; Bentley, J.K.; et al. Human rhinovirus 1B exposure induces phosphatidylinositol 3-kinase-dependent airway inflammation in mice. Am. J. Respir. Crit. Care Med. 2008, 177, 1111–1121. [Google Scholar] [CrossRef]
- Tuthill, T.J.; Papadopoulos, N.G.; Jourdan, P.; Challinor, L.J.; Sharp, N.A.; Plumpton, C.; Shah, K.; Barnard, S.; Dash, L.; Burnet, J.; et al. Mouse respiratory epithelial cells support efficient replication of human rhinovirus. J. Gen. Virol. 2003, 84, 2829–2836. [Google Scholar] [CrossRef]
- Triantafilou, K.; Vakakis, E.; Richer, E.A.J.; Evans, G.L.; Villiers, J.P.; Triantafilou, M. Human rhinovirus recognition in non-immune cells is mediated by Toll-like receptors and MDA-5, which trigger a synergetic pro-inflammatory immune response. Virulence 2011, 2, 22–29. [Google Scholar] [CrossRef]
- Slater, L.; Bartlett, N.W.; Haas, J.J.; Zhu, J.; Message, S.D.; Walton, R.P.; Sykes, A.; Dahdaleh, S.; Clarke, D.L.; Belvisi, M.G.; et al. Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium. PLoS Pathog. 2010, 6, e1001178. [Google Scholar] [CrossRef]
- Padovan, E.; Spagnoli, G.C.; Ferrantini, M.; Heberer, M. IFN-alpha2a induces IP-10/CXCL10 and MIG/CXCL9 production in monocyte-derived dendritic cells and enhances their capacity to attract and stimulate CD8+ effector T cells. J. Leukoc. Boil. 2002, 71, 669–676. [Google Scholar]
- Contoli, M.; Message, S.D.; Laza-Stanca, V.; Edwards, M.R.; Wark, P.A.B.; Bartlett, N.W.; Kebadze, T.; Mallia, P.; Stanciu, L.A.; Parker, H.L.; et al. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat. Med. 2006, 12, 1023–1026. [Google Scholar] [CrossRef]
- Kohlmeier, J.E.; Cookenham, T.; Roberts, A.D.; Miller, S.C.; Woodland, D.L. Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity 2010, 33, 96–105. [Google Scholar] [CrossRef]
- Khaitov, M.R.; Laza-Stanca, V.; Edwards, M.R.; Walton, R.P.; Rohde, G.; Contoli, M.; Papi, A.; Stanciu, L.A.; Kotenko, S.V.; Johnston, S.L. Respiratory virus induction of alpha-, beta- and lambda-interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy 2009, 64, 375–386. [Google Scholar] [CrossRef]
- Bartlett, N.W.; Slater, L.; Glanville, N.; Haas, J.J.; Caramori, G.; Casolari, P.; Clarke, D.L.; Message, S.D.; Aniscenko, J.; Kebadze, T.; et al. Defining critical roles for NF-κB p65 and type I interferon in innate immunity to rhinovirus. EMBO Mol. Med. 2012, 4, 1244–1260. [Google Scholar] [CrossRef]
- Triantafilou, K.; Vakakis, E.; Kar, S.; Richer, E.; Evans, G.L.; Triantafilou, M. Visualisation of direct interaction of MDA5 and the dsRNA replicative intermediate form of positive strand RNA viruses. J. Cell Sci. 2012, 125, 4761–4769. [Google Scholar] [CrossRef]
- Mogensen, T.H. IRF and STAT Transcription Factors—From Basic Biology to Roles in Infection, Protective Immunity, and Primary Immunodeficiencies. Front. Immunol. 2018, 9, 3047. [Google Scholar] [CrossRef]
- Mears, H.V.; Sweeney, T.R. Better together: The role of IFIT protein-protein interactions in the antiviral response. J. Gen. Virol. 2018, 99, 1463–1477. [Google Scholar] [CrossRef]
