Genetic Variation for Seed Metabolite Levels in Brachypodium distachyon

 ,
, 
Abstract
:
1. Introduction
2. Results
2.1. Genetic and Metabolic Variation among Forty B. distachyon Inbred Lines
2.2. Metabolotype Differences Between Bd21 and Bd3-1
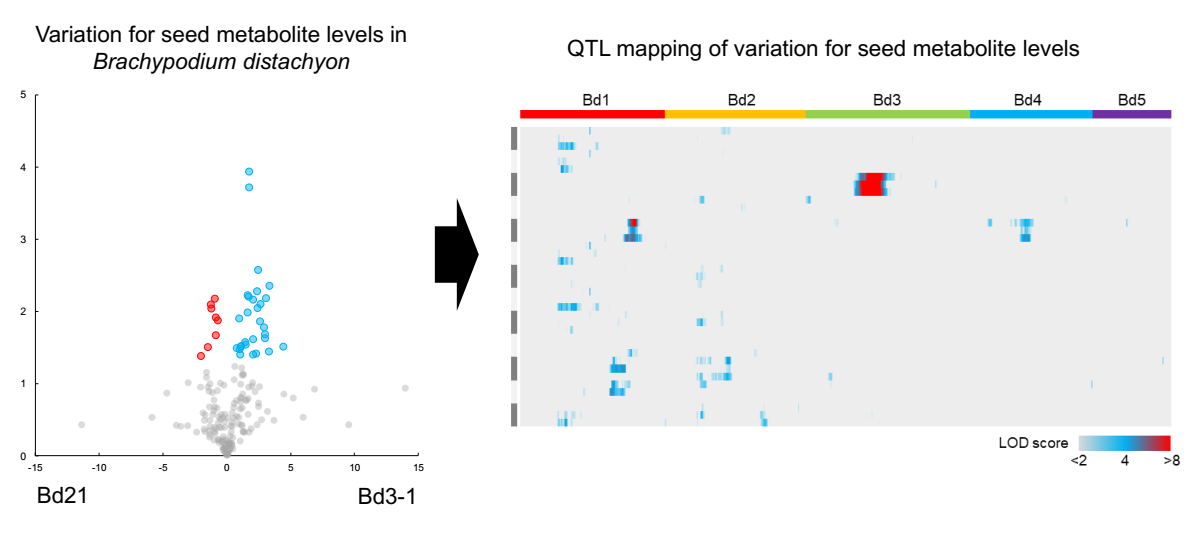
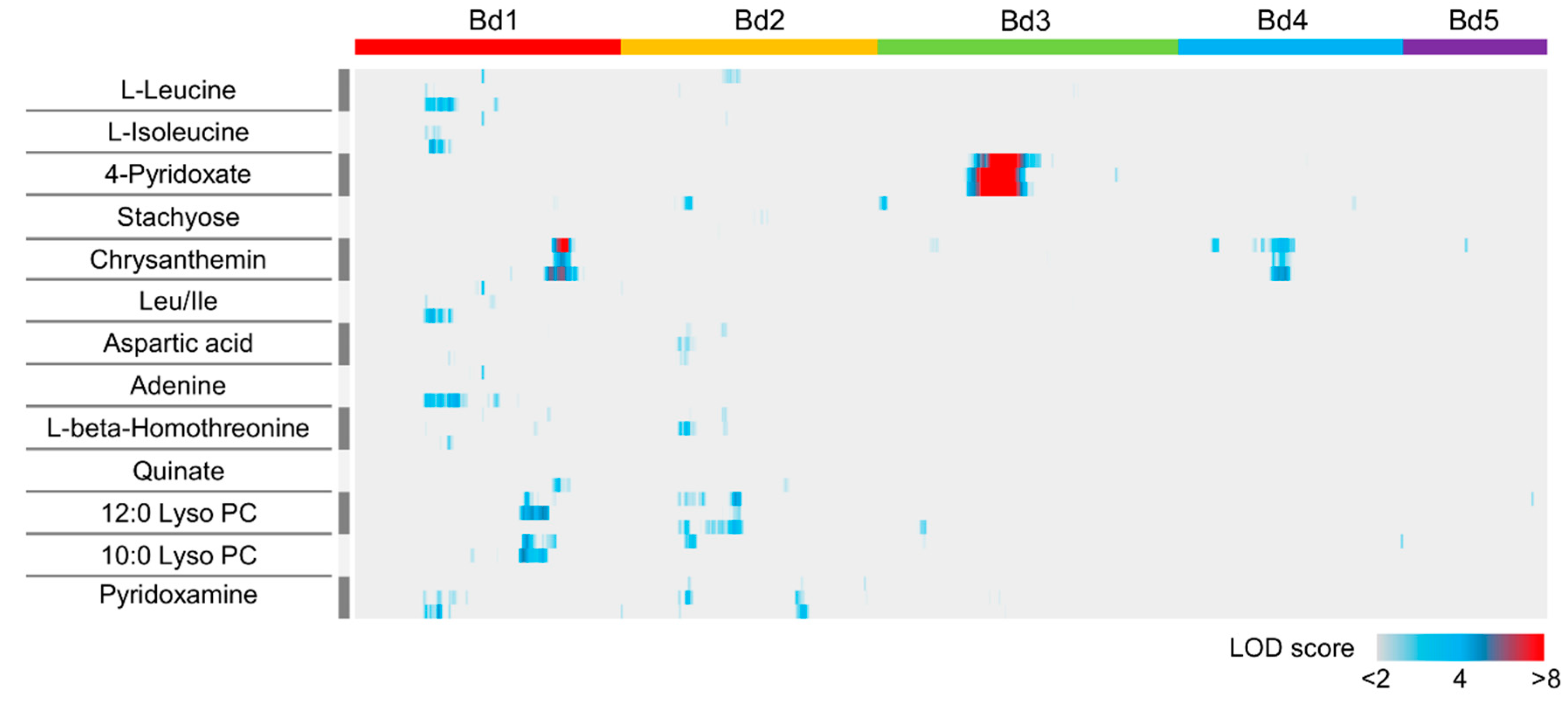
2.3. QTL Mapping of Metabolite Accumulation Using RILs Derived from a Cross Between Bd21 and Bd3-1
2.4. QTLs Related to Vitamin B6 Metabolism in B. distachyon
2.5. Interaction of QTLs for Accumulation of Chrysanthemin in B. distachyon
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Metabolome Analysis
4.3. Genetic Map Construction and QTL Analysis
4.4. Genome-Wide SNPs
4.5. Data Accessibility
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bruinsma, J. World Agriculture: Towards 2015/2030. Summary Report; Food and Agriculture Organization of the United Nations: Rome Italy, 2002. [Google Scholar]
- McKevith, B. Nutritional aspects of cereals. Nutr. Bulletin 2004, 29, 111–142. [Google Scholar] [CrossRef]
- Harrigan, G.G.; Martino-Catt, S.; Glenn, K.C. Metabolomics, metabolic diversity and genetic variation in crops. Metabolomics 2007, 3, 259–272. [Google Scholar] [CrossRef]
- Bhullar, N.K.; Gruissem, W. Nutritional enhancement of rice for human health: the contribution of biotechnology. Biotechnol. Adv. 2013, 31, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Tian, L. Breeding Major Cereal Grains through the Lens of Nutrition Sensitivity. Molecular. Plant 2018, 11, 23–30. [Google Scholar] [CrossRef]
- Luo, J. Metabolite-based genome-wide association studies in plants. Curr. Opin. Plant. Biol. 2015, 24, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Yang, L.; Zhang, D.; Shi, J. Plant Metabolomics: An Indispensable System Biology Tool for Plant Science. Int. J. Mol. Sci. 2016, 17, 767. [Google Scholar] [CrossRef]
- Keurentjes, J.J.B.; Fu, J.Y.; de Vos, C.H.R.; Lommen, A.; Hall, R.D.; Bino, R.J.; van der Plas, L.H.W.; Jansen, R.C.; Vreugdenhil, D.; Koornneef, M. The genetics of plant metabolism. Nat. Genet. 2006, 38, 842–849. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Matsuda, F. Metabolomics for functional genomics, systems biology and biotechnology. Annu. Rev. Plant. Biol. 2010, 61, 463–489. [Google Scholar] [CrossRef]
- Davey, J.W.; Hohenlohe, P.A.; Etter, P.D.; Boone, J.Q.; Catchen, J.M.; Blaxter, M.L. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat. Rev. Genet. 2011, 12, 499–510. [Google Scholar] [CrossRef]