- Saba, T.G.; Chung, Y.; Hong, J.Y.; Sajjan, U.S.; Bentley, J.K.; Hershenson, M.B. Rhinovirus-induced macrophage cytokine expression does not require endocytosis or replication. Am. J. Respir. Cell Mol. Boil. 2014, 50, 974–984. [Google Scholar] [CrossRef]
- Han, M.; Chung, Y.; Young Hong, J.; Rajput, C.; Lei, J.; Hinde, J.L.; Chen, Q.; Weng, S.P.; Bentley, J.K.; Hershenson, M.B. Toll-like receptor 2-expressing macrophages are required and sufficient for rhinovirus-induced airway inflammation. J. Allergy Clin. Immunol. 2016, 138, 1619–1630. [Google Scholar] [CrossRef]
- Jacobs, S.E.; Lamson, D.M.; St George, K.; Walsh, T.J. Human rhinoviruses. Clin. Microbiol. Rev. 2013, 26, 135–162. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Liang, Y.; Zhou, Y.; Shen, P. NF-κB and Its Regulation on the Immune System. Cell. Mol. Immunol. 2004, 1, 343–350. [Google Scholar]
- Doyle, W.J.; Skoner, D.P.; Gentile, D. Nasal cytokines as mediators of illness during the common cold. Curr. Allergy Asthma Rep. 2005, 5, 173–181. [Google Scholar] [CrossRef]
- Gentile, D.A.; Villalobos, E.; Angelini, B.; Skoner, D. Cytokine levels during symptomatic viral upper respiratory tract infection. Ann. Allergy Asthma Immunol. 2003, 91, 362–367. [Google Scholar] [CrossRef]
- Casanova, V.; Sousa, F.H.; Stevens, C.; Barlow, P.G. Antiviral therapeutic approaches for human rhinovirus infections. Future Virol. 2018, 13, 505–518. [Google Scholar] [CrossRef]
- Mousa, H.A.-L. Prevention and Treatment of Influenza, Influenza-Like Illness, and Common Cold by Herbal, Complementary, and Natural Therapies. J. Evid.-Based Complement. Altern. Med. 2017, 22, 166–174. [Google Scholar] [CrossRef]
- Nahas, R.; Balla, A. Complementary and alternative medicine for prevention and treatment of the common cold. Can. Fam. Physician 2011, 57, 31–36. [Google Scholar]
- Switalla, S.; Lauenstein, L.; Prenzler, F.; Knothe, S.; Förster, C.; Fieguth, H.-G.; Pfennig, O.; Schaumann, F.; Martin, C.; Guzman, C.A.; et al. Natural innate cytokine response to immunomodulators and adjuvants in human precision-cut lung slices. Toxicol. Appl. Pharmacol. 2010, 246, 107–115. [Google Scholar] [CrossRef]
- Danov, O.; Jiménez Delgado, S.M.; Obernolte, H.; Seehase, S.; Dehmel, S.; Braubach, P.; Fieguth, H.-G.; Matschiner, G.; Fitzgerald, M.; Jonigk, D.; et al. Human lung tissue provides highly relevant data about efficacy of new anti-asthmatic drugs. PLoS ONE 2018, 13, e0207767. [Google Scholar] [CrossRef]
- Neuhaus, V.; Schaudien, D.; Golovina, T.; Temann, U.-A.; Thompson, C.; Lippmann, T.; Bersch, C.; Pfennig, O.; Jonigk, D.; Braubach, P.; et al. Assessment of long-term cultivated human precision-cut lung slices as an ex vivo system for evaluation of chronic cytotoxicity and functionality. J. Occup. Med. Toxicol. (Lond. Engl.) 2017, 12, 13. [Google Scholar] [CrossRef]
- Neuhaus, V.; Schwarz, K.; Klee, A.; Seehase, S.; Förster, C.; Pfennig, O.; Jonigk, D.; Fieguth, H.-G.; Koch, W.; Warnecke, G.; et al. Functional testing of an inhalable nanoparticle based influenza vaccine using a human precision cut lung slice technique. PLoS ONE 2013, 8, e71728. [Google Scholar] [CrossRef]
- Koch, A.; Saran, S.; Tran, D.D.H.; Klebba-Färber, S.; Thiesler, H.; Sewald, K.; Schindler, S.; Braun, A.; Klopfleisch, R.; Tamura, T. Murine precision-cut liver slices (PCLS): A new tool for studying tumor microenvironments and cell signaling ex vivo. Cell Commun. Signal. 2014, 12, 73. [Google Scholar] [CrossRef]
- Neuhaus, V.; Chichester, J.A.; Ebensen, T.; Schwarz, K.; Hartman, C.E.; Shoji, Y.; Guzmán, C.A.; Yusibov, V.; Sewald, K.; Braun, A. A new adjuvanted nanoparticle-based H1N1 influenza vaccine induced antigen-specific local mucosal and systemic immune responses after administration into the lung. Vaccine 2014, 32, 3216–3222. [Google Scholar] [CrossRef]
- Neuhaus, V.; Danov, O.; Konzok, S.; Obernolte, H.; Dehmel, S.; Braubach, P.; Jonigk, D.; Fieguth, H.-G.; Zardo, P.; Warnecke, G.; et al. Assessment of the Cytotoxic and Immunomodulatory Effects of Substances in Human Precision-cut Lung Slices. J. Vis. Exp. JoVE 2018, e57042. [Google Scholar] [CrossRef]
- Rochlitzer, S.; Hoymann, H.-G.; Müller, M.; Braun, A. No exacerbation but impaired anti-viral mechanisms in a rhinovirus-chronic allergic asthma mouse model. Clin. Sci. (Lond. Engl. 1979) 2014, 126, 55–65. [Google Scholar] [CrossRef]
- Henjakovic, M.; Sewald, K.; Switalla, S.; Kaiser, D.; Müller, M.; Veres, T.Z.; Martin, C.; Uhlig, S.; Krug, N.; Braun, A. Ex vivo testing of immune responses in precision-cut lung slices. Toxicol. Appl. Pharmacol. 2008, 231, 68–76. [Google Scholar] [CrossRef]
- Muller, U.; Steinhoff, U.; Reis, L.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.; Aguet, M. Functional role of type I and type II interferons in antiviral defense. Science 1994, 264, 1918–1921. [Google Scholar] [CrossRef]
- Hayney, M.S.; Henriquez, K.M.; Barnet, J.H.; Ewers, T.; Champion, H.M.; Flannery, S.; Barrett, B. Serum IFN-gamma-induced protein 10 (IP-10) as a biomarker for severity of acute respiratory infection in healthy adults. J. Clin. Virol. 2017, 90, 32–37. [Google Scholar] [CrossRef]
- Ganesan, S.; Pham, D.; Jing, Y.; Farazuddin, M.; Hudy, M.H.; Unger, B.; Comstock, A.T.; Proud, D.; Lauring, A.S.; Sajjan, U.S. TLR2 Activation Limits Rhinovirus-Stimulated CXCL-10 by Attenuating IRAK-1-Dependent IL-33 Receptor Signaling in Human Bronchial Epithelial Cells. J. Immunol. (Baltimore MD 1950) 2016, 197, 2409–2420. [Google Scholar] [CrossRef]
- Parsons, K.S.; Hsu, A.C.; Wark, P.A.B. TLR3 and MDA5 signalling, although not expression, is impaired in asthmatic epithelial cells in response to rhinovirus infection. Clin. Exp. Allergy 2014, 44, 91–101. [Google Scholar] [CrossRef]
- Lötzerich, M.; Roulin, P.S.; Boucke, K.; Witte, R.; Georgiev, O.; Greber, U.F. Rhinovirus 3C protease suppresses apoptosis and triggers caspase-independent cell death. Cell Death Dis. 2018, 9, 272. [Google Scholar] [CrossRef]
- Girkin, J.L.; Hatchwell, L.M.; Collison, A.M.; Starkey, M.R.; Hansbro, P.M.; Yagita, H.; Foster, P.S.; Mattes, J. TRAIL signaling is proinflammatory and proviral in a murine model of rhinovirus 1B infection. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2017, 312, L89–L99. [Google Scholar] [CrossRef]
- Catrysse, L.; Vereecke, L.; Beyaert, R.; van Loo, G. A20 in inflammation and autoimmunity. Trends Immunol. 2014, 35, 22–31. [Google Scholar] [CrossRef]