- Scossa, F.; Brotman, Y.; Lima, F.D.E.; Willmitzer, L.; Nikoloski, Z.; Tohge, T.; Fernie, A.R. Genomics-based strategies for the use of natural variation in the improvement of crop metabolism. Plant Sci. 2016, 242, 47–64. [Google Scholar] [CrossRef]
- Riewe, D.; Jeon, H.J.; Lisec, J.; Heuermann, M.C.; Schmeichel, J.; Seyfarth, M.; Meyer, R.C.; Willmitzer, L.; Altmann, T. A naturally occurring promoter polymorphism of the Arabidopsis FUM2 gene causes expression variation and is associated with metabolic and growth traits. Plant J. 2016, 88, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Angelovici, R.; Lipka, A.E.; Deason, N.; Gonzalez-Jorge, S.; Lin, H.; Cepela, J.; Buell, R.; Gore, M.A.; Dellapenna, D. Genome-wide analysis of branched-chain amino acid levels in Arabidopsis seeds. Plant Cell 2013, 25, 4827–4843. [Google Scholar] [CrossRef]
- Schauer, N.; Semel, Y.; Roessner, U.; Gur, A.; Balbo, I.; Carrari, F.; Pleban, T.; Perez-Melis, A.; Bruedigam, C.; Kopka, J.; et al. Comprehensive metabolic profiling and phenotyping of interspecific introgression lines for tomato improvement. Nat. Biotechnol. 2006, 24, 447–454. [Google Scholar] [CrossRef]
- Wen, W.W.; Li, D.; Li, X.; Gao, Y.Q.; Li, W.Q.; Li, H.H.; Liu, J.; Liu, H.J.; Chen, W.; Luo, J.; et al. Metabolome-based genome-wide association study of maize kernel leads to novel biochemical insights. Nat. Commun. 2014, 5, 3438. [Google Scholar] [CrossRef] [Green Version]
- International Brachypodium, I. Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 2010, 463, 763–768. [Google Scholar] [Green Version]
- Bevan, M.W.; Garvin, D.F.; Vogel, J.P. Brachypodium distachyon genomics for sustainable food and fuel production. Curr. Opin. Biotechnol. 2010, 21, 211–217. [Google Scholar] [CrossRef]
- Girin, T.; David, L.C.; Chardin, C.; Sibout, R.; Krapp, A.; Ferrario-Mery, S.; Daniel-Vedele, F. Brachypodium: a promising hub between model species and cereals. J. Exp. Bot. 2014, 65, 5683–5696. [Google Scholar] [CrossRef] [Green Version]
- Shewry, P.R.; Hey, S.J. The contribution of wheat to human diet and health. Food Energy Secur. 2015, 4, 178–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalmais, M.; Antelme, S.; Ho-Yue-Kuang, S.; Wang, Y.; Darracq, O.; d’Yvoire, M.B.; Cezard, L.; Legee, F.; Blondet, E.; Oria, N.; et al. A TILLING Platform for Functional Genomics in Brachypodium distachyon. PloS ONE 2013, 8, e65503. [Google Scholar] [CrossRef] [PubMed]
- de Bang, L.; Torp, A.M.; Rasmussen, S.K. TILLING in Brachypodium distachyon. Methods Mol. Biol. 2018, 1667, 173–186. [Google Scholar] [PubMed]
- Thole, V.; Alves, S.C.; Worland, B.; Bevan, M.W.; Vain, P. A protocol for efficiently retrieving and characterizing flanking sequence tags (FSTs) in Brachypodium distachyon T-DNA insertional mutants. Nat. Protoc. 2009, 4, 650–661. [Google Scholar] [CrossRef]
- Hsia, M.M.; O’Malley, R.; Cartwright, A.; Nieu, R.; Gordon, S.P.; Kelly, S.; Williams, T.G.; Wood, D.F.; Zhao, Y.; Bragg, J.; et al. Sequencing and functional validation of the JGI Brachypodium distachyon T-DNA collection. Plant J. 2017, 91, 361–370. [Google Scholar] [CrossRef]
- Vogel, J.P.; Tuna, M.; Budak, H.; Huo, N.; Gu, Y.Q.; Steinwand, M.A. Development of SSR markers and analysis of diversity in Turkish populations of Brachypodium distachyon. BMC Plant Biol. 2009, 9, 88. [Google Scholar] [CrossRef]
- Tyler, L.; Fangel, J.U.; Fagerstrom, A.D.; Steinwand, M.A.; Raab, T.K.; Willats, W.G.; Vogel, J.P. Selection and phenotypic characterization of a core collection of Brachypodium distachyon inbred lines. BMC Plant Biol. 2014, 14, 25. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, C.J.; Doyle, M.R.; Manzaneda, A.J.; Rey, P.J.; Mitchell-Olds, T.; Amasino, R.M. Natural Variation of Flowering Time and Vernalization Responsiveness in Brachypodium distachyon. Bioenerg. Res. 2010, 3, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Ream, T.S.; Woods, D.P.; Schwartz, C.J.; Sanabria, C.P.; Mahoy, J.A.; Walters, E.M.; Kaeppler, H.F.; Amasino, R.M. Interaction of Photoperiod and Vernalization Determines Flowering Time of Brachypodium distachyon. Plant Physiol. 2014, 164, 694–709. [Google Scholar] [CrossRef] [PubMed]
- Bettgenhaeuser, J.; Corke, F.M.; Opanowicz, M.; Green, P.; Hernandez-Pinzon, I.; Doonan, J.H.; Moscou, M.J. Natural Variation in Brachypodium Links Vernalization and Flowering Time Loci as Major Flowering Determinants. Plant Physiol. 2017, 173, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Villalobos, D.; Hardtke, C.S. Natural genetic variation of root system architecture from Arabidopsis to Brachypodium: towards adaptive value. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 1552–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Lee, M.Y.; Huo, N.; Bragg, J.; Yan, L.; Yuan, C.; Li, C.; Holditch, S.J.; Xie, J.; Luo, M.C.; et al. Fine mapping of the Bsr1 barley stripe mosaic virus resistance gene in the model grass Brachypodium distachyon. PloS ONE 2012, 7, e38333. [Google Scholar] [CrossRef]