- Coornaert, B.; Carpentier, I.; Beyaert, R. A20: Central Gatekeeper in Inflammation and Immunity. J. Biol. Chem. 2009, 284, 8217–8221. [Google Scholar] [CrossRef]
- Hatesuer, B.; Hoang, H.T.T.; Riese, P.; Trittel, S.; Gerhauser, I.; Elbahesh, H.; Geffers, R.; Wilk, E.; Schughart, K. Deletion of Irf3 and Irf7 Genes in Mice Results in Altered Interferon Pathway Activation and Granulocyte-Dominated Inflammatory Responses to Influenza A Infection. J. Innate Immun. 2017, 9, 145–161. [Google Scholar] [CrossRef]
- Krausgruber, T.; Blazek, K.; Smallie, T.; Alzabin, S.; Lockstone, H.; Sahgal, N.; Hussell, T.; Feldmann, M.; Udalova, I.A. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat. Immunol. 2011, 12, 231–238. [Google Scholar] [CrossRef]
- Shaheen, Z.R.; Christmann, B.S.; Stafford, J.D.; Moran, J.M.; Buller, R.M.L.; Corbett, J.A. CCR5 is a required signaling receptor for macrophage expression of inflammatory genes in response to viral double-stranded RNA. Am. J. Physiol.-Regul. Integr. Comp. Physiol. 2019, 316, R525–R534. [Google Scholar] [CrossRef]
- Vijay, R.; Fehr, A.R.; Janowski, A.M.; Athmer, J.; Wheeler, D.L.; Grunewald, M.; Sompallae, R.; Kurup, S.P.; Meyerholz, D.K.; Sutterwala, F.S.; et al. Virus-induced inflammasome activation is suppressed by prostaglandin D2/DP1 signaling. Proc. Natl. Acad. Sci. USA 2017, 114, E5444–E5453. [Google Scholar] [CrossRef]
- Dahl, T.B.; Holm, S.; Aukrust, P.; Halvorsen, B. Visfatin/NAMPT: A multifaceted molecule with diverse roles in physiology and pathophysiology. Annu. Rev. Nutr. 2012, 32, 229–243. [Google Scholar] [CrossRef]
- Jacques, C.; Holzenberger, M.; Mladenovic, Z.; Salvat, C.; Pecchi, E.; Berenbaum, F.; Gosset, M. Proinflammatory actions of visfatin/nicotinamide phosphoribosyltransferase (Nampt) involve regulation of insulin signaling pathway and Nampt enzymatic activity. J. Biol. Chem. 2012, 287, 15100–15108. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb. Perspect. Boil. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Matsushita, K.; Takeuchi, O.; Standley, D.M.; Kumagai, Y.; Kawagoe, T.; Miyake, T.; Satoh, T.; Kato, H.; Tsujimura, T.; Nakamura, H.; et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009, 458, 1185–1190. [Google Scholar] [CrossRef]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef]
- Wolf, J.; Rose-John, S.; Garbers, C. Interleukin-6 and its receptors: A highly regulated and dynamic system. Cytokine 2014, 70, 11–20. [Google Scholar] [CrossRef]
- Rose-John, S. The Soluble Interleukin 6 Receptor: Advanced Therapeutic Options in Inflammation. Clin. Pharmacol. Ther. 2017, 102, 591–598. [Google Scholar] [CrossRef]
- Schumacher, N.; Schmidt, S.; Schwarz, J.; Dohr, D.; Lokau, J.; Scheller, J.; Garbers, C.; Chalaris, A.; Rose-John, S.; Rabe, B. Circulating Soluble IL-6R but Not ADAM17 Activation Drives Mononuclear Cell Migration in Tissue Inflammation. J. Immunol. (Baltimore MD 1950) 2016, 197, 3705–3715. [Google Scholar] [CrossRef]
- Switalla, S.; Knebel, J.; Ritter, D.; Dasenbrock, C.; Krug, N.; Braun, A.; Sewald, K. Determination of genotoxicity by the Comet assay applied to murine precision-cut lung slices. Toxicol. In Vitro 2013, 27, 798–803. [Google Scholar] [CrossRef] [PubMed]