- Kouzai, Y.; Kimura, M.; Watanabe, M.; Kusunoki, K.; Osaka, D.; Suzuki, T.; Matsui, H.; Yamamoto, M.; Ichinose, Y.; Toyoda, K.; et al. Salicylic acid-dependent immunity contributes to resistance against Rhizoctonia solani, a necrotrophic fungal agent of sheath blight, in rice and Brachypodium distachyon. New Phytol. 2018, 217, 771–783. [Google Scholar] [CrossRef]
- Des Marais, D.L.; Razzaque, S.; Hernandez, K.M.; Garvin, D.F.; Juenger, T.E. Quantitative trait loci associated with natural diversity in water-use efficiency and response to soil drying in Brachypodium distachyon. Plant Sci. 2016, 251, 2–11. [Google Scholar] [CrossRef]
- Colton-Gagnon, K.; Ali-Benali, M.A.; Mayer, B.F.; Dionne, R.; Bertrand, A.; Do Carmo, S.; Charron, J.B. Comparative analysis of the cold acclimation and freezing tolerance capacities of seven diploid Brachypodium distachyon accessions. Ann. Bot. 2014, 113, 681–693. [Google Scholar] [CrossRef]
- Luo, N.; Liu, J.; Yu, X.; Jiang, Y. Natural variation of drought response in Brachypodium distachyon. Physiol. Plant 2011, 141, 19–29. [Google Scholar] [CrossRef]
- Fisher, L.H.; Han, J.; Corke, F.M.; Akinyemi, A.; Didion, T.; Nielsen, K.K.; Doonan, J.H.; Mur, L.A.; Bosch, M. Linking Dynamic Phenotyping with Metabolite Analysis to Study Natural Variation in Drought Responses of Brachypodium distachyon. Front. Plant Sci. 2016, 7, 1751. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, X.; Yu, X.; Zhao, X.; Luo, N.; Pei, Z.; Liu, H.; Garvin, D.F. Quantitative Trait Loci Associated with Drought Tolerance in Brachypodium distachyon. Front. Plant Sci. 2017, 8, 811. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.P.; Contreras-Moreira, B.; Woods, D.P.; Des Marais, D.L.; Burgess, D.; Shu, S.; Stritt, C.; Roulin, A.C.; Schackwitz, W.; Tyler, L.; et al. Extensive gene content variation in the Brachypodium distachyon pan-genome correlates with population structure. Nat. Commun. 2017, 8, 2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onda, Y.; Hashimoto, K.; Yoshida, T.; Sakurai, T.; Sawada, Y.; Hirai, M.Y.; Toyooka, K.; Mochida, K.; Shinozaki, K. Determination of growth stages and metabolic profiles in Brachypodium distachyon for comparison of developmental context with Triticeae crops. Proc. Biol. Sci. 2015, 282, 1811. [Google Scholar] [CrossRef] [PubMed]
- Wilson, P.B.; Streich, J.C.; Murray, K.D.; Eichten, S.R.; Cheng, R.; Aitken, N.C.; Spokas, K.; Warthmann, N.; Gordon, S.P.; Accession, C.; et al. Global Diversity of the Brachypodium Species Complex as a Resource for Genome-Wide Association Studies Demonstrated for Agronomic Traits in Response to Climate. Genetics 2019, 211, 317–331. [Google Scholar] [CrossRef]
- Korte, A.; Farlow, A. The advantages and limitations of trait analysis with GWAS: a review. Plant Methods 2013, 9, 29. [Google Scholar] [CrossRef]
- Woods, D.P.; Bednarek, R.; Bouche, F.; Gordon, S.P.; Vogel, J.P.; Garvin, D.F.; Amasino, R.M. Genetic Architecture of Flowering-Time Variation in Brachypodium distachyon. Plant Physiol. 2017, 173, 269–279. [Google Scholar] [CrossRef]
- Onda, Y.; Mochida, K. Exploring Genetic Diversity in Plants Using High-Throughput Sequencing Techniques. Curr. Genom. 2016, 17, 358–367. [Google Scholar] [CrossRef]
- Mochida, K.; Koda, S.; Inoue, K.; Hirayama, T.; Tanaka, S.; Nishii, R.; Melgani, F. Computer vision-based phenotyping for improvement of plant productivity: a machine learning perspective. Gigascience 2019, 8. [Google Scholar] [CrossRef]
- Handakumbura, P.P.; Stanfill, B.; Rivas-Ubach, A.; Fortin, D.; Vogel, J.P.; Jansson, C. Metabotyping as a Stopover in Genome-to-Phenome Mapping. Sci. Rep. 2019, 9, 1858. [Google Scholar] [CrossRef] [PubMed]
- Lisec, J.; Meyer, R.C.; Steinfath, M.; Redestig, H.; Becher, M.; Witucka-Wall, H.; Fiehn, O.; Torjek, O.; Selbig, J.; Altmann, T.; et al. Identification of metabolic and biomass QTL in Arabidopsis thaliana in a parallel analysis of RIL and IL populations. Plant J. 2008, 53, 960–972. [Google Scholar] [CrossRef]
- Knoch, D.; Riewe, D.; Meyer, R.C.; Boudichevskaia, A.; Schmidt, R.; Altmann, T. Genetic dissection of metabolite variation in Arabidopsis seeds: evidence for mQTL hotspots and a master regulatory locus of seed metabolism. J. Exp. Bot. 2017, 68, 1655–1667. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, F.; Okazaki, Y.; Oikawa, A.; Kusano, M.; Nakabayashi, R.; Kikuchi, J.; Yonemaru, J.I.; Ebana, K.; Yano, M.; Saito, K. Dissection of genotype-phenotype associations in rice grains using metabolome quantitative trait loci analysis. Plant J. 2012, 70, 624–636. [Google Scholar] [CrossRef] [Green Version]