- Niehof, M.; Hildebrandt, T.; Danov, O.; Arndt, K.; Koschmann, J.; Dahlmann, F.; Hansen, T.; Sewald, K. RNA isolation from precision-cut lung slices (PCLS) from different species. BMC Res. Notes 2017, 10, 121. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | Total DEGs * | Up Regulated | Down Regulated | |
|---|---|---|---|---|
| BRO effect under baseline condition | ||||
| (Medium/Vehicle) | (Medium/Medium) | 37 | 14 | 23 |
| (Medium/BRO 1:10) | (Medium/Vehicle) | 6693 | 2497 | 4196 |
| RV induced gene regulation | ||||
| (RV/Vehicle) | (RV/Medium) | 1 | 1 | 0 |
| (RV/Vehicle) | (Medium/Vehicle) | 692 | 631 | 61 |
| BRO effect on RV infection | ||||
| (RV/BRO 1:10) | (RV/Vehicle) | 5665 | 2220 | 3445 |
| (RV/BRO 1:100) | (RV/Vehicle) | 1257 | 446 | 811 |
| (RV/BRO 1:1000) | (RV/Vehicle) | 71 | 9 | 62 |
| Gene | Fold Change | Gene | Fold Change | ||
|---|---|---|---|---|---|
| RV vs. Med | RV+BRO vs. RV | RV vs. Med | RV+BRO vs. RV | ||
| Proinflammatory chemokines/cytokines | Leukocyte activation | ||||
| Ccl2 | 5.08 * | −6.20 * | Cd40 | 7.77 * | 1.40 |
| Ccl5 | 34.23 * | −1.50 | Cd69 | 5.99 * | −1.52 |
| Cxcl10 | 178.71 * | −6.27 * | Icos/Icosl | 1.03/2.43 | 2.77 */5.90 * |
| Il1b | 1.61 | −2.49 * | Apoptosis | ||
| Casp1 | 2.99 * | −2.83 * | Tnfsf10 | 14.66 * | −61.82 * |
| Il6 | 15.56 * | −1.68 | Tnfaip3 | 3.71 * | 23.7 * |
| Il6r | −1.19 | 4.30 * | Casp8 | 1.43 | −2.14 * |
| Il6st(gp130) | −1.34 | 2.98 * | Traf1/2 | 1.67/1.38 | 6.4 */6.5 * |
| Adam17 | −1.10 | 2.43 * | Inducible enzymes and prostaglandin | ||
| Tnf | 4.05 * | 2.60 * | Nos2 | 3.76 * | −28.68 * |
| Leukocyte infiltration | Ptgs2 | 2.88 * | 2.94 * | ||
| Icam1 | 2.25 * | 7.66 * | Ptges | 1.13 | −4.40 * |
| Vcam1 | 8.36 * | 2.16 * | Namp | 5.03 * | −3.02 * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reamon-Buettner, S.M.; Niehof, M.; Hirth, N.; Danov, O.; Obernolte, H.; Braun, A.; Warnecke, J.; Sewald, K.; Wronski, S. Transcriptomic Analysis Reveals Priming of The Host Antiviral Interferon Signaling Pathway by Bronchobini® Resulting in Balanced Immune Response to Rhinovirus Infection in Mouse Lung Tissue Slices. Int. J. Mol. Sci. 2019, 20, 2242. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092242
Reamon-Buettner SM, Niehof M, Hirth N, Danov O, Obernolte H, Braun A, Warnecke J, Sewald K, Wronski S. Transcriptomic Analysis Reveals Priming of The Host Antiviral Interferon Signaling Pathway by Bronchobini® Resulting in Balanced Immune Response to Rhinovirus Infection in Mouse Lung Tissue Slices. International Journal of Molecular Sciences. 2019; 20(9):2242. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092242
Chicago/Turabian StyleReamon-Buettner, Stella Marie, Monika Niehof, Natalie Hirth, Olga Danov, Helena Obernolte, Armin Braun, Jürgen Warnecke, Katherina Sewald, and Sabine Wronski. 2019. "Transcriptomic Analysis Reveals Priming of The Host Antiviral Interferon Signaling Pathway by Bronchobini® Resulting in Balanced Immune Response to Rhinovirus Infection in Mouse Lung Tissue Slices" International Journal of Molecular Sciences 20, no. 9: 2242. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092242