- Garvin, D.F.; Gu, Y.Q.; Hasterok, R.; Hazen, S.P.; Jenkins, G.; Mockler, T.C.; Mur, L.A.J.; Vogel, J.P. Development of genetic and genomic research resources for Brachypodium distachyon, a new model system for grass crop research. Crop Sci. 2008, 48, S69–S84. [Google Scholar] [CrossRef]
- Fitzpatrick, T.B.; Basset, G.J.; Borel, P.; Carrari, F.; DellaPenna, D.; Fraser, P.D.; Hellmann, H.; Osorio, S.; Rothan, C.; Valpuesta, V.; et al. Vitamin deficiencies in humans: can plant science help? Plant Cell 2012, 24, 395–414. [Google Scholar] [CrossRef]
- Mooney, S.; Hellmann, H. Vitamin B6: Killing two birds with one stone? Phytochemistry 2010, 71, 495–501. [Google Scholar] [CrossRef]
- Santos-Buelga, C.; Mateus, N.; De Freitas, V. Anthocyanins. Plant pigments and beyond. J. Agric. Food Chem. 2014, 62, 6879–6884. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, P.; Luo, Y.; Zhao, M.; Chen, F. Health benefits of anthocyanins and molecular mechanisms: Update from recent decade. Crit. Rev. Food Sci. Nutr. 2017, 57, 1729–1741. [Google Scholar] [CrossRef] [PubMed]
- Fudge, J.; Mangel, N.; Gruissem, W.; Vanderschuren, H.; Fitzpatrick, T.B. Rationalising vitamin B6 biofortification in crop plants. Curr. Opin. Biotechnol. 2017, 44, 130–137. [Google Scholar] [CrossRef]
- Hong, M.J.; Kim, D.Y.; Nam, B.M.; Ahn, J.W.; Kwon, S.J.; Seo, Y.W.; Kim, J.B. Characterization of novel mutants of hexaploid wheat (Triticum aestivum L.) with various depths of purple grain color and antioxidant capacity. J. Sci. Food Agric. 2019, 99, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.; Alderman, S.; Garvin, D.F.; Pfender, W.F. Infection of Brachypodium distachyon by formae speciales of Puccinia graminis: early infection events and host-pathogen incompatibility. Plos One 2013, 8, e56857. [Google Scholar] [CrossRef] [PubMed]
- Onda, Y.; Takahagi, K.; Shimizu, M.; Inoue, K.; Mochida, K. Multiplex PCR Targeted Amplicon Sequencing (MTA-Seq): Simple, Flexible and Versatile SNP Genotyping by Highly Multiplexed PCR Amplicon Sequencing. Front. Plant Sci. 2018, 9, 201. [Google Scholar] [CrossRef]
- Sawada, Y.; Akiyama, K.; Sakata, A.; Kuwahara, A.; Otsuki, H.; Sakurai, T.; Saito, K.; Hirai, M.Y. Widely targeted metabolomics based on large-scale MS/MS data for elucidating metabolite accumulation patterns in plants. Plant Cell Physiol. 2009, 50, 37–47. [Google Scholar] [CrossRef]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018, 46, W486–W494. [Google Scholar] [CrossRef] [PubMed]
- Broman, K.W.; Wu, H.; Sen, S.; Churchill, G.A. R/qtl: QTL mapping in experimental crosses. Bioinformatics 2003, 19, 889–890. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolite | Replicate | Chromosome | QTLs (cM) | Maximum LOD | Percentage of Additive Effect (%) | Percentage of Variance Explained (%) | LOD Support Interval (1.5-LOD Dropping from the Maximum LOD) | Closest SNP Maker | Physical Location of Closest SNP Marker * |
|---|---|---|---|---|---|---|---|---|---|
| 4-Pyridoxate | 1 | 3 | 171.0 | 17.3 | −62.8 | 38.4 | 168.3–173.0 cM (15.3–15.6) | BD1742_2 | 22,818,945 |
| 2 | 3 | 171.0 | 20.7 | −64.7 | 44.1 | 170.0–173.4 cM (19.1–18.2) | |||
| 3 | 3 | 171.0 | 21.4 | −67.2 | 45.2 | 168.3–174.0 cM (19.2–19.6) | |||
| Pyridoxamine | 3 | 1 | 108.4 | 4.7 | −34.3 | 12.3 | 98.0–113.0 (2.8–2.7) | BD1723_1 | 30,380,964 |
| Replicate | Chromosome | QTLs (cM) | Maximum LOD | Percentage of additive effect (%) | Percentage of Variance Explained (%) | LOD Support Interval (1.5-LOD Dropping from the Maximum LOD) | Closest SNP Maker | Physical Location of Closest SNP Marker * |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | 262.0 | 8.3 | 45.6 | 20.9 | 258.0–268.0 (6.7–5.9) | BD3636_1 | 63,763,612 |
| 2 | 1 | 261.0 | 4.9 | 35.4 | 13.0 | 256.0–266.0 (3.4–3.1) | BD4076_1 | 63,379,714 |
| 3 | 1 | 261.0 | 6.2 | 39.5 | 16.1 | 242.0–265 (4.7–4.6) | ||
| 1 | 4 | 136.0 | 4.0 | –33.3 | 10.7 | 122.0–146.0 (2.3–2.0) | BD1893_1 | 23,806,368 |
| 3 | 4 | 138.4 | 4.9 | –35.7 | 12.9 | 127–141.7 (3.4–3.0) | BD3589_1 | 26,658,442 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onda, Y.; Inoue, K.; Sawada, Y.; Shimizu, M.; Takahagi, K.; Uehara-Yamaguchi, Y.; Hirai, M.Y.; Garvin, D.F.; Mochida, K. Genetic Variation for Seed Metabolite Levels in Brachypodium distachyon. Int. J. Mol. Sci. 2019, 20, 2348. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092348
Onda Y, Inoue K, Sawada Y, Shimizu M, Takahagi K, Uehara-Yamaguchi Y, Hirai MY, Garvin DF, Mochida K. Genetic Variation for Seed Metabolite Levels in Brachypodium distachyon. International Journal of Molecular Sciences. 2019; 20(9):2348. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092348
Chicago/Turabian StyleOnda, Yoshihiko, Komaki Inoue, Yuji Sawada, Minami Shimizu, Kotaro Takahagi, Yukiko Uehara-Yamaguchi, Masami Y. Hirai, David F. Garvin, and Keiichi Mochida. 2019. "Genetic Variation for Seed Metabolite Levels in Brachypodium distachyon" International Journal of Molecular Sciences 20, no. 9: 2348. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20092348